Enzymatic Systems for Cellulose Acetate Degradation


 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
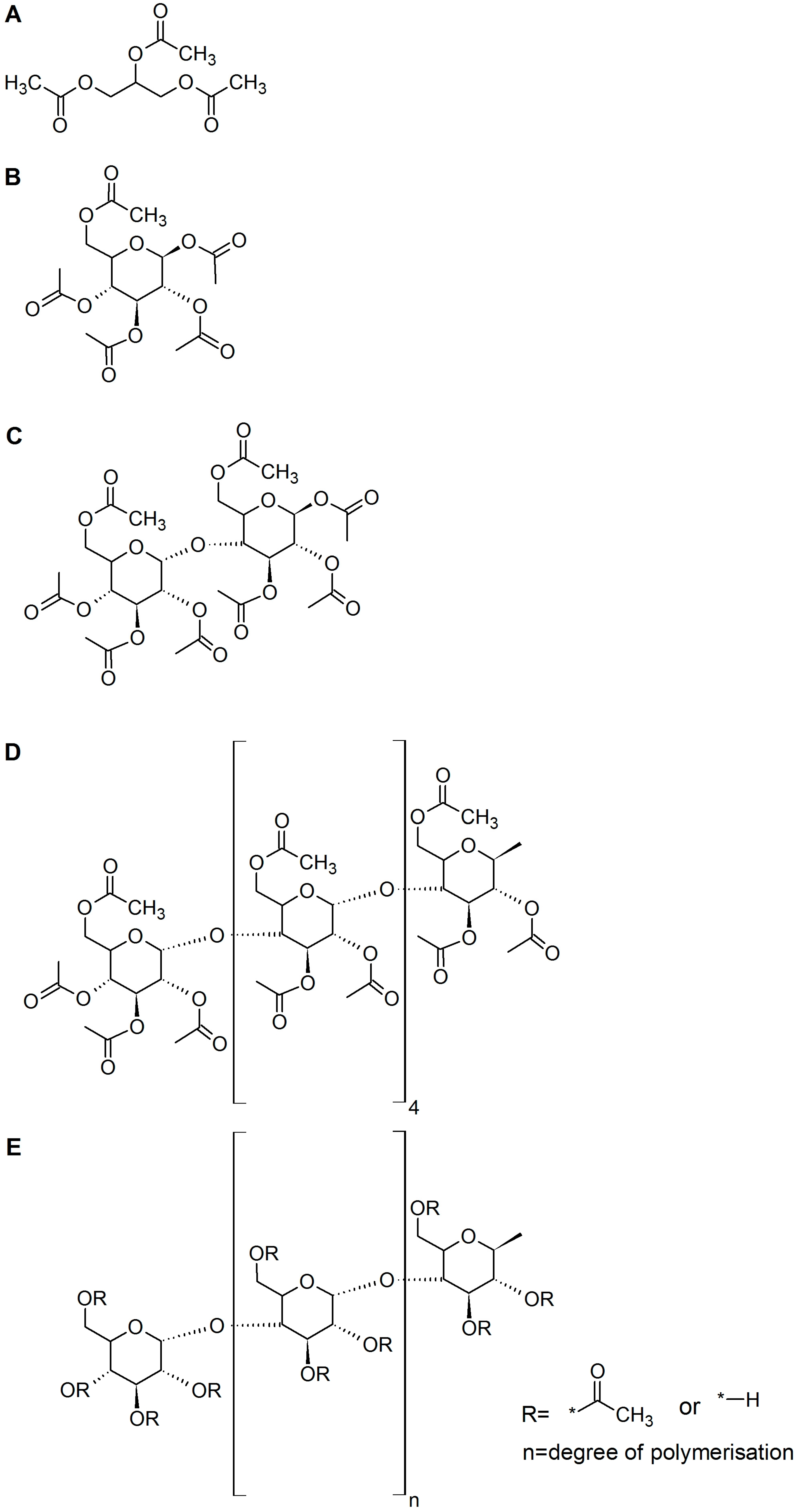
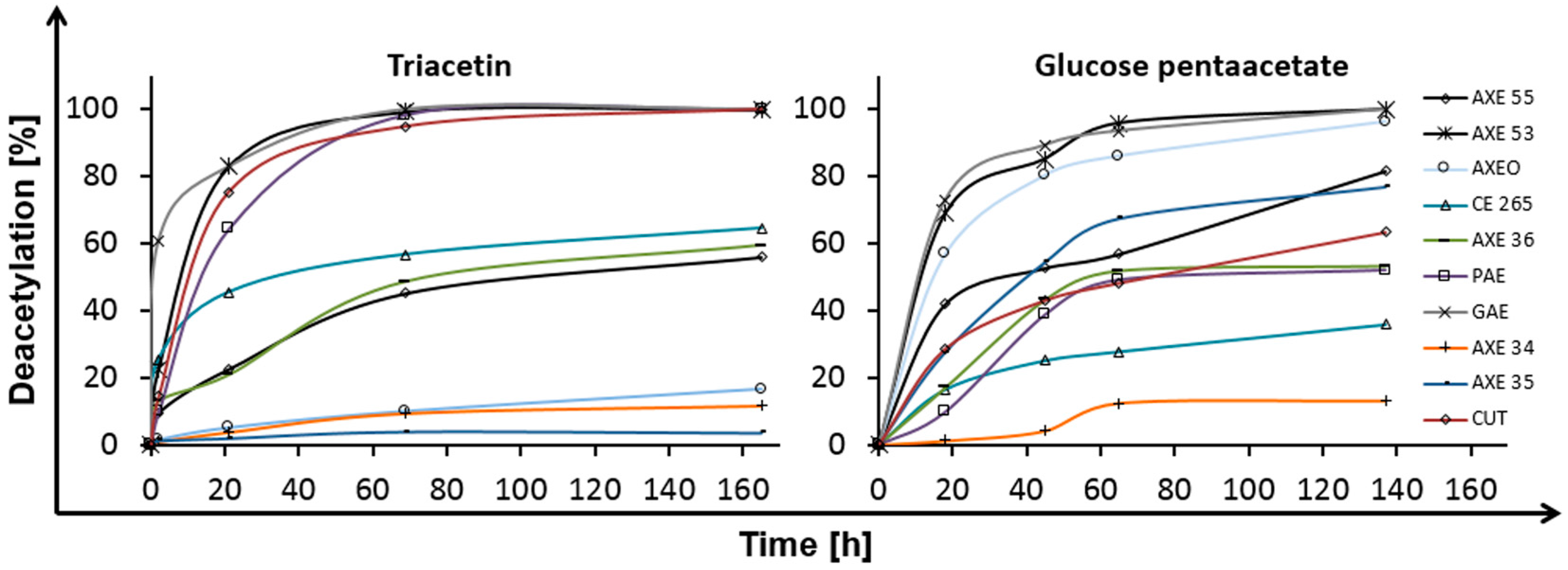
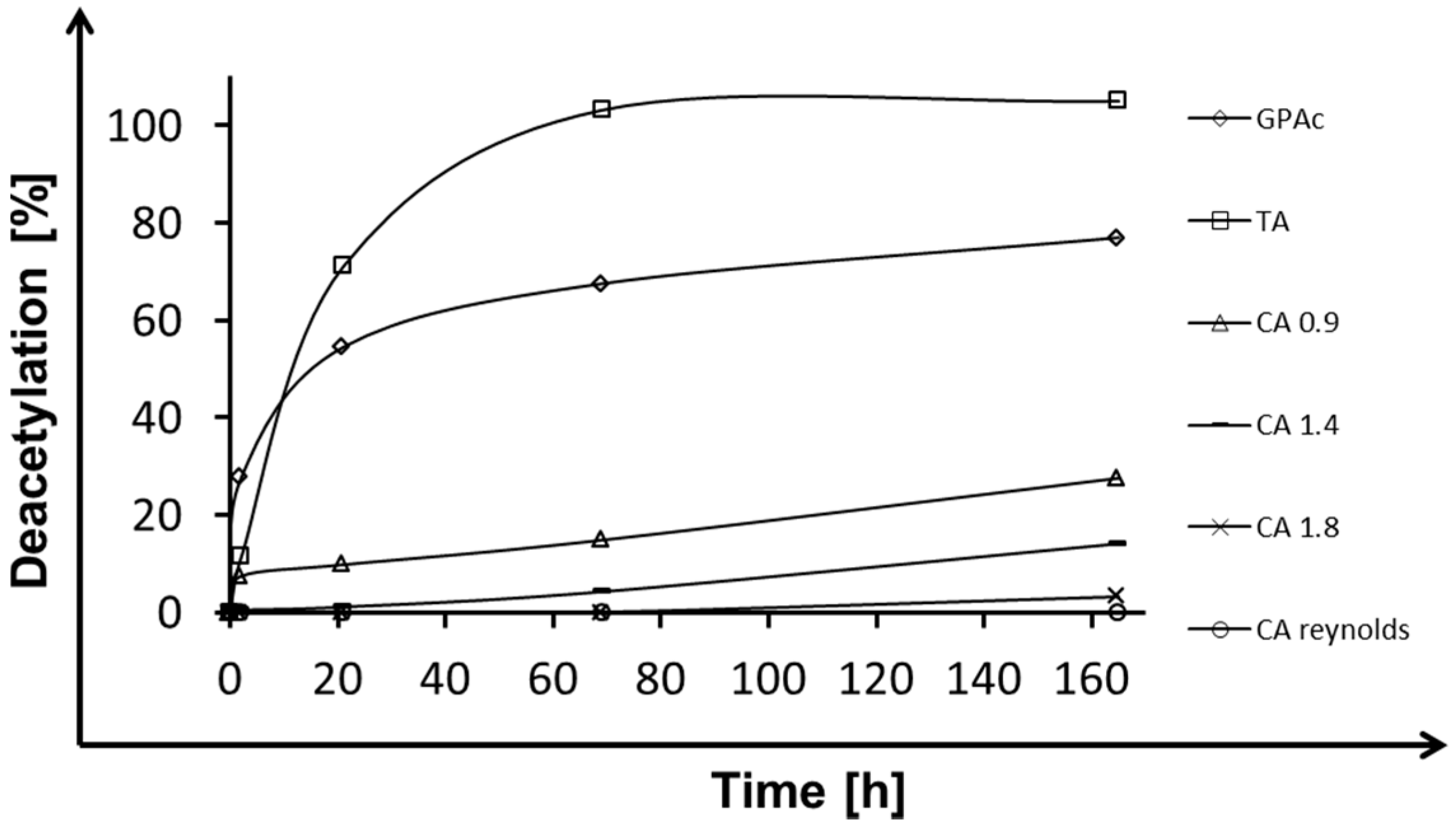
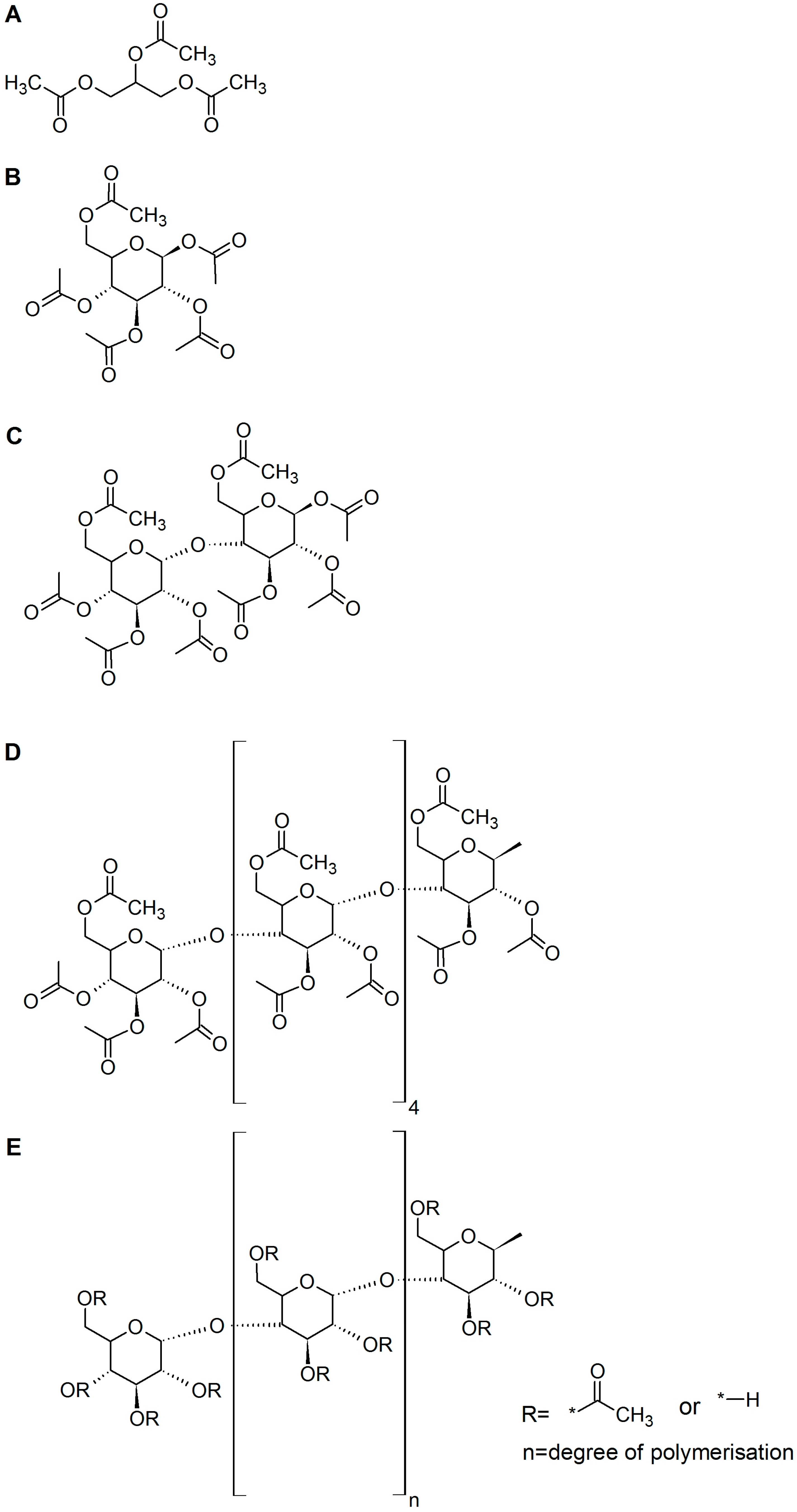
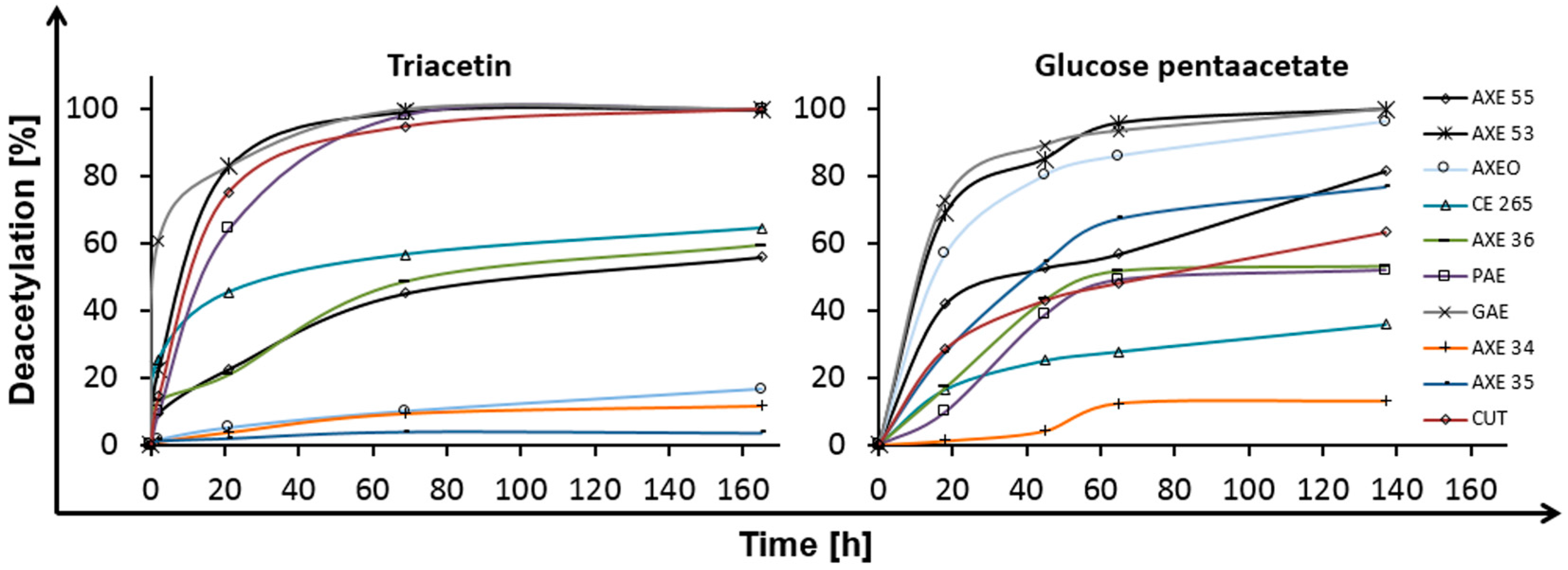
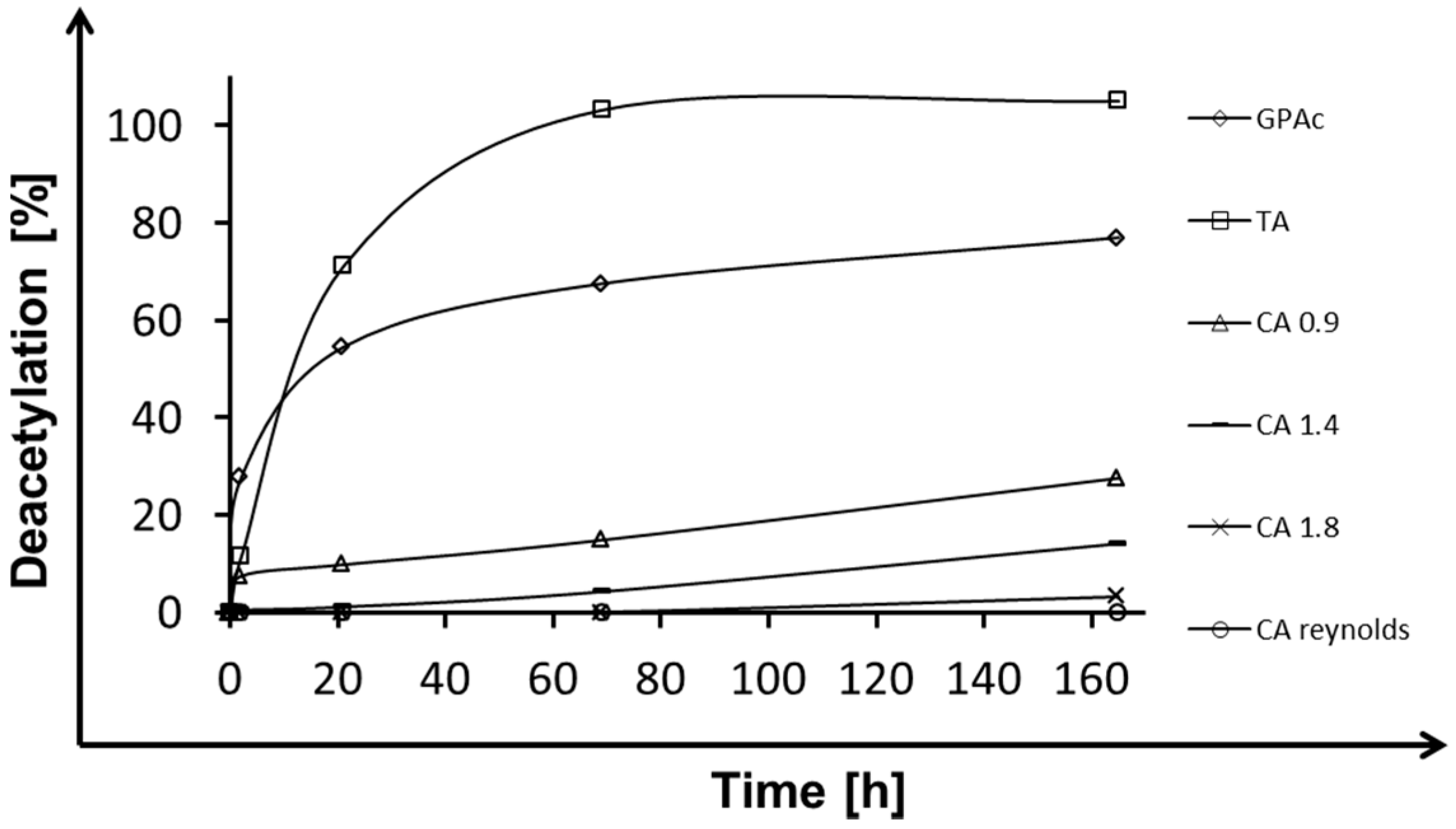
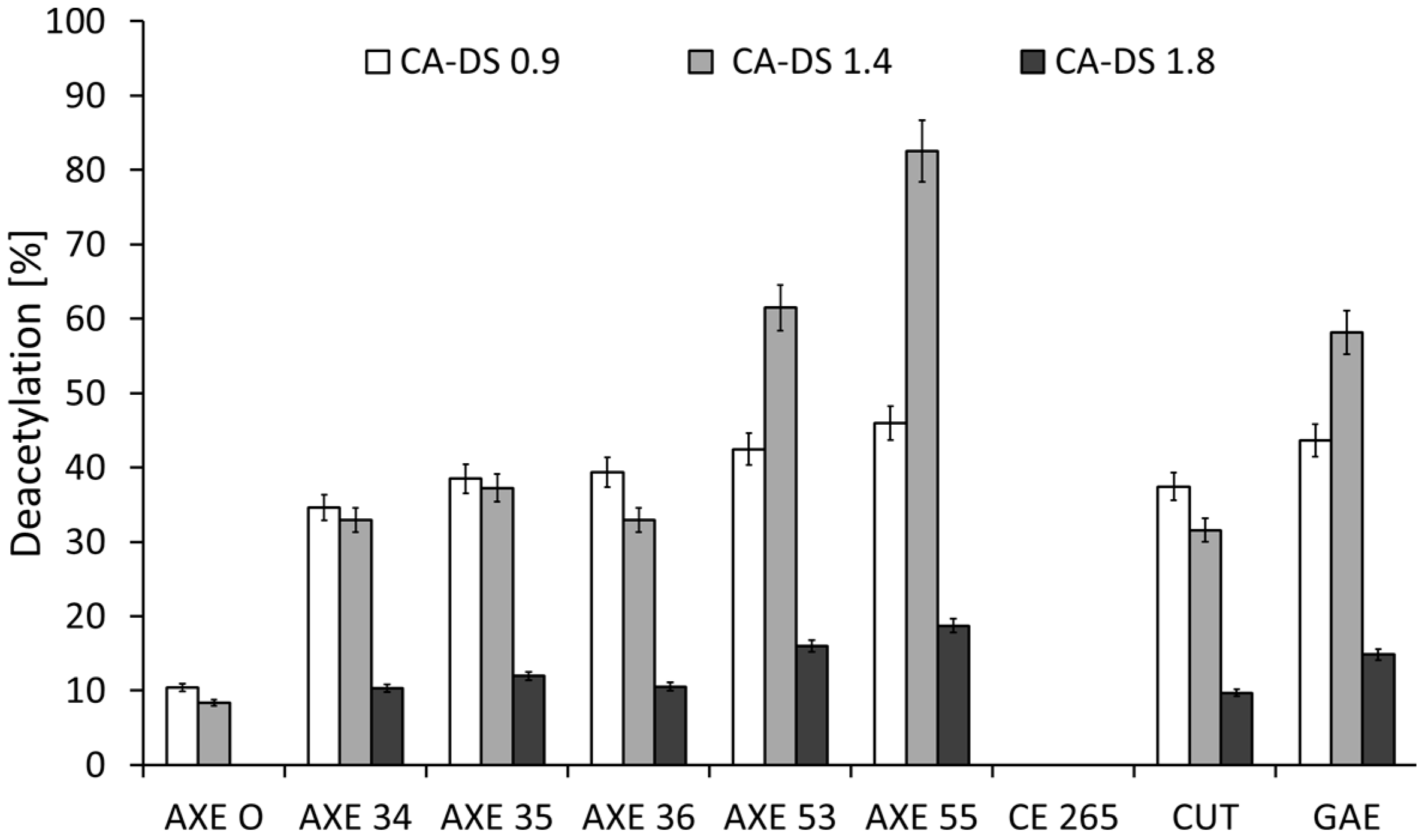
2.1. Deacetylation
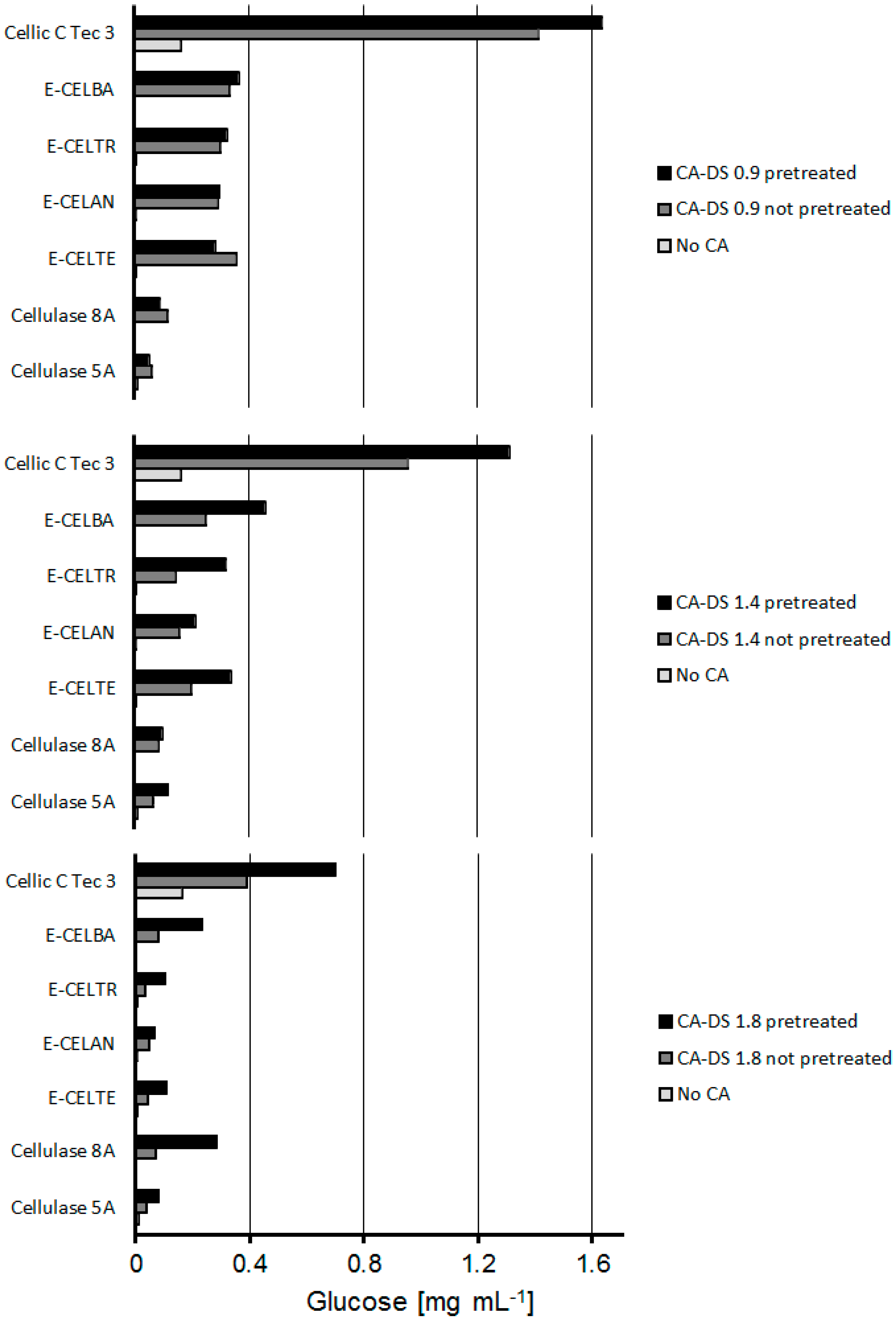
2.2. Combined CA Degradation with Esterases and Cellulases
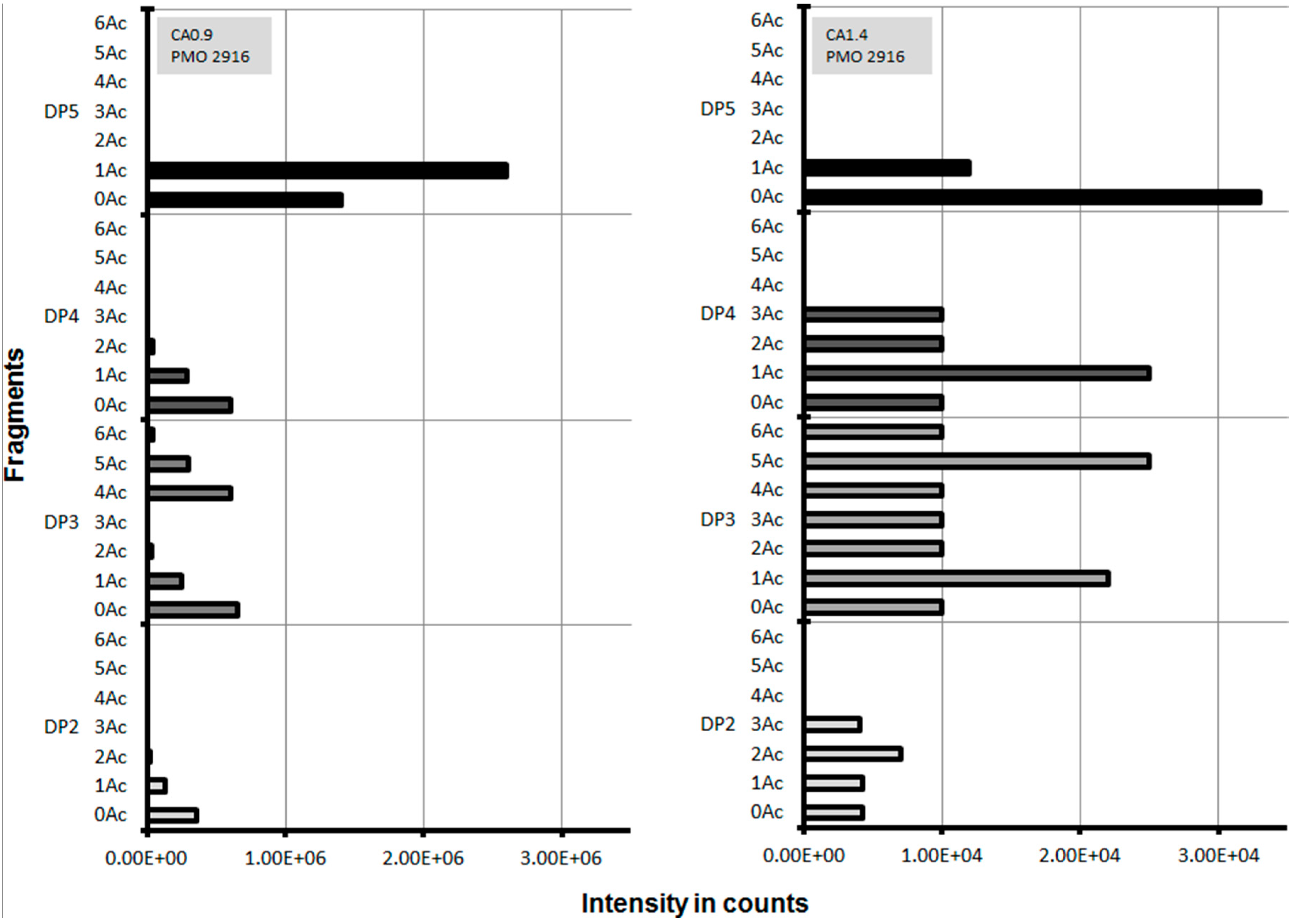
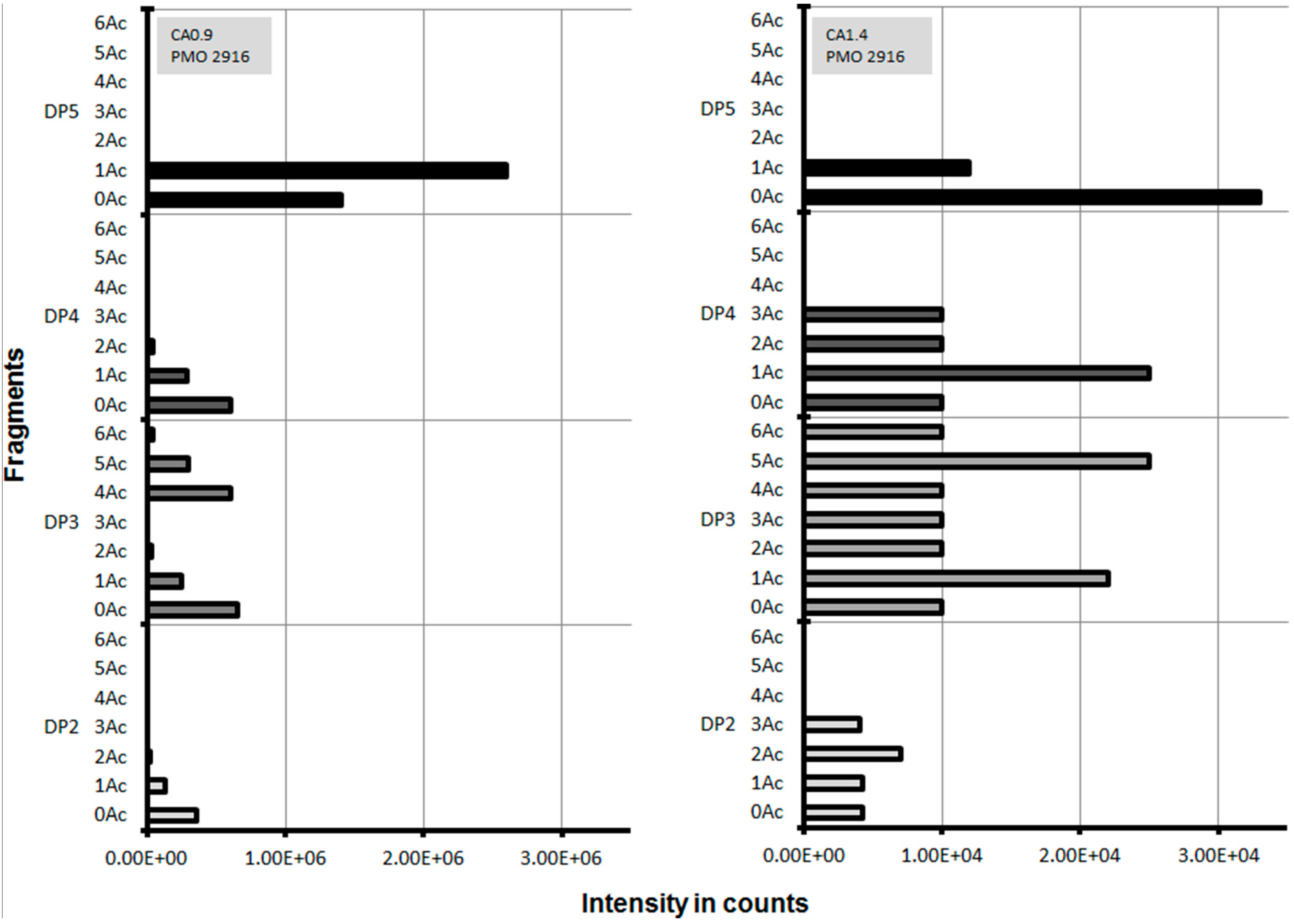
2.3. Lytic Polysaccharide Monooxygenase (LPMO) Hydrolysis of CA
3. Materials and Methods
3.1. Enzymes, Substrates, and Other Chemicals
3.2. Deacetylation of CA and Acetylated Oligomer
3.3. Combined Deacetylation and Hydrolysis of CA
3.4. Direct Enzymatic Hydrolysis of CA
3.5. Monitoring Deacetylation of CA and Oligomers
3.6. Monitoring Enzymatic Cleavage of CA
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Quintana, R.; Persenaire, O.; Lemmouchi, Y.; Bonnaud, L.; Dubois, P. Compatibilization of Co-Plasticized Cellulose Acetate/water Soluble Polymers Blends by Reactive Extrusion. Polym. Degrad. Stabil. 2016, 126, 31–38. [Google Scholar] [CrossRef]
- Rosa, D.S.G.; Uedes, C.G.F.; Casarin, F.; Braganca, F.C. The Effect of the Mw of PEG in PCL/CA Blends. Polym. Test. 2005, 24, 542–548. [Google Scholar] [CrossRef]
- Daud, W.W.R.; Djuned, F.M. Cellulose Acetate from Oil Palm Empty Fruit Bunch via a One Step Heterogeneous Acetylation. Carbohydr. Polym. 2015, 132, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Puls, J.; Wilson, S.A.; Hölter, D. Degradation of Cellulose Acetate-Based Materials: A Review. J. Polym. Environ. 2011, 19, 152–165. [Google Scholar] [CrossRef]
- Novotny, T.E.; Lum, K.; Smith, E.; Wang, V.; Barnes, R. Cigarettes Butts and the Case for an Environmental Policy on Hazardous Cigarette Waste. Int. J. Envrion. Res. Public Health 2009, 6, 1691–1705. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.M.; Thomas, W.C.; Suthar, J.N.; Brown, D.M. Accelerated Degradation of Cellulose Acetate Cigarette Filters Using Controlled-Release Acid Catalysis. Green Chem. 2012, 14, 2266–2272. [Google Scholar] [CrossRef]
- Buchanan, C.M.; Gardner, R.M.; Komarek, R. Aerobic Biodegradation of Cellulose Acetate. Appl. Polym. 1972, 47, 1709–1719. [Google Scholar] [CrossRef]
- Hon, N. Photodegradation of Cellulose Acetate Fibers. Polym. Chem. 1977, 15, 725–744. [Google Scholar] [CrossRef]
- Sternberg, R.; Bindra, D.S.; Wilson, G.S.; Thévenot, D.R. Covalent Enzyme Coupling on Cellulose Acetate Membranes for Glucose Sensor Developmen. Anal. Chem. 1988, 12, 2781–2786. [Google Scholar] [CrossRef] [Green Version]
- Yudanova, T.N.; Skokova, I.F.; Gal, L.S. Fabrication of Biologically Active Fibre Materilas with Predetermined Properties. Fibre Chem. 2000, 6, 411–413. [Google Scholar] [CrossRef]
- Biely, P. Microbial Carbohydrate Esterases Deacetylating Plant Polysaccharides. Biotechnol. Adv. 2012, 30, 1575–1588. [Google Scholar] [CrossRef] [PubMed]
- Abrusci, C.; Marquina, D.; Santos, A.; Del Amo, A.; Corrales, T.; Catalina, F. Biodeterioration of Cinematographic Cellulose Triacetate by Sphingomonas Paucimobilis Using Indirect Impedance and Chemiluminescence Techniques. Int. Biodeterior. Biodegrad. 2009, 63, 759–764. [Google Scholar] [CrossRef]
- Sakai, K.; Yamauchi, T.; Nakasu, F.; Ohe, T. Biodegradation of Cellulose Acetate by Neisseria sicca. Biosci. Biotechnol. Biochem. 1996, 60, 1617–1622. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Gilkes, N.; Saddler, J.N. Effect of Acetyl Groups on Enzymatic Hydrolysis of Cellulosic Substrates. Int. J. Biol. Chem. Phys. Technol. Wood 2006, 60, 398–401. [Google Scholar] [CrossRef]
- Olaru, L.; Olaru, N.; Popa, V.I. On Enzymatic Degradation of Cellulose Acetate. Iran. Polym. J. 2004, 13, 235–240. [Google Scholar]
- Crawford, R.R.; Esmerian, O.K. Effect of Plasticizers on Some Physical Properties of Cellulose Acetate Phthalate Films. J. Pharm. Sci. 1971, 60, 312–314. [Google Scholar] [CrossRef] [PubMed]
- Perz, V.; Bleymaier, K.; Sinkel, C.; Kueper, U.; Bonnekessel, M.; Ribitsch, D.; Guebitz, G.M. Substrate Specificities of Cutinases on Aliphatic-Aromatic Polyesters and on Their Model Substrates. N. Biotechnol. 2016, 33, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Matamá, T.; Casal, M.; Cavaco-Paulo, A. Direct Enzymatic Esterification of Cotton and Avicel with Wild-Type and Engineered Cutinases. Cellulose 2013, 20, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Morley, K.L.; Chauve, G.; Kazlauskas, R.; Dupont, C.; Shareck, F.; Marchessault, R.H. Acetyl Xylan Esterase-Catalyzed Deacetylation of Chitin and Chitosan. Carbohydr. Polym. 2006, 63, 310–315. [Google Scholar] [CrossRef]
- Krastanova, I.; Guarnaccia, C.; Zahariev, S.; Degrassi, G.; Lamba, D. Heterologous Expression, Purification, Crystallization, X-ray Analysis and Phasing of the Acetyl Xylan Esterase from Bacillus Pumilus. Biochim. Biophys. Acta 2005, 1748, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Altaner, C.; Saake, B.; Tenkanen, M.; Eyzaquirre, J.; Faulds, C.B.; Biely, P.; Viikari, L.; Siika-aho, M.; Puls, J. Regioselective Deacetylation of Cellulose Acetates by Acetyl Xylan Esterases of Different CE-Families. J. Biotechnol. 2003, 105, 95–104. [Google Scholar] [CrossRef]
- Willför, S.; Sundberg, K.; Tenkanen, M.; Holmbom, B. Spruce-Derived Mannans—A Potential Raw Material for Hydrocolloids and Novel Advanced Natural Materials. Carbohydr. Polym. 2008, 72, 197–210. [Google Scholar] [CrossRef]
- Xu, C.; Leppanen, A.-S.; Eklund, P.; Holmlund, P.; Sjoholm, R.; Sundberg, K.; Willfor, S. Acetylation and Characterization of Spruce (Picea abies) Galactoglucomannans. Carbohydr. Res. 2010, 345, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Poutanen, K.; Sundberg, M.; Korte, H.; Puls, J. Deacetylation of Xylans by Acetyl Esterases of Trichoderma Reesei. Appl. Microbiol. Biotechnol. 1990, 33, 506–510. [Google Scholar] [CrossRef]
- Rao, L.; Xue, Y.; Zhou, C.; Tao, J.; Li, G.; Lu, G.R.; Ma, Y. A Thermostable Esterase from Thermoanaerobacter Tengcongensis Opening up a New Family of Bacterial Lipolytic Enzymes. Biochim. Biophys. Acta 2011, 1814, 1695–1702. [Google Scholar] [CrossRef] [PubMed]
- Chepyshko, H.; Lai, C.-P.; Huang, L.-M.; Liu, J.-H.; Shaw, J.-F. Multifunctionality and Diversity of GDSL Esterase/lipase Gene Family in Rice (Oryza sativa L. Japonica) Genome: New Insights from Bioinformatics Analysis. BMC Genom. 2012, 13, 309. [Google Scholar] [CrossRef] [PubMed]
- Akoh, C.C.; Lee, G.C.; Liaw, Y.C.; Huang, T.H.; Shaw, J.F. GDSL Family of Serine Esterases/lipases. Prog. Lipid Res. 2004, 43, 534–552. [Google Scholar] [CrossRef] [PubMed]
- Moriyoshi, K.; Koma, D.; Yamanaka, H.; Sakai, K.; Ohmoto, T. Expression and Characterization of a Thermostable Acetylxylan Esterase from Caldanaerobacter subterraneus Subsp. Tengcongensis Involved in the Degradation of Insoluble Cellulose Acetate. Biosci. Biotechnol. Biochem. 2013, 77, 2495–2498. [Google Scholar] [CrossRef] [PubMed]
- Frandsen, K.E.H.; Simmons, T.J.; Dupree, P.; Poulsen, J.-C.N.; Hemsworth, J.-C.N.; Ciano, L.; Johnston, E.M.; Tovborg, M.; Johansen, K.S.; von Freiesleben, P.; et al. The molecular basis of polysaccharide cleavage by lytic polysaccharide monooxygenases. Nat. Chem. Biol. 2016, 12, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Dimarogona, M.; Topakas, E.; Christakopoulos, P. Cellulose degradation by oxidative enzymes. Comput. Struct. Biotechnol. J. 2012, 2, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Isaksen, T.; Westereng, B.; Aachmann, F.L.; Agger, J.W.; Kracher, D.; Kittl, R.; Ludwig, R.; Haltrich, D.; Eijsink, V.G.H.; Horn, S.J. A C4-oxidizing lytic polysaccharide monooxygenase cleaving both cellulose and cello-oligosaccharides. J. Biol. Chem. 2014, 289, 2632–2642. [Google Scholar] [CrossRef] [PubMed]
- Žifčáková, L.; Baldrian, P. Fungal polysaccharide monooxygenases: New players in the decomposition of cellulose. Fungal Ecol. 2012, 5, 481–489. [Google Scholar] [CrossRef]
- Ghose, T.K. International Union of Pure Commission on Biotechnology. Measurement of cellulase activity. Pure Appl. Chem. 1987, 59, 257–268. [Google Scholar] [CrossRef]
- Liu, Y.S.; Baker, J.O.; Zeng, Y.; Himmel, M.E.; Haas, T.; Ding, S.Y. Cellobiohydrolase Hydrolyzes Crystalline Cellulose on Hydrophobic Faces. J. Biol. Chem. 2011, 286, 11195–11201. [Google Scholar] [CrossRef] [PubMed]
- Ike, M.; Ko, Y.; Yokohama, K.; Sumitani, J.-I.; Kawaguchi, T.; Ogasawara, W.; Okada, H.; Morikawa, Y. Cellobiohydrolase I (Cel7A) from Trichoderma Reesei Has Chitosanase Activity. J. Mol. Catal. B Enzym. 2007, 47, 159–163. [Google Scholar] [CrossRef]
- Textor, L.C.; Colussi, F.; Silveira, R.L.; Serpa, V.; de Mello, B.L.; Muniz, J.R.; Squina, F.M.; Pereira, N., Jr.; Skaf, M.S.; Polikarpov, I. Joint X-ray Crystallographic and Molecular Dynamics Study of Cellobiohydrolase I from Trichoderma Harzianum: Deciphering the Structural Features of Cellobiohydrolase Catalytic Activity. FEBS J. 2013, 280, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Saake, B.; Horner, S.; Puls, J. Progress in the Enzymatic Hydrolysis of Cellulose Derivatives. Cellul. Deriv. 1998, 15, 201–216. [Google Scholar] [CrossRef]
- Kikkawa, Y.; Fukuda, M.; Kashiwada, A.; Matsuda, K.; Kanesato, M.; Wada, M.; Imanaka, T.; Tanaka, T. Binding Ability of Chitinase onto Cellulose: An Atomic Force Microscopy Study. Polym. J. 2011, 43, 742–744. [Google Scholar] [CrossRef]
- Lee, S.-J.; Altaner, C.; Puls, J.; Saake, B. Determination of the Substituent Distribution along Cellulose Acetate Chains as Revealed by Enzymatic and Chemical Methods. Carbohydr. Polym. 2003, 54, 353–362. [Google Scholar] [CrossRef]
- Pellis, A.; Haernvall, K.; Pichler, C.M.; Ghazaryan, G.; Breinbauer, R.; Guebitz, G.M. Enzymatic Hydrolysis of Poly(ethylene Furanoate). J. Biotechnol. 2015, 235, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Kittl, R.; Kracher, D.; Burgstaller, D.; Haltrich, D.; Ludwig, R. Production of Four Neurospora Crassa Lytic Polysaccharide Monooxygenases in Pichia Pastoris Monitored by a Fluorimetric Assay. Biotechnol. Biofuels 2012, 5, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Esterase | Triacetin | Glucose Pentaacetate | Cellobiose Octaacetate | Cellohexaose Eicosaacetate | CA-DS 0.9 | CA-DS 1.4 | CA-DS 1.8 | CA-DS 2.3 | CA-DS 2.5 |
|---|---|---|---|---|---|---|---|---|---|
| AXE O | +++ | +++ | + | + | + | - | - | - | - |
| AXE 34 | + | + | + | + | ++ | ++ | + | - | - |
| AXE 35 | + | ++ | + | - | ++ | ++ | + | - | - |
| AXE 36 | + | ++ | + | + | ++ | ++ | + | - | - |
| AXE 53 | ++++ | ++++ | + | + | +++ | +++ | ++ | - | - |
| AXE 55 | +++ | +++ | + | + | +++ | ++++ | + | - | - |
| CE 265 | + | + | - | - | - | - | - | - | - |
| CUT | ++++ | ++++ | + | + | +++ | +++ | + | - | - |
| GAE | ++++ | ++++ | +++ | ++ | +++ | +++ | ++ | - | - |
| PAE | ++++ | +++ | +++ | ++ | +++ | +++ | ++ | - | - |
| Substrate | % Glucose (w/w) | % Acetic Acid (w/w) | Glucose Recovery Pretreated (%) | Glucose Recovery not Pretreated (%) |
|---|---|---|---|---|
| CA-DS 0.9 | 80.6 | 19.4 | 53.8 | 46.5 |
| CA-DS 1.4 | 72.7 | 27.3 | 47.8 | 34.9 |
| CA-DS 1.8 | 67.4 | 32.6 | 27.6 | 15.3 |
| Enzyme | Origin | pH | Temp. (°C) | Family |
|---|---|---|---|---|
| Acetyl xylan esterase (AXE O) | Orpinomyces sp. | 6.7–7 | 40 | 6 |
| Acetyl xylan esterase (AXE 34) | Clostridium thermocellum | 7 | 50 | 3 |
| Acetyl xylan esterase (AXE 35) | Clostridium thermocellum | 6.5 | 50 | 4 |
| Acetyl xylan esterase (AXE 36) | Clostridium thermocellum | 6.5 | 50 | 4 |
| Acetyl xylan esterase (AXE 53) | Cellvibrio japonicus | 8.5 | 25 | 2 |
| Acetyl xylan esterase (AXE 55) | Cellvibrio japonicus | 8.5 | 25 | 2 |
| Carboxylesterase (CE 265) | Bacillus subtilis | 7 | 37 | N.A. |
| Cutinase (CUT) | Thermobifida cellulosilytica | 7 | 50 | N.A. |
| Glucomannan acetyl esterase (GAE) | Clostridium thermocellum | 7 | 50 | 2 |
| Pectin acetyl esterase (PAE) | Clostridium thermocellum | 6.5 | 50 | 12 |
| Enzyme | Origin | pH | Temp. (°C) | Family |
|---|---|---|---|---|
| Cellulase E-CELAN | Aspergillus niger | 4.5 | 55 | GH12 |
| Cellulase E-CELTR | Trichoderma longibrachiatum | 4.5 | 70 | GH7 |
| Cellulase E-CELTE | Talaromyces emersonii. | 4.5 | 70 | GH5 |
| Cellulase E-CELBA | Bacillus amyloliquefaciens | 6.5 | 55 | GH5 |
| Cellulase 8A | Escherichia coli | 7 | 40 | GH8 |
| Cellulase 5A | Bacillus subtilis | 7.5 | 55 | GH5 |
| Cellulase Cellic C Tec 3 | N.A. | 5 | 55 | N.A. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haske-Cornelius, O.; Pellis, A.; Tegl, G.; Wurz, S.; Saake, B.; Ludwig, R.; Sebastian, A.; Nyanhongo, G.S.; Guebitz, G.M. Enzymatic Systems for Cellulose Acetate Degradation. Catalysts 2017, 7, 287. https://doi.org/10.3390/catal7100287
Haske-Cornelius O, Pellis A, Tegl G, Wurz S, Saake B, Ludwig R, Sebastian A, Nyanhongo GS, Guebitz GM. Enzymatic Systems for Cellulose Acetate Degradation. Catalysts. 2017; 7(10):287. https://doi.org/10.3390/catal7100287
Chicago/Turabian StyleHaske-Cornelius, Oskar, Alessandro Pellis, Gregor Tegl, Stefan Wurz, Bodo Saake, Roland Ludwig, Andries Sebastian, Gibson S. Nyanhongo, and Georg M. Guebitz. 2017. "Enzymatic Systems for Cellulose Acetate Degradation" Catalysts 7, no. 10: 287. https://doi.org/10.3390/catal7100287