New Insights on the Oxidation of Unsaturated Fatty Acid Methyl Esters Catalyzed by Niobium(V) Oxide. A Study of the Catalyst Surface Reactivity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
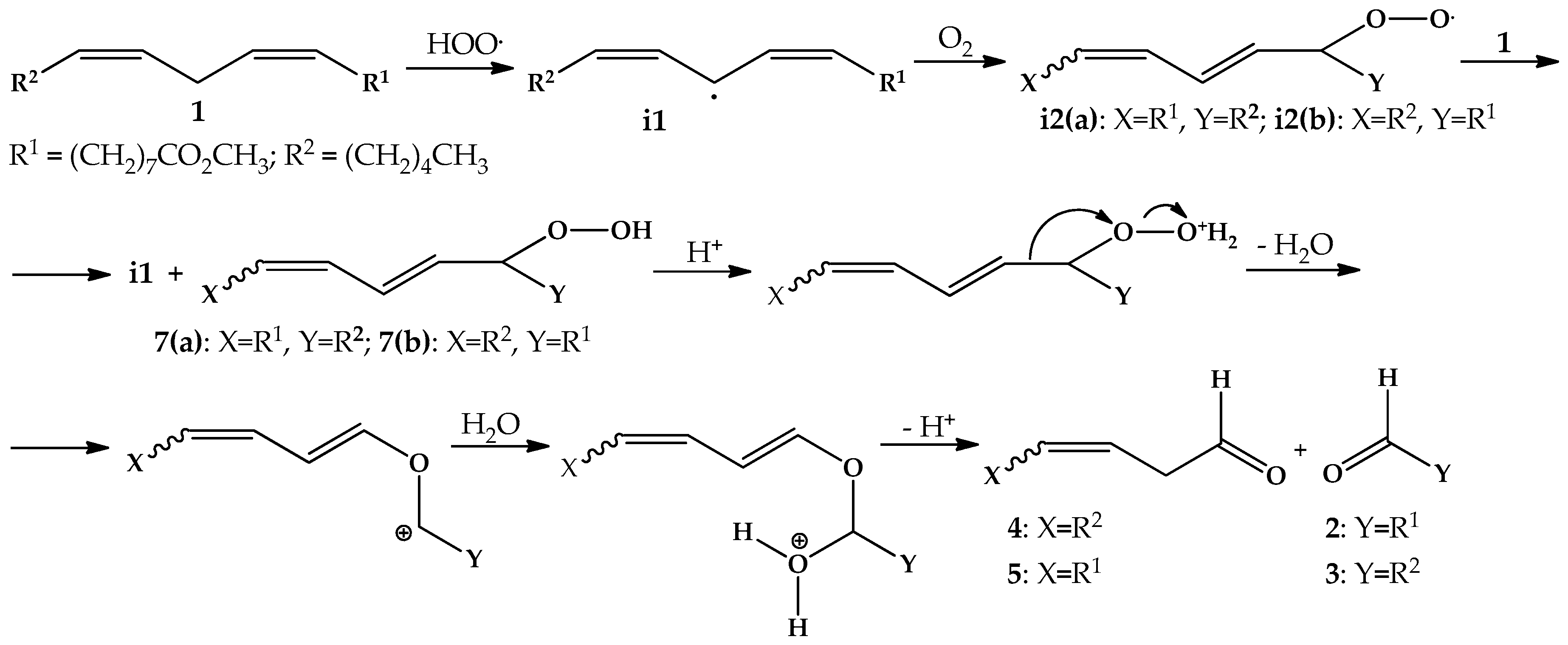
:1. Introduction
2. Results and Discussion
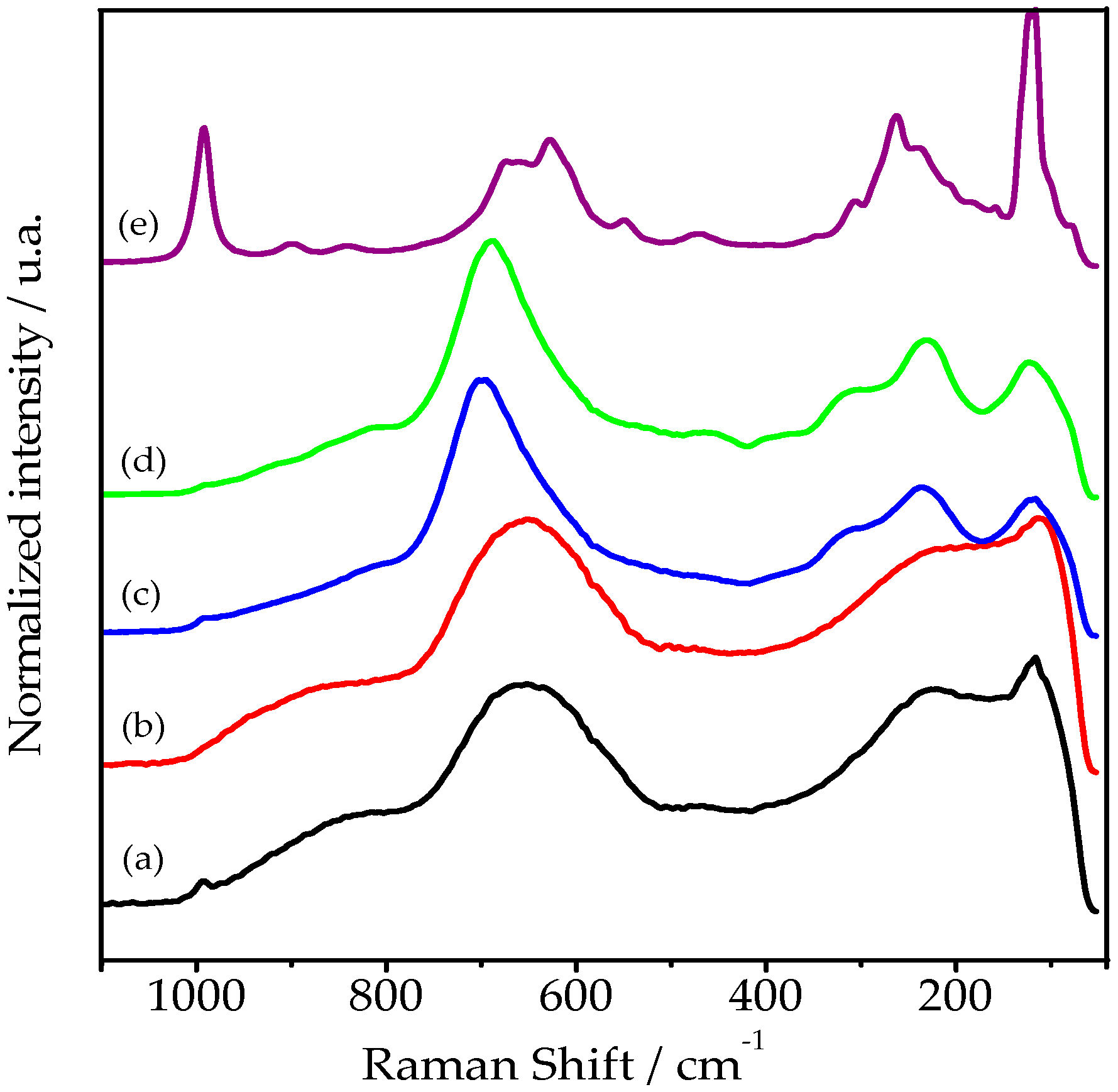
2.1. Analysis by Raman Spectroscopy
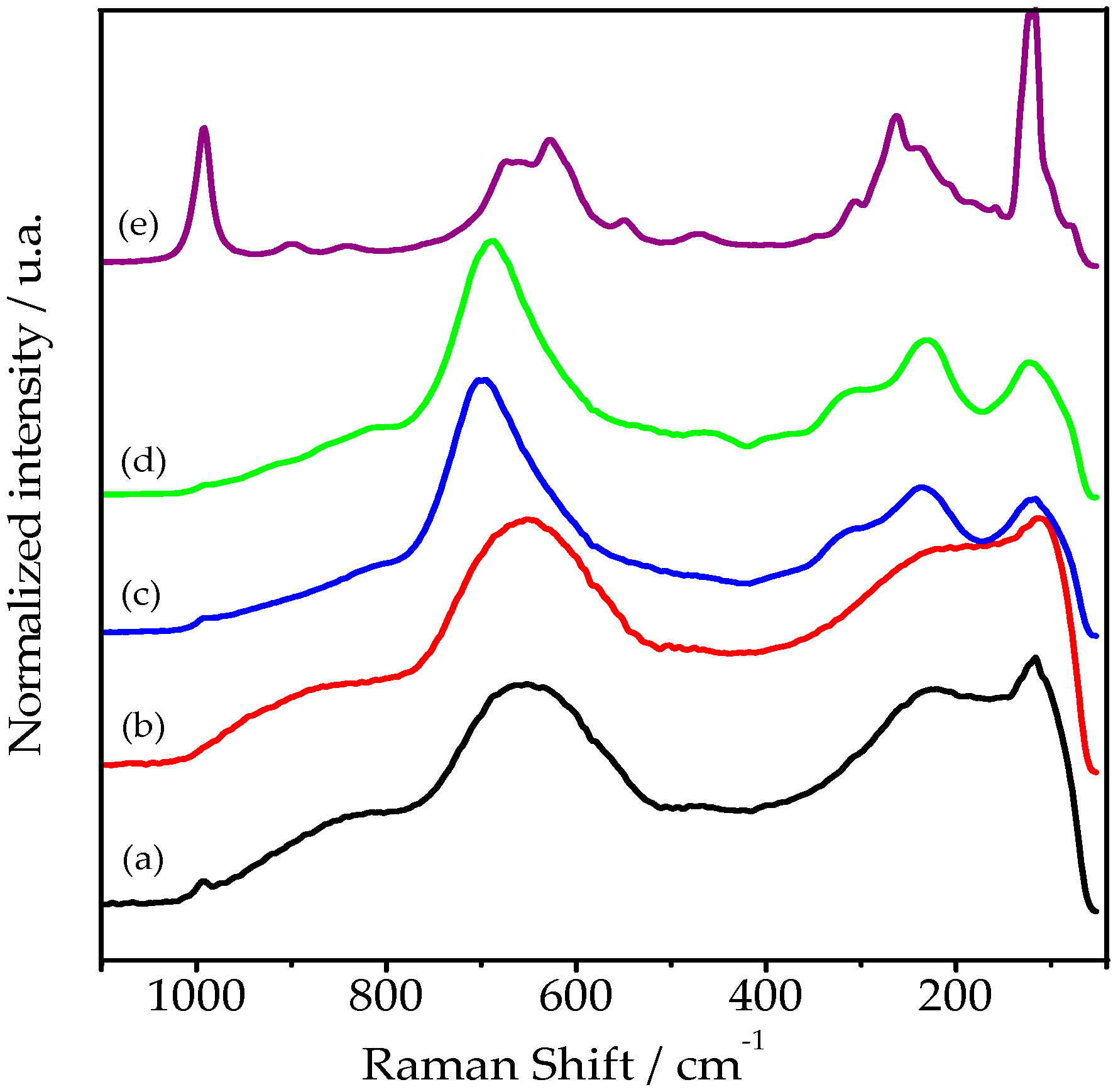
2.1.1. HY340 Set: Before H2O2 Addition
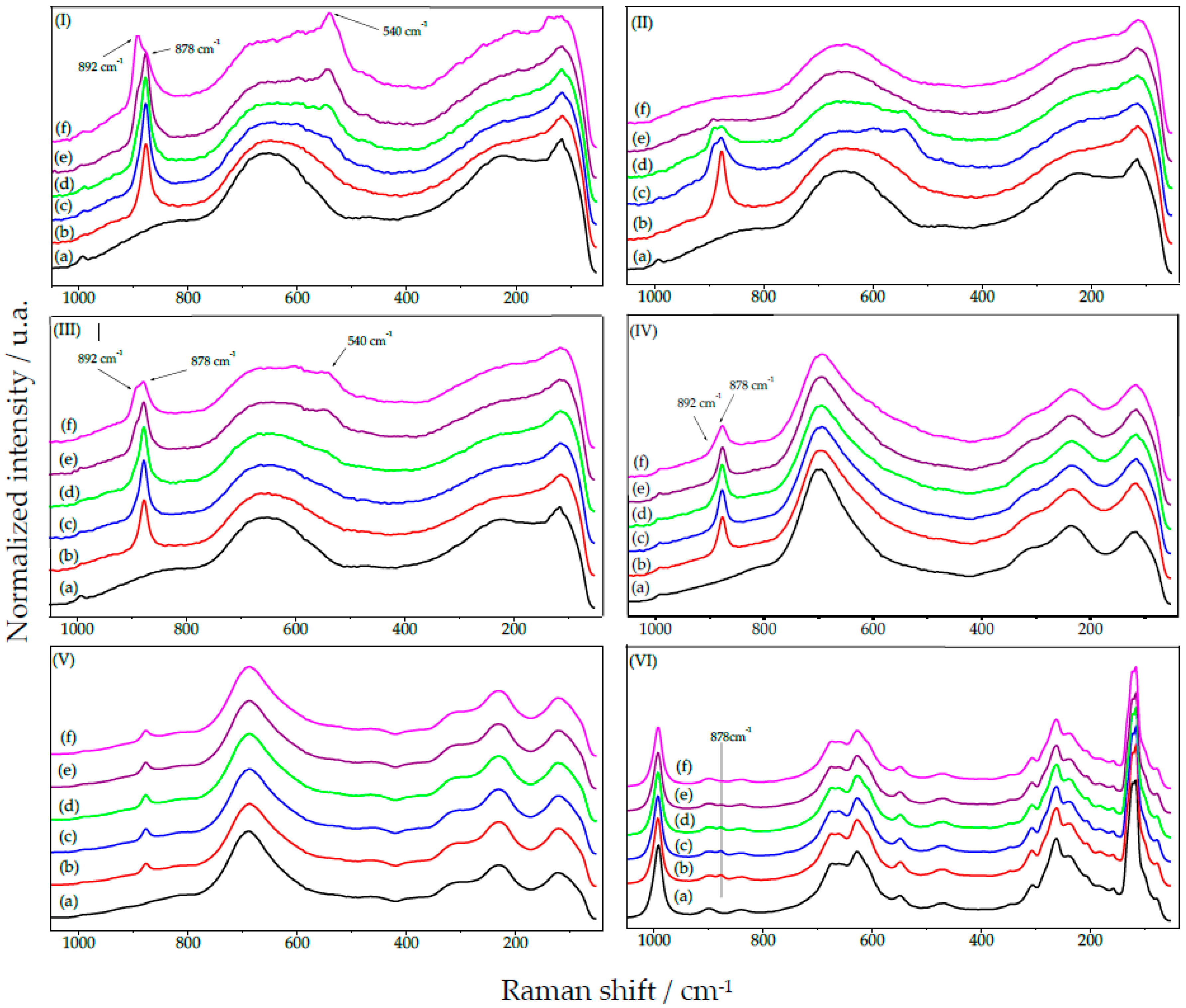
2.1.2. HY340 Set: After H2O2 Addition
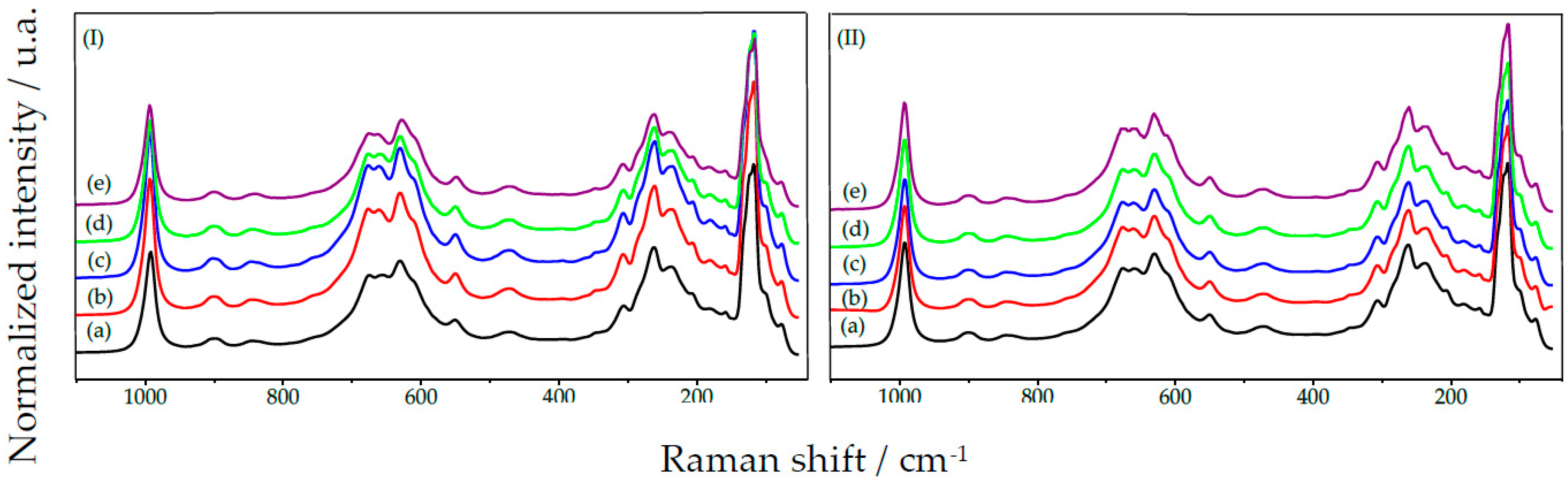
2.1.3. HY340 Set: Comparison between before and after H2O2 Addition
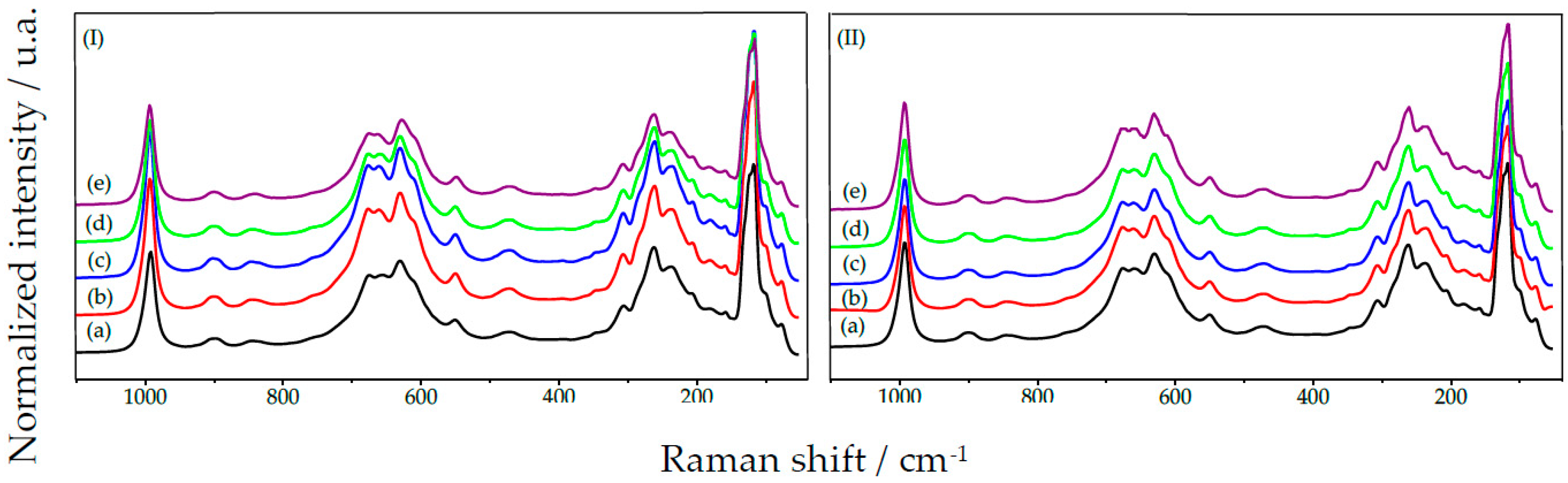
2.1.4. GO Set: Before H2O2 Addition
2.1.5. GO Set: After H2O2 Addition
2.1.6. GO Set: Comparison between before and after H2O2 Addition
2.1.7. UP Set: Before and after H2O2 Addition
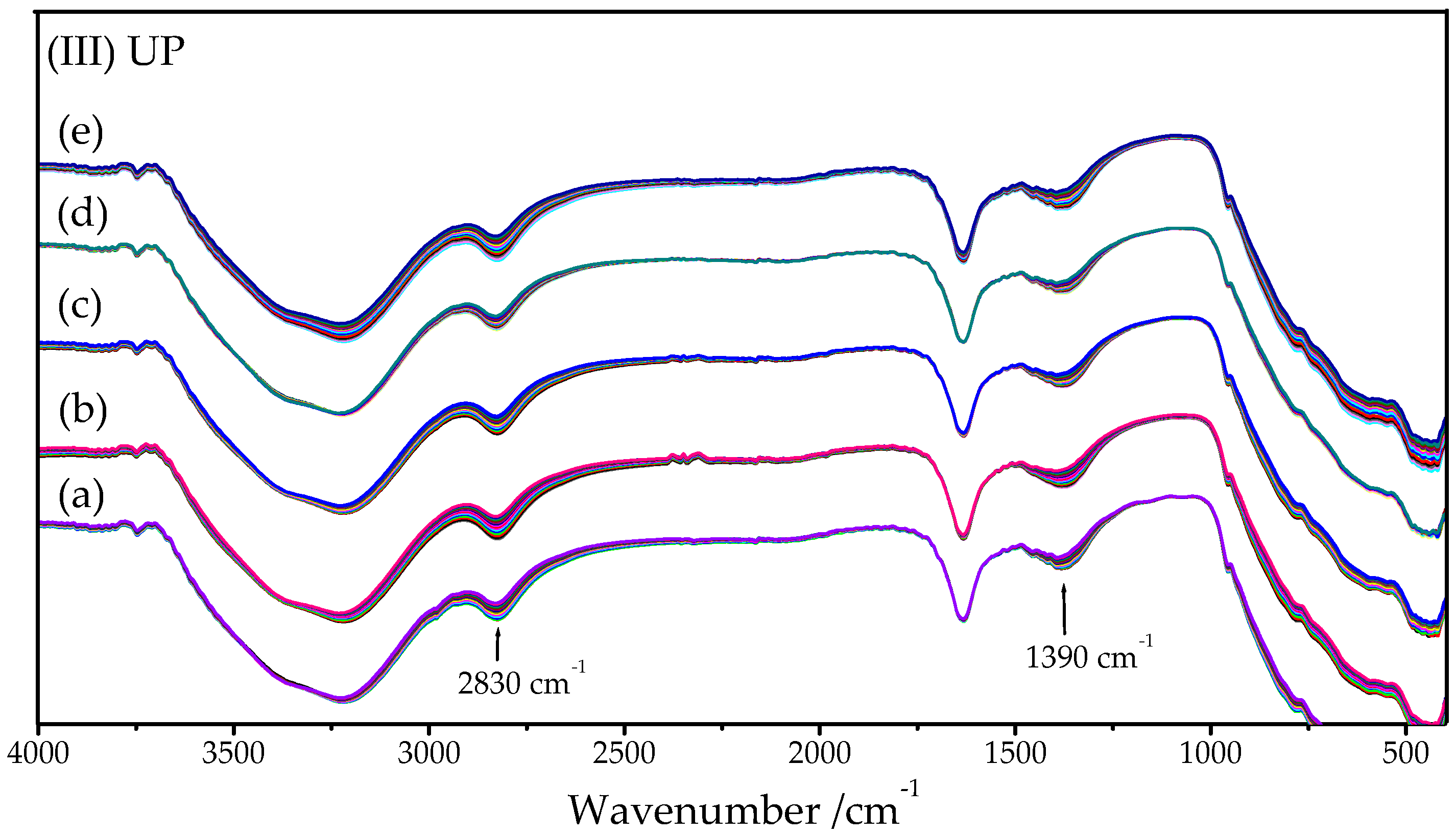
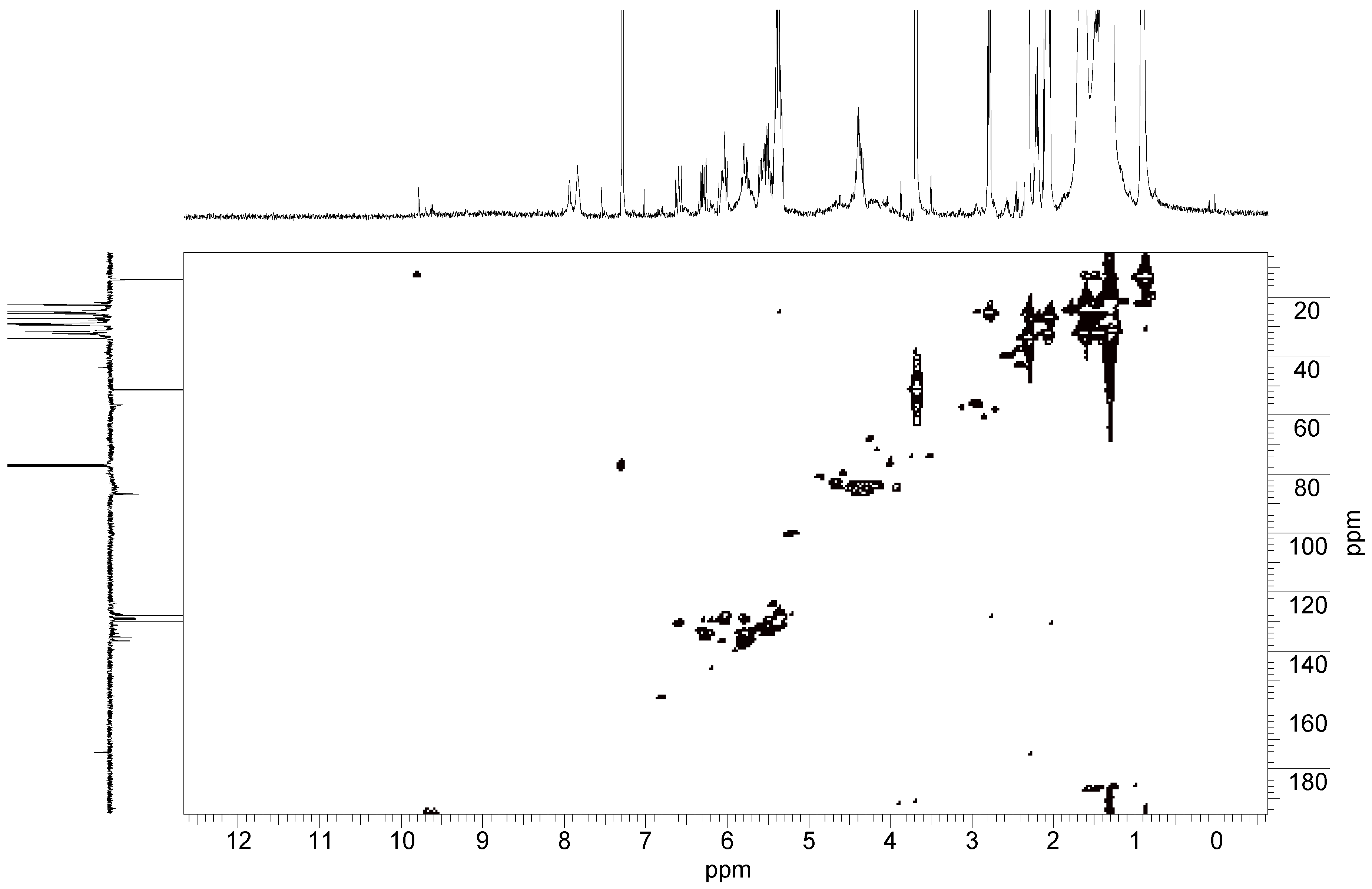
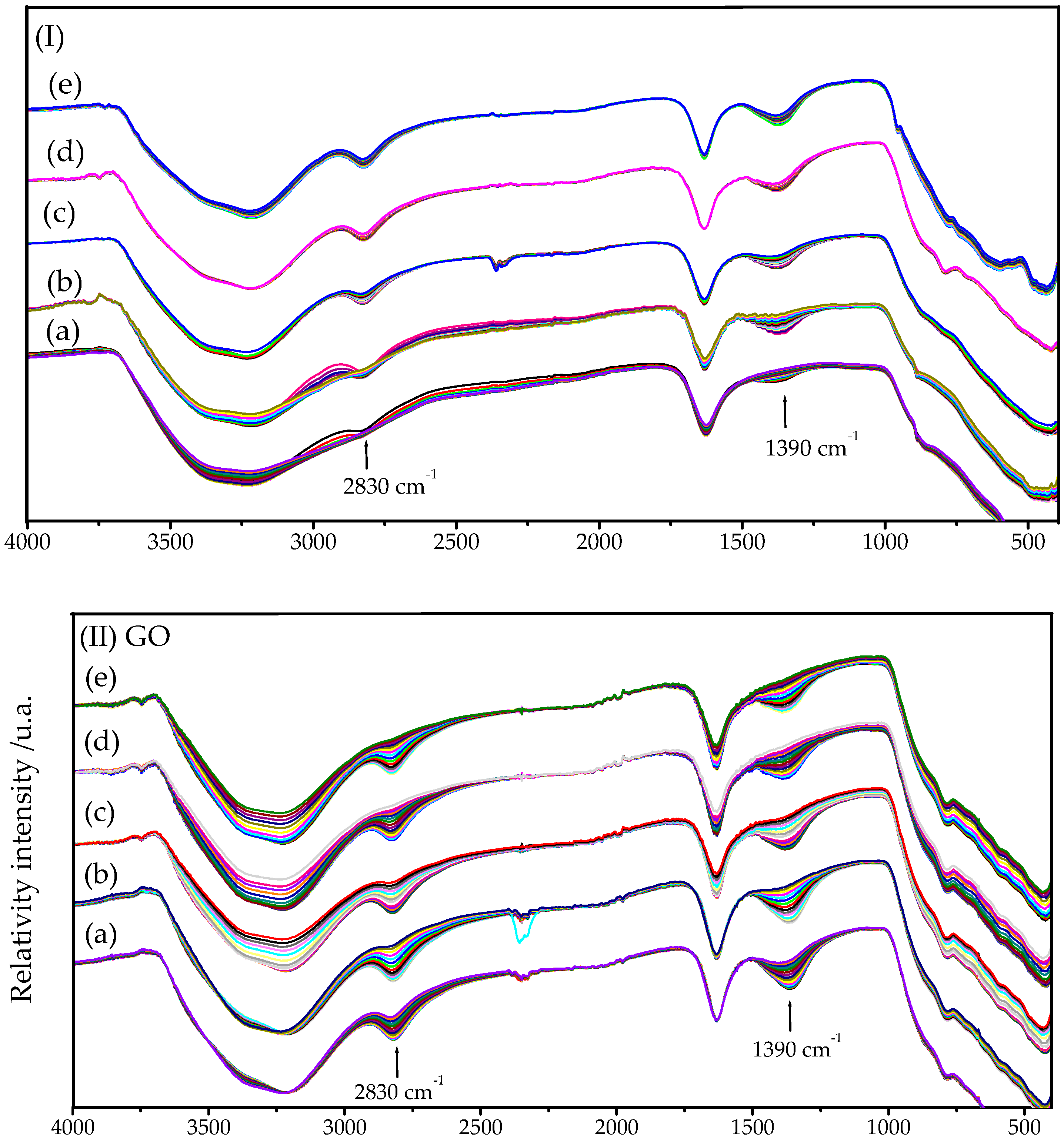
2.2. FTIR-ATR Spectroscopy
2.2.1. HY340 Set: After H2O2 Addition
2.2.2. GO Set: After H2O2 Addition
2.2.3. UP Set: After H2O2 Addition
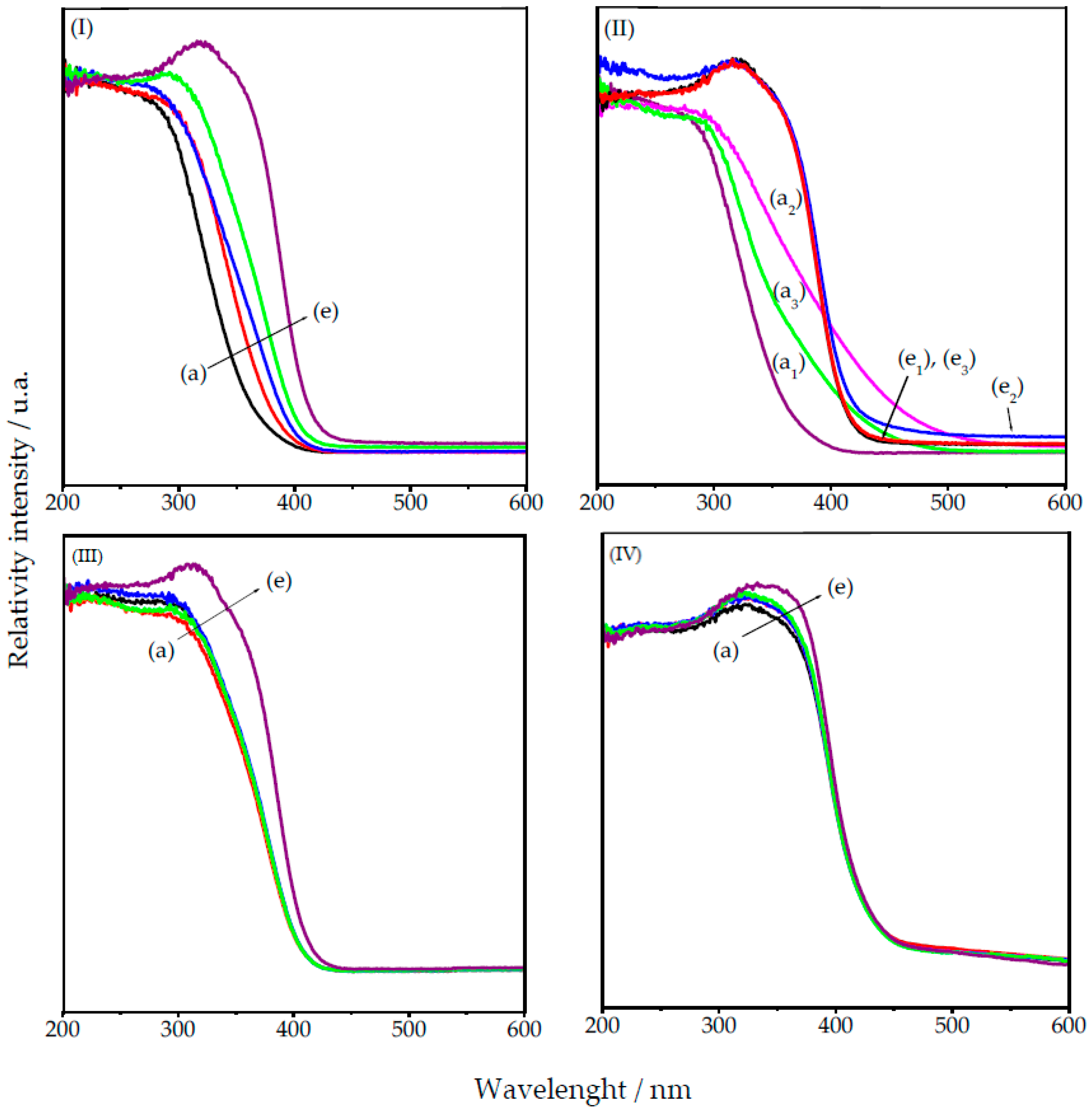
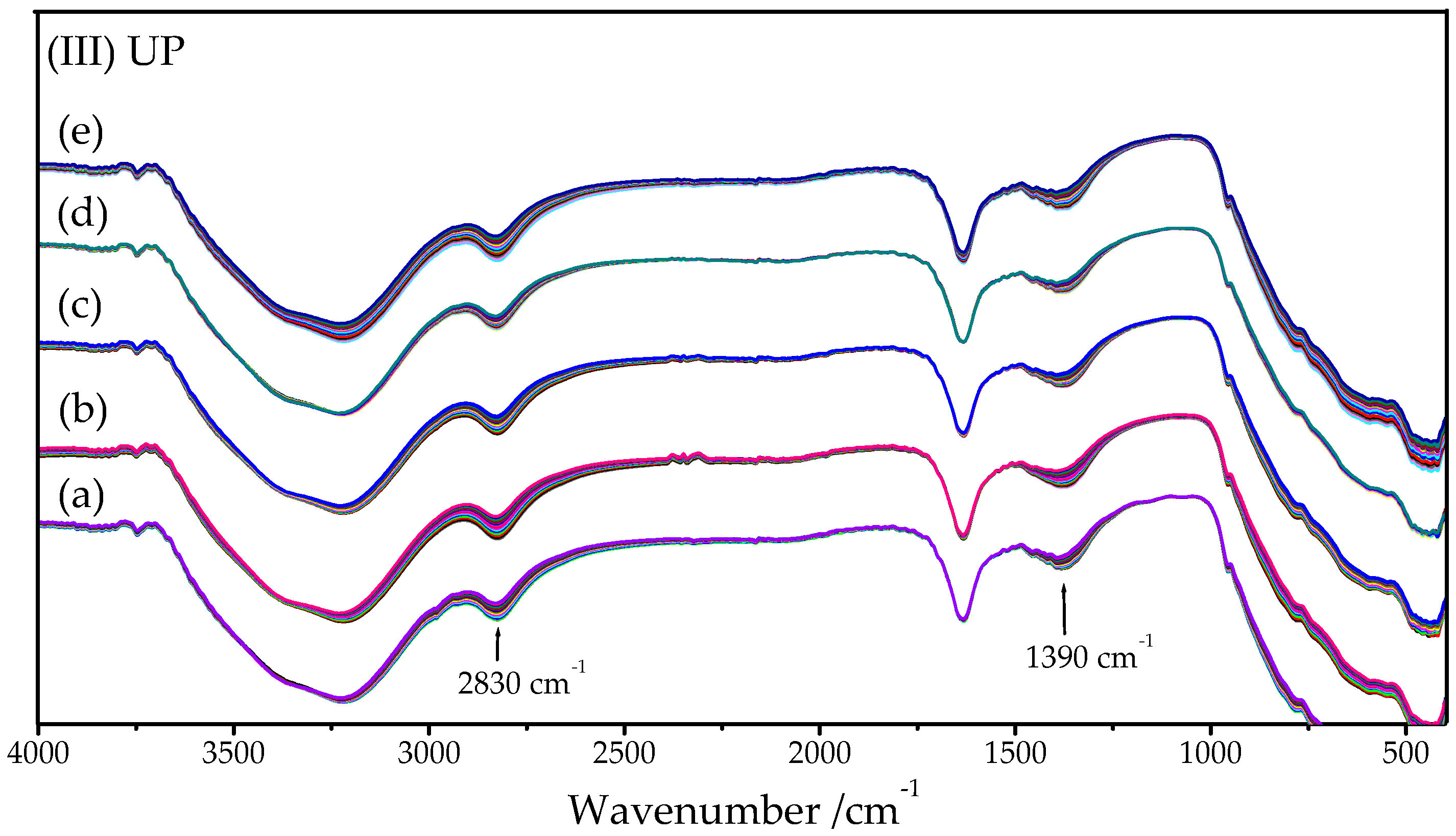
2.3. UV-Visible Spectroscopy
2.3.1. HY340 Set: Before and after H2O2 Addition
2.3.2. GO Set
2.3.3. UP Set
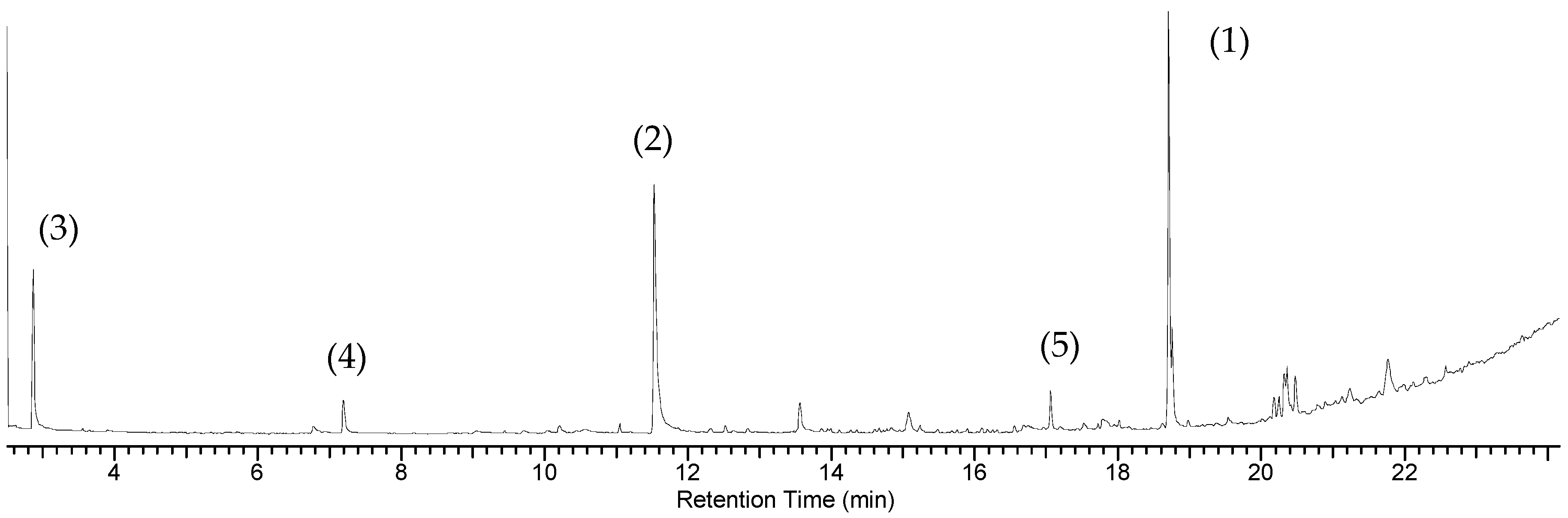
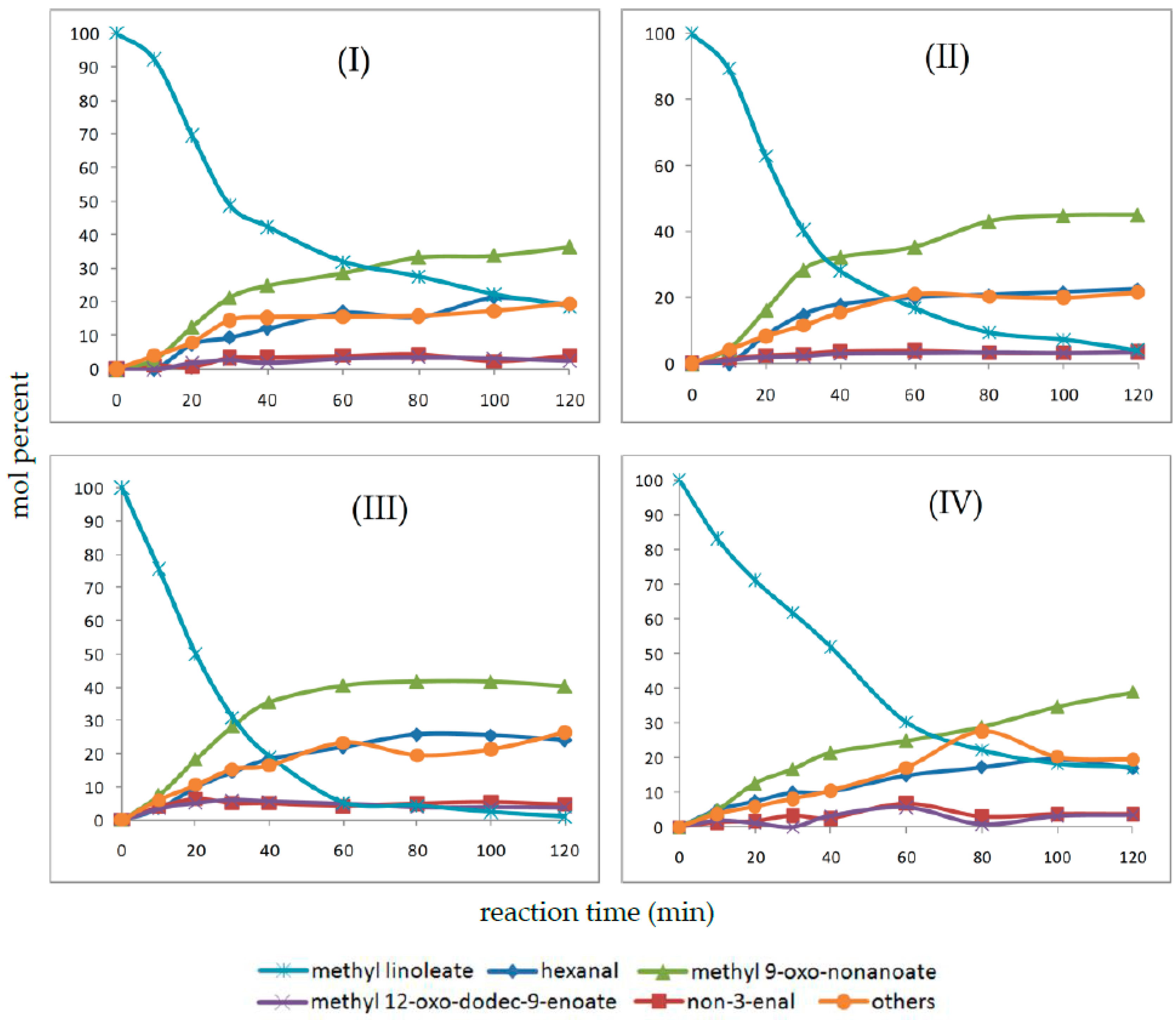
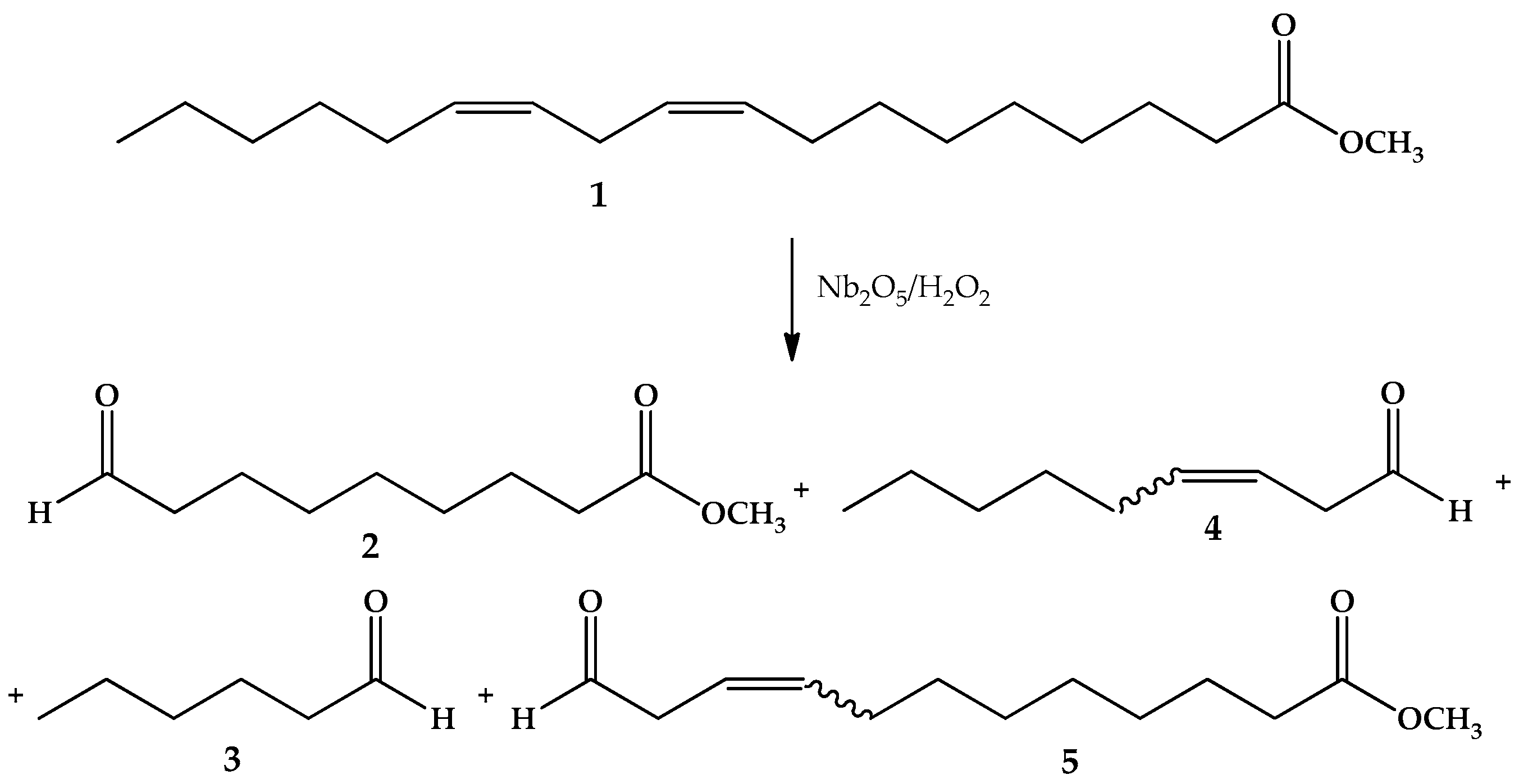
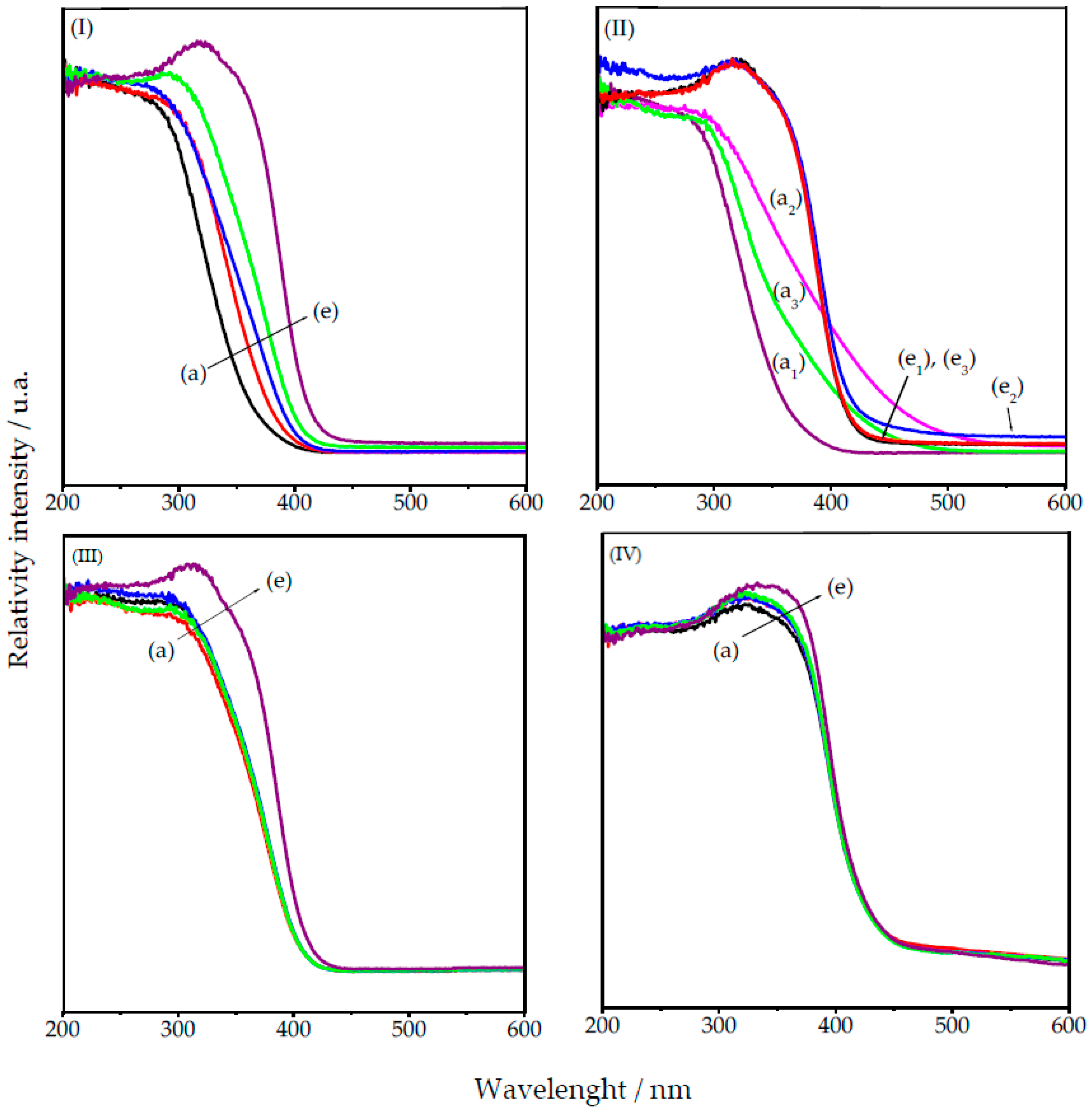
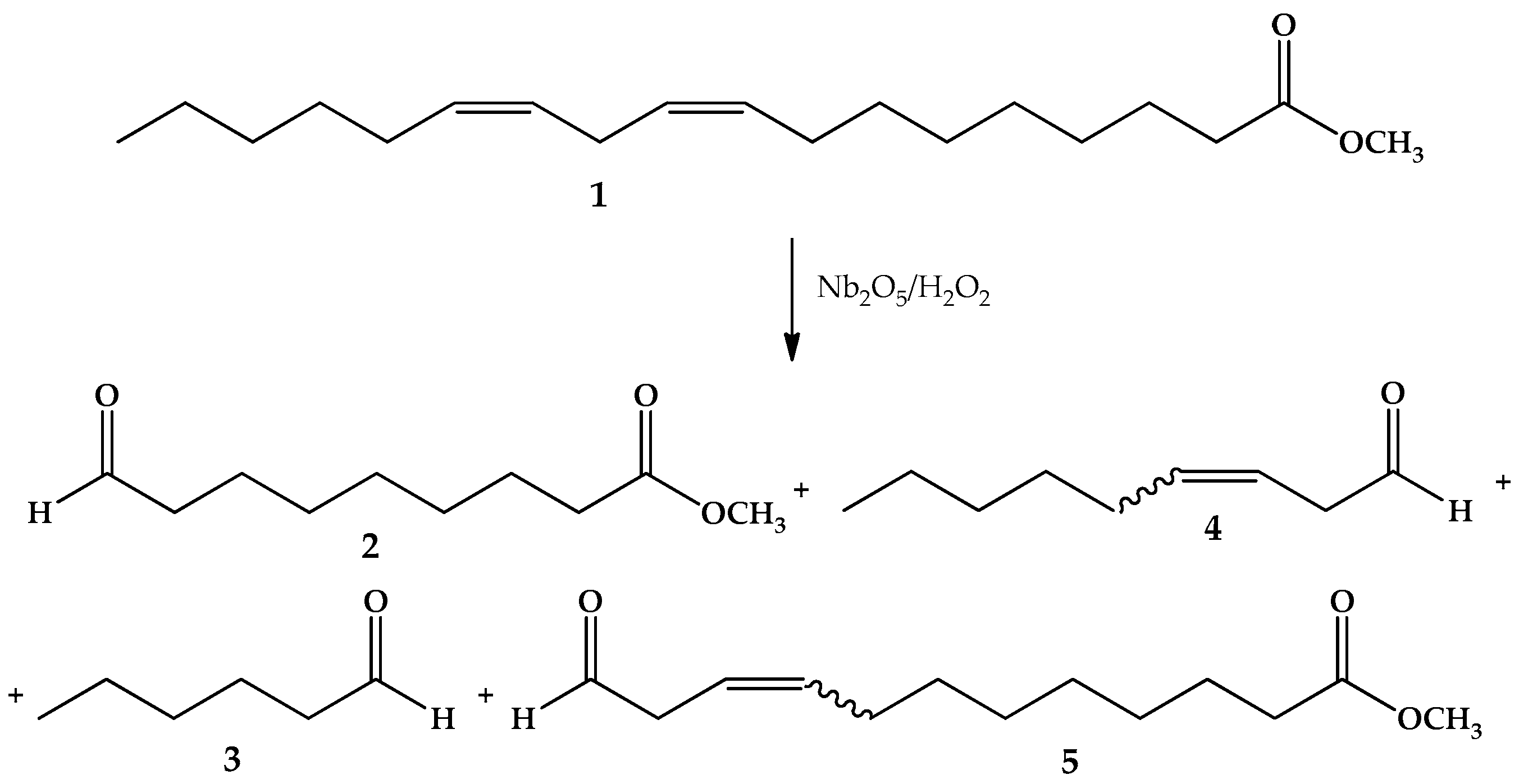
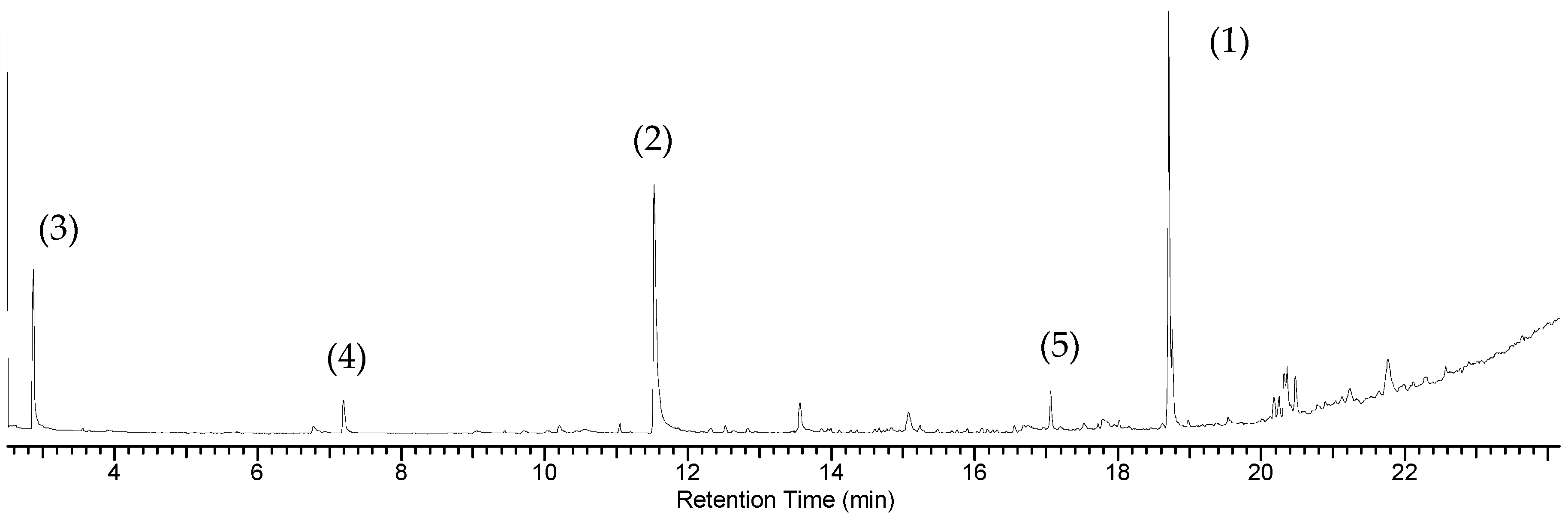
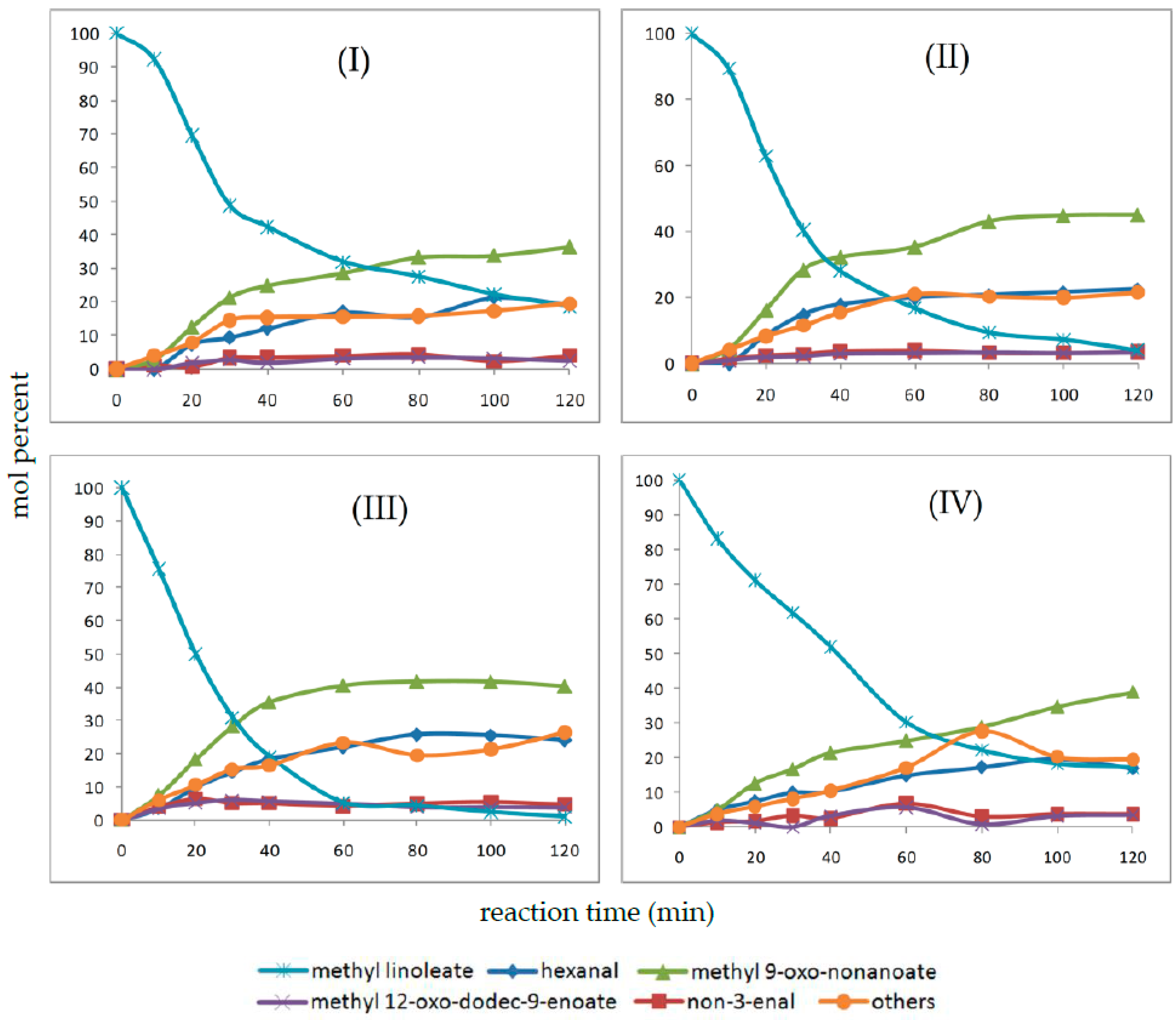
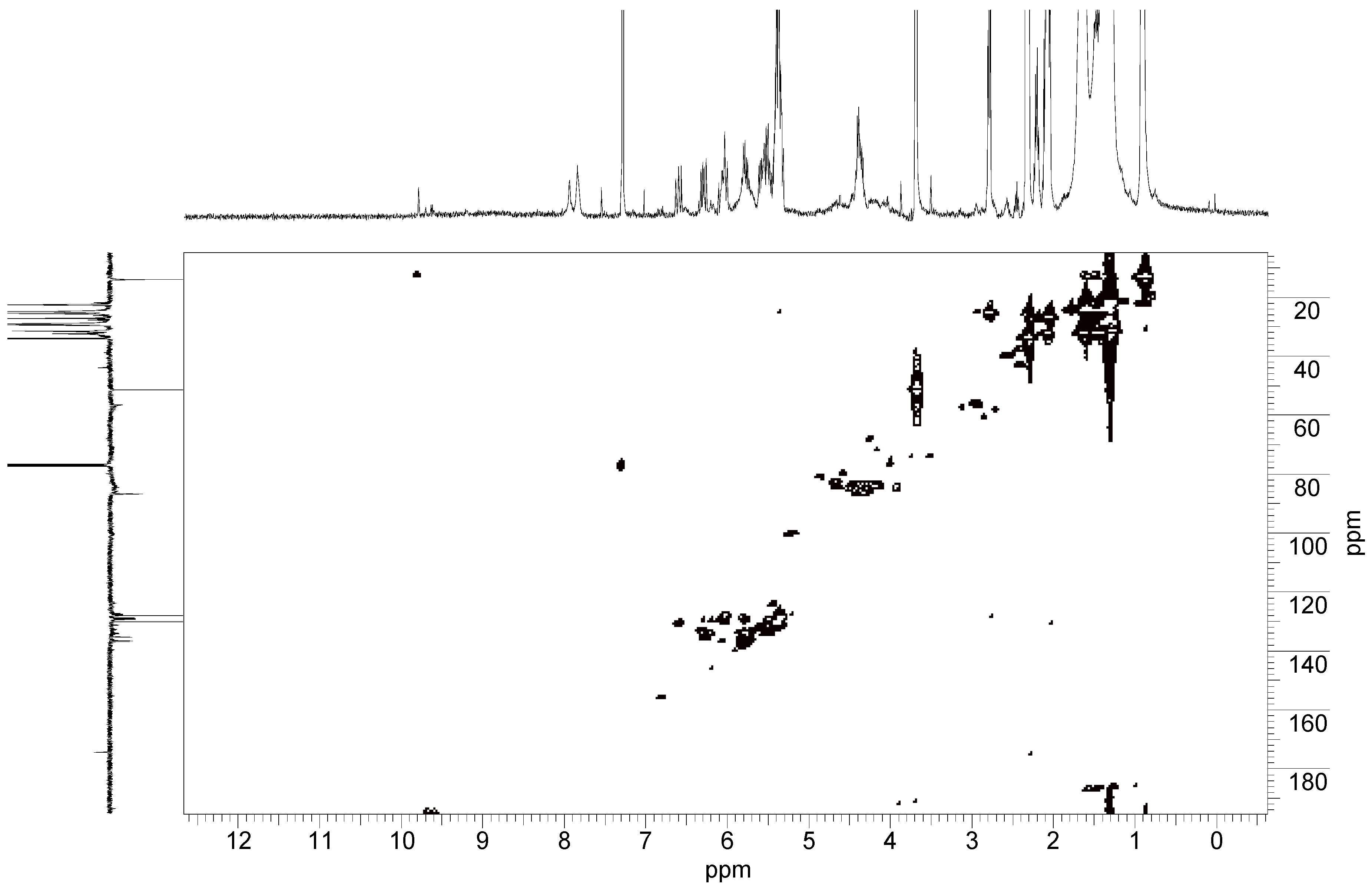
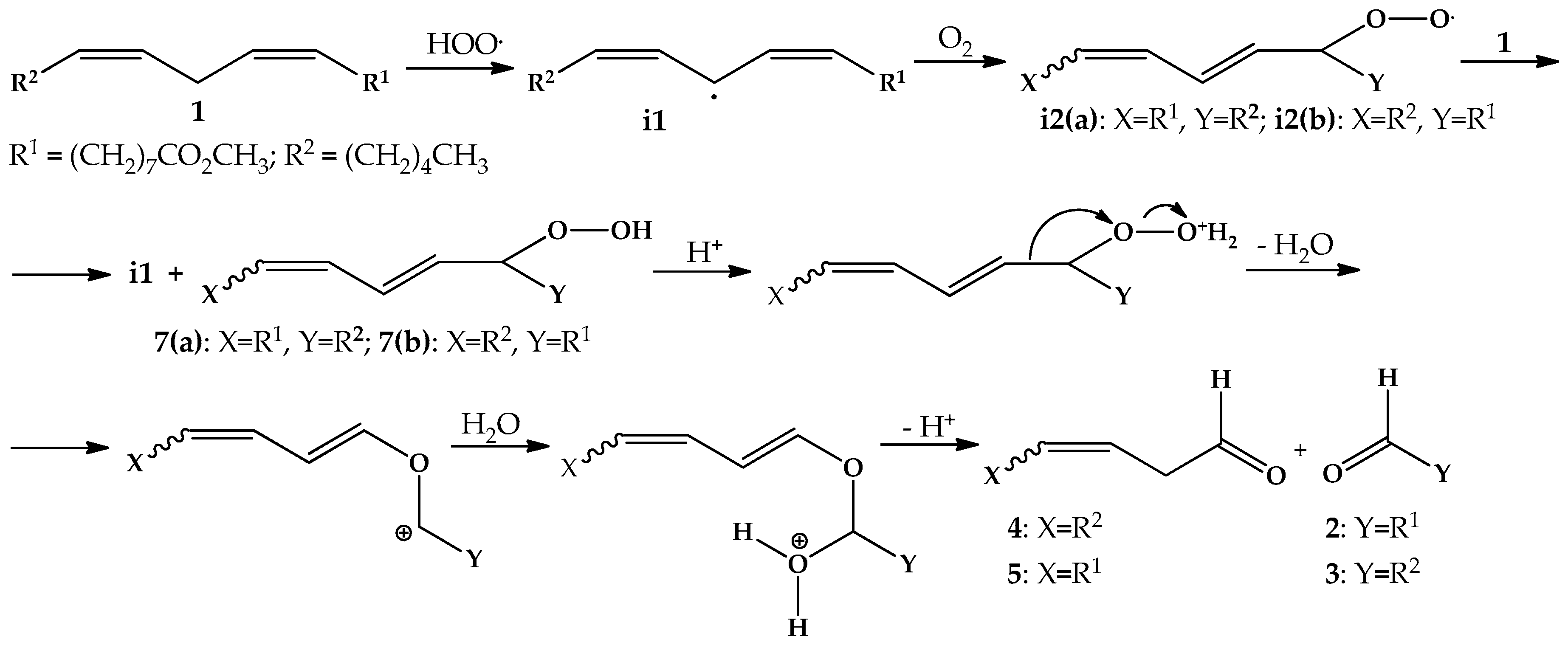
2.4. Oxidation Reaction of Linoleic Acid Methyl Ester Catalyzed by Different Types of Niobium(V) Oxide
3. Materials and Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Iizuka, T.; Ogasawara, K.; Tanabe, K. Acidic and Catalytic Properties of Niobium Pentaoxide. Bull. Chem. Soc. Jpn. 1983, 56, 2927–2931. [Google Scholar] [CrossRef]
- Nakajima, K.; Baba, Y.; Noma, R.; Kitano, M.; Kondo, J.N.; Hayashi, S.; Hara, M. Nb2O5·nH2O as a Heterogeneous Catalyst with Water-Tolerant Lewis Acid Sites. J. Am. Chem. Soc. 2011, 133, 4224–4227. [Google Scholar] [CrossRef] [PubMed]
- Nico, C.; Monteiro, T.; Graça, M.P.F. Niobium oxides and niobates physical properties: Review and prospects. Prog. Mater. Sci. 2016, 80, 1–37. [Google Scholar] [CrossRef]
- Valencia-Balvín, C.; Pérez-Walton, S.; Dalpian, G.M.; Osorio-Guillén, J.M. First-principles equation of state and phase stability of niobium pentoxide. Comp. Mater. Sci. 2014, 81, 133–140. [Google Scholar] [CrossRef]
- Aleshina, L.A.; Malinenko, V.P.; Phouphanov, A.D.; Jakovleva, N.M. The short-range order of anodic amorphous oxide films of Ta and Nb. J. Non-Cryst. Solids 1986, 87, 350–360. [Google Scholar] [CrossRef]
- Frevel, L.K.; Rlnn, H.W. Powder Diffraction Standards for Niobium Pentoxide and Tantalum Pentoxide. Anal. Chem. 1955, 1329–1330. [Google Scholar] [CrossRef]
- Holtzberg, F.; Reisman, A.; Berry, M.; Berkenblit, M. Chemistry of the Group VB Pentoxides. VI. The Polymorphism of Nb2O5. J. Am. Chem. Soc. 1957, 79, 2039–2043. [Google Scholar] [CrossRef]
- Kato, V.K.; Tamura, S. Die Kristallstruktur von T-Nb2O5. Acta Cryst. 1975, B31, 673–677. [Google Scholar] [CrossRef]
- Gatehouse, B.M.; Wadsley, A.D. The Crystal Structure of the High Temperature form of Niobium Pentoxide. Acta Cryst. 1964, 17, 1545–1554. [Google Scholar] [CrossRef]
- Tanabe, K. Niobic acid as an unusual acidic solid material. Mater. Chem. Phys. 1987, 17, 217–225. [Google Scholar] [CrossRef]
- Ushikubo, T.; Koike, Y.; Wada, K.; Xie, L.; Wang, D.; Guo, X. Study of the structure of niobium oxide by X-ray absorption fine structure and surface science techniques. Catal. Today 1996, 28, 59–69. [Google Scholar] [CrossRef]
- Jehng, J.-M.; Wachs, I.E. Structural Chemistry and Raman Spectra of Niobium Oxides. Chem. Mater. 1991, 3, 100–107. [Google Scholar] [CrossRef]
- Lebarbier, V.; Houalla, M.; Onfroy, T. New insights into the development of Brønsted acidity of niobic acid. Catal. Today 2012, 192, 123–129. [Google Scholar] [CrossRef]
- Ko, E.I.; Weissman, J.G. Structures of niobium pentoxide and their implications on chemical behavior. Catal. Today 1990, 8, 27–36. [Google Scholar] [CrossRef]
- Weissman, J.G.; Ko, E.I.; Wynblatt, P.; Howe, J.M. High-resolution electron microscopy and image simulation of TT-,T-, and H-niobia and model silica-supported niobium surface oxides. Chem. Mater. 1989, 1, 187–193. [Google Scholar] [CrossRef]
- Ikeya, T.; Senna, M. Change in the structure of niobium pentoxide due to mechanical and thermal treatments. J. Non-Cryst. Solids 1988, 105, 243–250. [Google Scholar] [CrossRef]
- Vaska, L. Dioxygen-Metal Complexes: Toward a Unified View. Acc. Chem. Res. 1975, 9, 175–183. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Kochi, J.K. Chapter 4, Activation of Molecular Oxygen by Metal Complexes. In Metal-Catalyzed Oxidations of Organic Compounds, Mechanistic Principles and Synthetic Methodology Including Biochemical Processes, 1st ed.; Academic Press: New York, NY, USA, 1981; pp. 71–119. ISBN 978-0-12-639380-4. [Google Scholar]
- Bayot, D.; Devillers, M.; Peeters, D. Vibrational Spectra of Eight-Coordinate Niobium and Tantalum Complexes with Peroxo Ligands: A Theoretical Simulation. Eur. J. Inorg. Chem. 2005, 2005, 4118–4123. [Google Scholar] [CrossRef]
- Oliveira, L.C.A.; Ramalho, T.C.; Gonçalves, M.; Cereda, F.; Carvalho, K.T.; Nazarro, M.S.; Sapag, K. Pure niobia as catalyst for the oxidation of organic contaminants: Mechanism study via ESI-MS and theoretical calculations. Chem. Phys. Lett. 2007, 446, 133–137. [Google Scholar] [CrossRef]
- Ziolek, M.; Sobczak, I.; Decyk, P.; Sobanska, K.; Pietrzyk, P.; Sojka, Z. Search for reactive intermediates in catalytic oxidation with hydrogen peroxide over amorphous niobium(V) and tantalum(V) oxides. Appl. Catal. B Environ. 2015, 164, 288–296. [Google Scholar] [CrossRef]
- Fan, W.; Zhang, Q.; Deng, W.; Wang, Y. Niobic Acid Nanosheets Synthesized by a Simple Hydrothermal Method as Efficient Brønsted Acid Catalysts. Chem. Mater. 2013, 25, 3277–3287. [Google Scholar] [CrossRef]
- Paulis, M.; Martin, M.; Soria, D.B.; Diaz, A.; Odriozola, J.A.; Montes, M. Preparation and characterization of niobium oxide for the catalytic aldol condensation of acetone. Appl. Catal. A 1999, 180, 411–420. [Google Scholar] [CrossRef]
- Pittman, R.M.; Bell, A.T. Raman studies of the structure of niobium oxide/titanium oxide (Nb2O5·TiO2). J. Phys. Chem. 1993, 97, 12178–12185. [Google Scholar] [CrossRef]
- Izumi, F.; Kodama, H. Crystallization and relative stabilities of Polymorphs of Niobium(V) Oxide under hydrothermal conditions. Z. Anorg. Allg. Chem. 1978, 440, 155–167. [Google Scholar] [CrossRef]
- Jimenez-Morales, I.; Teckchandani-Ortiz, A.; Santamaria-Gonzalez, J.; Maireles-Torres, P.; Jimenez-Lopez, A. Selective dehydration of glucose to 5-hydroxymethylfurfural on acidic mesoporous tantalum phosphate. Appl. Catal. B Environ. 2014, 144, 22–28. [Google Scholar] [CrossRef]
- Ali, M.; Siddiki, S.M.A.; Kon, K.; Shimizu, K.I. A Heterogeneous Niobium(V) Oxide Catalyst for the Direct Amidation of Esters. ChemCatChem 2015, 7, 2705–2710. [Google Scholar] [CrossRef]
- Gupta, N.K.; Fukuoka, A.; Nakajima, K. Amorphous Nb2O5 as a Selective and Reusable Catalyst for Furfural Production from Xylose in Biphasic Water and Toluene. ACS Catal. 2017, 7, 2430–2436. [Google Scholar] [CrossRef]
- Mbaraka, I.K.; Shanks, B.H. Conversion of oils and fats using advanced mesoporous heterogeneous catalysts. J. Am. Oil Chem. Soc. 2006, 83, 79–91. [Google Scholar] [CrossRef]
- Biermann, U.; Bornscheuer, U.; Meier, M.A.; Metzger, J.O.; Schäfer, H.J. Oils and Fats as Renewable Raw Materials in Chemistry. Angew. Chem. Int. Ed. Engl. 2011, 50, 3854–3871. [Google Scholar] [CrossRef] [PubMed]
- Anneken, D.J.; Both, S.; Christoph, R.; Fieg, G.; Steinberner, U.; Westfechtel, A. Fatty Acids. Ullmann’s Encycl. Ind. Chem. 2006, 14, 73–116. [Google Scholar] [CrossRef]
- Godard, A.; De Caro, P.; Thiebaud-Roux, S.; Vedrenne, E.; Mouloungui, Z. New Environmentally Friendly Oxidative Scission of Oleic Acid into Azelaic Acid and Pelargonic Acid. J. Am. Oil Chem. Soc. 2013, 90, 133–140. [Google Scholar] [CrossRef]
- Samarth, N.B.; Mahanwar, P.A. Modified Vegetable Oil Based Additives as a Future Polymeric Material—Review. Open J. Org. Polym. Mater. 2015, 5, 1–22. [Google Scholar] [CrossRef]
- Kerenkan, A.E.; Béland, F.; Do, T.-O. Chemically catalyzed oxidative cleavage of unsaturated fatty acids and their derivatives into valuable products for industrial applications: A review and perspective. Catal. Sci. Technol. 2016, 6, 971–987. [Google Scholar] [CrossRef]
- Holleben, M.L.A.V.; Schuch, C.M. Ativação do peróxido de hidrogênio para a epoxidação de olefinas não-funcionalizadas. Quim. Nova 1997, 20, 58–71. [Google Scholar] [CrossRef]
- Spannring, P.; Bruijnincx, P.C.A.; Weckhuysen, B.M.; Gebbink, R.J.M.K. Transition metal-catalyzed oxidative double bond cleavage of simple and bio-derived alkenes and unsaturated fatty acids. Catal. Sci. Technol. 2014, 4, 2182–2209. [Google Scholar] [CrossRef]
- Khlebnikova, T.B.; Pai, Z.P.; Fedoseeva, L.A.; Mattsat, Y.V. Catalytic oxidation of fatty acids. II. Epoxidation and oxidative cleavage of unsaturated fatty acid esters containing additional functional groups. React. Kinet. Catal. Lett. 2009, 98, 9–17. [Google Scholar] [CrossRef]
- Pérez, J.E.; Haagenson, D.M.; Pryor, S.W.; Ulven, C.A.; Wiesenborn, D.P. Production and Characterization of Epoxidized Canola Oil. Trans. ASABE 2009, 52, 1289–1297. [Google Scholar] [CrossRef]
- Mungroo, R.; Goud, V.V.; Pradhan, N.C.; Dalai, A.K. Modification of epoxidised canola oil. Asia-Pac. J. Chem. Eng. 2011, 6, 14–22. [Google Scholar] [CrossRef]
- Tan, S.G.; Chow, W.S. Biobased Epoxidized Vegetable Oils and Its Greener Epoxy Blends: A Review. Polym.-Plast. Technol. Eng. 2010, 49, 1581–1590. [Google Scholar] [CrossRef]
- Santacesaria, E.; Renken, A.; Russo, V.; Turco, R.; Tesser, R.; Di Serio, M. Biphasic Model Describing Soybean Oil Epoxidation with H2O2 in Continuous Reactors. Ind. Eng. Chem. Res. 2012, 51, 8760–8767. [Google Scholar] [CrossRef]
- Oakley, M.A.; Woodward, S.; Coupland, K.; Parker, D.; Temple-Heald, C. Practical dihydroxylation and C–C cleavage of unsaturated fatty acids. J. Mol. Catal. A Chem. 1999, 150, 105–111. [Google Scholar] [CrossRef]
- Köckritz, A.; Martin, A. Synthesis of azelaic acid from vegetable oil-based feedstocks. Eur. J. Lipid Sci. Technol. 2011, 113, 83–91. [Google Scholar] [CrossRef]
- Kulik, A.; Janz, A.; Pohl, M.M.; Martin, A.; Köckritz, A. Gold-catalyzed synthesis of dicarboxylic and monocarboxylic acids. Eur. J. Lipid Sci. Technol. 2012, 114, 1327–1332. [Google Scholar] [CrossRef]
- Santacesaria, E.; Sorrentino, A.; Rainone, F.; Di Serio, M.; Speranza, F. Oxidative Cleavage of the Double Bond of Monoenic Fatty Chains in Two Steps: A New Promising Route to Azelaic Acid and Other Industrial Products. Ind. Eng. Chem. Res. 2000, 39, 2766–2771. [Google Scholar] [CrossRef]
- Zaldman, B.; Kisilev, A.; Sasson, Y.; Garti, N. Double bond oxidation of unsaturated fatty acids. J. Am. Oil Chem. Soc. 1988, 65, 611–615. [Google Scholar] [CrossRef]
- Turnwald, S.E.; Lorier, M.A.; Wright, L.J.; Mucalo, M.R. Oleic Acid Oxidation Using Hydrogen Peroxide in Conjunction with Transition Metal Catalysis. J. Mater. Sci. Lett. 1998, 17, 1305–1307. [Google Scholar] [CrossRef]
- Schaich, K.M.; Borg, D.C. Fenton reactions in lipid phases. Lipids 1988, 23, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Noureddini, H.; Kanabur, M. Liquid-phase catalytic oxidation of unsaturated fatty acids. J. Am. Oil Chem. Soc. 1999, 76, 305–312. [Google Scholar] [CrossRef]
- Rup, S.; Zimmermann, F.; Meux, E.; Schneider, M.; Sindt, M.; Oget, N. The ultrasound-assisted oxidative scission of monoenic fatty acids by ruthenium tetroxide catalysis: Influence of the mixture of solvents. Ultrason. Sonochem. 2009, 16, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Otte, K.B.; Kirtz, M.; Nestl, B.M.; Hauer, B. Synthesis of 9-Oxononanoic Acid, a Precursor for Biopolymers. ChemSusChem 2013, 6, 2149–2156. [Google Scholar] [CrossRef] [PubMed]
- Liguori, A.; Belsito, E.L.; Gioia, M.L.; Leggio, A.; Malagrinò, F.; Romio, E.; Siciliano, C.; Tagarelli, A. GC/MS Analysis of Fatty Acids in Italian Dry Fermented Sausages. Open Food Sci. J. 2015, 9, 5–13. [Google Scholar] [CrossRef]
- Sun, Y.-E.; Wang, W.-D.; Chen, H.-W.; Li, C. Autoxidation of Unsaturated Lipids in Food Emulsion. Crit. Rev. Food Sci. Nutr. 2011, 51, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Min, D.B.; Boff, J.M. Chapter 11, Lipid Oxidation of Edible Oil. In Food Lipids: Chemistry, Nutrition and Biotechnology, 2nd ed.; Akoh, C.C., Min, D.B., Eds.; CRC Press: New York, NY, USA, 2002; pp. 335–364. ISBN 0-8247-0749-4. [Google Scholar]
- Mattila, H.; Khorobrykh, S.; Havurinne, V.; Tyystjärvi, E. Reactive oxygen species: Reactions and detection from photosynthetic tissues. J. Photochem. Photobiol. B 2015, 152, 176–214. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.-R. Comprehensive Handbook of Chemical Bond Energies; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Choe, E.; Min, D.B. Mechanisms and Factors for Edible Oil Oxidation. Compr. Rev. Food Sci. Food Saf. 2006, 5, 169–186. [Google Scholar] [CrossRef]
- Guerrero-Perez, M.O.; Banares, M.A. Niobium as promoting agent for selective oxidation reactions. Catal. Today 2009, 142, 245–251. [Google Scholar] [CrossRef]
- Chen, C.; Zhao, X.; Chen, J.; Hua, L.; Zhang, R.; Guo, L.; Hou, Z. Niobium Peroxide-Catalyzed Selective Epoxidation of Allylic Alcohols. ChemCatChem 2014, 6, 3231–3238. [Google Scholar] [CrossRef]
- Marchetti, F.; Pampaloni, G.; Zacchini, S. Epoxide ring opening and insertion into the M–X bond of niobium and tantalum pentahalides: Synthesis of dihalide-tris(2-haloalcoholato) complexes. Polyhedron 2009, 28, 1235–1240. [Google Scholar] [CrossRef]
- Marin-Astorga, N.; Martinez, J.J.; Suarez, D.N.; Cubillos, J.; Rojas, H.; Ortiz, C.A. Nb2O5 as Heterogeneous Catalysts for the Selective Oxidation of Geraniol. Curr. Org. Chem. 2012, 16, 2797–2801. [Google Scholar] [CrossRef]
- Nowak, I.; Kilos, B.; Ziolek, M.; Lewandowska, A. Epoxidation of cyclohexene on Nb-containing meso- and macroporous materials. Catal. Today 2003, 78, 487–498. [Google Scholar] [CrossRef]
- Tiozzo, C.; Bisio, C.; Carniato, F.; Guidotti, M. Grafted non-ordered niobium-silica materials: Versatile catalysts for the selective epoxidation of various unsaturated fine chemicals. Catal. Today 2014, 235, 49–57. [Google Scholar] [CrossRef]
- Tiozzo, C.; Palumbo, C.; Psaro, R.; Bisio, C.; Carniato, F.; Gervasini, A.; Guidotti, M. The stability of niobium-silica catalysts in repeated liquid-phase epoxidation tests: A comparative evaluation of in-framework and grafted mixed oxides. Inorg. Chim. Acta 2015, 431, 190–196. [Google Scholar] [CrossRef]
- Turco, R.; Aronne, A.; Carniti, P.; Gervasini, A.; Minieri, L.; Pernice, P.; Di Serio, M. Influence of preparation methods and structure of niobium oxide-based catalysts in the epoxidation reaction. Catal. Today 2015, 254, 99–103. [Google Scholar] [CrossRef]
- Bayot, D.; Tinant, B.; Devillers, M. Water-soluble niobium peroxo complexes as precursors for the preparation of Nb-based oxide catalysts. Catal. Today 2003, 78, 439–447. [Google Scholar] [CrossRef]
- Bayot, D.; Tinant, B.; Mathieu, B.; Declercq, J.P.; Devillers, M. Spectroscopic and Structural Characterizations of Novel Water-Soluble Peroxo[polyaminocarboxylato bis(N-oxido)]niobate(V) Complexes. Eur. J. Inorg. Chem. 2003, 4, 737–743. [Google Scholar] [CrossRef]
- Voraberger, H.; Ribitsch, V.; Janotta, M.; Mizaikoff, B. Application of Mid-Infrared Spectroscopy: Measuring Hydrogen Peroxide Concentrations in Bleaching Baths. Appl. Spectrosc. 2003, 57, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, K.T.; Silva, A.C.; Oliveira, L.C.; Gonçalves, M.; Magriotis, Z.M. Nióbia sintética modificada como catalisador na oxidação de corante orgânico: Utilização de H2O2 e O2 atmosférico como oxidantes. Quim. Nova 2009, 32, 1373–1377. [Google Scholar] [CrossRef]
- Ziolek, M.; Sobczak, I.; Decyk, P.; Wolski, L. The ability of Nb2O5 and Ta2O5 to generate active oxygen in contact with hydrogen peroxide. Catal. Commun. 2013, 37, 85–91. [Google Scholar] [CrossRef]
- Grechkin, A.N.; Kuramshin, R.A.; Latypov, S.K.; Safonova, Y.Y.; Gafarova, T.E.; Ilyasov, A.V. Hydroperoxides of α-ketols. FEBS J. 1991, 199, 451–457. [Google Scholar] [CrossRef]
- Miccichè, F.; van Haveren, J.; Oostveen, E.; Ming, W.; van der Linde, R. Oxidation and oligomerization of ethyl linoleate under the influence of the combination of ascorbic acid 6-palmitate/iron-2-ethylhexanoate. Appl. Catal. A Gen. 2006, 297, 174–181. [Google Scholar] [CrossRef]
- Juita; Dlugogorski, B.Z.; Kennedy, E.M.; Mackie, J.C. Low temperature oxidation of linseed oil: A review. Fire Sci. Rev. 2012, 1, 1–36. [Google Scholar] [CrossRef]
- Brash, A.R. Autoxidation of methyl linoleate: Identification of the bis-allylic 11-hydroperoxide. Lipids 2000, 35, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Yanez, J.; Sevilla, C.L.; Becker, D.; Sevilla, M.D. Low-temperature autoxidation in unsaturated lipids: An electron spin resonance study. J. Phys. Chem. 1987, 91, 487–491. [Google Scholar] [CrossRef]
- Venkataraman, S.; Schafer, F.Q.; Buettner, G.R. Detection of Lipid Radicals Using EPR. Antioxid. Redox Signal. 2004, 6, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Ramalho, T.C.; Oliveira, L.C.A.; Carvalho, K.T.G.; Souza, E.F.; Cunha, E.F.F.; Nazzaro, M. The molecular basis for the behaviour of niobia species in oxidation reaction probed by theoretical calculations and experimental techniques. Mol. Phys. 2009, 107, 171–179. [Google Scholar] [CrossRef]
- Kremer, M.L. “Complex” versus “free radical” mechanism for the catalytic decomposition of H2O2 by ferric ions. Int. J. Chem. Kinet. 1985, 17, 1299–1314. [Google Scholar] [CrossRef]
- Salem, I.A.; El-Maazawi, M.; Zaki, A.B. Kinetics and mechanisms of decomposition reaction of hydrogen peroxide in presence of metal complexes. Int. J. Chem. Kinet. 2000, 32, 643–666. [Google Scholar] [CrossRef]
- Wang, Z. Hock Rearrangement. In Comprehensive Organic Name Reactions and Reagents; John Wiley & Sons: New York, NY, USA, 2010; pp. 1438–1442. ISBN 9780470638859. [Google Scholar]
- Li, Z.; Tran, V.H.; Duke, R.K.; Ng, M.C.; Yang, D.; Duke, C.C. Synthesis and biological activity of hydroxylated derivatives of linoleic acid and conjugated linoleic acids. Chem. Phys. Lipids 2009, 158, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Mungroo, R.; Pradhan, N.C.; Goud, V.V.; Dalai, A.K. Epoxidation of Canola Oil with Hydrogen Peroxide Catalyzed by Acidic Ion Exchange Resin. J. Am. Oil Chem. Soc. 2008, 85, 887–896. [Google Scholar] [CrossRef]
- Farias, M.; Martinelli, M. Epoxidation of soybean oil with [MoO2(acac)2]/TBHP Catalytic system in [bmim][PF6]. Quim. Nova 2012, 35, 1538–1541. [Google Scholar] [CrossRef]
- Mimoun, H.; De Roch, I.S.; Sajus, L. Epoxydation des olefines par les complexes peroxydiques covalents du molybdene-VI. Tetrahedron 1970, 26, 37–50. [Google Scholar] [CrossRef]
- Mimoun, H.; Saussine, L.; Daire, E.; Postel, M.; Fischer, J.; Weiss, R. Vanadium(V) peroxy complexes. New versatile biomimetic reagents for epoxidation of olefins and hydroxylation of alkanes and aromatic hydrocarbons. J. Am. Chem. Soc. 1983, 105, 3101–3110. [Google Scholar] [CrossRef]
- Bregante, D.T.; Flaherty, D.W. Periodic Trends in Olefin Epoxidation over Group IV and V Framework-Substituted Zeolite Catalysts: A Kinetic and Spectroscopic Study. J. Am. Chem. Soc. 2017, 139, 6888–6898. [Google Scholar] [CrossRef] [PubMed]
- Pai, Z.P.; Tolstikov, A.G.; Berdnikova, P.V.; Kustova, G.N.; Khlebnikova, T.B.; Selivanova, N.V.; Kostrovskii, V.G. Catalytic oxidation of olefins and alcohols with hydrogen peroxide in a two-phase system giving mono- and dicarboxylic acids. Russ. Chem. Bull. 2005, 54, 1847–1854. [Google Scholar] [CrossRef]
- Machulek, A.; Quina, F.H.; Gozzi, F.; Silva, V.O.; Friedrich, L.C.; Moraes, J.E. Chapter 11, Fundamental Mechanistic Studies of the Photo-Fenton Reaction for the Degradation of Organic Pollutants. In Organic Pollutants Ten Years after the Stockholm Convention—Environmental and Analytical Update; Intech: London, UK, 2012; pp. 271–293. ISBN 978-953-307-917-2. [Google Scholar]
- Oliveira, L.C.A.; Portilho, M.F.; Silva, A.C.; Taroco, H.A.; Souza, P.P. Modified niobia as a bifunctional catalyst for simultaneous dehydration and oxidation of glycerol. Appl. Catal. B Environ. 2012, 117–118, 29–35. [Google Scholar] [CrossRef]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paraguassú Cecchi, C.M.; Cesarín-Sobrinho, D.; Buarque Ferreira, A.B.; Netto-Ferreira, J.C. New Insights on the Oxidation of Unsaturated Fatty Acid Methyl Esters Catalyzed by Niobium(V) Oxide. A Study of the Catalyst Surface Reactivity. Catalysts 2018, 8, 6. https://doi.org/10.3390/catal8010006
Paraguassú Cecchi CM, Cesarín-Sobrinho D, Buarque Ferreira AB, Netto-Ferreira JC. New Insights on the Oxidation of Unsaturated Fatty Acid Methyl Esters Catalyzed by Niobium(V) Oxide. A Study of the Catalyst Surface Reactivity. Catalysts. 2018; 8(1):6. https://doi.org/10.3390/catal8010006
Chicago/Turabian StyleParaguassú Cecchi, Christian Marcelo, Darí Cesarín-Sobrinho, Aurélio Baird Buarque Ferreira, and José Carlos Netto-Ferreira. 2018. "New Insights on the Oxidation of Unsaturated Fatty Acid Methyl Esters Catalyzed by Niobium(V) Oxide. A Study of the Catalyst Surface Reactivity" Catalysts 8, no. 1: 6. https://doi.org/10.3390/catal8010006