1. Introduction
Catalysis, by making chemical and biochemical reactions kinetically feasible, is fundamental in different fields, from inorganic, organic, and biochemical industrial synthesis to material and food sciences. A catalyst can be homogeneous or heterogeneous if it is present in the same phase of the reagents or in a different phase, respectively. A heterogeneous catalyst is usually adsorbed on a surface or encapsulated in a solid, porous support. It offers several advantages over homogeneous catalysts, particularly allowing its separation and recovery to be used for several reaction cycles. Additionally, immobilization can increase its stability, improving the process from an economic and ecological point of view, minimizing costs and limiting waste products and solvent use. There are different methods to immobilize heterogeneous catalysts that are classified on the bases of its interaction and the supporting matrix: i.e., covalent binding, electrostatic binding, absorption, and encapsulation.
Enzymes are biological catalysts, and their features are increasingly important for industrial processes in several fields (chemistry, pharmaceutics, biotechnology, etc.). Moreover, the fact that they are able to catalyze reactions in a very specific and selective way makes them an essential tool in the development of biosensors. As they are typically more costly and less stable than small-molecule catalysts, the need to stabilize and separate enzymes from the reagents’ phase is particularly relevant for processes involving large scale applications and requiring evaluation of costs and efficiency. Hence, enzyme immobilization has been tackled in different ways to obtain their compartmentalization and an improved stability. It is possible to distinguish between surface and volume confinement. The former realizes when the protein is adsorbed or covalently attached to a surface or an interface, avoiding free diffusion, but not altering significantly the protein relationship to its immediate environment. Besides avoiding protein leaching, surface confinement is useful to build optimized enzymatic machinery by binding different proteins in a selected arrangement to enhance the efficiency of a given process. Volume confinement corresponds to true entrapment, when a protein is completely embedded in a confining matrix: this might alter both the protein structure and (thermo)dynamics and provides a different environment, altering the solvent properties and the availability of substrates for the enzyme, which can be ultimately exploited to obtain compartmentalized nanoreactors whose properties can be shaped, altering the supporting matrix [
1,
2,
3].
As enzymes are proteins, they are usually stable in physiological or near-physiological conditions, but denaturation occurs upon temperature increase or pH changes. To overcome these limits, different immobilization methods are used to retain, or even improve, their stability and performance. Several methods are traditionally used for enzyme immobilization: covalent binding, either direct or with a linker fragment, as well as adsorption are generally employed for surface confinement, while absorption/coprecipitation, embedding within preformed compartments (such as within zeolites), or aggregating the matrix around the protein (either covalently bound or not) are utilized for volume confinement. Methods involving covalent binding guarantee a stable interaction with the solid support, thus preventing leaching, but require chemical reactions that might alter enzyme function [
4]. A peculiar type of volume confinement by covalent binding can be obtained by cross-linking of enzymes together with linker molecules. The resulting bioconjugate aggregate has an elevated concentration in functional units, with respect to those produced by other immobilized techniques, and provides good enzyme activity retention, preventing heat and thermal denaturation or proteolysis. Cross-linking techniques were initially applied to stabilize and immobilize protein crystals, but their diffusion was hampered by the inherent difficulty of obtaining enzyme crystals. More recently, cross-linked aggregates are produced by precipitation of the insoluble bioconjugate: the resulting material is not highly pure and homogeneous, but easy to produce and suitable for several application purposes [
2].
More recently, various types of smart or new materials have been used for enzyme immobilization. Among them, we can recall magnetic and light active materials, which, because of their ability to “move” in response to external stimuli, can be used to facilitate product recovery or the release of the enzyme itself from the immobilization matrix [
2] or active materials able to add interesting functionalities such as graphene derivatives or metal-organic frameworks (MOF). Nanoparticles of graphene can bind enzymes by absorption, showing a great biocompatibility and improving enzyme stability, as shown with the use of graphene quantum dots, which are able to stabilize and protect the active sites of trypsin, while altering their optical properties in the process [
5]. Furthermore, graphene oxide has been exploited due to its ability to bind and inactivate pro-tumorigenic enzymes, which increase the risk of metastasis, suggesting a possible application in formulating non-invasive cancer prevention treatments [
6].
MOF have been recently proposed as carriers for immobilized enzymes. MOF–enzyme interactions can be obtained by different techniques: adsorption, covalent binding, or diffusion within a preformed MOF, as well as co-precipitation or in situ MOF synthesis, which allows the immobilization of enzymes larger than the MOF pores. With respect to more traditional support materials, MOFs have been proposed because of their tunability, both physical (pore size and volume) and chemical (modification on both metal and linker sites), their high thermomechanical stability and good optoelectronic properties (including the possibility to be magnetic), and their ability to embed “difficult” molecules, such as small gaseous molecules (H
2, CH
4, CO
2, NO). Studies on MOF supported enzymes demonstrated significant improvements of enzyme stability, but not of enzyme activity, shifting the interest from their use as a simple enzyme support to components of multifunctional composites [
7].
In this review, we focus on encapsulation or entrapment methods. An ideal matrix for immobilizing enzymes should be chemically inert, but physically strong and stable, requiring preparation conditions compatible with the preservation of the enzyme’s native state and function. With respect to the latter aspect, it is worth noticing that enzymes follow the protein structure, function, and dynamics paradigm, which directly correlates the three-dimensional structure and the tertiary and quaternary conformations of a protein with its function. Hence, protein encapsulation can be not only a way to separate the biological catalyst from the solution reagents’ phase, but also an active support that can modulate enzyme structure and conformations, its dynamics, and the local environment, with a direct effect on its function.
2. Hard and Soft Entrapment: Host Matrices’ Effects on Biomolecules
Biological systems are highly compartmentalized, with various levels of confinement within living organisms [
8,
9,
10]. The main difference between biological compartmentalization and artificial confinement is that while the latter is generally hard, i.e., obtained with chemical structures scarcely alterable by biomolecules and, generally, in a truly different phase, the former is characterized by soft structures capable of intermingling with biomolecules, which may or may not be in the same phase. The occurrence of these structures is one of the difficulties in studying biological matrices, as these compartmentalized systems are often overwhelmingly complex and they need to be modelled by more simple confined systems, capable of taking into account the lack of the thermodynamic ideality of natural biological structures. As for the confinement in biological/biomimetic systems, the distinction between hard and soft confinement must be borne in mind. Usually, hard confinement is defined as the one in which the walls of the confining matrix are fixed, whereas, in soft confinement, the motions of the matrix are possible. In this respect, “fixed” matrices have to be considered as a crude approximation, since atomic motions are always present in physical systems, even at very low, cryogenic, temperatures, due to zero-point atomic vibrations. From the dynamical point of view, therefore, hard confinement refers to a situation in which matrix motions are of high frequency (picoseconds/nanoseconds time scale) and low amplitude (less than 0.1 Å) and therefore influence protein motions in these time/space scales while almost completely hindering large amplitude, slow, movements. On the other hand, in soft confinement, matrix movements occur in the timescale from microseconds to seconds and in the 1 Å up to 100 Å space scale. Therefore, protein tertiary or even quaternary conformational changes occurring in the above space–time scales are allowed. The key concept, therefore, is that confinement is not only geometric, but also dynamic (stereodynamic confinement).
Both confinement methods, hard and soft, work mostly by excluded-volume effects. However, while soft confinement (which can be obtained either with a simple increase of biological macromolecule concentration (crowding or even self-crowding) or with the addition of cosolutes able to coat and encapsulate biomolecules) refers to effects by one soluble component (usually a macromolecule) to one other, hard confinement (usually found when a biomolecule is trapped within the pores of a solid, crystalline, or glassy matrix) refers to effects by a fixed (or confining) boundary to a soluble macromolecule. In both cases, there is a reduction of possible available conformations, either due to the presence of a high volume fraction of other molecules or to a static barrier to movement. An important qualitative difference is that macromolecular crowding favors compact conformations, with the minimal free energy cost compatible with the interactions with the crowder (usually the one with the smaller radius of gyration), while confinement favors conformations having a shape that is complementary to the shape of the confining volume [
8,
11]. This applies also to disaccharide matrices trehalose (
Figure 1a) [
12], as well as to silica gel matrices generated by templating the macromolecule via a sol-gel process: the embedded macromolecule adopts the conformation that better complies with the confining matrix when the original template cavity is produced. An alternative view is that the macromolecule shapes the matrix while it is forming, like a hand with a glove. In both views, the net result is that, after the gel formation, every conformation incompatible with the shape of that cavity becomes unfavored.
Moreover, crowding nonspecifically enhances processes leading to the reduction of total excluded volume, like the formation of complexes or molecule binding or the formation of aggregates, and it is generally expected to increase the rate of slow, transition state limited association reactions and to decrease the rate of fast, diffusion limited association reactions. Actually, in biomimetic systems, it is possible to obtain intermediate forms ranging from soft to hard confinement, which are often modulated by simple variables, such as temperature or water content [
8,
11].
The main differences among hard, artificial matrices, as silica gels, and soft, biological ones, can be summarized in two main points. Firstly, silica matrices, which are covalently bound porous solids, once prepared, have a scarcely tunable rigidity, because the latter stems from chemical events such as polymerization or condensation reactions, which determine the physical properties of the resulting matrix. A low degree of condensation, which in the case of silica gels corresponds to a high amount of free hydroxyl groups, high ability of water binding, and large pores, yields looser matrices; a high degree of condensation yields more rigid systems with smaller pores. This is the result of a different average numbers of covalent bonds per molecular unit, which generates a discontinuity in the behavior at the molecular scale: the rigidity increases stepwise and is defined at the moment of the gel formation, and any modulation arises from the presence of a mixed population of pores with different diameters or hydration [
13].
Conversely, saccharide confining matrices generally have either covalently bound spongy structures with highly deformable pores or lack altogether a structured scaffold (
Figure 2a). Their structural properties stem only from intermolecular interactions and therefore are fully dependent on the content of co-solutes (salts, small molecules) or of water. In this case, the strength of confinement can be largely modulated by altering the relative composition of the matrix itself (or of the “solvent” fraction embedded within the confining scaffold, if present), even though it generally falls within the scope of the so-called soft confinement. In this confinement, the matrix is in a jammed state, i.e., a state where the small thermal fluctuations do not allow the correct sampling of the equilibrium state. Here, the embedded protein is unable to escape the cage formed by the jammed neighbor molecules [
10].
From an applicative point of view, the large modulability of saccharide hydrogels has also been used when it is necessary to recover the enzyme from the matrix, as in cases of preparations thought to overcome the animal digestive system and after releasing their proteinic payload [
14].
For the same structural reasons (prevailing covalent bonds and low flexibility), in silica gel or other more structured matrices (e.g., zeolites), the confining environment is geometrically characterized: the dimensions and shapes of the pores that form in the rigid scaffold determine the confining ability, the type of macromolecules that can be embedded, and the properties of the “solvent” layer trapped within the pores [
13]. The larger flexibility in saccharide confinement makes porosity not the primary measure of the confining ability: polysaccharide matrices with defined porosity do exist, but the macromolecular scaffold is in any case less rigid and allows a certain degree of adaptability, allowing also the hosting of macromolecules with dimensions or shapes not properly matching those of the pores. In some cases, specific interactions can be observed between the sugar hydroxyls and definite parts of the embedded protein, resulting in an increased efficiency in preservation [
15,
16]. Low MW saccharide matrices (oligo or disaccharides) lack entirely the concept of a pore, as they are true amorphous matrices without a regular supramolecular organization. In both poly- and oligo-saccharide matrices, the impact of water and co-solutes is strong, and this allows an easy modulation of the hosting matrix properties. Although an effect can be noticed also in silica gels, this does not alter to a high degree the confining properties, which are modulated mainly by the degree of polymerization.
A form of organization within trehalose matrices has been proposed by proton annihilation lifetime spectroscopy studies, which showed that in amorphous trehalose matrices holes are present, with a size dependent on the water content, at variance with crystalline trehalose, where fixed sized channels through which water is able to diffuse are present [
17]. These water dependent holes are thought to help the preservation of embedded structures acting as buffering regions able to accommodate excess water molecules. Similar functional features were detected by SAXS and EPR on matrices containing different proteins: in this case, the “holes” have much larger volumes and are characterized by either a lack or scarcity of embedded protein. For this reason, water can be absorbed and released during dehydration-rehydration cycles [
18,
19,
20]. Moreover, water dependent holes (in amorphous solids) and fixed sized channels (in crystals) were reported from results from various techniques (DSC, NMR, X-ray, and Brillouin scattering and X-ray powder diffraction) [
17,
21]. Due to the occurrence of polymorphism both in amorphous and crystalline phases, water molecules would leave and reenter the structure without substantially altering the network of hydrogen bonds existing in the solution environment: in this way, any embedded biostructure would not experience abrupt alteration in its environment, and the preservation would be favored even in a crystallized or partly crystallized matrix [
21]. This occurs at least until strong dehydration conditions are met, where the structure is preserved by the inhibition of water induced translational motions hampering conformational changes, leading to denaturation [
22].
These studies showed that even in saccharide matrices, where a structural, porous scaffold is lacking, the formation of local structures with holes or areas with different water absorption properties play a key role in the preservation of embedded protein, provided that a H-bond connected network exists, as its presence is mandatory for thermal protein stabilization at low water content and for slowing down protein conformational dynamics [
20]. Forms of local phase separation are sometimes fundamental for the preservation process, even if this is not immediately evident, as has been demonstrated in the case of beta-lactoglobulin/trehalose solid mixtures (
Figure 2a).
The second main difference between hard and soft biomatrices concerns the embedded solvent, which can be unequivocally identified only in structured matrices with clearly built scaffolds and has distinctive characteristics in the two types of confinement. In hard matrices, as silica gels or zeolites, the scaffold “phase” is distinct from the protein + solvent “phase”. The dynamics of the two phases are always decoupled, while the effect of the scaffold on the embedded phase is usually described as “slaving” (the idea is that the scaffold induces a constrained dynamics on the solvent, which in turn slaves the protein dynamics). In soft, poly- to disaccharide matrices, even in case one can identify a structured scaffold, there is not a true decoupling, as the distinction between the two phases is blurred. In this type of matrix, it is possible to identify a changing behavior with water content ranging from a slaving similar to what was put in evidence with silica gels to a reciprocating coupling, up to conditions where the embedded component induces a behavior on the embedding one (protein that slaves the matrix; see
Section 3.6). A decoupling may also happen in saccharides and has been reported and described as “nanophase separation”, i.e., a separation of the dynamics of the two components, which is not distinct enough to make up a true phase separation [
23].
2.1. Silica Gels as an Example of Hard Confinement: Advantages and Modulation of Protein Structure and Function
Nanoporous and mesoporous silica gels are an encapsulation matrix widely explored for proteins, and particularly for enzymes, in biotechnological applications or biochemical and biophysical investigations. As a protein immobilization matrix, silica gel has several advantages [
24,
25,
26,
27,
28,
29,
30]: (a) the preparation protocol based on sol-gel chemistry requires mild conditions in terms of temperature and pH, compatible with the protein’s structural and functional integrity; (b) the matrix precursors allow an easy and versatile chemistry, allowing a tunable environment to be adapted to each protein; (c) the matrix can be molded to create solids with any shape and geometry, depending on the final application; (d) the matrix has tunable tightness and an average size of the pores; (e) the matrix is transparent, which allows the optical monitoring of the reactions.
The fundamental condition making silica gel an effective way to immobilize enzymes is the possibility to prepare it under mild conditions, compatible with the integrity of biological macromolecules. This process, optimized in the 1990s, is called sol-gel chemistry [
31,
32,
33]. Besides the chemical and physical compatibility of the process, it has been observed, based on a large number of silica gel encapsulated proteins, that the native conformation is conserved upon encapsulation, with a full, partially, or in some cases, even a largely increased enzymatic activity [
25,
29]. Caging of protein molecules in the pores of the silica gel can have crowding and hydration effects on the enzyme conformations [
34,
35] or isolate a single tertiary or quaternary protein conformation [
4,
36,
37,
38,
39,
40,
41].
Given the interesting properties of silica as an encapsulation matrix for biocatalysts, several biotechnological devices have been conceived and developed, such as bioreactors and biosensors [
24,
25,
29] and biomedical devices [
42,
43,
44,
45,
46]. Moreover, the physicochemical properties of silica gels make them particularly suitable for the biochemical and biophysical characterization of encapsulated proteins, thanks to the inertness, stability, and optical transparency, which allows applying most of the most common spectroscopic techniques. In addition, the tunable immobilization tightness, as well as polarity depending on the precursors mixed silicates allow isolating single protein conformations or slow down conformational transitions by several orders of magnitude [
41]. This latter aspect, while it has relevance for basic research studies on protein function, including enzymes, can descend to practical applications, since the modulation of the structure through the equilibrium among different conformations generates functional modulations.
2.2. Sol-Gel Process: Chemistry and Parameters Affecting Matrix Features
The sol-gel process attracted great interest in the field of protein encapsulation, with an easy chemistry accessible not only to inorganic and organic chemists, but also to other researchers expert in handling biological macromolecules. By selecting the silica precursors and controlling the mechanism and the kinetics of gelification, it is possible to modulate the final structure of the matrix, i.e., pore size, layer thickness, hydrophilicity/hydrophobicity inside the pores, degree of hydration, etc.
The reagents used as a matrix precursor are tetraalkoxysilanes or mono-, di-, or tri-alkyl alkoxysilanes (organically modified silica gels, or Ormosils). Tetramethoxysilane (TMOS) and tetraethoxysilane (TEOS) are the most commonly used. The hydrolysis reaction leads to the substitution of the alkoxy groups (-OR) with hydroxy groups (-OH) by a water nucleophilic attack on silicon atoms (
Scheme 1).
Methanol and ethanol released from TMOS and TEOS, respectively, have a less harmful effect on the protein than that of long chain alcohols. The product of hydrolysis, the so-called sol, has colloidal properties [
47].
The following step is a condensation reaction, leading to the formation of siloxane bonds (Si-O-Si), with the production of water (
Scheme 2) or alcohol (
Scheme 3). The viscosity of the sol increases during the condensation reactions until a gel is formed.
In the immobilization of a catalyst, an important parameter is the nature of its interactions with the matrix. For biological catalysts such as enzymes, another crucial element to be considered is the influence of the matrix on their conformation and dynamics. For the control of the interactions between biomolecules and silica gel, it is important to consider the microstructure of the gel and the chemical nature of the surface of the pores that contain the protein. To guarantee an efficient and stable catalysis over turnovers, the pores should have a size suitable to allow the free diffusion of solvent molecules and ligands (such as ionic buffers, substrates, products of the catalyzed reaction), but at the same time avoid the release of encapsulated catalyst.
Silica gel morphology, encapsulating tightness and composition, thanks to the easy chemistry, can be properly tuned by controlling the key parameters of the reaction, mainly the H
2O:Si ratio, pH, and solvent [
48]. The H
2O: Si ratio has a different effect on hydrolysis and condensation reactions: water is a reagent in the former, while a product in the latter; hence, a higher H
2O:Si ratio promotes hydrolysis, while it has a more complex behavior on condensation, with the alcohol-producing condensation predominant at ratio <<4 and a water producing condensation favored with ratio >>4.
Under acidic conditions, the protonation of the alkoxide makes it a better leaving group to SN
2 nucleophilic attack from water molecules, while under basic conditions, alkoxides’ hydrolysis occurs by hydroxyl anions’ attack and following alkoxide anion release [
48,
49]. Given this mechanism, a pH dependence of the hydrolysis process is observed, with a minimum at pH 7. The hydrolysis rate increases at each hydrolysis step on the same tetraalkoxysilane molecule in alkaline conditions, while it decreases under acidic conditions. However, inorganic acids are considered more efficient catalysts for the hydrolysis reaction than bases since the latter can be neutralized by the formed silanol groups. The condensation reactions, which can be monitored macroscopically by observing gelling time, have a more complex trend, with a maximum rate around pH 4–5 and a minimum at pH below 2.5. Strongly basic conditions do not allow gelification [
50,
51]. The presence of solvents or co-solvents can affect the catalysts activity depending on their polarity and their capability to bind protons or hydroxyl ions.
The modulation of the reaction rates affects the properties of the silica network and, hence, the immobilization capacity and its effect on the immobilized enzymes. Growth models have been applied to describe and simulate the effect of reaction rates on silica structure. A low pH and low H
2O:Si ratio favor a higher gel tightness, with smaller pores generated by a cluster-cluster polymerization, within a highly branched matrix. In alkaline conditions, condensation reactions are limited by the availability of hydrolyzed precursors, and the polymer growth is driven by a monomer-cluster process, resulting in a looser silica matrix [
48,
49,
52]. A different gel tightness can differently block or slow down conformational transitions necessary to achieve enzyme functions or isolate single protein conformations with higher or lower activity.
The possibility that the microenvironment of the pores can differ from bulk solution has been evaluated. Recent experiments demonstrated that, even though pores show a hydrophobic environment, the presence of OH groups as from the hybrid gel formed by TMOS/
n-butyltrimetoxysilane (BTMS) precursors allowed slowing down the exchange of polar solvents and hence the preservation of enzymes’ activity [
53].
2.3. Enzyme Encapsulation in Silica Gels for Protein Delivery, Biosensors, and Bioreactors
The mild preparation conditions and the optical properties of silica gels and their biocompatibility [
54] were exploited to encapsulate enzymes for biosensing, biocatalysis, and biomedical applications back in the 1990s. Since then, a wealth of experimental reports has appeared in the literature, often with emphasis on practical applications and, sometimes, on the chemical and biophysical basis of the observed effects on protein structure, function, and dynamics. Several excellent reviews have been published over the years [
25,
47,
55,
56,
57,
58,
59,
60,
61]. We focus here on some of the most recent reports.
The effect of the conformational distribution on enzyme activity was investigated for β-galactosidase encapsulated in silica gels, obtained from TEOS. Circular dichroism, fluorescence, and activity were monitored as a function of the temperature range 25–65 °C [
62]. The α-helix content of the encapsulated enzyme was higher with respect to the enzyme in solution both at 25 (9.5 vs. 5.4%) and 65 °C (11 vs. 2.7%). The tryptophan fluorescence spectrum of the encapsulated enzyme showed a red shift with respect to the soluble form, indicating a more polar environment surrounding the 34 residues and possibly a less compact structure. Moreover, the denaturation midpoint was found to be 59 and 48 °C for the soluble and encapsulated enzyme, respectively. The activity of β-galactosidase gels at 0.5 mM ortho-nitrophenyl-β-galactoside (ONPG) was the same as the soluble form between 35 and 50 °C, but two-fold lower at 1.5 mM, likely due to the rate limitation of diffusion of the substrate within the gels. Overall, data indicate that the encapsulated β-galactosidase is less stable than the enzyme in solution, likely due to alteration of the conformational distribution. A decreased stability for an encapsulated enzyme with respect to the soluble form is unusual as the gel matrix tends to prevent unfolding, which is accompanied by volume expansion [
63,
64].
The pyridoxal 5’-phosphate dependent methionine γ-lyase was incapsulated in either TMOS or TEOS derived gels as a reference system of enzyme delivery for biopharmaceutical applications. The UV-Vis, circular dichroism, and fluorescence spectra of the soluble and encapsulated enzyme were compared and found to be very similar, indicating that entrapment did not alter protein conformation. However, the enzyme stability toward chemical denaturation with guanidinium hydrochloride was higher for the encapsulated enzyme, indicating that silica matrix restrains unfolding events. The activity was assayed under conditions where substrate diffusion was calculated to be not rate limiting the catalytic reaction, i.e., on gel particles with reduced dimensions as obtained upon macro-gels’ sonication. K
M was slightly higher for the encapsulated enzyme, whereas k
cat was about four-fold lower, with the resulting catalytic efficiency of the encapsulated enzyme about six-fold lower [
65].
The comparison of the stability of tyrosinase from black wild truffle in gel and in solution showed that at 60 °C, the soluble form was completely inactive, whereas the enzyme encapsulated in TEOS gels was still 40% active. The enzymatic activity using catechol as the substrate was dramatically affected by encapsulation with the apparent K
M and V
max found to be 0.35 mM and 6 μM min
−1, respectively. These values are significantly different than those obtained for the soluble enzyme (K
M = 2.5 mM and V
max = 102 μM⋅min
−1). These findings suggest that the gel matrix has stabilized an enzyme conformation that binds tightly the substrate, but is less able to catalyze the oxidation of catechol [
66]. A bi-bilayer of encapsulated enzyme was used for the development of a biosensor for detecting catechol, which is a well known pollutant. The biosensor exhibited a linear response on catechol, but a decreased efficiency upon repetitive use, likely due to enzyme leaching or inactivation. Furthermore, the biosensor was not sensitive enough to the concentration of catechol as the limit of detection was 52 μM, in the upper limit of the acceptable concentration of phenolic compounds in wastewater [
66].
A biosensor for detecting phenolic compounds was also developed based on laccase gels. Encapsulation was carried out using both TMOS and MTMOS to increase the matrix hydrophobicity [
67]. Enzyme encapsulation was proven by a careful comparative analysis of FT-IR spectra with and without entrapped protein. The laccase biosensor was probed using resorcinol, catechol, or hydroquinone as a substrate. A better linear range was obtained for resorcinol, whereas the best sensitivity and limit of detection was obtained for catechol. Due to the very slow reaction rate of hydroquinone, only resorcinol and catechol reactions were fully kinetically characterized. It was found that K
M of resorcinol for laccase gels was two-fold higher than the soluble enzyme, with the V
max of the reaction almost four-fold higher, whereas both K
M and V
max of catechol were close to those determined for the soluble enzyme. The laccase biosensor showed good stability for at least two months. Furthermore, the laccase biosensor was able to determine correctly the concentrations of solutions of resorcinol and catechol spiked tap water [
67].
Lipase encapsulated in silica gels was supported on Celite
® for the development of a bioreactor for the transesterification of triolein to methyl oleate. The resulting bioreactor exhibited good stability without loss of activity over a five day period, suggesting a good possibility for the efficient production of biodiesel [
68].
An interesting example of enzyme sol-gel encapsulation for the development of a bioreactor was reported by Muderrisouglu and colleagues [
69]. β-glucuronidase was immobilized into TEOS-poly ethylene oxide (PEO) derived gels. The enzyme activity was found to be 21% of the soluble form, whereas K
M decreased from 1.58 to 0.42 mM, indicating a tighter apparent substrate binding to the entrapped enzyme conformation. β-glucuronidase gels were used in a microfluidic system for the removal of glucuronic acid from plant derived β-glucuronides for pharmaceutical applications. The efficiency of the bioreactor with respect to a gel batch preparation was assessed by monitoring spectrophotometrically the hydrolysis of
p-nitrophenyl-β-
d-glucuronide. It was found that the system was more efficient than the batch at a flux rate of 1 μL/min, but not at higher flux rate. However, the microfluidic platform maintained continuous catalytic activity for seven days [
69].
Marchetti and colleagues exploited the specific enzymatic activity of allantoinase encapsulated in TMOS derived wet nanoporous silica gels in a fluorimetric assay for the quantification of allantoin in biological fluids [
70]. Allantoin, an oxidation product of urate, has long been proposed as a marker of oxidative stress, and its level increases in the plasma of patients undergoing chemotherapy and in several pathological conditions. However, current standards for its quantification rely on costly and time consuming techniques. In the proposed assay, enzymatic conversion of allantoin into allantoate is followed by acid hydrolysis of the latter into glyoxylate in the presence of resorcinol, yielding a stable lactone emitting fluorescence in the visible range. Encapsulated allantoinase retained full catalytic activity and gave fluorescence readouts comparable to the enzyme in solution for allantoin quantification in serum, urine, and saliva samples. The device, where the allantoin doped gel is deposited into the caps of 200 µL plastic vials, could be reused for at least three times with no apparent loss of functionality, suggesting that the method might be suitable for routine clinical determination of allantoin levels in biological fluids [
70].
2.4. Trapping Single Conformations of Allosteric Proteins for Tuning Their Function: Hemoglobin and Enzymes
Besides the development of bioreactors and biosensors, immobilization in silica gels of allosteric enzymes and, in general, proteins has provided a wealth of information on tertiary and quaternary transitions. In fact, silica matrices lead to an enormous slowing down of quaternary conformational changes, which allows separating in time the large scale motions from more local tertiary rearrangements. The molecular origin of these kinetic effects can be traced to direct interaction of the gel matrix with the embedded proteins and/or by the altered dynamics of the protein hydration shell. The properties of protein transient states or scarcely populated conformations are in many cases not directly or easily studied in aqueous solutions, but they become accessible when trapped in silica gels.
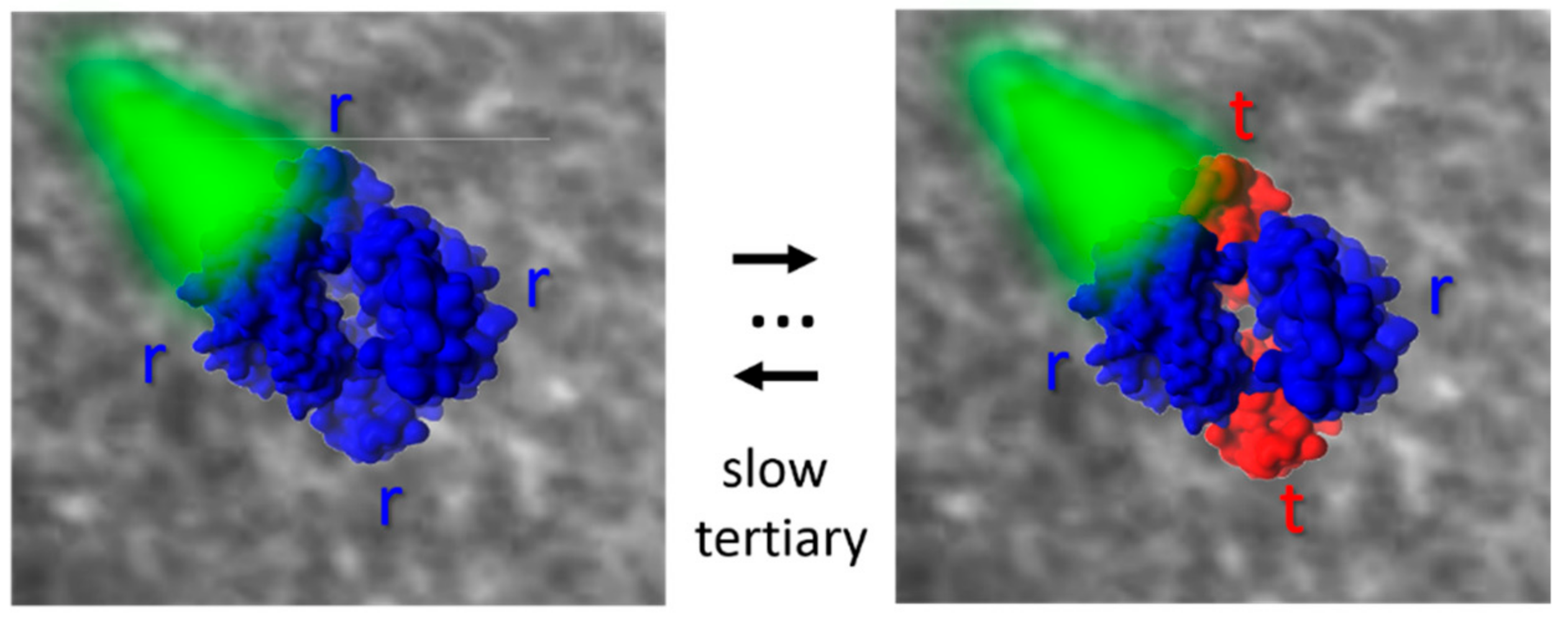
The most studied case is certainly that of the quaternary transition in human hemoglobin (Hb), which has served as a paradigmatic system for allosteric transitions [
71]. For no other protein, the fine details of allosteric regulation and associated dynamics and how these are affected by encapsulation in polymeric matrices have been characterized with comparable effort. This, together with multiple functions other than oxygen transport, including a role in redox intracellular processes that motivated the definition of Hb as an “honorary enzyme” [
72], justifies some room within this review. Indeed, a biophysical characterization of how encapsulation regulates tertiary and quaternary equilibria, and the rate of protein dynamics is of great help in understanding, controlling, and optimizing the enzyme functions required for biosensing and biocatalysis. Hemoglobin is a tetrameric protein, where assembly is made of a dimer of dimers, and each dimer contains two different chains, termed α and β. In solution, ligand release triggers a quaternary transition in Hb, with a timescale of ~1 μs [
73,
74], but this transition is dramatically slowed down by silica gel encapsulation [
75]. A 15.5° rotation of the α1β1 dimer vs. α2β2 appears as the main change from the crystal structures of R and T states. The main changes are evident at the α1β2 interface, at the so called “hinge” and “switch” contacts. The R−T switch was reported to involve separate stages, characterized by 3 and 20 μs time constants, related to the “hinge” and the “switch” contacts, respectively [
28,
76]. Wide-angle X-ray scattering indicated that most of the change observed in the shape was associated with the faster phase [
73].
The R → T transition for the fully unliganded Hb can be slowed down by 10 orders of magnitude, as judged by optical absorption and resonance Raman spectroscopy [
29,
31,
32,
34,
77]. Thanks to this, the low-affinity T quaternary structure can be trapped by gels prepared from fully deoxygenated Hb, whereas the high-affinity R state is trapped inside gels of the fully oxygenated Hb [
29,
75,
78]. By preventing the quaternary switching, gel entrapment of Hb results in non-cooperative oxygen binding both for the R and T state [
29,
75,
78]. These results had a key role in settling the long standing controversy between concerted (e.g., Monod–Wyman–Changeux) and other models for protein allosteric regulation, with the latter predicting significant cooperative behavior even in the absence of quaternary relaxations. In this respect, it is worth noting that hard confinement in silica gels, while being able to almost completely prevent the protein quaternary conformational changes (i.e., the protein dynamics in microseconds or slower time scale), has a much lower effect on the short time (picoseconds/nanoseconds) dynamics. In fact, neutron scattering experiments showed that the methyl groups’ dynamics in myoglobin encapsulated in silica gel is unaffected, while the side chains’ anharmonic motions are reduced (but not prevented) in a hydration dependent extent [
79,
80,
81].
Spiro and coworkers [
82] used a combined spectroscopic and computational approach to monitor conformational transitions in the α and β chains of Hb, using hybrid hemoglobins, in which Fe-heme was replaced by Ni-heme in either the α or β subunits. The use of hybrid Hb allowed separate monitoring of the α or β hemes along the allosteric pathway. Using resonance Raman spectroscopy of hybrid Hb in silica gel, which greatly slows protein motions, they observed that the Fe−histidine stretching frequency, related to heme reactivity, moves from frequencies characteristic of the R to those typical of the T state, for both α or β chains, before the quaternary R−T (and T−R) transition occurs.
Simulations using protein energy landscape exploration (PELE) [
83] allowed highlighting remarkable differences between α and β chains. While in β chains, the movements of E and F helices are in the same direction, thus producing no change in the orientation of the Fe-His bond and only a small reduction of the frequency, in α chains, there is a heme distortion and a tilting of the Fe-His bond due to the orthogonal movement of E and F helices, leading to a larger frequency decrease. These differences were attributed to a different configuration of the EF corner and to a different orientation of the hydrogen bonds and salt bridges connecting H with F and A helices.
From this combined analysis, the authors could gain important information on the coupling between tertiary and quaternary transitions and on the different response of α and β chains. The two steps in the R to T transition do not have the same properties for the two chains. A significant tertiary change precedes the second step only in β chains, while the first step is accompanied by a more important change only in α chains. Moreover, this approach confirms the tertiary two-state (TTS) model by Eaton and coworkers [
84], according to which to tertiary conformations, r and t, endowed with high and low reactivity, respectively, can populate R and T quaternary states, and demonstrates that the r and t tertiary states are structurally/dynamically different in the two quaternary forms, even if they are functionally undistinguishable. The observed time courses are consistent with computational mapping of the R−T transition by Karplus and coworkers [
35].
Eaton and coworkers have recently analyzed studies on Hb immobilized in single crystals and silica gels to test the predictions of the four major allosteric models that have been proposed for Hb: the quaternary two-state model of Monod, Wyman, and Changeux; the TTS of Henry et al., which is the simplest extension of the Monod–Wyman–Changeux model to include pre-equilibria of tertiary, as well as quaternary conformations; the structure based model of Szabo and Karplus; and the modification of the latter model by Lee and Karplus [
85]. Based on their thorough analysis, the authors showed that a quantitative description of all available results was only possible with the tertiary two-state (
Figure 3). The inclusion of the inequivalence of the tertiary equilibrium constants for the α and β subunits in both the T and R quaternary structures gave the best agreement with the experimental results.
A different system where wet silica gels were used to trap unstable conformations is the allosteric enzyme glucosamine-6-phosphate deaminase from
Escherichia coli. [
86] The enzyme deaminates glucosamine-6-phosphate to yield fructose 6-phosphate and ammonia and is subject to allosteric regulation by
n-acetylglucosamine 6-phosphate. Since encapsulation in wet silica gels prevents quaternary, but not tertiary switching, the authors were able to block the enzyme in the T state and study the role of intrasubunit local mobility in allosteric activation. Under these conditions, homotropic cooperativity was lost while catalytic properties and allosteric activation by glucosamine-6-phosphate were retained. The local, tertiary relaxation kinetics showed a remarkable reduction in rate, as previously reported for other systems, and the relaxation kinetics was interpreted with an induced-fit mechanism. Assuming a model with two tertiary states of low and high affinity, t and r, and fitting the velocity-substrate plots as a linear combination of two hyperbolic functions with K
t and K
r as K
M values, results from solution experiments analyzed with the Monod–Wyman–Changeux model could be reproduced. The reported case was considered consistent with the tertiary two-state model by Eaton and coworkers originally proposed for hemoglobin (see above).
The effect of isolating different conformations is a limiting case in encapsulating proteins. In a proper condition, the matrix can be exploited for slowing down without blocking conformational transitions and folding events. Direct observation of early intermediates is fundamental to understand the folding mechanism of a protein [
87]. Yet, the very short time scale of folding and unfolding often results in experimentally difficult to resolve burst phases. Several experimental methods were proposed to achieve the demanding resolution that is needed to access early events like chain collapse and formation of secondary structure [
87,
88,
89]. The folding kinetics can be substantially slowed down by trapping native proteins in silica gels that are rich in solvent and preserve the solution structure of the molecule. In a recent work, Okabe et al. encapsulated five different globular proteins in silica gels containing 80% (w/w) water and monitored folding by CD spectroscopy [
90]. The decreased folding rates allowed the direct observation of the initial transitions, which would be otherwise inaccessible in water. Kinetics were described by single exponential relaxations and were characterized by activation energies of 14−31 kJ/mol.
3. Entrapment in Saccharides, Modulation of Properties with Hydration, Type of Sugar, Molecular Weight
Saccharide matrices, from highly concentrated, viscous solutions to rubbery amorphous solids and up to dehydrated glasses, are utilized by some organisms to shield themselves from the effects of adverse environmental conditions, as extreme temperatures, or a lack of water, or even irradiation. Upon exposure to conditions unfavorable to life, they start to synthesize specific sugars within the cells that produce a protective environment propaedeutic to a following state of suspended animation needed for survival. This triggered a strong interest for the study of saccharide matrices, not only for the potential applications that their protective ability forewarns, but also for their extreme tunability, which in many cases is as simple as varying the environmental moisture.
Many sugars were shown to have such preserving properties, among them disaccharides, oligosaccharides, and polysaccharides, but in the literature, a special importance is bestowed to trehalose, whose peculiar effectiveness has triggered a widespread interest from both fundamental and applicative points of view.
Within saccharide matrices, the main types of interactions that can occur are those naturally present in biological matter: saccharides are medium-to-large, hydrophilic organic molecules, with the surface rich in hydroxyl groups; hence, they are likely to be hydrated and to absorb a layer of solvent, and possibly salts and co-solutes, at the surface. The capability of saccharides to absorb water can be exceptionally large in some charged sugars, like in the case of hyaluronic acid, pectin, and the lateral chains of proteoglycans, the two latter being able to swell and constitute a significant part of soft and compressible structures of plant and animal tissues. Conversely, hydrophobic patches in, e.g., proteins tend to be excluded from the interaction with sugars, this being one of the reasons for the stabilizing effectiveness of sugars toward globular proteins.
When used as confining matrices, saccharides are assumed not to bind directly (covalently) to the protein, and one could identify a confining agent and a confined subject. However, we should remind that proteins covalently bound to sugars are very common in nature (e.g., glycoproteins and proteoglycans), with sugar chains performing various roles. Some proteins as collagen, lens crystalline, ferritin, apolipoprotein, and albumin are normally glycated in vivo. Glycoproteins are usually carefully controlled products by selective modification that is generally associated with a gain of function (or stabilization, e.g., against proteolytic enzymes) of the target protein.
The occurrence of covalent protein-sugar bonds can also be detrimental, in particular if the saccharide coating is carried out with reducing sugars: in this case, uncontrolled, nonselective glycation occurs, producing a generalized loss of function of the embedded protein and increased unfolding and aggregation. This side effect makes saccharides with reducing ends not useful in building confining matrices [
91].
Conversely, in the absence of reactions leading to covalent binding, the establishment of non-covalent interactions between sugars and proteins results in protein stabilization, in particular against the effects of extreme temperatures or excessive drought. This triggered the use of saccharide matrices to improve long term storage in the food, medicine, pharmaceutics, and biotechnology industries, besides the diffuse use in fundamental research. Among disaccharides, trehalose imposes itself because of its preservation effectiveness, useful also to avoid discoloration, loss of aroma, and alteration due to cooking, and its lower and longer lasting sweetness with respect to sucrose (
Figure 1b) and can be found in a long list of processed food, from confectionery to vegetables, beverages and baked goods [
92]. The peculiar effectiveness of trehalose has been a subject of intensive studies with the aim to elucidate its working mechanism, then to exploit industrially its preserving properties. Among the various hypotheses proposed for saccharide based biopreservation, many are based on direct interaction between sugar, protein, and water, differentiating only the reciprocal relationship of the three components and in different specificity of the interactions (water replacement or water entrapment hypothesis) [
93]. Some of these theories are not mutually exclusive and can be successfully applied to mono-/di-saccharide matrices, as well as to polysaccharide ones, although many reports point out a substantial difference between small saccharides, which act mostly by direct, specific interactions, and polysaccharides, where a significant crowding/confinement effect is superimposed on specific interactions [
93,
94]. No parameter could however be singled out as a measure of saccharide protective effectiveness [
95]. The effect of many variables has been studied for this aim, namely hydration, temperature, and composition [
23,
93,
96], addressing also kinetics and thermodynamics aspects, from the atomistic [
15,
97] to supra-molecular scale [
96,
98,
99].
3.1. Saccharide Mixed with Other Bio-Compatible Molecules
Due to the non-covalent nature of the saccharide confining matrices, it is possible to exploit the combined effect of mixtures of small saccharides and polysaccharides. Actually, the use of disaccharides, which are often presented as ideal biopreservers, presents some drawbacks in the case of industrial application: large hygroscopicity, coupled to easy morphological changes with easy crystallization upon hydration/dehydration cycles. These properties make disaccharides scarcely valuable in particular for food products, where stability in time and visual appeal are mandatory characteristics. At variance, oligo- and poly-saccharides have valued rheological properties and could be used to improve disaccharides’ shortcomings. Moreover, many long chain carbohydrates have biopreserving properties, mostly due to their high glass transition temperature (T
g) rather than to their direct interaction with an embedded biostructure, less effective than in disaccharides due to their steric hindrance and large free volume [
100,
101,
102]. However, notwithstanding their high T
g, the polysaccharides efficiency works only up to the complete vitrification of the system: a T
g much higher than the useful interval of temperatures does not improve the system protective ability [
103].
The matrix shaped by the polysaccharides appears as a flexible porous medium, with an embedded solvent fraction where the smaller components (glycerol, small saccharides) constitute a viscous fluid flowing within the pores and the protein; although sterically hindered, it is not strongly constrained [
104]. The presence of small components within the solvent part of the matrix enhances the ability of the whole matrix to interact with the embedded protein, resulting in a synergistic protection [
101,
105], in particular if the small component is able to bind directly to a significant extent [
98,
106]. Indeed, adding a small interacting component helps to reduce the residual mobility of the protein even in the solid state, improving the global system efficiency [
103]. The presence of small saccharides can also have a strengthening effect on the polysaccharide fraction, producing more stable networks both acting by cross-linking the polymer chains and by reducing the amount of free water around the polysaccharide [
107].
Combinations of disaccharides (or, less efficiently, oligosaccharides) and long chain saccharides such as dextran have been employed in stabilizing proteins like actin [
100] and in the food industry [
108]. In this type of matrix, it is possible to assist a change in the relative importance of the saccharide components: while at high hydration, the macromolecular scaffold is dominant in the process of immobilization, leading to the preservation and rise of T
g, upon dehydration, the small, highly interacting, disaccharides prevail, and protein stabilization through direct interaction becomes the dominant contribution [
94,
109]. Because of this interchange, the mixed matrices have been shown to be effective in a large hydration range [
109].
A similar behavior, with synergistic effects, can be observed if the macromolecular component is a protein, as put in evidence in the case of bovine serum albumin (BSA) or gelatin [
99,
110], or if it is composed by a mixture of polysaccharides and proteins: in the latter case, the two components interact among themselves, forming networks capable of high water retention and high viscosity [
111].
In mixed matrices of starch and small saccharides, the matrix stability appears to be related to the strength of the intermolecular H-bonds between low and high molecular components and to the mobility of the low MW component within the matrix [
112]. In this respect, trehalose, with its lesser ability to form intramolecular hydrogen bonds with respect to other disaccharides, appears very promising as it builds more easily large hydrogen bond (HB) networks involving also the protein and the high MW component, enhancing the reduction in mobility of the embedded protein [
113].
With the aim of obtaining new confining agents that conjugate the structural properties of polymers with the protective ability of trehalose, some glycopolymers, which embed the trehalose moiety, have been developed. Among them, two types have been distinguished. In one case, the trehalose moiety is not a part of the polymeric backbone, but it is simply attached to it as a lateral chain (
Figure 1d); in this respect, they are similar to natural glyco-derivatives such as glycoproteins. These polymers work exactly as polymer/trehalose mixed matrices, with the exception that trehalose is covalently attached to the surface, avoiding direct contact between the biomolecules and the polymer backbone (which often leads to adhesion and denaturation) and influencing the properties of the water molecules within the matrix. They have been used to protect various classes of biomolecules (enzymes, antibodies, hormones membranes, nucleic acids, even whole cells), where they have been shown to outperform the trehalose matrices themselves. In particular, they are most effective at low concentration and in conditions where there is a risk of depletion of the soluble disaccharide (e.g., for formulations to be used in diluted solutions) and show superior heat protecting properties [
14,
114,
115,
116,
117]. In the other type, the trehalose moiety is a part of the polymeric backbone (
Figure 1e); these glycopolymers have peculiar properties, different from the mixed matrices, and keep increased preservation properties with respect to analogous ones without the trehalose moiety; they have been found to be significantly effective in the protection of nucleic acid fragments [
118].
3.2. Enzymes Embedded in Trehalose, a Soft Matrix
Traditionally, embedding proteins (or other biostructures) in solid matrices have been exploited for studying general problems on protein–solvent, protein–cosolutes, or even protein–protein interactions. These matrices have been utilized to alter or tune the protein structure, dynamics, and function by making use of a specific or aspecific interaction with the confining surfaces, as well as altering of the physical properties of the external environment.
In this respect, the usual approach of fundamental research had been considerably “protein-centric”, while from an applicative point of view, a “stabilization procedure-centric” approach had been employed. Coexistence and mingling of these two approaches is beginning to appear in the recent literature, where the fundamental bases are investigated with a look toward the possible applications [
119].
Various possible applications have been proposed for saccharide matrices with entrapped proteins or enzymes. The typical applications of biopreserving sugars range from medicine (cryopreservation of cells or even whole organs, wound healing and blood substitutes) to the food/pharmaceutical industries where sugar solid matrices have been widely employed as a preserving excipient [
120,
121,
122,
123,
124,
125,
126].
Back to fundamental research, it is known that proteins need to undergo conformational changes while performing specific functions such as ligand binding or enzymatic catalysis. In this respect, trehalose has been used to study reactions that are strictly dependent on protein structure dynamics.
As for enzymes [
127,
128,
129], the possibility to slow down the conformational changes allows accessing the details of the reaction mechanism that otherwise could not be accessible due to their short time scales. Lactate dehydrogenase (LDH) has been used as a model enzyme in studies of protein stability. LDH catalyzes the reaction of the conversion of pyruvate to lactate by using nicotinamide adenine dinucleotide (NADH) in reduced form. It has been proposed that the catalytic mechanism depends on the dynamics of the protein and in particular on the local motions of a loop capping the substrate binding site [
130]. High temperatures induce large scale motions in the whole protein structure and in particular a structural rearrangement of the catalytic site [
131], leading to a state with low affinity for the ligand. It has been shown that trehalose prevented the temperature induced transition toward the state with low affinity for the ligand, by hampering the fluctuations of the catalytic loop by viscosity effects.
Other enzymes like E1E2 -ATPases undergo major conformational changes during their catalytic cycle, as they alternatively bind substrates, hydrolyze ATP, and pump protons. A relevant example is H
+-ATPase from the plasma membrane of fungi and yeasts, the primary pump needed to generate proton gradients across the cell, in particular during heat shock. Trehalose, at the same concentration present in the yeast cytoplasm during heat shock conditions, was found to protect the isolated
Kluyveromyces lactis H
+-ATPase from inactivation at high temperatures, by increasing the activation energy for ATP hydrolysis, but also by limiting the diffusion of the substrate across sugar networks surrounding the protein. The same effect was found in pyrophosphatase from yeast during incubation at 50 °C [
132,
133] and in yeast enzymes glucose-6-phosphate dehydrogenase and phosphoglucose-isomerase [
134]. In glucose oxidase, studied in the absence and in the presence of trehalose, it was found that trehalose stabilizes the protein by decreasing the inactivation rate constant and favoring the compact folded state. At high trehalose concentrations, the enzyme efficiency increases due to the inhibition of the exposure of hydrophobic regions allowing for ligand dissociation [
127]. Another interesting example is Renilla luciferase (Rluc) from
Renilla reniformis, a bioluminescent protein, important for the detection of specific molecular targets. The effects of trehalose and sucrose on the kinetic stability of Rluc were studied [
113], measuring the activity of the enzyme in the presence of sugars at high temperatures. Molecular dynamics (MD) simulations were carried out to complement the experimental data, providing a molecular basis for the high efficiency of trehalose in stabilizing the protein. In particular, it is suggested the trehalose networks interacting with the helices could keep the structure stable at high temperatures, by reducing the helix fluctuations, in line with results from MD simulations of carbonmonoxy-myoglobin (MbCO) in trehalose matrices [
15].
A dependence of residual trypsin activity on trehalose or sucrose concentration was found for dehydrated samples after prolonged thermal treatment, attributing the higher effectiveness of trehalose to its better interaction with the enzyme [
135].
The preservation and immobilization of enzymes have been exploited recently to produce immobilized biocatalysts that conjugate the advantages of the efficiency and selectivity of enzymes, with the easiness of separation and the ability to work at higher temperatures and with different solvents of the heterogeneous catalysts [
136]. A fundamental aspect needed for an enzyme to work is its thermodynamic stability in the harsher conditions needed for its use: non-covalent embedding in preserving matrices is a simple method to increase the stability without compromising the flexibility needed for the enzyme to work [
136].
3.3. Membrane Enzymes
A very interesting model to explore the relation between protein conformational dynamics and kinetic processes is the photosynthetic reaction center (RC) from the purple bacterium
Rhodobacter sphaeroides. In RC, light excitation gives rise to electron transfer reactions along a path of several protein bound co-factors, providing charge separated states with lifetimes sizably depending on the protein matrix structure and dynamics. At cryogenic temperatures, a distribution of rate constants for the charge recombination processes is observed; at room temperature, given that different protein conformations could be sampled on the charge recombination time scale (substates averaging), almost exponential kinetics are measured. In trehalose glasses at very low water content, it is observed that the recombination sizably accelerates, becoming broadly distributed as at cryogenic temperature [
137]. Moreover, embedding RC in dry trehalose matrixes preserves the structure and the activity of the protein. The anchorage hypothesis (see
Section 3.6) has been proposed for the RC–matrix interaction, also on the basis of different experiments performed at different water contents [
138]. Indeed, a rigid water HB network bridging protein groups and sugar molecules at the protein–matrix interface would hinder conformational rearrangements of side-chains, thus contributing to the stabilization of the charge separated states. This model could also rationalize results from continuous illumination experiments where small tier fluctuations could be recovered due to the breakage of H-bonds at the RC surface, at least temporarily [
139]. This model also helps in clarifying the differences observed in RC in dehydrated films in the absence of sugar with respect to trehalose matrices. Protein dynamics is strongly inhibited in both host environments, but protein fluctuations, which stabilize the charge separated state and also build up larger scale changes leading to thermal denaturation, are hindered in trehalose by increasing the sugar concentration in the matrix. Thermal denaturation is prevented already by a few trehalose molecules per RC, suggesting a percolative behavior, which again could be explained on the basis of the anchorage hypothesis. Denaturation is then totally hampered above a protein/sugar threshold; a rough calculation of protein and trehalose free volumes at that molar ratio leads to a picture in which trehalose molecules could fill the void in a completely dehydrated protein.
To have better insight into the details of the protein–matrix interaction and the role of H bonding, electron paramagnetic resonance (EPR) measurements have been performed at different protein/sugar ratios in trehalose or sucrose matrices [
20], by dissolving the nitroxide radical. Different effects have been observed in the two sugars. While in case of trehalose, the thermal stabilization has no dependence on the protein concentration, in the case of sucrose, only low sugar/protein ratio matrices could stabilize RC. EPR results suggested that the two homologous disaccharides form two different microscopic environments to immobilize the protein at low hydration. In sucrose, only at low sugar content, the matrix is a homogeneous network with long range connectivity, confirming that the ability of sugar in bioprotection by soft confinement relies on the specific intermolecular sugar–water–protein HB networks established in the system.
Coupling between protein dynamics and reaction transfer kinetics has been also studied in the Photosystem I (PS I) from the cyanobacterium
Synechocystis sp. [
140]. PS I is a trimeric protein composed by two subunits, PsaA and PsaB, with a chlorophyll placed on the lumenal side and a [4Fe-4S] cluster displaced on the opposite side of the complex. Each of the two branches contains two chlorophyll molecules and one phylloquinone. The terminal [4Fe-4S] clusters are both bound to the subunit PsaC. Following light excitation, the excited singlet state of the primary electron donor (the lumenal chlorophyll of one subunit) delivers an electron to an accepting branch chlorophyll forming a charge separated state. The electron is then transferred to the corresponding phylloquinone, to the iron-sulfur cluster within the subunit, and finally to a terminal iron-sulfur cluster bound to PsC. The rates of the electron reaction span from hundreds of femtoseconds to hundreds of milliseconds. The PS I complex has been embedded in trehalose matrices at low water content, to study the coupling between the different electron transfer reaction steps and the protein dynamics along the different time scales. The kinetics of charge recombination has been studied by time resolved EPR and optical NIR absorption spectroscopy after a pulsed excitation, as a function of the hydration of the matrix. Results show that the electron transfer process is coupled to the low frequency protein/solvent conformational motions and is suppressed at room temperature in dehydrated trehalose matrices. As in the case of trehalose-RC glasses, the interaction of the protein surface with trehalose would induce a strong dynamical constraint, which propagates over long distances down to the inner regions of these large protein complexes. The trehalose influence strongly depends on the hydration level, selectively inhibiting one or the other reaction transfer step. Most important, in very dry trehalose glasses, different PS I conformers could be trapped at room temperature, allowing kinetically distinguishing different recombination pathways along different branches in the protein.
3.4. Heme Proteins: Myoglobin and Hemoglobin
As further examples, ligand recombination processes have been studied in MbCO encapsulated in trehalose matrices with flash photolysis experiments [
141,
142]. Studies performed on carbonmonoxy Hb (HbCO) embedded in trehalose glasses at room temperature with different complementary techniques [
143] showed that the trehalose selectively perturbs the ligand binding kinetics, allowing for fast relaxation, but inhibiting large time–space scale motions. Fluorescence measurements indicate that the structural changes that usually lead to a transient opening of the distal heme site are damped. Moreover, from resonance Raman measurements, it is suggested how the trehalose could rigidify the Hb network involving the heme propionates, thus contributing to hampering the heme motions. It has also been shown that the ligand escape from the protein matrix is prevented even at room temperature by reducing tertiary helices motions, an effect with several important implications, for example in studies of ligand trapped in xenon cavities. Indeed, to trap the photoproducts, one should perform experiments at cryogenic temperatures, which however slows down the cavity population over time. On the other hand, at higher temperatures, the ligand would escape from the protein. Embedding proteins in trehalose glasses is a unique way to trap photoproducts, by limiting the conformational changes that allow the ligand to escape, but at the same time preserving low tier protein fluctuations that allow the ligand to access different xenon cavities [
144].
3.5. Enzyme Stabilization by Saccharide Mixed with Other Bio-Compatible Molecules
Mixtures of saccharide and polymers have been successfully used to stabilize enzymes. In particular, a freeze dried matrix of polyethylene glycol mixed with various sugars (trehalose, lactose, or mannitol) was able to preserve lactate dehydrogenase or phosphofructokinase better than analogous monocomponent matrices [
110].
With respect to the balance between the direct interaction and confinement effect, polysaccharide matrices have properties comparable to other high molecular weight matrices, i.e., their principal effect is due to crowding or confinement. Mechanisms of crowding are all alike irrespective of the crowder nature or chemical composition; hence, similar effects are in principle expected, be the crowder a biological macromolecule (a protein or a saccharide) or a synthetic oligo-/polymer [
145]. Crowding reduces the number of accessible extended conformations of the embedded biomolecules, favoring compact and associated states and reducing conformational entropy. Kinetically, the effect is the acceleration of any reaction passing through a transition state more compact than the initial state [
146], provided that the rate of encounter (which is usually reduced by encapsulation) is not a limiting step. A too strong confinement can however make most dynamical processes stop [
147]. It has also been postulated that a biomolecule in a confined environment spends most of its time in a compact molten globule state more than in a native fold, and it can be involved in the reactive processes as a molten globule [
148]. Crowding effects have successfully been exploited in agro-industrial and pharmaceutical fields by using edible or biocompatible saccharides, with the aim to obtain aspecific immobilizers that do not undergo direct interactions with the embedded molecules. These preserving agents have a major drawback: they lose effectiveness at very low hydration, in particular if they have a high molecular weight, even if some saccharides including specific, effective moieties (e.g., trehalosyl derivatives) have been shown to be able to partially overcome this disadvantage [
98,
145,
149,
150].
Examples of soft confinement have been obtained with dextrans (
Figure 1c) of several concentrations and sizes to mimic cellular crowding effects on the kinetics of several model enzymatic reactions: the hydrolysis of
N-succinyl-
l-phenyl-Ala-
p-nitroanilide catalyzed by α-chymotrypsin, the oxidation of 2,2′-azino-bis (3-ethylbenzothiazoline-6-sulphonic acid) (ABTS) by H
2O
2 catalyzed by horseradish peroxidase, and the oxidation of NADH in the presence of pyruvate catalyzed by LDH [
151]. Results show that in the case of small enzymes, the volume occupied by the crowding agent plays an important role in the initial velocity of reactions, but not its size. At variance, in the case of large enzymes, also the dimension of the crowding agent influences the initial velocity of the reaction.
Effects of solid matrices trehalose and inulin (a short chain polysaccharide of mainly fructose) were tested on the enzymatic activity of alkaline phosphatase upon redissolution, at various water contents, with the aim to modulate the T
g. In the most effective conditions (high T
g-low hydration), protein degradation may happen either due to insufficient interactions with the saccharide or to large local free volume: in both cases, trehalose is more likely to fit on the irregular protein surface than the rigid inulin in the absence of a suitable water layer, this independent of the protein/sugar ratio employed [
152].
3.6. Effect of Saccharide Matrices on Protein Structure and Dynamics
Saccharide matrices have been frequently employed as a tool to immobilize proteins for studies on protein conformational dynamics, specifically to slow down dynamical protein transitions [
153], usually choosing Mb and its derivative MbCO as the elective probe protein. Concentrated saccharide amorphous systems containing MbCO have been studied with various techniques, e.g., Mössbauer spectroscopy, elastic neutron scattering (ENS), Infrared spectroscopy (FTIR), and MD simulations [
15,
93,
96,
154,
155,
156,
157,
158,
159]. All the results concurred in pointing out a generalized reduction of protein dynamics, with the suppression of the commonly found protein dynamical transition (PDT, an increase in atomic mean square displacements amplitude at 200–220 K, [
160,
161]). The comparison with simulation data on trehalose solutions of lysozyme confirmed that solid, confining matrices are needed to suppress the high temperature large scale protein motions, while a sugar solution is almost ineffective [
162]. The analysis of solvent composition and HB patterns suggested that the sugar anchors a thin water layer at the protein surface, whose molecules bridge protein and matrix H-bond groups, reduce protein non-harmonic motions, and stabilize the protein conformation. Reducing the water content, a few direct protein–trehalose interactions start to emerge, indicating that the water replacement and water entrapment hypotheses are not mutually exclusive, but their occurrence depends only on external parameters, as hydration or sugar/protein ratio [
163]. MD simulations showed also that trehalose could form patches around the protein, reducing the protein backbone flexibility [
16,
164].
FTIR studies in a wide range of water contents and temperatures, at fixed sugar/protein ratio, in different sugars [
142,
154,
165,
166,
167] hint at a tight protein–matrix dynamical coupling, attributed to the presence of the protein-medium HB network evidenced by MD results (or anchorage hypothesis, [
137,
138,
154,
168,
169,
170]; see also
Section 3.3). Besides the hydration, which tunes the switch from a locally structured solid to a honeyed liquid, the structure of a saccharide matrix is sizably determined by the relative amounts of sugar and embedded protein.
FTIR measurements on various disaccharide matrices at different protein/sugar ratios, but at very low water content, indicate that there is a strong dependence of stability properties on the protein concentration [
23]. In these measurements, the stretching band of the bound CO is exploited as a marker for the protein structure; specific sub-components and their population are attributed to proteins in an unstressed, native conformation or to protein undergoing structural alterations. By adopting a common language in protein dynamics in reference to infrared measurements, we speak of slaving of one component on the other when the former is prevalent in shaping the equilibrium conformation properties of the latter. Conversely, we speak of coupling when the two components concur in shaping each other’s equilibrium states. Results pointed out that in the protein rich systems (in the case of Mb roughly when the mass of the protein is 1–2 times the mass of the sugar, depending on the sugar), the matrix is slaved to the protein, whose structure is however perturbed. In the sugar rich systems, when the mass of the protein is less than half the sugar one, the matrix is dominated by the sugar, which maintains the protein in an unstressed form. In the intermediate concentration range, the matrix is dominated by the protein, but enough sugar is now present to preserve the protein structure (protein/matrix coupling). This is the range at which the system’s properties are defined by both components. With respect to other saccharides, in trehalose, the system promptly switches between the two conditions, where one of the components dominates, by increasing or decreasing the sugar content. Above a certain sugar amount, the matrix becomes similar to a sugar-only matrix, to which the protein must adapt and that could contain also domains without (or with less) protein. Such types of domains have been already observed with small angle X-ray scattering (SAXS) measurements, in myoglobin/disaccharide samples at roughly equal mass ratios [
18,
19], as well as with electron paramagnetic resonance measurements in samples containing larger proteins [
169]. Furthermore, FTIR results showed how in sucrose, the protein is strongly perturbed at very high sugar content, likely due to sugar nanocrystallization, and that a similar, although weaker, destabilization occurs also for the other sugars tested. Protein alteration in sucrose matrices agrees with the results on both Mb [
96,
165] and larger proteins [
137,
169].
FTIR results point out the existence of an “optimal” sugar/protein ratio, which in most cases corresponds to the same mass of both components, where the surrounding matrix and embedded protein interact more strongly. At this ratio, the embedded protein appears to be stable at very low water content, although it is not necessarily unperturbed: a condition for almost unperturbed protein can be also identified, but at a higher sugar content, when a more hydrated matrix is more stable. The value of the optimal sugar/protein ratio bears also a strong dependence on protein size and preservation conditions (freeze-drying vs. full amorphization). Indeed, higher values have been reported in the case of a monoclonal antibody [
171] and of the reaction center of
Rhodobacter sphaeroides [
137,
169]. This could be expected since a complete coverage of the proteins is needed for preservation, and more sugar is needed for a larger protein; this is more evident if freeze-drying is employed instead of full amorphization, as in the former, the formation of some amount of crystals is easier. Indeed, in the case of the freeze dried antibody, a too high sugar content causes a reduction in protein stability [
171], a behavior similar to the one obtained in Mb.
Mb thermal stability in saccharide matrices has been investigated by differential scanning calorimetry (DSC) [
172], by using various disaccharides. A monotonic increase of the denaturation temperature (
Tden) upon dehydration has been observed in trehalose and sucrose, while in reducing sugars, regularity is lost at low hydrations, where the Maillard reaction might occur. Analogous studies with proteins of different sizes and charges in trehalose matrices indicated that at high to intermediate hydration, the presence of embedded proteins increases the
Tg of the encapsulating matrix, with a larger effect for a larger protein size, but no apparent influence of protein charge. This increase has been attributed to a “confining” effect by the large protein on the matrix. Overall, in these systems,
Tden increases, depending on both protein size and charge, up to about 60−70 °C at low hydrations [
173].
Mb thermal denaturation was also studied in matrices containing other preserving agents. DSC and UV-Vis spectrophotometry measurements were performed on samples of Mb embedded in amorphous gelatin and trehalose/gelatin matrixes or even only Mb matrices (self-crowding), at different hydrations, with the aim at disentangling the effect of saccharides and polypeptidic crowders on protein preservation. Furthermore, in these cases, two different regimes are observed, depending on water content. At high to intermediate hydration, samples appear inhomogeneous, with the coexistence of ice and a glassy fraction at low temperatures. A higher Tg is observed in the presence of a more proteinaceous matrix, but with a lower Tden: in the presence of enough water to allow Mb and gelatin mobility, the more hydrophobic nature of collagen surfaces would favor hydrophobic patching. Small saccharides as trehalose, at variance, can impair interaction among proteins, which would lead to denaturation, by interposition and preservation of protein native solvation. At low hydration, the systems are more homogeneous, with no formation of ice. Tg data follow the Gordon–Taylor curve, typical of homogeneous glasses, only in trehalose matrix, while proteinaceous systems exhibit an erratic behavior, likely due to component segregation, which prevents a true glassy behavior. In these conditions, gelatin matrices become more effective than trehalose in preservation, as showed by a very sharp rise of the Tden and an increase of ΔHden of Mb. Moreover, the effects of the two components appear to be roughly additive.
The self-crowding effects observed in Mb-only samples appear to be analogous to those in Mb-gelatin samples in both hydration regimes [
99]. These DSC results are confirmed by UV-visible spectroscopy measurements in thermally equilibrated samples at low hydration. Results allowed estimating the recovery fraction after thermal treatment at increasing heating times, which conveys information on the protein massive denaturation, immediately comparable with the results from DSC. According to both techniques, gelatin-only and gelatin–trehalose matrices perform similarly, both outdoing the trehalose-only matrix in the low hydration range. Moreover, spectroscopy also allows probing the ability of retaining bound CO molecules after thermal treatments, an event not necessarily implying the large conformational changes that lead to denaturation. The bound CO persistence can be taken as a measure of “functional preservation”, i.e., the preservation of the protein in native or native-like forms, which are still able to perform their original functions. In the low hydration range, CO retention is higher in trehalose than in gelatin, in particular at low heating times, when a large fraction of the probe protein is still in native-like form. This is consistent with their accepted working mechanisms: trehalose mainly interacts with the protein surface, thus impairing local structural deformation, while gelatin acts by crowding/confinement and is effective where large scale conformation fluctuations and/or volume variations occur. Conversely, at long heating times, when also massive denaturation is occurring, the mixed matrix appears to have the best performance, likely taking advantage of the combined effects of both components. From these results, it is possible to state that the polymeric and the saccharide components act differently on the different stages of the preservation process and to identify the best preserver, or the best mixture, for the degradation process to be contrasted [
99].
4. Outlook
Several methods are currently exploited to immobilize enzymes in compartmentalized matrices, with the aim of investigating catalysis in systems mimicking crowded and confining environments, which are known to improve the enzyme stability. The ultimate goal is to improve the enzymatic catalysis for applications that up to now have been the domain of metal based heterogeneous catalysis. Among these immobilization methods, absorption, covalent linking, enzyme cross-linking, or even “matrix controlled diffusion” techniques [
174] are all known to produce sizable effects on the enzymes, affecting their structure and altering, usually improving, their functional properties. In this review, we focused on enzyme immobilization by entrapment, by comparing two different families of encapsulating matrices, namely silica gels and saccharide matrices. The advantages and drawbacks of both methods were addressed, and several examples of biological catalysts were reported whose structure, dynamics, and function were fine tuned either by “soft confinement” as in saccharides matrices or by “hard confinement”, where an enzyme is trapped within the pores of a rigid matrix.
Results here presented show how these entrapment methods indeed provide activity retention and structure stability for a large variety of proteins, including both soluble and membrane enzymes. Compared with other immobilization protocols, silica and trehalose based systems are easily prepared at “gentle” experimental conditions, and their effects on enzymes can be controlled by varying hydration, co-solutes, and protein concentration. There is neither the requirement of chemical derivatization of the protein, which is not covalently bound to the support, nor exposition to the harsh conditions needed to co-precipitate aggregates, therefore promoting the preservation of enzyme structure and functions.
Silica gels or saccharide matrices, and in particular trehalose matrices, could help in formulating new, low cost, strategies for protein stability control, paving the way for further development of catalysis in biological applications with relevant implications in biotechnology, medicine, and pharmaceutics.

 ,
, 
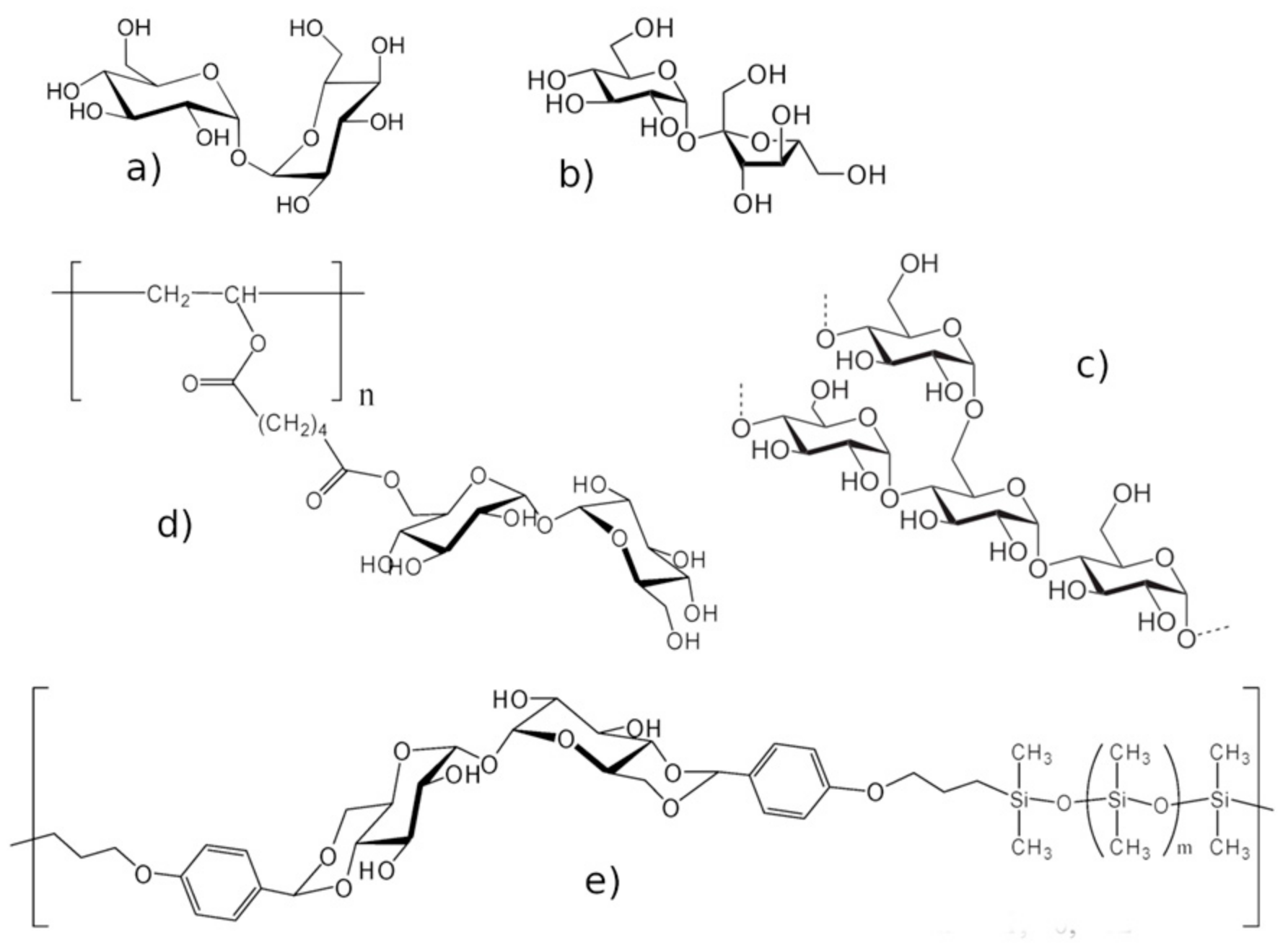{kind=link}
{kind=link}
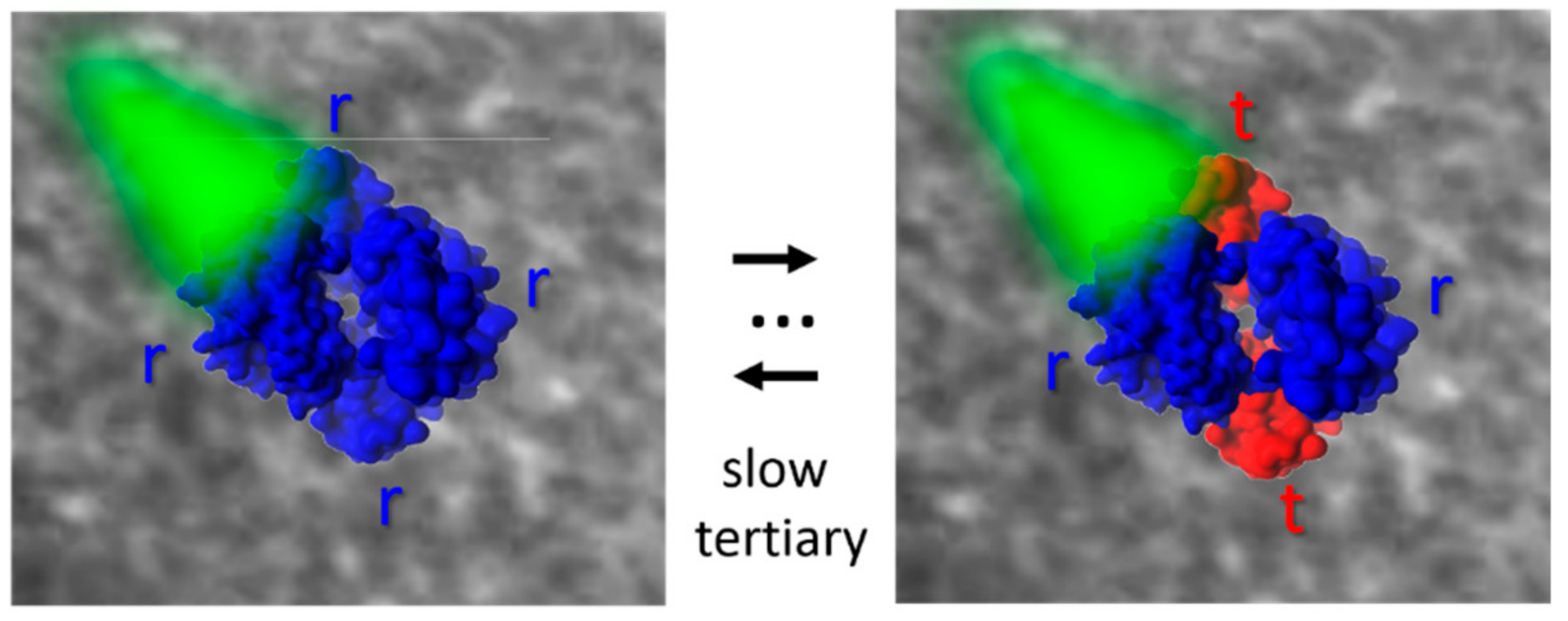{kind=link}
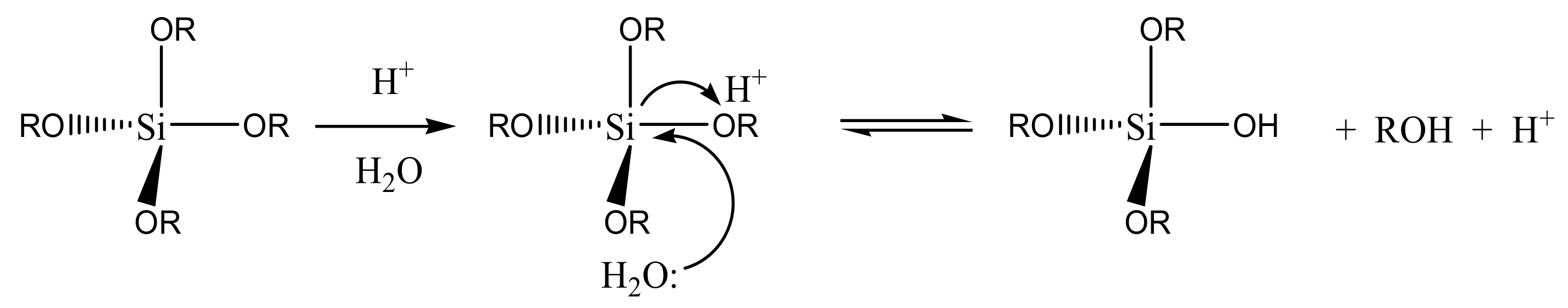{kind=link}
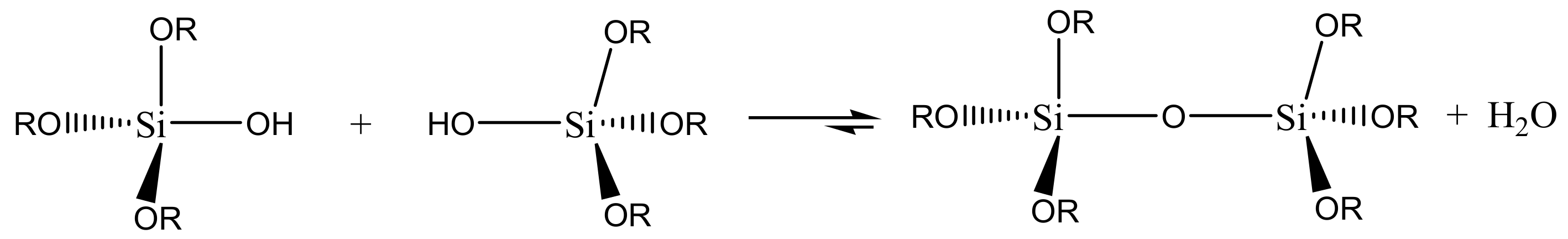{kind=link}
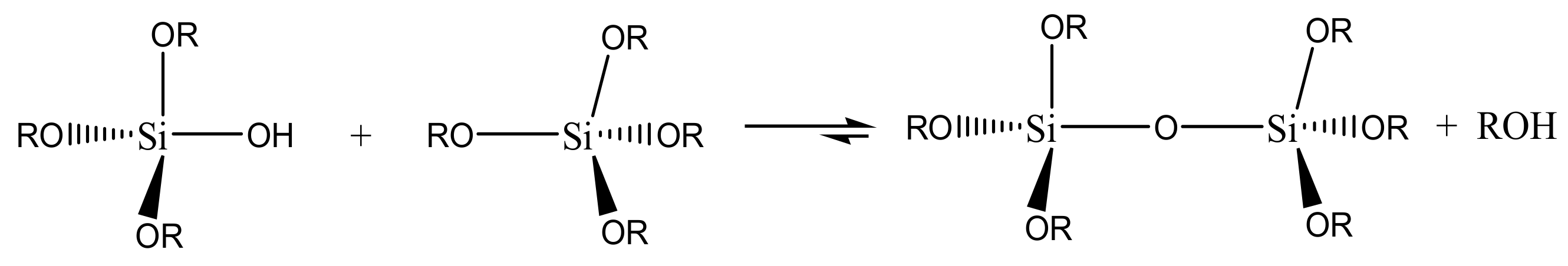{kind=link}