
Nature and Location of Carbonaceous Species in a Composite HZSM-5 Zeolite Catalyst during the Conversion of Dimethyl Ether into Light Olefins

 ,
, 


Abstract
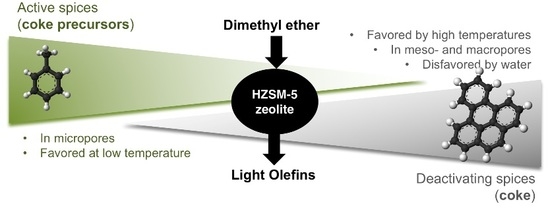
:
1. Introduction
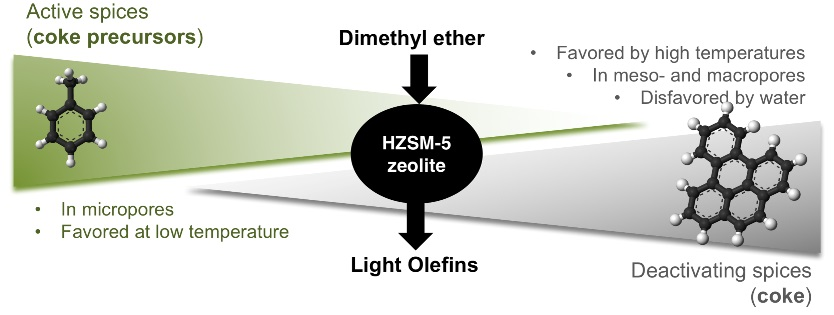
2. Results and Discussion
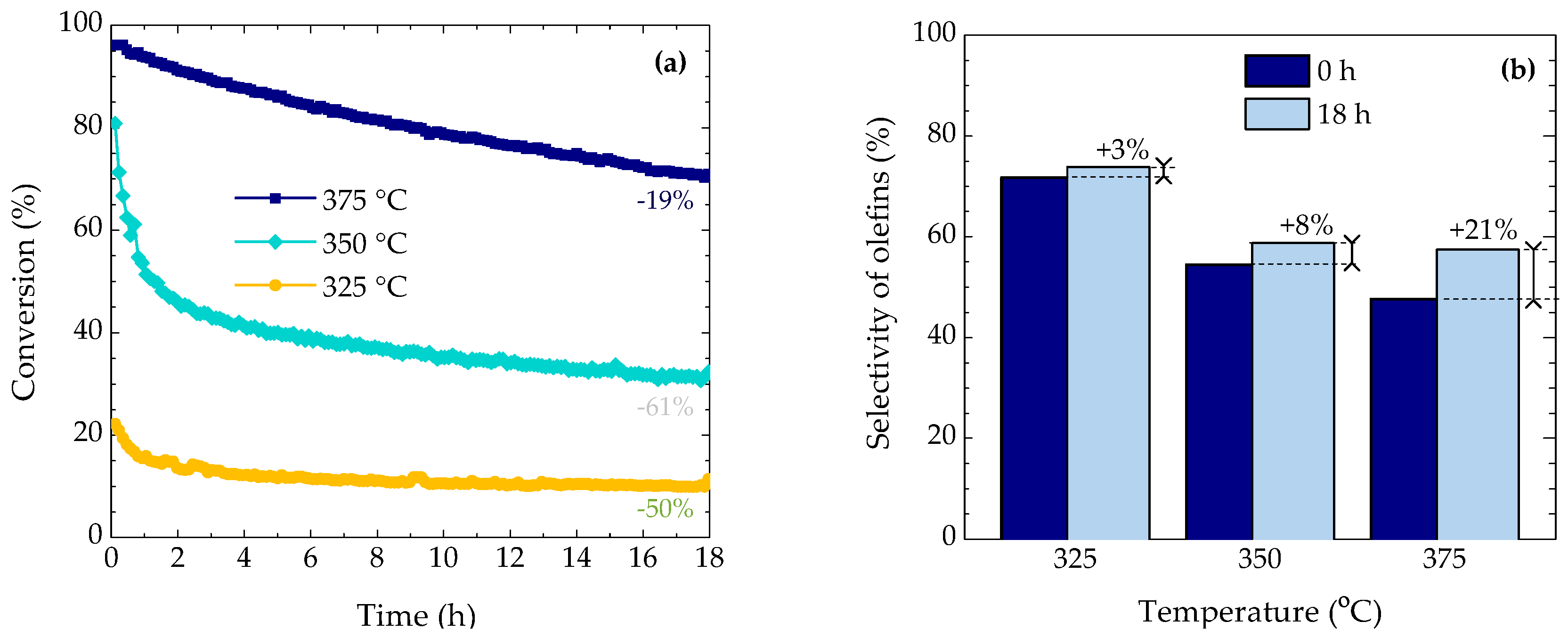
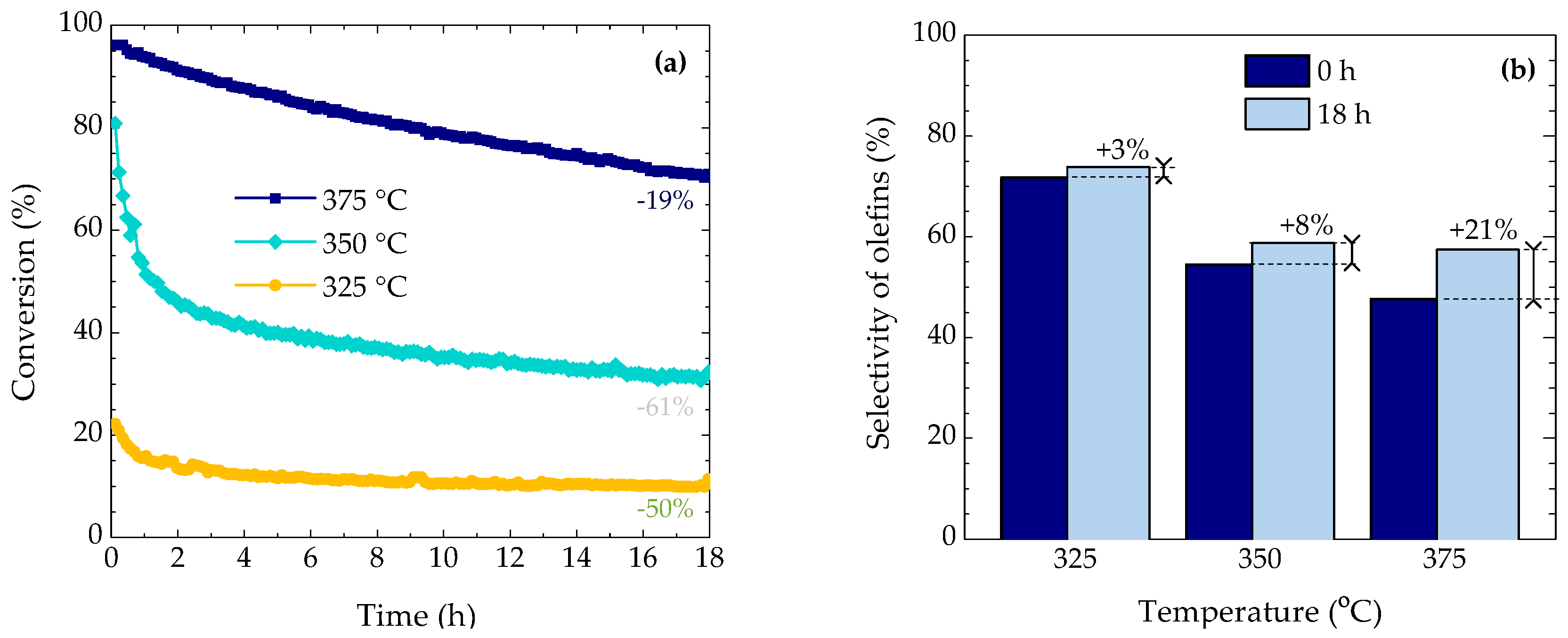
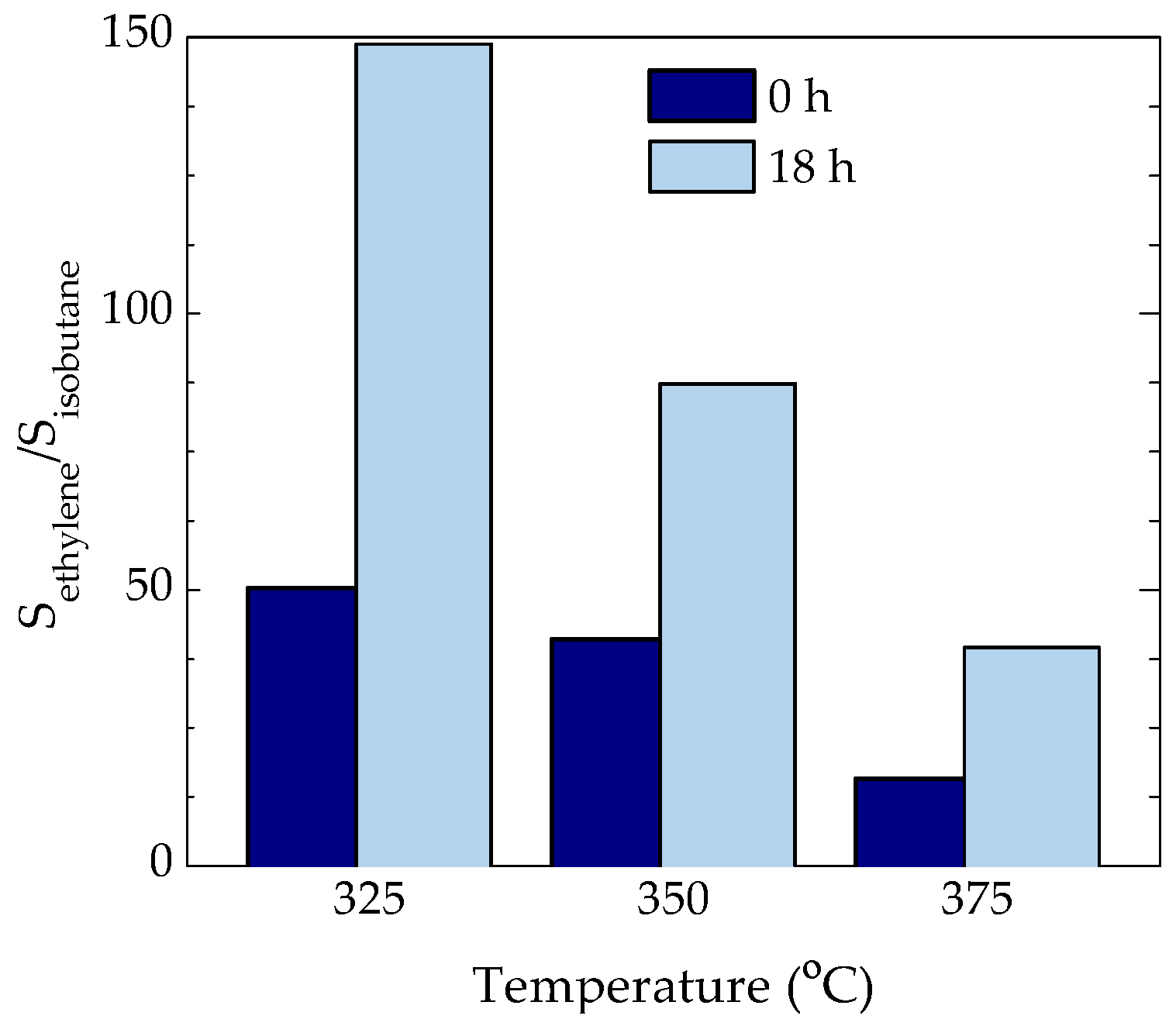
2.1. Evolution of Conversion and Selectivity
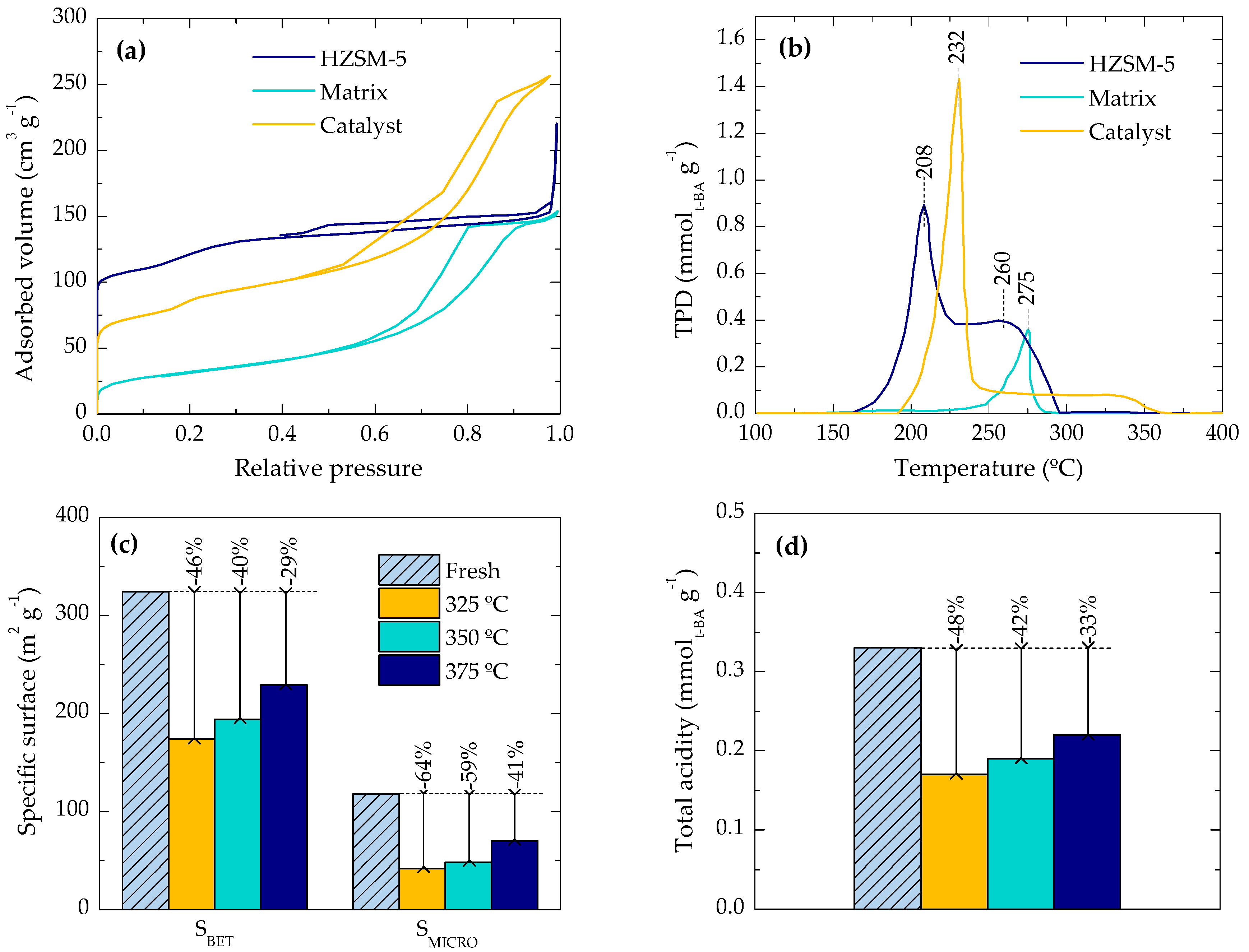
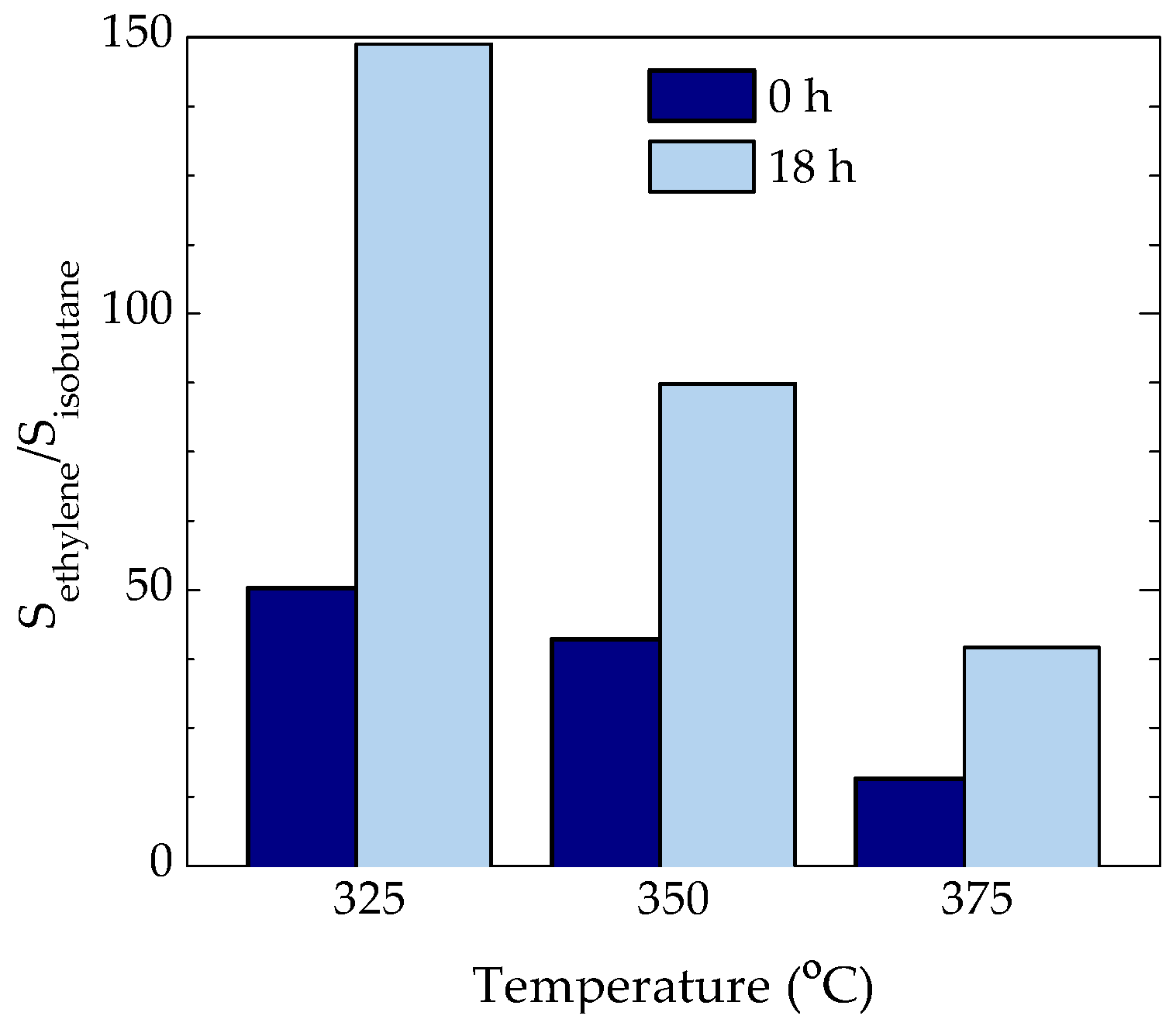
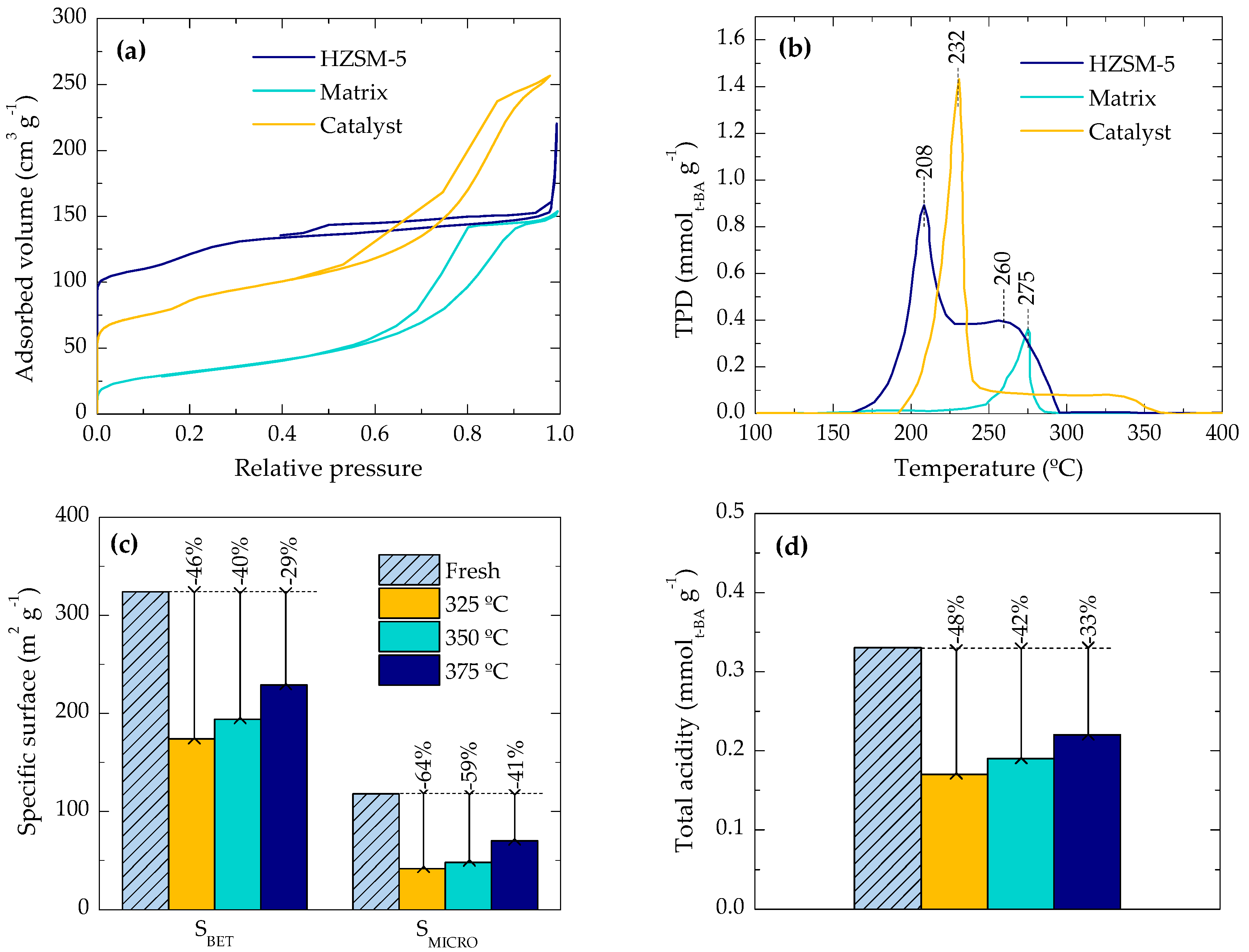
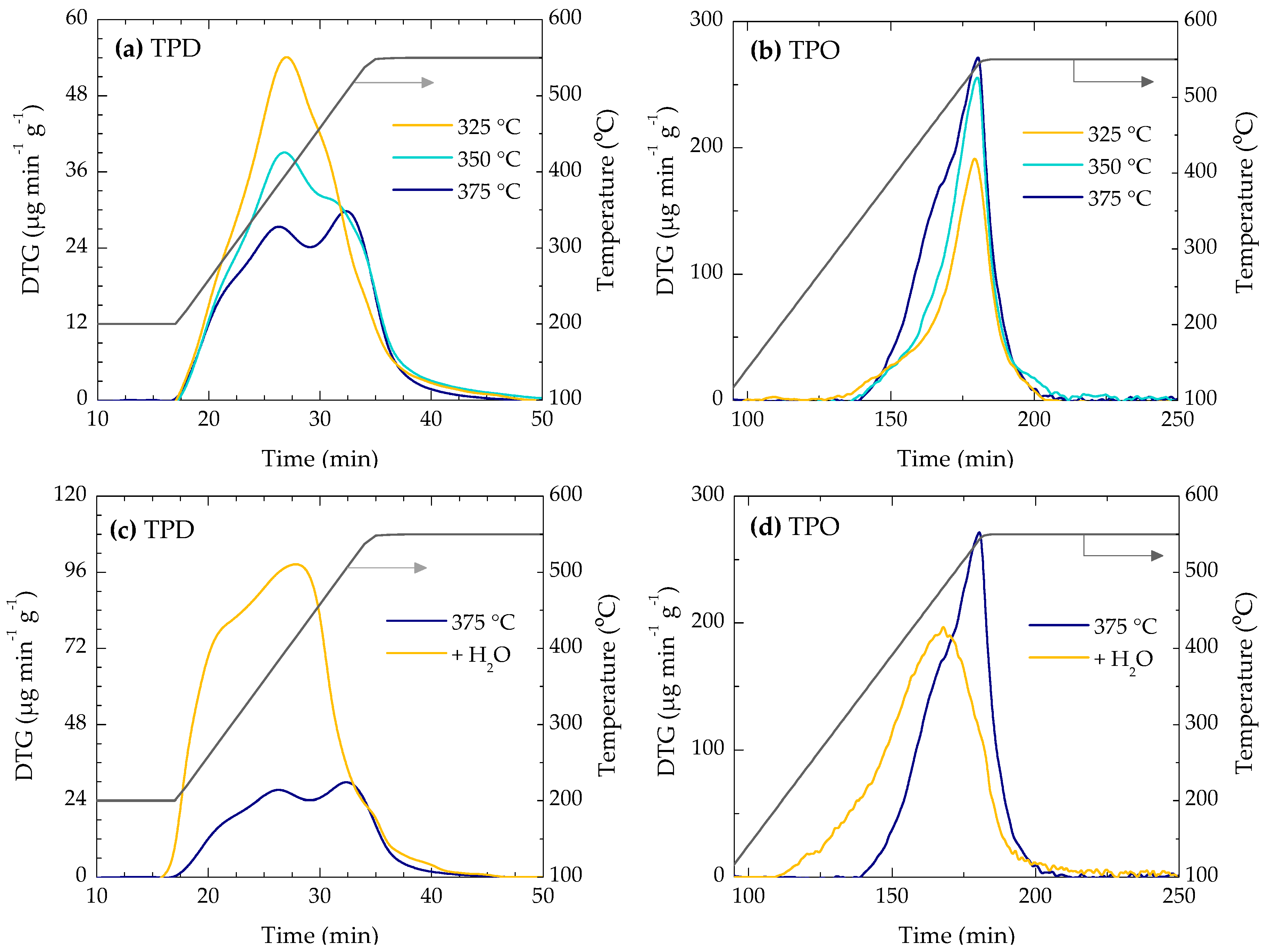
2.2. Location of Coke
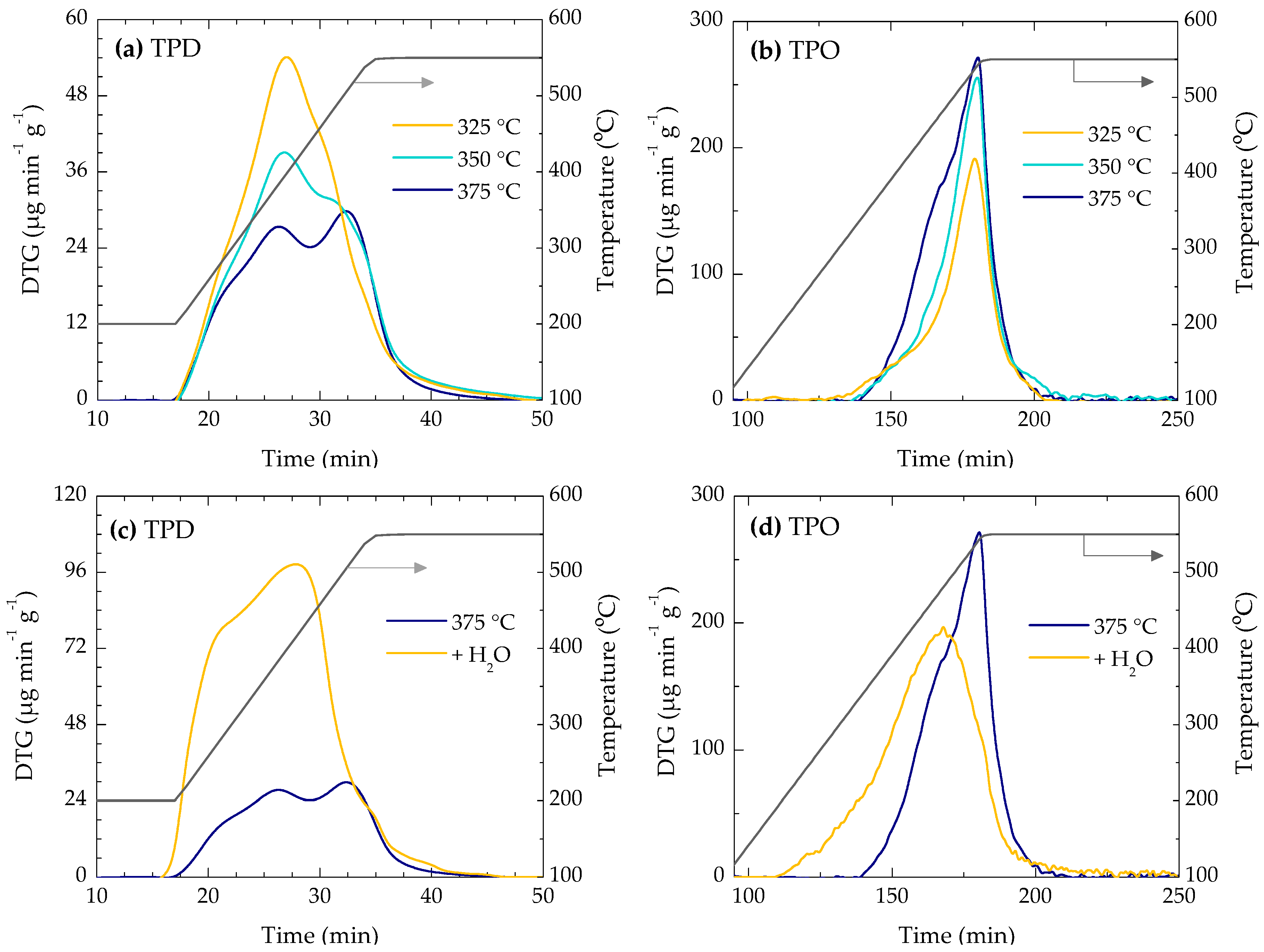
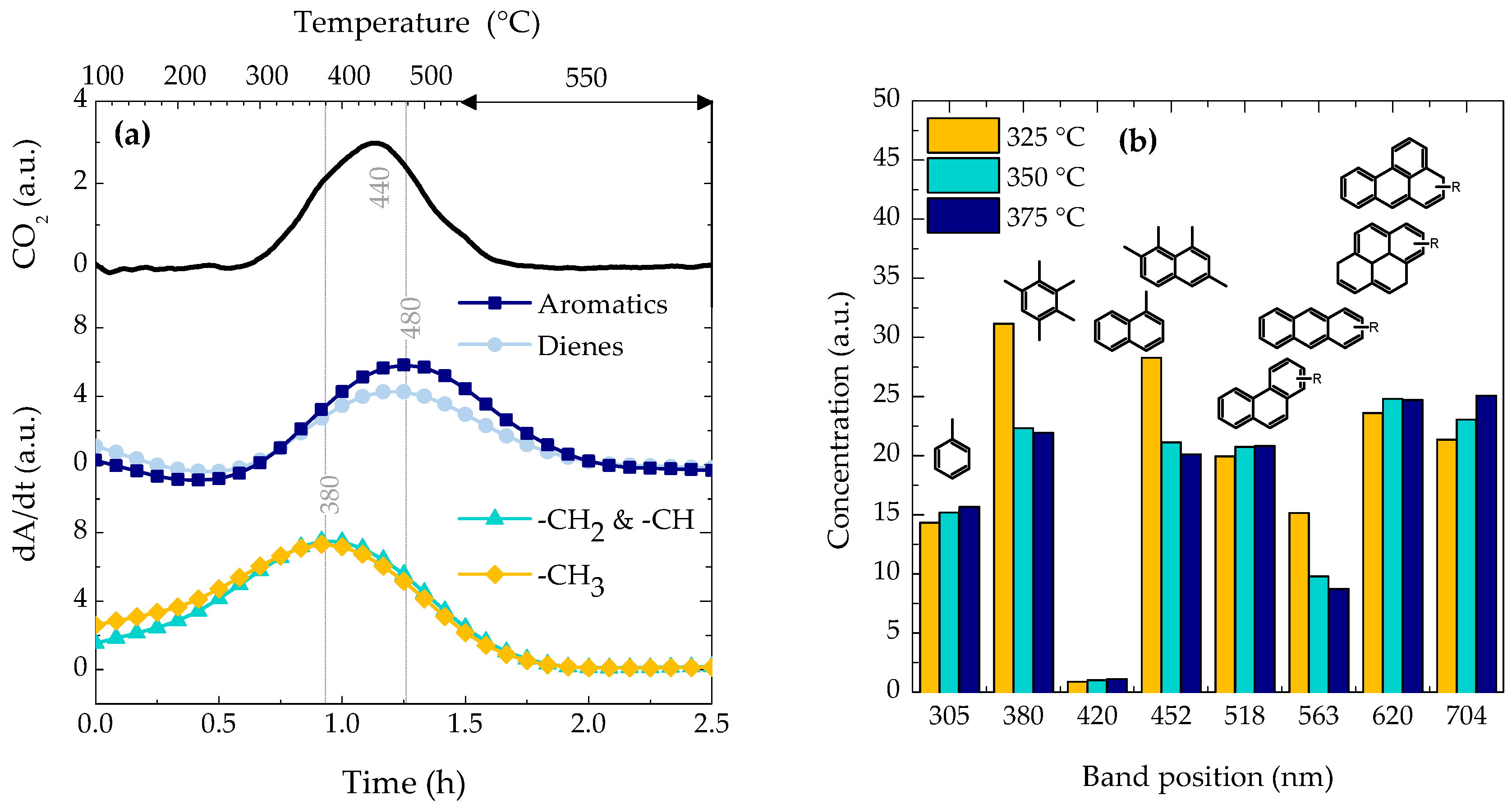
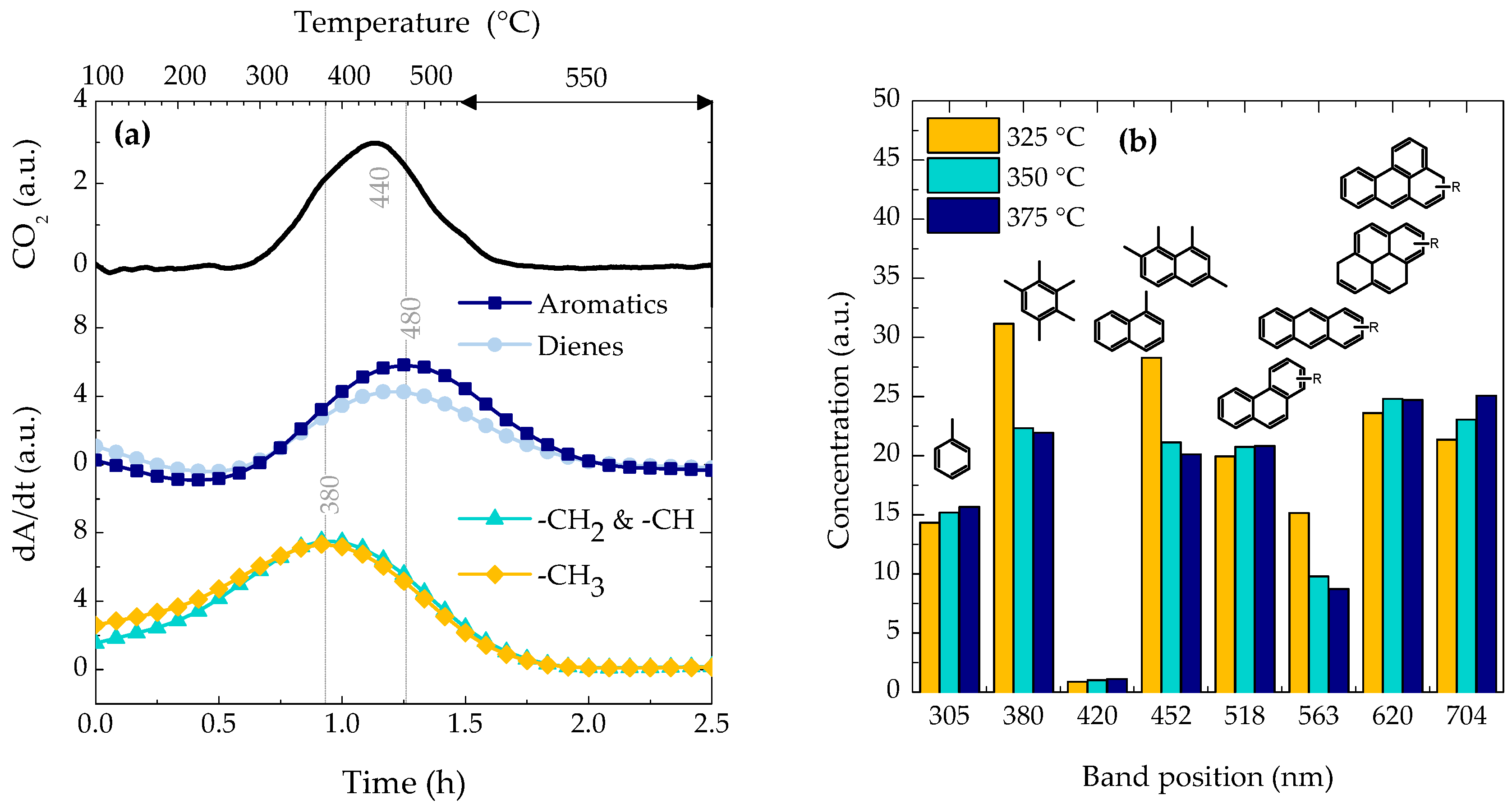
2.3. Coke Nature
3. Materials and Methods
3.1. Catalysis Preparation
3.2. Catalyst Characterization
3.3. Reaction and Analysis Equipment
3.4. Deactivated Catalyst Characterization
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Park, Y.K.; Lee, C.W.; Kang, N.Y.; Choi, W.C.; Choi, S.; Oh, S.H.; Park, D.S. Catalytic cracking of lower-valued hydrocarbons for producing light olefins. Catal. Surv. Asia 2010, 14, 75–84. [Google Scholar] [CrossRef]
- Park, S.; Inagaki, S.; Kubota, Y. Selective formation of light olefins from dimethyl ether over MCM-68 modified with phosphate species. Catal. Today 2016, 265, 218–224. [Google Scholar] [CrossRef]
- Sadrameli, S.M. Thermal/catalytic cracking of liquid hydrocarbons for the production of olefins: A state-of-the-art review II: Catalytic cracking review. Fuel 2016, 173, 285–297. [Google Scholar] [CrossRef]
- Chang, C.D.; Chu, C.T.W.; Socha, R.F. Methanol conversion to olefins over ZSM-5. I. Effect of temperature and zeolite SiO2/Al2O3. J. Catal. 1984, 86, 289–296. [Google Scholar] [CrossRef]
- Ateka, A.; Pérez-Uriarte, P.; Gamero, M.; Ereña, J.; Aguayo, A.T.; Bilbao, J. A comparative thermodynamic study on the CO2 conversion in the synthesis of methanol and of DME. Energy 2017, 120, 796–804. [Google Scholar] [CrossRef]
- Olah, G.A.; Goeppert, A.; Prakash, G.K.S. Chemical recycling of carbon dioxide to methanol and dimethyl ether: From greenhouse gas to renewable, environmentally carbon neutral fuels and synthetic hydrocarbons. J. Org. Chem. 2009, 74, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Ghavipour, M.; Behbahani, R.M.; Rostami, R.B.; Lemraski, A.S. Methanol/dimethyl ether to light olefins over SAPO-34: Comprehensive comparison of the products distribution and catalyst performance. J. Nat. Gas Sci. Eng. 2014, 21, 532–539. [Google Scholar] [CrossRef]
- Olsbye, U.; Svelle, S.; Bjørgen, M.; Beato, P.; Janssens, T.V.W.; Joensen, F.; Bordiga, S.; Lillerud, K.P. Conversion of methanol to hydrocarbons: How zeolite cavity and pore size controls product selectivity. Angew. Chem. 2012, 51, 5810–5831. [Google Scholar] [CrossRef] [PubMed]
- Abasov, S.I.; Babayeva, F.A.; Guliyev, B.B.; Piriyev, N.N.; Rustamov, M.I. Features of methanol and dimethyl ether conversion to hydrocarbons on modified zeolites Y and ZSM-5. Theor. Exp. Chem. 2013, 49, 58–64. [Google Scholar] [CrossRef]
- Pérez-Uriarte, P.; Ateka, A.; Aguayo, A.T.; Bilbao, J. Comparison of HZSM-5 zeolite and SAPO (-18 and -34) based catalysts for the production of light olefins from DME. Catal. Lett. 2016, 146, 1892–1902. [Google Scholar] [CrossRef]
- Hirota, Y.; Yamada, M.; Uchida, Y.; Sakamoto, Y.; Yokoi, T.; Nishiyama, N. Synthesis of SAPO-18 with low acidic strength and its application in conversion of dimethylether to olefins. Microporous Mesoporous Mater. 2016, 232, 65–69. [Google Scholar] [CrossRef]
- Zhao, D.; Zhang, Y.; Li, Z.; Wang, Y.; Yu, J. Synthesis of SAPO-18/34 intergrowth zeolites and their enhanced stability for dimethyl ether to olefins. RSC Adv. 2017, 7, 939–946. [Google Scholar] [CrossRef]
- Zhao, D.; Zhang, Y.; Peng, Y.; Yu, J. A novel preparation of SAPO-18 molecular sieve with enhanced stability for dimethyl ether to olefins. Catal. Lett. 2016, 146, 2261–2267. [Google Scholar] [CrossRef]
- Al-Dughaither, A.S.; De Lasa, H. HZSM-5 zeolites with different SiO2/Al2O3 ratios. Characterization and NH3 desorption kinetics. Ind. Eng. Chem. Res. 2014, 53, 15303–15316. [Google Scholar] [CrossRef]
- Pérez-Uriarte, P.; Gamero, M.; Ateka, A.; Díaz, M.; Aguayo, A.T.; Bilbao, J. Effect of the acidity of HZSM-5 zeolite and the binder in the DME transformation to olefins. Ind. Eng. Chem. Res. 2016, 55, 1513–1521. [Google Scholar] [CrossRef]
- Al-Dughaither, A.S.; De Lasa, H. Neat dimethyl ether conversion to olefins (DTO) over HZSM-5: Effect of SiO2/Al2O3 on porosity, surface chemistry, and reactivity. Fuel 2014, 138, 52–64. [Google Scholar] [CrossRef]
- Bleken, F.; Skistad, W.; Barbera, K.; Kustova, M.; Bordiga, S.; Beato, P.; Lillerud, K.P.; Svelle, S.; Olsbye, U. Conversion of methanol over 10-ring zeolites with differing volumes at channel intersections: Comparison of TNU-9, IM-5, ZSM-11 and ZSM-5. Phys. Chem. Chem. Phys. 2011, 13, 2539–2549. [Google Scholar] [CrossRef] [PubMed]
- Gil-Coba, J.; Marie-Rose, S.C.; Lavoie, J.M. Effect of water content and catalysts acidity in the products distribution during propylene synthesis with a mixture of DME and methanol. Catal. Lett. 2016, 146, 2534–2542. [Google Scholar] [CrossRef]
- Ilias, S.; Bhan, A. Mechanism of the catalytic conversion of methanol to hydrocarbons. ACS Catal. 2013, 3, 18–31. [Google Scholar] [CrossRef]
- Khare, R.; Liu, Z.; Han, Y.; Bhan, A. A mechanistic basis for the effect of aluminum content on ethene selectivity in methanol-to-hydrocarbons conversion on HZSM-5. J. Catal. 2017, 348, 300–305. [Google Scholar] [CrossRef]
- Liang, T.; Chen, J.; Qin, Z.; Li, J.; Wang, P.; Wang, S.; Wang, G.; Dong, M.; Fan, W.; Wang, J. Conversion of methanol to olefins over H-ZSM-5 zeolite: Reaction pathway is related to the framework aluminum siting. ACS Catal. 2016, 6, 7311–7325. [Google Scholar] [CrossRef]
- Tian, P.; Wei, Y.; Ye, M.; Liu, Z. Methanol to olefins (MTO): From fundamentals to commercialization. ACS Catal. 2015, 5, 1922–1938. [Google Scholar] [CrossRef]
- Hao, J.; Zhao, Y.; Ye, M.; Liu, Z. Influence of temperature on fluidized-bed catalyst attrition behavior. Chem. Eng. Technol. 2016, 39, 927–934. [Google Scholar] [CrossRef]
- Vu, X.H.; Armbruster, U.; Martin, A. Micro/mesoporous zeolitic composites: Recent developments in synthesis and catalytic applications. Catalysts 2016, 6, 183. [Google Scholar] [CrossRef]
- Michels, N.L.; Mitchell, S.; Pérez-Ramírez, J. Effects of binders on the performance of shaped hierarchical MFI zeolites in methanol-to-hydrocarbons. ACS Catal. 2014, 4, 2409–2417. [Google Scholar] [CrossRef]
- Ibáñez, M.; Artetxe, M.; López, G.; Elordi, G.; Bilbao, J.; Olazar, M.; Castaño, P. Identification of the coke deposited on an HZSM-5 zeolite catalyst during the sequenced pyrolysis-cracking of HDPE. Appl. Catal. B 2014, 148, 436–445. [Google Scholar] [CrossRef]
- Guisnet, M.; Magnoux, P. Organic chemistry of coke formation. Appl. Catal. A Gen. 2001, 212, 83–96. [Google Scholar] [CrossRef]
- Schulz, H. “Coking” of zeolites during methanol conversion: Basic reactions of the MTO-, MTP- and MTG processes. Catal. Today 2010, 154, 183–194. [Google Scholar] [CrossRef]
- Olsbye, U.; Svelle, S.; Lillerud, K.P.; Wei, Z.H.; Chen, Y.Y.; Li, J.F.; Wang, J.G.; Fan, W.B. The formation and degradation of active species during methanol conversion over protonated zeotype catalysts. Chem. Soc. Rev. 2015, 44, 7155–7176. [Google Scholar] [CrossRef] [PubMed]
- Castaño, P.; Ruiz-Martinez, J.; Epelde, E.; Gayubo, A.G.; Weckhuysen, B.M. Spatial distribution of zeolite ZSM-5 within catalyst bodies affects selectivity and stability of methanol-to-hydrocarbons conversion. ChemCatChem 2013, 5, 2827–2831. [Google Scholar] [CrossRef]
- Castaño, P.; Elordi, G.; Olazar, M.; Bilbao, J. Imaging the profiles of deactivating species on the catalyst used for the cracking of waste polyethylene by combined microscopies. ChemCatChem 2012, 4, 631–635. [Google Scholar] [CrossRef]
- Aguayo, A.T.; Castaño, P.; Mier, D.; Gayubo, A.G.; Olazar, M.; Bilbao, J. Effect of cofeeding butane with methanol on the deactivation by coke of a HZSM-5 zeolite catalyst. Ind. Eng. Chem. Res. 2011, 50, 9980–9988. [Google Scholar] [CrossRef]
- Gayubo, A.G.; Alonso, A.; Valle, B.; Aguayo, A.T.; Olazar, M.; Bilbao, J. Kinetic modelling for the transformation of bioethanol into olefins on a hydrothermally stable Ni-HZSM-5 catalyst considering the deactivation by coke. Chem. Eng. J. 2011, 167, 262–277. [Google Scholar] [CrossRef]
- Corma, A.; Melo, F.V.; Sauvanaud, L. Attempts to improve the product slate quality: Influence of coke-on-catalyst content. Ind. Eng. Chem. Res. 2007, 46, 4100–4109. [Google Scholar] [CrossRef]
- Epelde, E.; Santos, J.I.; Florian, P.; Aguayo, A.T.; Gayubo, A.G.; Bilbao, J.; Castaño, P. Controlling coke deactivation and cracking selectivity of MFI zeolite by H3PO4 or KOH modification. Appl. Catal. A Gen. 2015, 505, 105–115. [Google Scholar] [CrossRef]
- Epelde, E.; Ibañez, M.; Aguayo, A.T.; Gayubo, A.G.; Bilbao, J.; Castaño, P. Differences among the deactivation pathway of HZSM-5 zeolite and SAPO-34 in the transformation of ethylene or 1-butene to propylene. Microporous Mesoporous Mater. 2014, 195, 284–293. [Google Scholar] [CrossRef]
- Epelde, E.; Gayubo, A.G.; Olazar, M.; Bilbao, J.; Aguayo, A.T. Modified HZSM-5 zeolites for intensifying propylene production in the transformation of 1-butene. Chem. Eng. J. 2014, 251, 80–91. [Google Scholar] [CrossRef]
- Gayubo, A.G.; Aguayo, A.T.; Morán, A.L.; Olazar, M.; Bilbao, J. Role of water in the kinetic modeling of catalyst deactivation in the MTG process. AIChE J. 2002, 48, 1561–1571. [Google Scholar] [CrossRef]
- Aguayo, A.T.; Gayubo, A.G.; Atutxa, A.; Olazar, M.; Bilbao, J. Catalyst deactivation by coke in the transformation of aqueous ethanol into hydrocarbons. Kinetic modeling and acidity deterioration of the catalyst. Ind. Eng. Chem. Res. 2002, 41, 4216–4224. [Google Scholar] [CrossRef]
- Mier, D.; Gayubo, A.G.; Aguayo, A.T.; Olazar, M.; Bilbao, J. Olefin production by cofeeding methanol and n-butane: Kinetic modeling considering the deactivation of HZSM-5 zeolite. AIChE J. 2011, 57, 2841–2853. [Google Scholar] [CrossRef]
- Chen, S.; Manos, G. Study of coke and coke precursors during catalytic cracking of n-hexane and 1-hexene over ultrastable y zeolite. Catal. Lett. 2004, 96, 195–200. [Google Scholar] [CrossRef]
- Chen, S.; Manos, G. In situ thermogravimetric study of coke formation during catalytic cracking of normal hexane and 1-hexene over ultrastable y zeolite. J. Catal. 2004, 226, 343–350. [Google Scholar] [CrossRef]
- Bauer, F.; Karge, H.G. Characterization of coke on zeolites. Mol. Sieves 2007, 5, 249–364. [Google Scholar]
- Ibáñez, M.; Gamero, M.; Ruiz-Martínez, J.; Weckhuysen, B.M.; Aguayo, A.T.; Bilbao, J.; Castaño, P. Simultaneous coking and dealumination of zeolite H-ZSM-5 during the transformation of chloromethane into olefins. Catal. Sci. Technol. 2016, 6, 296–306. [Google Scholar] [CrossRef]
- Pérez-Uriarte, P.; Ateka, A.; Gayubo, A.G.; Cordero-Lanzac, T.; Aguayo, A.T.; Bilbao, J. Deactivation kinetics for the conversion of dimethyl ether to olefins over a HZSM-5 zeolite catalyst. Chem. Eng. J. 2017, 311, 367–377. [Google Scholar] [CrossRef]
- Mukarakate, C.; McBrayer, J.D.; Evans, T.J.; Budhi, S.; Robichaud, D.J.; Iisa, K.; Ten Dam, J.; Watson, M.J.; Baldwin, R.M.; Nimlos, M.R. Catalytic fast pyrolysis of biomass: The reactions of water and aromatic intermediates produces phenols. Green Chem. 2015, 17, 4217–4227. [Google Scholar] [CrossRef]
- Pérez-Uriarte, P.; Ateka, A.; Aguayo, A.T.; Gayubo, A.G.; Bilbao, J. Kinetic model for the reaction of DME to olefins over a HZSM-5 zeolite catalyst. Chem. Eng. J. 2016, 302, 801–810. [Google Scholar] [CrossRef]
- Karge, H.G.; Nießen, W.; Bludau, H. In-situ FTIR measurements of diffusion in coking zeolite catalysts. Appl. Catal. A Gen. 1996, 146, 339–349. [Google Scholar] [CrossRef]
- Arsenova, N.; Bludau, H.; Haag, W.O.; Karge, H.G. In situ IR spectroscopic study of the adsorption behaviour of ethylbenzene and diethylbenzenes related to ethylbenzene disproportionation over hy zeolite. Microporous Mesoporous Mater. 1998, 23, 1–10. [Google Scholar] [CrossRef]
- Castaño, P.; Elordi, G.; Olazar, M.; Aguayo, A.T.; Pawelec, B.; Bilbao, J. Insights into the coke deposited on HZSM-5, Hβ and HY zeolites during the cracking of polyethylene. Appl. Catal. B Environ. 2011, 104, 91–100. [Google Scholar] [CrossRef]
- Aguayo, A.T.; Gayubo, A.G.; Ereña, J.; Atutxa, A.; Bilbao, J. Coke aging and its incidence on catalyst regeneration. Ind. Eng. Chem. Res. 2003, 42, 3914–3921. [Google Scholar] [CrossRef]
- Mores, D.; Stavitski, E.; Kox, M.H.F.; Kornatowski, J.; Olsbye, U.; Weckhuysen, B.M. Space- and time-resolved in-situ spectroscopy on the coke formation in molecular sieves: Methanol-to-olefin conversion over H-ZSM-5 and H-SAPO-34. Chem. Eur. J. 2008, 14, 11320–11327. [Google Scholar] [CrossRef] [PubMed]
- Mores, D.; Kornatowski, J.; Olsbye, U.; Weckhuysen, B.M. Coke formation during the methanol-to-olefin conversion: In situ microspectroscopy on individual H-ZSM-5 crystals with different brønsted acidity. Chem. A Eur. J. 2011, 17, 2874–2884. [Google Scholar] [CrossRef] [PubMed]
- Benito, P.L.; Aguayo, A.T.; Gayubo, A.G.; Bilbao, J. Catalyst equilibration for transformation of methanol into hydrocarbons by reaction-regeneration cycles. Ind. Eng. Chem. Res. 1996, 35, 2177–2182. [Google Scholar] [CrossRef]
- Aguayo, A.T.; Gayubo, A.G.; Vivanco, R.; Olazar, M.; Bilbao, J. Role of acidity and microporous structure in alternative catalysts for the transformation of methanol into olefins. Appl. Catal. A Gen. 2005, 283, 197–207. [Google Scholar] [CrossRef]
- Pérez-Uriarte, P.; Ateka, A.; Gamero, M.; Aguayo, A.T.; Bilbao, J. Effect of the operating conditions in the transformation of DME to olefins over a HZSM-5 zeolite catalyst. Ind. Eng. Chem. Res. 2016, 55, 6569–6578. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature (°C) | 325 | 350 | 375 |
| TPD Species (wt %) | 3.64 | 3.33 | 2.57 |
| Coke (wt %) | 1.33 | 1.41 | 1.42 |
| Total (wt %) | 4.98 | 4.75 | 3.99 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibáñez, M.; Pérez-Uriarte, P.; Sánchez-Contador, M.; Cordero-Lanzac, T.; Aguayo, A.T.; Bilbao, J.; Castaño, P. Nature and Location of Carbonaceous Species in a Composite HZSM-5 Zeolite Catalyst during the Conversion of Dimethyl Ether into Light Olefins. Catalysts 2017, 7, 254. https://doi.org/10.3390/catal7090254
Ibáñez M, Pérez-Uriarte P, Sánchez-Contador M, Cordero-Lanzac T, Aguayo AT, Bilbao J, Castaño P. Nature and Location of Carbonaceous Species in a Composite HZSM-5 Zeolite Catalyst during the Conversion of Dimethyl Ether into Light Olefins. Catalysts. 2017; 7(9):254. https://doi.org/10.3390/catal7090254
Chicago/Turabian StyleIbáñez, María, Paula Pérez-Uriarte, Miguel Sánchez-Contador, Tomás Cordero-Lanzac, Andrés T. Aguayo, Javier Bilbao, and Pedro Castaño. 2017. "Nature and Location of Carbonaceous Species in a Composite HZSM-5 Zeolite Catalyst during the Conversion of Dimethyl Ether into Light Olefins" Catalysts 7, no. 9: 254. https://doi.org/10.3390/catal7090254
APA StyleIbáñez, M., Pérez-Uriarte, P., Sánchez-Contador, M., Cordero-Lanzac, T., Aguayo, A. T., Bilbao, J., & Castaño, P. (2017). Nature and Location of Carbonaceous Species in a Composite HZSM-5 Zeolite Catalyst during the Conversion of Dimethyl Ether into Light Olefins. Catalysts, 7(9), 254. https://doi.org/10.3390/catal7090254