Strategies of Coping with Deactivation of NH3-SCR Catalysts Due to Biomass Firing


Center for Catalysis and Sustainable Chemistry, Department of Chemistry, Building 207, Technical University of Denmark, DK-2800 Kongens Lyngby, Denmark
*
Author to whom correspondence should be addressed.
Catalysts 2018, 8(4), 135; https://doi.org/10.3390/catal8040135
Submission received: 28 February 2018
/
Revised: 22 March 2018
/
Accepted: 22 March 2018
/
Published: 30 March 2018
(This article belongs to the Special Issue Selective Catalytic Reduction of NOx)
Abstract
:Firing of biomass can lead to rapid deactivation of the vanadia-based NH3-SCR catalyst, which reduces NOx to harmless N2. The deactivation is mostly due to the high potassium content in biomasses, which results in submicron aerosols containing mostly KCl and K2SO4. The main mode of deactivation is neutralization of the catalyst’s acid sites. Four ways of dealing with high potassium contents were identified: (1) potassium removal by adsorption, (2) tail-end placement of the SCR unit, (3) coating SCR monoliths with a protective layer, and (4) intrinsically potassium tolerant catalysts. Addition of alumino silicates, often in the form of coal fly ash, is an industrially proven method of removing K aerosols from flue gases. Tail-end placement of the SCR unit was also reported to result in acceptable catalyst stability; however, flue-gas reheating after the flue gas desulfurization is, at present, unavoidable due to the lack of sulfur and water tolerant low temperature catalysts. Coating the shaped catalysts with thin layers of, e.g., MgO or sepiolite reduces the K uptake by hindering the diffusion of K+ into the catalyst pore system. Intrinsically potassium tolerant catalysts typically contain a high number of acid sites. This can be achieved by, e.g., using zeolites as support, replacing WO3 with heteropoly acids, and by preparing highly loaded, high surface area, very active V2O5/TiO2 catalyst using a special sol-gel method.
{kind=link}
{kind=link}
{kind=link}
1. Introduction
The amount of electricity generated from firing solid biomass has been rising steeply in Europe over the last decades and is expected to continue to do so [1]. Similar trends are seen in other regions of the world [2,3]. Replacing fossil fuels, especially coal, by biomass aims at reducing the CO2 emissions associated with thermal power plants [2,4,5,6,7]. Even though renewable energy sources like solar and wind power are more and more cost competitive [8] and make up an increasing share of power generation in most regions [9], some thermal power plant capacity is still needed due to the renewables’ fluctuating nature and the current lack of sufficient storage capacity [10]. Firing and co-firing of biomass can cause several problems in the power plant like slagging and fouling problems in boilers [11], ash deposition on heat exchangers, and increased catalyst deactivation in the NOx removing unit [12,13,14,15,16,17,18]. This review deals with the last-mentioned problem.
NOx gases cause formation of photochemical smog, acid rain (HNO3), and ground level ozone formation. These conditions in turn have adverse consequences on human life and ecosystems. NOx emissions from power plants can be reduced by modifications to the combustion process (primary measures) or post-combustion techniques (secondary measures). Secondary measures are typically more expensive but also afford a higher degree of NOx removal. Due to ever stricter environmental regulations, secondary measures are increasingly needed for power plants to be compliant. The highest degree of NOx removal is achieved with selective catalytic reduction (SCR) using ammonia as the reductant [18,19]. The most widespread kind of catalyst is V2O5-WO3/TiO2 (VWT) [20,21]. The loading of the active species vanadia is typically between 1 and 5 wt.%, depending on the temperature of operation and the SO2 content in the flue gas. Tungsta adds acid sites, reduces SO2 oxidation, and reduces rutilization of anatase. The typical loading is between 5 and 10 wt.%.
The increased rate of catalyst deactivation experienced in biomass-fired plants is mostly caused by the relatively high alkali- and alkali-earth metal contents in most biomasses [11,17,20,21,22,23,24]. Alkaline metals cause deactivation by neutralizing the catalyst’s acid sites, hence reducing the adsorption of NH3 [13,25,26,27,28,29,30]. Potassium, in the form of submicron aerosols of mainly KCl and K2SO4 [31,32,33], is the most important poison due to both its relative abundance and high basicity [24,34]. Equation (1) gives a simplified neutralization reaction with M being any metal.
Other modes of deactivation like change in redox properties [35,36] and pore plugging [31] were reported to be of minor importance.
We have identified four kinds of strategies to deal with the high potassium content in biomasses: (1) potassium removal by adsorption; (2) tail-end placement of the SCR unit; (3) alkali barrier materials on the catalyst surface; and (4) intrinsically potassium resistant catalysts.
2. Strategies Coping with Potassium Rich Fuels
2.1. Potassium Removal by Adsorption
One way of reducing the impact of potassium salts is to minimize the amount taken up by the catalyst bed(s). An obvious strategy is to use an acidic guard bed in front of the catalyst modules. However, due to the high space velocities (5000–10,000 h−1) in SCR units and the high KCl content of about 0.2−1 g Nm−3 of the flue gas [37,38], such a guard bed would probably be saturated too rapidly and require substantial space. Assuming a KCl concentration of 0.2 g Nm−3 in the flue gas, a “guard bed space velocity” of 20,000 h−1, and a monolith density of 300 kg m−3, 1 h of exposure translates into about 180 μmol K per gram. Even highly acidic substances like H-type zeolites with low Si/Al ratios only possess around 5000 μmol of acid sites per gram [39]. To the best of our knowledge, no guard beds have been implemented so far.
Wang et al. [24] have published a critical review on additives mitigating ash related problems. They have grouped the additives by the following four capture mechanisms: (1) chemical absorption and reaction; (2) physical absorption; (3) dilution and inert elements enrichment and (4) restraining and powdering effects. The first mentioned mechanism was singled out to be the most effective and is based on converting troublesome ash elements into high temperature stable compounds. Additives causing chemical binding can be based on alumino silicates such as, e.g., kaolin, coal fly ash, cat litter, clay minerals, and detergent zeolites. Alumino silicates bind potassium according to the simplified Equation (2).
Addition of fly ash obtained from coal-fired plants is an industrially used strategy [40] to bind potassium. Coal fly-ash contains high levels of alumino silicates, which can bind potassium [14,40,41]. Coal fly-ash has the advantage of being abundant and low-cost. Diarmaid et al. [11] have very recently studied the efficacy of coal-fly ash in reducing the release of potassium from various biomass (white wood pellets, straw, and olive cake) pellets suspended in a methane flame. Additive loadings of 5, 15, and 25 wt.% were used. Olive cake requires larger amounts of alumino silicates to minimize potassium release, probably because it contains more potassium than the other two biomasses. In the presence of additive, up to 100% of K is retained, and in the wood and olive cake ash up to 80% is retained, demonstrating the effectiveness of alumino silicates even when burning pure biomasses with high potassium contents.
Firing coal with up to 10% [42,43] or even 20% [44] of biomass has also been reported to result in acceptable catalyst stability, probably because the resulting coal fly ash adsorbs released potassium compounds.
Sulfates of, e.g., ammonia, iron, aluminum, and phosphates of ammonia and calcium, as well as phosphoric acid, have also been listed by Wang et al. A possible issue with using sulfates is an increased formation of SO3. Injection of phosphorous-based “K-getter” compounds leads to the formation of, e.g., K3PO4 and K4P2O7. Dahlin et al. [27] performed a multivariate analysis of six catalyst poisons (Na, K, Mg, P, S, and Zn) by impregnating monolithic VWT catalysts with corresponding metal precursor solutions. The obtained model showed that P dampens the deactivating effect of K and was explained by the formation of phosphates, preventing the interaction of potassium with vanadia. The effect of K3PO4 on the stability of a vanadia-based catalyst was investigated by Castellino et al. [45] by exposing full length monoliths to a flue gas containing between 100 mg of K3PO4 per Nm3. 720 h of exposure caused almost 40% deactivation, which was mainly ascribed to potassium neutralizing the catalyst’s acid sites and thereby resembling the deactivation by KCl. The authors concluded that binding K by P is not advantageous to the SCR unit.
2.2. Tail-End Placement of the SCR Unit
Wieck-Hansen et al. [15] studied the catalyst stability using a slip stream from a 150 MW coal-straw (80%/20%) fired power plant. The catalyst was exposed to the flue gas at 350 °C without prior de-dusting, simulating high-dust placement, and at 280 °C downstream of a baghouse filter, which reduced the particulate concentration from 100 to a few mg Nm−3, simulating low-dust placement of the SCR unit. 2860 h of high-dust exposure caused about 35% activity loss, while 2350 h of low-dust exposure only caused 15% activity loss. The difference in stability can probably be explained by the removal of, e.g., KCl particles by the dust-filter. Tail-end placement would probably lead to an even higher stability because of the desulphurization unit further reducing the potassium content in the flue gas. Tail-end operation at the biomass co-fired Amager plant in Denmark indeed showed promising results between 2010 and 2012 [44]. Laboratory studies by Putluru et al. [46] have furthermore shown that heteropoly acid (instead of WO3)-promoted catalysts with a high (3 and 5 wt.%) vanadia loading can retain more than 90% of their activity at 225 °C when poisoned with 100 μmol K . A corresponding WO3 promoted catalyst lost almost 50% of its activity. At 400 °C, the loss was reported to be around 70% [47]. Generally, potassium poisoning has a stronger relative effect at high temperatures [23,48], which is reflected by a lower apparent activation energy upon potassium poisoning [23], which is consistent with acid neutralization being the main mode of deactivation.
Kristensen et al. [49] reported excellent potassium tolerance and activity of sol-gel prepared 20 wt.% V2O5/TiO2 at temperatures below 250 °C. The potassium loading introduced by KNO3 impregnation was 280 µmol K . A commercial reference catalyst got completely deactivated.
The major drawback with tail-end placement is that wet and dry SO2 scrubbers typically reduce the flue gas temperature to about 50 and 150 °C, respectively. The VWT catalyst is not active enough at these temperatures, making costly reheating to 180–280 °C necessary. Over the last 10 to 15 years, a high number of reports on low-temperature SCR catalysts have appeared [50]. The aim of these studies is to make re-heating redundant. However, most of the reported catalysts are based on manganese, making them extremely sulfur and water sensitive. In 2014, we summarized literature findings on the effects of SO2 and H2O and could not find any convincing reports on sulfur and water-resistant manganese-based catalysts [51]. Here we only give some examples of reports on catalysts being severely affected by SO2 and H2O. Casapu et al. [52] studied MnCeOx and reported a 79% activity reduction at 150 °C by adding 5 vol.% of water to the simulated flue gas. Flue gases typically contain at least 5 vol.% of water. Exposing the same catalyst to 50 ppm of SO2 for 30 min at 250 °C reduced the NO conversion from about 70 to 25%. Our group has experienced rapid and severe deactivation of MnFe/TiO2 and MnFeCe/TiO2 at 150 °C by SO2 levels as low as 5 ppm [51,53]. The modes of deactivation were formation of (NH4)2Mn2(SO4)3 and ammonium sulfates. Regeneration by heating to 400 °C was only effective with prior washing with base. 20 vol.% of water in the flue gas reduced the NO conversion over a MnFe/TiO2 from over 90% to 30.6%. Doping with ceria did not improve the water tolerance. In 2018, Gao et al. [54] reviewed the sulfur and water tolerance of Mn-based catalysts at low temperature and concluded, among other things, that more long term studies are needed to validate the viability of this kind of catalyst under realistic conditions.
2.3. Coating Monoliths with Basic Substances
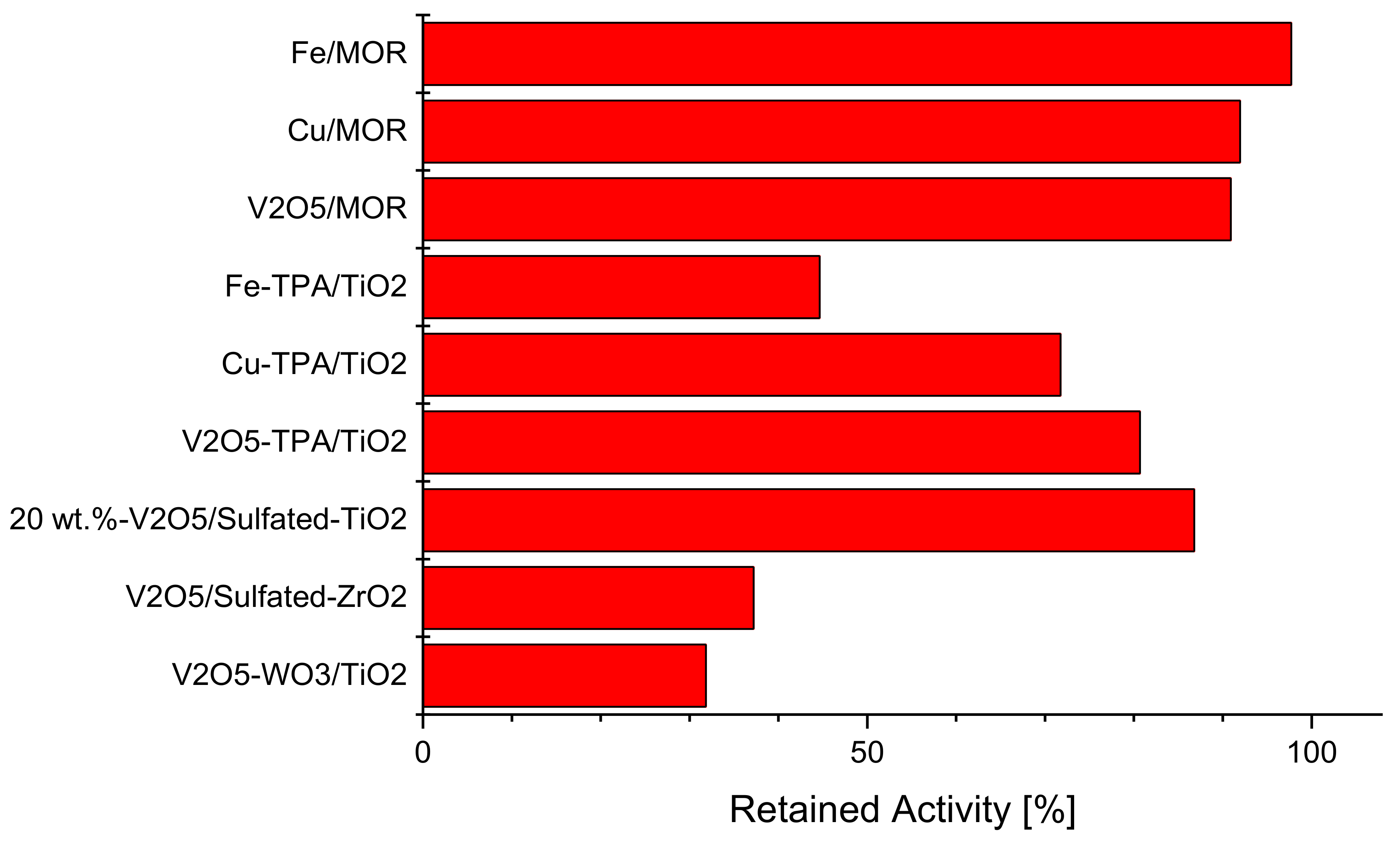
In order to reach the catalyst’s acid sites, potassium, typically originating from submicron aerosols of KCl and K2SO4, first needs to be deposited on the external catalyst (monolith) surface [48]. From there, potassium needs to separate from its counter-ion and diffuse into the catalyst pores, most likely through a surface transport mechanisms involving acid sites [31,55]. In other words, potassium mobility becomes a determining factor in the poisoning mechanism of monolithic samples. A pilot plant study performed by Jensen et al. [48] investigated the potassium uptake and the resulting deactivation of plate type samples with various WO3 (0, 7 wt.%) and V2O5 (1, 3, 6 wt.%) contents. According to ammonia chemisorptions measurements, both tungsta and vanadia add acid sites to the fresh samples, thereby favoring the potassium uptake. This, in turn, leads to an increased rate of deactivation, e.g., 600 h of KCl aerosol (0.12 μm) at 350 °C leads to 76, 81, 89, and 98% relative deactivation for 1%V2O5–0% WO3, 3%V2O5–0% WO3, 1%V2O5–7% WO3, and 3%V2O5–7% WO3, respectively. Based on these results, it is highly questionable if the commonly used strategy of simply increasing the number of surface acid sites is realistic under real life conditions. Despite the just quoted deactivation data, tungsta-free catalysts are not an option for biomass fired plants, because they start from a significantly lower base activity and probably suffer from rutilization over time.
Since the potassium uptake relies on acid sites on the outer monolith surface, it can be reduced by coating this surface with a basic material, thus reducing the relative rate of deactivation [23,56,57]. MgO and Sepiolite (Mg4Si6O15(OH)2·6H2O) have been reported as effective barrier materials. These substances are, on the one hand, basic enough to hinder potassium from penetrating the catalyst wall, and, on the other hand, they do not cause deactivation on their own. Olsen et al. [56] coated a plate type catalyst with composition of 3 wt.% V2O5–7 wt.% WO3/TiO2 with 8.06 wt.% MgO resulting in a roughly 200 μm thick layer and performed a pilot plant exposure campaign with KCl aerosols for several hundred hours at 350 °C. The coating layer reduced the rate of deactivation from 0.91% to 0.24% per day. These percentages refer to the initial activity of the uncoated sample. However, the decreased rate of deactivation comes at the cost of an initial activity reduction of about 42%. This activity reduction was ascribed to increased gas phase diffusion limitations introduced by the MgO layer, slight poisoning by MgO on the outer layer of the catalyst, or a combination thereof. SEM-EDS measurements confirmed that the outer MgO layer very effectively prevented potassium from diffusing into the catalyst and that magnesium did not diffuse into the catalyst. Kristensen [23] very successfully used sepiolite as a binder material for making plate type catalysts from 20 wt.% V2O5/TiO2 powder, reinforced silica sheets, and 20 wt.% sepiolite as binder. The resulting catalyst was exposed to a KCl aerosol for 632 h at 380 °C and thereafter crushed to a powder for lab scale activity measurements. A commercial of 3 wt.% V2O5–7 wt.% WO3/TiO2 plate type catalyst was used as reference. When tested at 400 °C after KCl exposure, the 20 wt.% V2O5/TiO2-Sepiolite composite retained 68% of its activity, translating into a first order rate constant of about 1650 cm3·g−1·s−1. The activity loss of the reference catalyst was 84%, and the resulting first order rate constant was reported to be only about 200 cm3·g−1·s−1. These activity losses were compared with data from a corresponding incipient wetness (KNO3) poisoning study. The losses experienced by the 20 wt.% V2O5/TiO2-Sepiolite composite and the reference translate into impregnated K loadings of 75 and 172 μmol K , strongly suggesting that sepiolite acts as a barrier material. This was confirmed by SEM-EDS measurements, showing that potassium mainly accumulated on the outer surface of the plate.
2.4. Intrinsically Potassium Resistant Catalysts
In this review “intrinsically potassium resistant” refers to catalysts that retain a high share of their activity, even when potassium is taken up from the flue gas and diffuses into the catalysts pore system. To the best of our knowledge, there is up to now no review on potassium tolerant catalysts.
The majority of studies mimic potassium poisoning by impregnation with potassium salts like e.g., KNO3, K2CO3, and KCl followed by calcination. The resulting K-loaded catalysts are typically tested in powder form in lab scale reactors. Studies performed by different laboratories are often difficult to compare due to vastly different experimental conditions and benchmark catalysts. For example, using different potassium loadings and activity testing in different temperature regimes might lead to different conclusions. Benchmarking against catalyst of different potassium tolerance might also lead to different conclusions. Because of these shortcomings in comparability, we start this section with results from our laboratory, which tested a high number of alternative catalysts using identical or very similar experimental conditions.
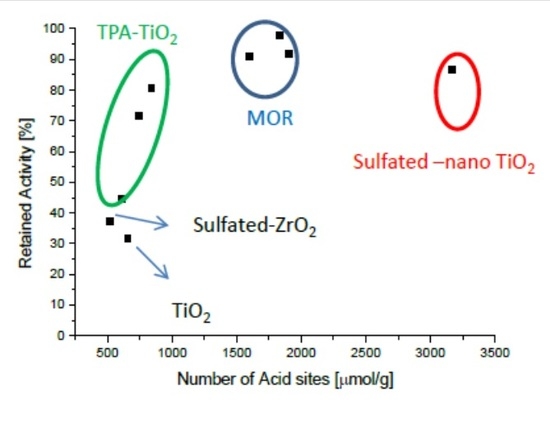
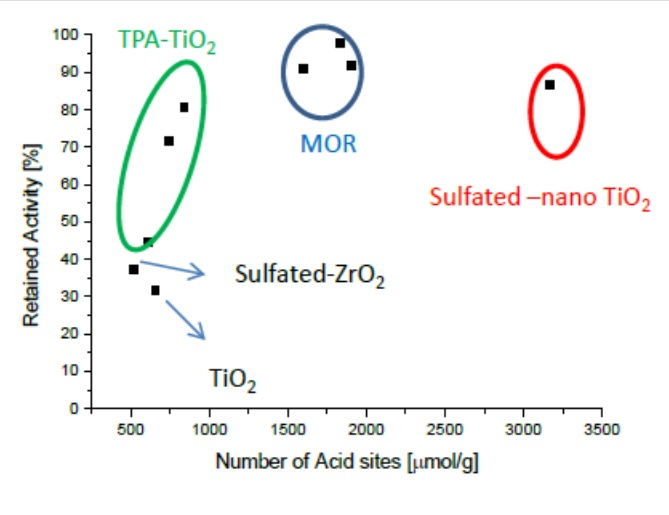
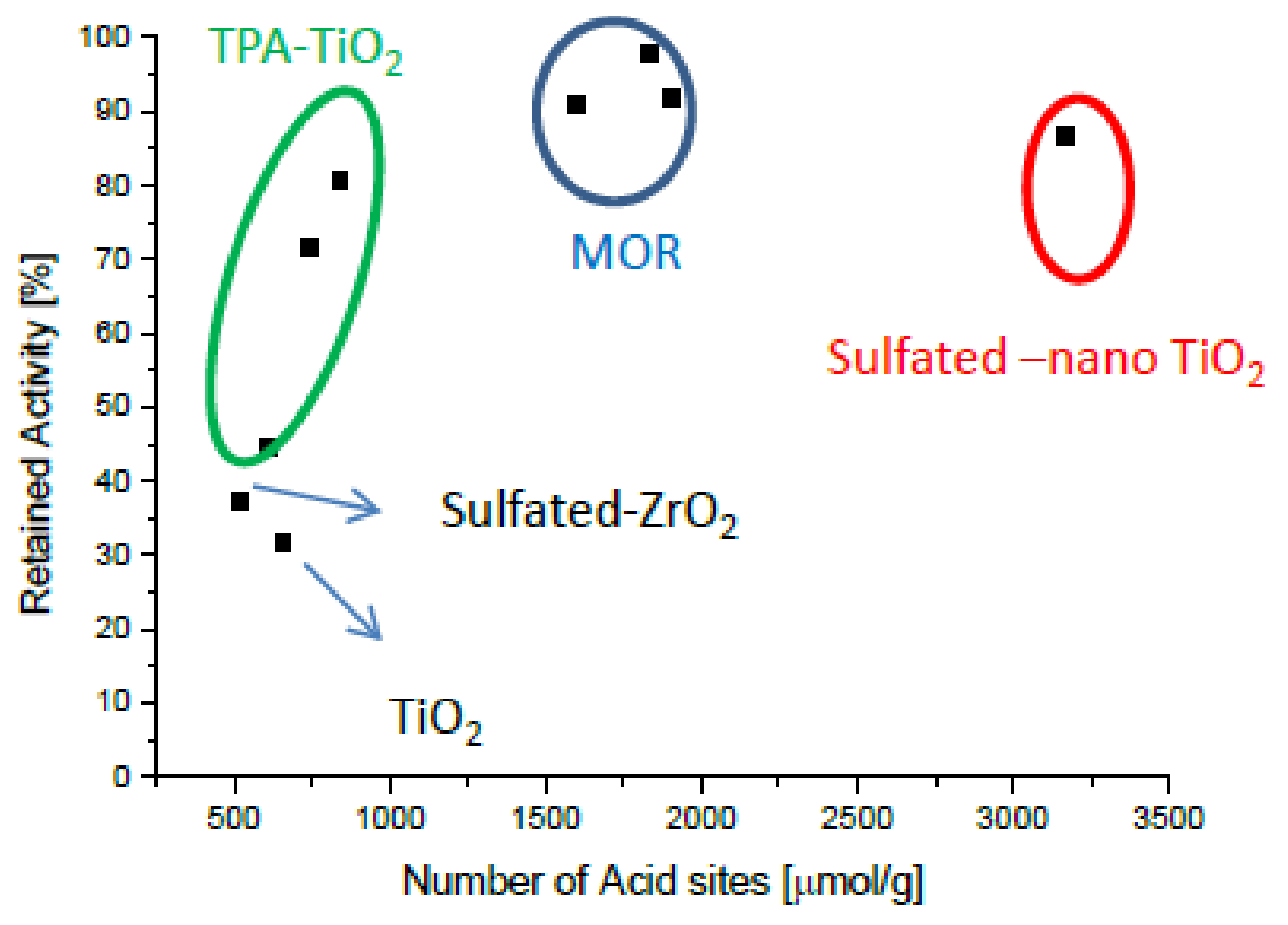
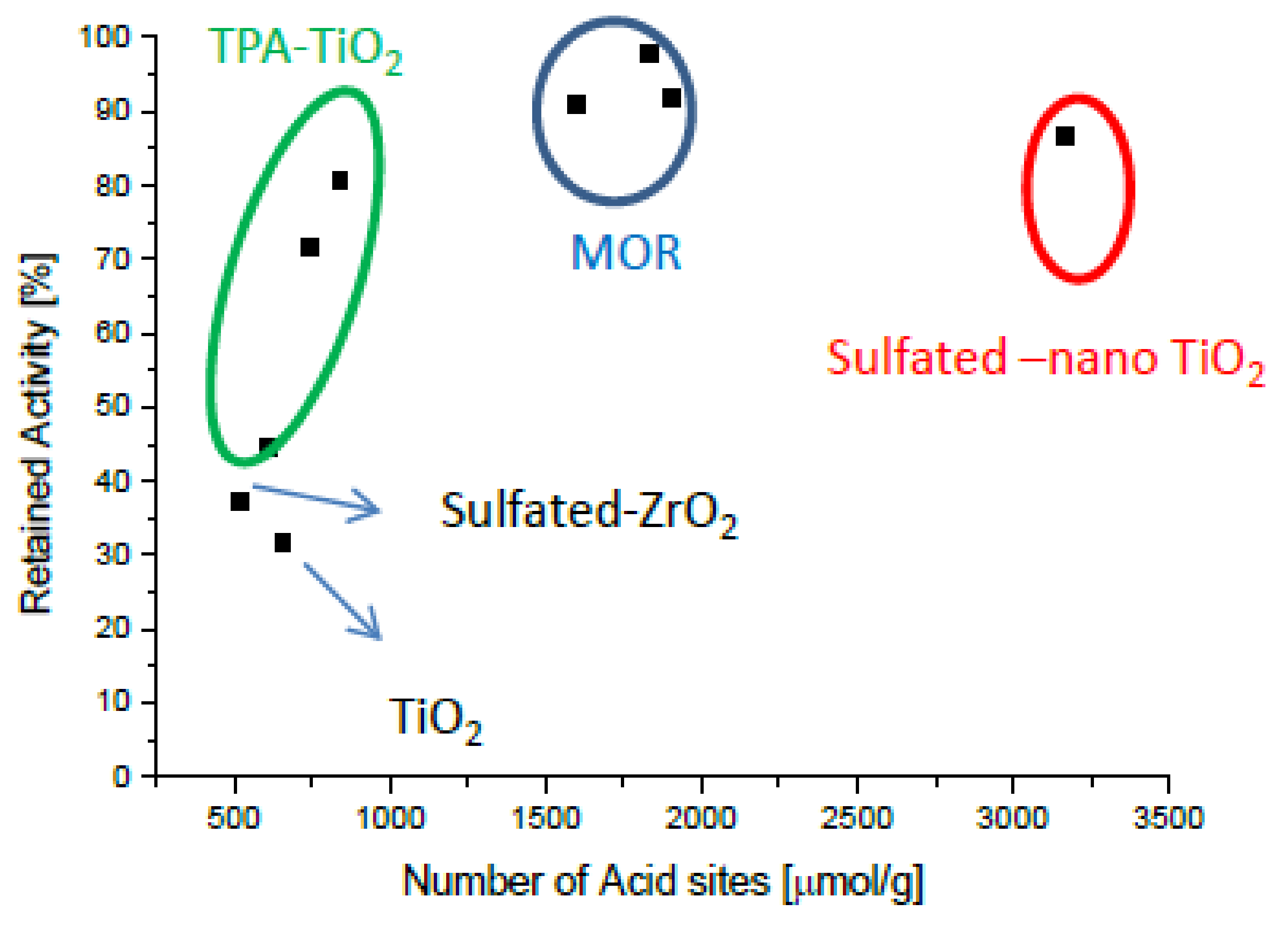
Figure 1 and Figure 2 present the potassium tolerance for an assortment of catalyst with various active metals (Fe, Cu, and V) and support materials (TiO2, tungsto phosphoric acid (TPA) promoted TiO2, mordernite (MOR), and sulfated ZrO2). The retained activity clearly depends on the number of acid sites of the fresh catalysts, which in turn is very much a function of the support material.
In this study, the highest alkali tolerance was obtained with MOR (Si/Al = 10)-based catalysts. Putluru et al. [58] optimized the Cu loading and tested the effect of 0, 250, and 500 µmol K/gcatalyst. 4 wt.% Cu/MOR retains about 60% of its initial activity after poisoning with 500 µmol K , while only half that potassium loading causes more than 80% deactivation on a reference catalyst containing 3 wt.% vanadia and 7 wt.% tungsta. Cu/BEA (Si/Al = 25) and Cu/ZSM5 (Si/Al = 15) exhibit only slightly lower potassium tolerance than Cu/MOR does. Cu/Zeolite catalysts are not only very potassium resistant but also very active at 400 °C with first order rate constants of up to 1800 cm3g−1s−1, while this value is only about 1000 cm3g−1s−1 for the VWT reference catalyst [49]. Since the high potassium tolerance is at least in part due to the high number of acid sites on the zeolites, these materials will probably have to be protected by a thin layer of, e.g., MgO in order to avoid increased uptake of potassium containing particles. Another issue with Cu-based catalyst is their sulfur intolerance [59,60]. Vanadia supported on zeolites are very potassium tolerant but suffer from relative low activities. Likewise, iron-zeolite catalysts show comparatively low activities below 400 °C.
Putluru et al. [61] also demonstrated that the WO3 component of the VWT catalyst can be replaced by heteropoly acids such as H3PW12O40, H4SiW12O40, H3PMo12O40, and H4SiMo12O40. Heteropoly acids contain more acid sites than WO3, and these can probably serve as sacrificial sites, which is reflected by a higher potassium tolerance. Tungsto phosphoric acid (TPA, H3PW12O40) resulted in the highest activity and the highest number of acid sites and is thermally more stable than the other heteropoly acids. Note that preparation of HPA-promoted catalyst is entirely based on impregnation and could therefore relatively easily be upscaled. A corresponding study on HPA-promoted Cu/TiO2 and Fe/TiO2 delivered similar results regarding activity and potassium tolerance [62]. The best HPAs were reported to be H3PW12O40 and H3PMo12O40. Another study by Putluru et al. [46] showed the effect of vanadia loading (3–6 wt.%) on the activity, and potassium tolerance of HPA promoted V2O5/TiO2 catalysts at temperatures below 300 °C. The optimum vandia loading was 5 wt.%, and the resulting catalysts were almost unaffected by 100 µmol K when tested at 225 °C.
The most active and potassium-tolerant catalyst published by our laboratory is a 20 wt.% V2O5/TiO2 prepared by a sol-gel route [23,47,49]. This catalyst contains about 5 times as many acid sites as the VWT reference and is at least twice as active. The conversion of SO2 to SO3 at 380 and 420 °C was reported to be less pronounced than over the VWT reference. This is probably due to the amorphous nature of vanadia, which is a result of the special sol-gel method of preparation. Impregnation with 500 µmol K resulted in the catalyst being about as active as the VWT reference loaded with only 150 µmol K . Pilot scale exposure to KCl aerosols has demonstrated that a 20 wt.% V2O5/TiO2—sepiolite composite catalyst suffers relatively little deactivation under more realistic conditions because of sepiolite impeding the surface diffusion of potassium.
Other research groups have also made many contributions over the last 10 years. Peng et al. [63] reported on the effect of doping V2O5-WO3/TiO2 with Ce. V0.4Ce5W5/Ti and V0.4W10/Ti loaded with 1% K convert 30 and 18% NO, respectively, when tested at 400 °C. Du et al. [64] investigated the effect of Sb and Nb additives to V2O5/TiO2. Both Sb and Nb have promotional effects on their own and can act synergistically. At 300 °C, potassium loaded VTi and VSb0.5NbTi show NO conversions of 22 and 43%, respectively. Gao et al. [65] reported on CeV mixed oxides supported on sulfated zirconia showing resistance to both SO2 and potassium. The formation of CeVO4 hinders the formation of Ce2(SO4)2, and vanadia suppresses the absorption of SO2, thus inhibiting NH4HSO4 formation. The potassium-loaded CeV mixed oxide catalyst maintains more than 95% NO conversion over 400 min of exposure to 600 ppm SO2, while the conversion over the V free catalyst drops to about 65%.
To the best of our knowledge, very few reports exist on the potassium tolerance of hydrocarbon-SCR. Ethanol-SCR using Ag/Al2O3 is comparable in activity to NH3-SCR over a 3 wt.% V2O5–7wt.% WO3/TiO2, however, is almost equally affected by potassium [66]. The mechanism of poisoning is not well understood but involves oxidation of ethanol to CO2. Another problem with using ethanol instead of NH3 as reductant is its much higher price. Furthermore, Ag/Al2O3 suffers from poor sulfur tolerance.
3. Conclusions
Different strategies of dealing with high concentrations of potassium in flue gases, typically present in biomass fired plants, were discussed. Addition of coal fly ash or other substances rich in alumino silicates like, e.g., kaolin is already an industrial practice and can very effectively bind potassium-containing aerosols. Lab scale experiments have demonstrated that this approach can be applied to various biomasses. The drawback of these additives is an increased concentration of particulates that need to be filtered off the flue gas. Tail-end placement of the SCR unit has also been demonstrated to work industrially. The major disadvantage of the tail-end placement, the expensive flue gas reheating to at least 180 °C, can, at present, not be avoided due to lack of catalysts that are sufficiently active, as well as due to sulfur and water tolerant at the outlet temperature of the desulfurization unit. Coating of shaped (monolith, plates) catalysts with thin layers of MgO or sepiolite was demonstrated to strongly reduce the rate of deactivation in pilot plant studies. The mildly basic nature of the protective layer impedes the diffusion of potassium ions into catalyst pores. Some of the studies report that the protective layer reduces the base activity by almost 50%, whereas others report a much lower penalty. Also, catalysts designed to tolerate higher loadings of potassium have been developed on a lab scale and include V, Cu, and Fe as active metals and heteropoly acid-promoted TiO2, sulfated ZrO2, and zeolites with a low Si/Al ratio as support materials. Most of the alternative catalysts gain their increased potassium tolerance from the addition of sacrificial acid sites. Since an increased number of acid sites was demonstrated to increase the potassium uptake from the flue gas, the addition of sacrificial sites probably only makes sense in conjunction with a protective layer of, e.g., MgO. The most promising results in this regard were obtained with a sol-gel prepared 20 wt.% V2O5/TiO2 in combination with sepiolite. This composite material is about twice as active as the commercial, takes up less potassium from the flue gas, and experiences less deactivation per amount of adsorbed potassium. Avoiding the issue of reduced ammonia adsorption due to potassium uptake by using hydrocarbons as reductants has so far not been promising. We believe that mitigating the effect of potassium in biomass-fired units requires a multidimensional approach. For example, researchers should, if possible, demonstrate, using pilot plant studies, that promising catalyst formulations are also combinable with effective barrier materials that can minimize potassium uptake. Cost benefit analyses should also compare the use of alumino silicate addition with the use of potentially more expensive catalysts and tail-end placement of the SCR unit.
Acknowledgments
Energinet.dk, Denmark, LAB S.A., Lyon, France and DONG Energy, Denmark are acknowledged for financial contribution to the ForskEL project 12096 “Low temperature deNOx technologies in waste incineration and power plants”.
Author Contributions
Leonhard Schill carried out the experimental work, design of figures and drafted the manuscript with further contribution from Rasmus Fehrmann who also managed the project.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bjerg, J.; Aden, R.; Ogand, J.A.; Arrieta, J.A.; Hahlbrock, A.; Holmquist, L.; Kellberg, C.; KIp, W.N.; Koch, J.; Langnickel, U.; et al. Biomass 2020: Opportunities, Challenges and Solutions. Eurelectric 2001, 72, 18. [Google Scholar]
- Agbor, E.; Zhang, X.; Kumar, A. A review of biomass co-firing in North America. Renew. Sustain. Energy Rev. 2014, 40, 930–943. [Google Scholar] [CrossRef]
- Livingston, W.R. The Status of Large Scale Biomass Firing: The Milling and Combustion of Biomass Materials in Large Pulverised Coal Boilers; IEA Bioenergy Report 2016; IEA Bioenergy: Paris, France, 2006; ISBN 9781910154267. Available online: http://www.ieabcc.nl/publications/IEA_Bioenergy_T32_cofiring_2016.pdf (accessed on 27 February 2018).
- Khorshidi, Z.; Ho, M.T.; Wiley, D.E. Techno-economic study of biomass co-firing with and without CO2 capture in an Australian black coal-fired power plant. Energy Procedia 2013, 37, 6035–6042. [Google Scholar] [CrossRef]
- Nuamah, A.; Malmgren, A.; Riley, G.; Lester, E. Biomass co-firing. Compr. Renew. Energy 2012, 5, 55–73. [Google Scholar] [CrossRef]
- Mann, M.; Spath, P. A life cycle assessment of biomass cofiring in a coal-fired power plant. Clean Prod. Process. 2001, 3, 81–91. [Google Scholar] [CrossRef]
- Kadiyala, A.; Kommalapati, R.; Huque, Z. Evaluation of the Life Cycle Greenhouse Gas Emissions from Different Biomass Feedstock Electricity Generation Systems. Sustainability 2016, 8, 1181. [Google Scholar] [CrossRef]
- Renewable Energy Agency. Renewable Power Generation Costs in 2017; International Renewable Energy Agency: Abu Dhabi, UAE, 2018. [Google Scholar]
- British Petroleum. BP Statistical Review of World Energy 2017; British Petroleum: London, UK, 2017; pp. 1–52. Available online: http://www.bp.com/content/dam/bp/en/corporate/pdf/energy-economics/statistical-review-2017/bp-statistical-review-of-world-energy-2017-full-report.pdf (accessed on 26 February 2018).
- Lüdge, S. The value of flexibility for fossil-fired power plants under the conditions of the Strommark 2.0. VGB PowerTech J. 2017, 3, 212–214. [Google Scholar]
- Clery, D.S.; Mason, P.E.; Rayner, C.M.; Jones, J.M. The effects of an additive on the release of potassium in biomass combustion. Fuel 2018, 214, 647–655. [Google Scholar] [CrossRef]
- Klimczak, M.; Kern, P.; Heinzelmann, T.; Lucas, M.; Claus, P. High-throughput study of the effects of inorganic additives and poisons on NH3-SCR catalysts-Part I: V2O5-WO3/TiO2 catalysts. Appl. Catal. B Environ. 2010, 95, 39–47. [Google Scholar] [CrossRef]
- Kern, P.; Klimczak, M.; Heinzelmann, T.; Lucas, M.; Claus, P. High-throughput study of the effects of inorganic additives and poisons on NH3-SCR catalysts. Part II: Fe-zeolite catalysts. Appl. Catal. B Environ. 2010, 95, 48–56. [Google Scholar] [CrossRef]
- Jensen-holm, H.; Lindenhoff, P.; Safronov, S. SCR Design Issues in Reduction of NOx Emissions from Thermal Power Plants; Haldor Topsøe A/S: Kongens Lyngby, Denmark, 2007. [Google Scholar]
- Wieck-Hansen, K.; Overgaard, P.; Larsen, O.H. Cofiring coal and straw in a 150 MWe power boiler experiences. Biomass Bioenergy 2000, 19, 395–409. [Google Scholar] [CrossRef]
- Baxter, L. Biomass Impacts on SCR Catalyst Performance; IEA Task 32 Bioenergy Report; IEA Bioenergy: Paris, France, 2005; Available online: http://task32.ieabioenergy.com/wp-content/uploads/2017/03/Combined_Final_Report_SCR.pdf (accessed on 25 February 2018).
- Argyle, M.; Bartholomew, C. Heterogeneous Catalyst Deactivation and Regeneration: A Review. Catalysts 2015, 5, 145–269. [Google Scholar] [CrossRef]
- Kiełtyka, M.A. Influence of Biomass Co-Firing on SCR Catalyst Deactivation 2; Techniques of NOx Emission Reduction; Instituto Superior Técnico, Universidade de Lisboa: Lisbon, Portugal, 2010. [Google Scholar]
- Mladenović, M.; Paprika, M.; Marinković, A. Denitrification techniques for biomass combustion. Renew. Sustain. Energy Rev. 2018, 82, 3350–3364. [Google Scholar] [CrossRef]
- Forzatti, P. Present status and perspectives in de-NOx SCR catalysis. Appl. Catal. A Gen. 2003, 222, 221–236. [Google Scholar] [CrossRef]
- Busca, G.; Lietti, L.; Ramis, G.; Berti, F. Chemical and mechanistic aspects of the selective catalytic reduction of NOx by ammonia over oxide catalysts: A review. Appl. Catal. B Environ. 1998, 18. [Google Scholar] [CrossRef]
- Knudsen, J.N.; Jensen, P.A.; Dam-Johansen, K. Transformation and release to the gas phase of Cl, K, and S during combustion of annual biomass. Energy Fuels 2004, 18, 1385–1399. [Google Scholar] [CrossRef]
- Kristensen, S.B. deNOx Catalysts for Biomass Combustion. Ph.D. Thesis, Technical University of Denmark, Kongens Lyngby, Denmark, April 2013. [Google Scholar]
- Wang, L.; Hustad, J.E.; Skreiberg, Ø.; Skjevrak, G.; Grønli, M. A critical review on additives to reduce ash related operation problems in biomass combustion applications. Energy Procedia 2012, 20, 20–29. [Google Scholar] [CrossRef]
- Shao, Y.; Wang, J.; Preto, F.; Zhu, J.; Xu, C. Ash deposition in biomass combustion or co-firing for power/heat generation. Energies 2012, 5, 5171–5189. [Google Scholar] [CrossRef]
- Davidsson, K.O.; Steenari, B.M.; Eskilsson, D. Kaolin addition during biomass combustion in a 35 MW circulating fluidized-bed boiler. Energy Fuels 2007, 21, 1959–1966. [Google Scholar] [CrossRef]
- Dahlin, S.; Nilsson, M.; Bäckström, D.; Bergman, S.L.; Bengtsson, E.; Bernasek, S.L.; Pettersson, L.J. Multivariate analysis of the effect of biodiesel-derived contaminants on V2O5-WO3/TiO2SCR catalysts. Appl. Catal. B Environ. 2016, 183, 377–385. [Google Scholar] [CrossRef]
- Khodayari, R.; Odenbrand, C.U.I. Regeneration of commercial SCR catalysts by washing and sulphation: Effect of sulphate groups on the activity. Appl. Catal. B Environ. 2001, 33, 277–291. [Google Scholar] [CrossRef]
- Forzatti, P.; Nova, I.; Tronconi, E.; Kustov, A.; Thøgersen, J.R. Effect of operating variables on the enhanced SCR reaction over a commercial V2O5-WO3/TiO2 catalyst for stationary applications. Catal. Today 2012, 184, 153–159. [Google Scholar] [CrossRef]
- Lisi, L.; Lasorella, G.; Malloggi, S.; Russo, G. Single and combined deactivating effect of alkali metals and HCl on commercial SCR catalysts. Appl. Catal. B Environ. 2004, 50, 251–258. [Google Scholar] [CrossRef]
- Zheng, Y.; Jensen, A.D.; Johnsson, J.E.; Thøgersen, J.R. Deactivation of V2O5-WO3-TiO2SCR catalyst at biomass fired power plants: Elucidation of mechanisms by lab- and pilot-scale experiments. Appl. Catal. B Environ. 2008, 83, 186–194. [Google Scholar] [CrossRef]
- Zeuthen, J.H.; Jensen, P.A.; Jensen, J.P.; Livbjerg, H. Aerosol Formation during the Combustion of Straw with Addition of Sorbents. Energy Fuels 2007, 860–870. [Google Scholar] [CrossRef]
- Sippula, O.; Lind, T.; Jokiniemi, J. Effects of chlorine and sulphur on particle formation in wood combustion performed in a laboratory scale reactor. Fuel 2008, 87, 2425–2436. [Google Scholar] [CrossRef]
- Chen, L.; Li, J.; Ge, M. The poisoning effect of alkali metals doping over nano V2O5-WO3/TiO2catalysts on selective catalytic reduction of NOx by NH3. Chem. Eng. J. 2011, 170, 531–537. [Google Scholar] [CrossRef]
- Wu, X.; Yu, W.; Si, Z.; Weng, D. Chemical deactivation of V2O5-WO3/TiO2 SCR catalyst by combined effect of potassium and chloride. Front. Environ. Sci. Eng. 2013, 7, 420–427. [Google Scholar] [CrossRef]
- Peng, Y.; Li, J.; Huang, X.; Li, X.; Su, W.; Sun, X.; Wang, D.; Hao, J. Deactivation mechanism of potassium on the V2O 5/CeO2 catalysts for SCR reaction: Acidity, reducibility and adsorbed-NOx. Environ. Sci. Technol. 2014, 48, 4515–4520. [Google Scholar] [CrossRef] [PubMed]
- Putluru, S.S.R.; Jensen, A.D. Alternative Alkali Resistant deNOx Technologies; Appendix-I: PSO Project 7318; Department of Chemical and Biochemical Engineering, Technical University of Denmark: Kongens Lyngby, Denmark, 2011; Available online: http://orbit.dtu.dk/files/6454472/NEI-DK-5569.pdf (accessed on 25 February 2018).
- Sorvajävi, T.; Maunula, J.; Silvennoinen, J.; Toivone, J. Optical Monitoring of KCl Vapor in 4 MW CFB Boiler during Straw Comubstion and Ferric Sulfate Injection; Optics Laboratory, Department of Physics, Tampere University of Technology: Tampere, Finnland, 2014. [Google Scholar]
- Niwa, M.; Katada, N.; Okumura, K. Characterization and Design of Zeolite Catalysts (Solid Acidity, Shape Selectivity and Loading Properties); Springer: Berlin/Heidelberg, Germany, 2010; p. 25. ISBN 9783642126192. [Google Scholar]
- Wu, H.; Shafique, M.; Arendt, P.; Sander, B.; Glarborg, P. Impact of coal fly ash addition on ash transformation and deposition in a full-scale wood suspension-firing boiler. Fuel 2013, 113, 632–643. [Google Scholar] [CrossRef]
- Steenari, B.M.; Lindqvist, O. High-temperature reactions of straw ash and the anti-sintering additives kaolin and dolomite. Biomass Bioenergy 1998, 14, 67–76. [Google Scholar] [CrossRef]
- Overgaard, P.; Sander, B.; Junker, H.; Friborg, K.; Larsen, O.H. Small-scale CHP Plant based on a 75 kWel Hermetic Eight Cylinder Stirling Engine for Biomass Fuels—Development, Technology and Operating Experiences. In Proceedings of the 2nd World Conference on Biomass for Energy, Industry and Climate Protection, Rome, Italy, 10–14 May 2004; pp. 1261–1264. [Google Scholar]
- Henderson, C. Cofiring of biomass in coal-fired power plants—European experience The role of biomass in Europe. Presented at the FCO/IEA CCC Workshops on Policy and Investment Frameworks to Introduce CCT in Hebei and Shandong Provinces, China, 8–9 and 13–14 January 2015. [Google Scholar]
- Jensen-holm, H.; Castellino, F.; White, T.N. SCR DeNOx catalyst considerations when using biomass in power generation. In Proceedings of the Power Plant Air Pollutant Control “MEGA” Symposium, Baltimore, MD, USA, 20–23 August 2012; Available online: https://www.topsoe.com/sites/default/files/scr_denox_catalyst_considerations_when_using_biomass_in_power_generation_2012.ashx__0.pdf (accessed on 23 February 2018).
- Castellino, F.; Jensen, A.D.; Johnsson, J.E.; Fehrmann, R. Influence of reaction products of K-getter fuel additives on commercial vanadia-based SCR catalysts. Part I. Potassium phosphate. Appl. Catal. B Environ. 2009, 86, 196–205. [Google Scholar] [CrossRef]
- Putluru, S.S.R.; Schill, L.; Godiksen, A.; Poreddy, R.; Mossin, S.; Jensen, A.D.; Fehrmann, R. Promoted V2O5/TiO2 catalysts for selective catalytic reduction of NO with NH3 at low temperatures. Appl. Catal. B Environ. 2016, 183, 282–290. [Google Scholar] [CrossRef] [Green Version]
- Putluru, S.S.R.; Kristensen, S.B.; Due-Hansen, J.; Riisager, A.; Fehrmann, R. Alternative alkali resistant deNOx catalysts. Catal. Today 2012, 184, 192–196. [Google Scholar] [CrossRef]
- Olsen, B.K.; Kügler, F.; Castellino, F.; Jensen, A.D. Poisoning of vanadia based SCR catalysts by potassium: Influence of catalyst composition and potassium mobility. Catal. Sci. Technol. 2016, 6, 2249–2260. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, S.B.; Kunov-Kruse, A.J.; Riisager, A.; Rasmussen, S.B.; Fehrmann, R. High performance vanadia-anatase nanoparticle catalysts for the Selective Catalytic Reduction of NO by ammonia. J. Catal. 2011, 284, 60–67. [Google Scholar] [CrossRef]
- Fu, M.; Li, C.; Lu, P.; Qu, L.; Zhang, M.; Zhou, Y.; Yu, M.; Fang, Y. A review on selective catalytic reduction of NOx by supported catalysts at 100–300 °C—Catalysts, mechanism, kinetics. Catal. Sci. Technol. 2014, 4, 14–25. [Google Scholar] [CrossRef]
- Schill, L. Alternative Catalysts and Technologies for NOx Removal from Biomass- and Waste-Fired Plants. Ph.D. Thesis, Technical University of Denmark, Kongens Lyngby, Denmark, April 2014. [Google Scholar]
- Casapu, M.; Kröcher, O.; Elsener, M. Screening of doped MnOx-CeO2 catalysts for low-temperature NO-SCR. Appl. Catal. B Environ. 2009, 88, 413–419. [Google Scholar] [CrossRef]
- Fehrmann, R.; Jensen, A.D. Low Temperature deNOx Technologies for Biomass and Waste Fired Power Plants; Forskel Energinet.dk Project No. 12096; Department of Chemistry, Technical University of Denmark: Kongens Lyngby, Denmark, 2017; Available online: https://energiforskning.dk/sites/energiteknologi.dk/files/slutrapporter/12096_slutrapport.pdf (accessed on 20 February 2018).
- Gao, C.; Shi, J.-W.; Fan, Z.; Gao, G.; Niu, C. Sulfur and Water Resistance of Mn-Based Catalysts for Low-Temperature Selective Catalytic Reduction of NOx: A Review. Catalysts 2018, 8, 11. [Google Scholar] [CrossRef]
- Zheng, Y.; Jensen, A.D.; Johnsson, J.E. Deactivation of V2O5-WO3-TiO2SCR catalyst at a biomass-fired combined heat and power plant. Appl. Catal. B Environ. 2005, 60, 253–264. [Google Scholar] [CrossRef]
- Olsen, B.J.; Kügler, F.; Castellino, F.; Schill, L.; Fehrmann, R.; Jensen, A.D. Deactivation of SCR Catalysts by Potassium: A Study of Potential Alkali Barrier Materials. VGB PowerTech J. 2017, 3, 56–64. [Google Scholar]
- Jensen, A.D.; Castellino, F.; Rams, P.D.; Pedersen, J.B.; Putluru, S.S.R. Deactivation Resistant Catalyst for Selective Catalytic Reduction of NOx. U.S. Patent 2012/0315206A1, 13 December 2012. [Google Scholar]
- Putluru, S.S.R.; Riisager, A.; Fehrmann, R. Alkali resistant Cu/zeolite deNOxcatalysts for flue gas cleaning in biomass fired applications. Appl. Catal. B Environ. 2011, 101, 183–188. [Google Scholar] [CrossRef]
- Cheng, Y.; Lambert, C.; Kim, D.H.; Kwak, J.H.; Cho, S.J.; Peden, C.H.F. The different impacts of SO2 and SO3 on Cu/zeolite SCR catalysts. Catal. Today 2010, 151, 266–270. [Google Scholar] [CrossRef]
- Kumar, A.; Smith, M.A.; Kamasamudram, K.; Currier, N.W.; Yezerets, A. Chemical deSOx: An effective way to recover Cu-zeolite SCR catalysts from sulfur poisoning. Catal. Today 2016, 267, 10–16. [Google Scholar] [CrossRef]
- Putluru, S.S.R.; Jensen, A.D.; Riisager, A.; Fehrmann, R. Heteropoly acid promoted V2O5/TiO2 catalysts for NO abatement with ammonia in alkali containing flue gases. Catal. Sci. Technol. 2011, 1, 631. [Google Scholar] [CrossRef]
- Putluru, S.S.R.; Mossin, S.; Riisager, A.; Fehrmann, R. Heteropoly acid promoted Cu and Fe catalysts for the selective catalytic reduction of NO with ammonia. Catal. Today 2011, 176, 292–297. [Google Scholar] [CrossRef]
- Peng, Y.; Li, J.; Shi, W.; Xu, J.; Hao, J. Design strategies for development of SCR catalyst: Improvement of alkali poisoning resistance and novel regeneration method. Environ. Sci. Technol. 2012, 46, 12623–12629. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Gao, X.; Fu, Y.; Gao, F.; Luo, Z.; Cen, K. The co-effect of Sb and Nb on the SCR performance of the V2O5/TiO2catalyst. J. Colloid Interface Sci. 2012, 368, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Wang, P.; Yu, F.; Wang, H.; Wu, Z. Dual resistance to alkali metals and SO2: Vanadium and cerium supported on sulfated zirconia as an efficient catalyst for NH3-SCR. Catal. Sci. Technol. 2016, 6, 8148–8156. [Google Scholar] [CrossRef]
- Schill, L.; Sankar, S.; Putluru, R.; Funk, C.; Houmann, C.; Fehrmann, R.; Degn, A. Applied Catalysis B: Environmental Ethanol-selective catalytic reduction of NO by Ag/Al2O3 catalysts: Activity and deactivation by alkali salts. Appl. Catal. B Environ. 2012, 127, 323–329. [Google Scholar] [CrossRef]
Figure 1.
Retained activity at 400 °C upon impregnation with 100 μmol K . (130 μmol K for V2O5/sulfated-ZrO2). Reproduced from [47].
Figure 1.
Retained activity at 400 °C upon impregnation with 100 μmol K . (130 μmol K for V2O5/sulfated-ZrO2). Reproduced from [47].

Figure 2.
Retained activity at 400 °C upon impregnation with 100 μmol K (130 μmol K for V2O5/sulfated-ZrO2) as a function of the number of acid sites of fresh catalysts. Generated with data from [47].
Figure 2.
Retained activity at 400 °C upon impregnation with 100 μmol K (130 μmol K for V2O5/sulfated-ZrO2) as a function of the number of acid sites of fresh catalysts. Generated with data from [47].
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Schill, L.; Fehrmann, R. Strategies of Coping with Deactivation of NH3-SCR Catalysts Due to Biomass Firing. Catalysts 2018, 8, 135. https://doi.org/10.3390/catal8040135
AMA Style
Schill L, Fehrmann R. Strategies of Coping with Deactivation of NH3-SCR Catalysts Due to Biomass Firing. Catalysts. 2018; 8(4):135. https://doi.org/10.3390/catal8040135
Chicago/Turabian StyleSchill, Leonhard, and Rasmus Fehrmann. 2018. "Strategies of Coping with Deactivation of NH3-SCR Catalysts Due to Biomass Firing" Catalysts 8, no. 4: 135. https://doi.org/10.3390/catal8040135
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.