
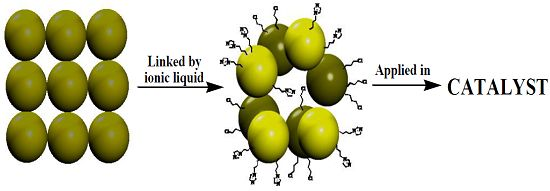
Phosphotungstate-Based Ionic Silica Nanoparticles Network for Alkenes Epoxidation

Abstract
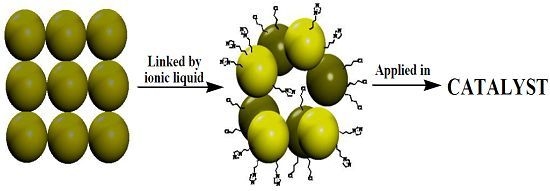
:
1. Introduction
2. Results and Discussion
2.1. Characterization
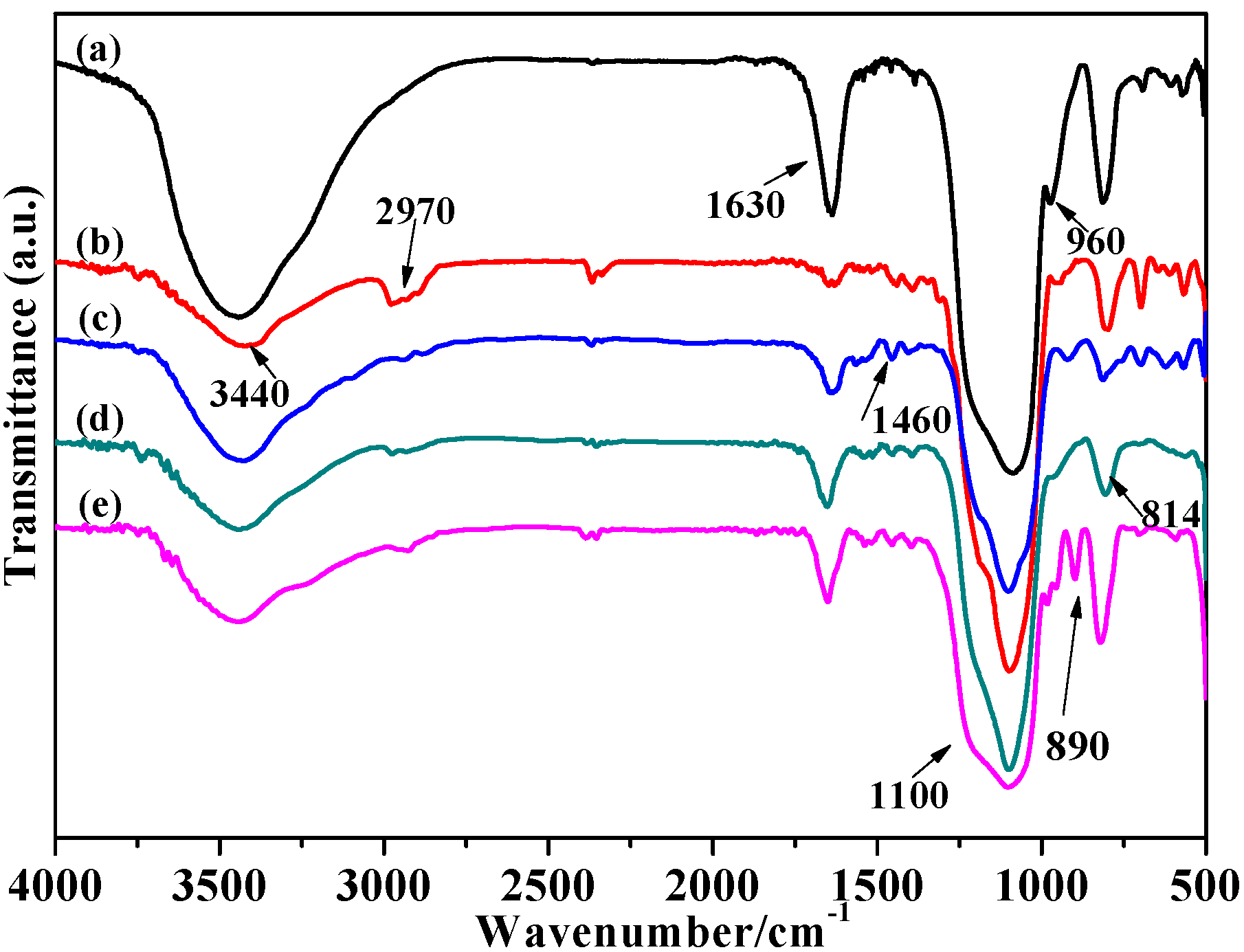
2.1.1. FT-IR Analysis
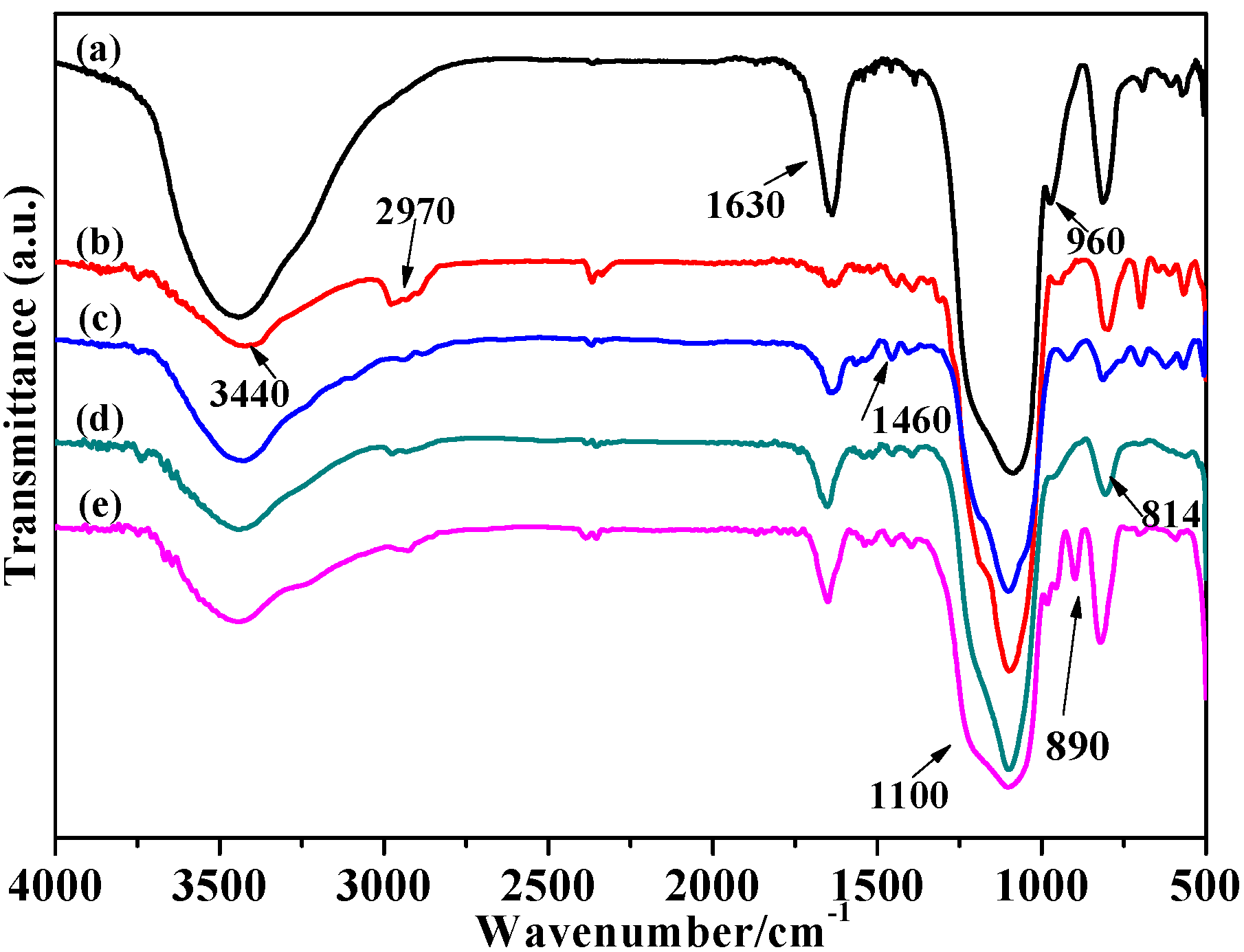
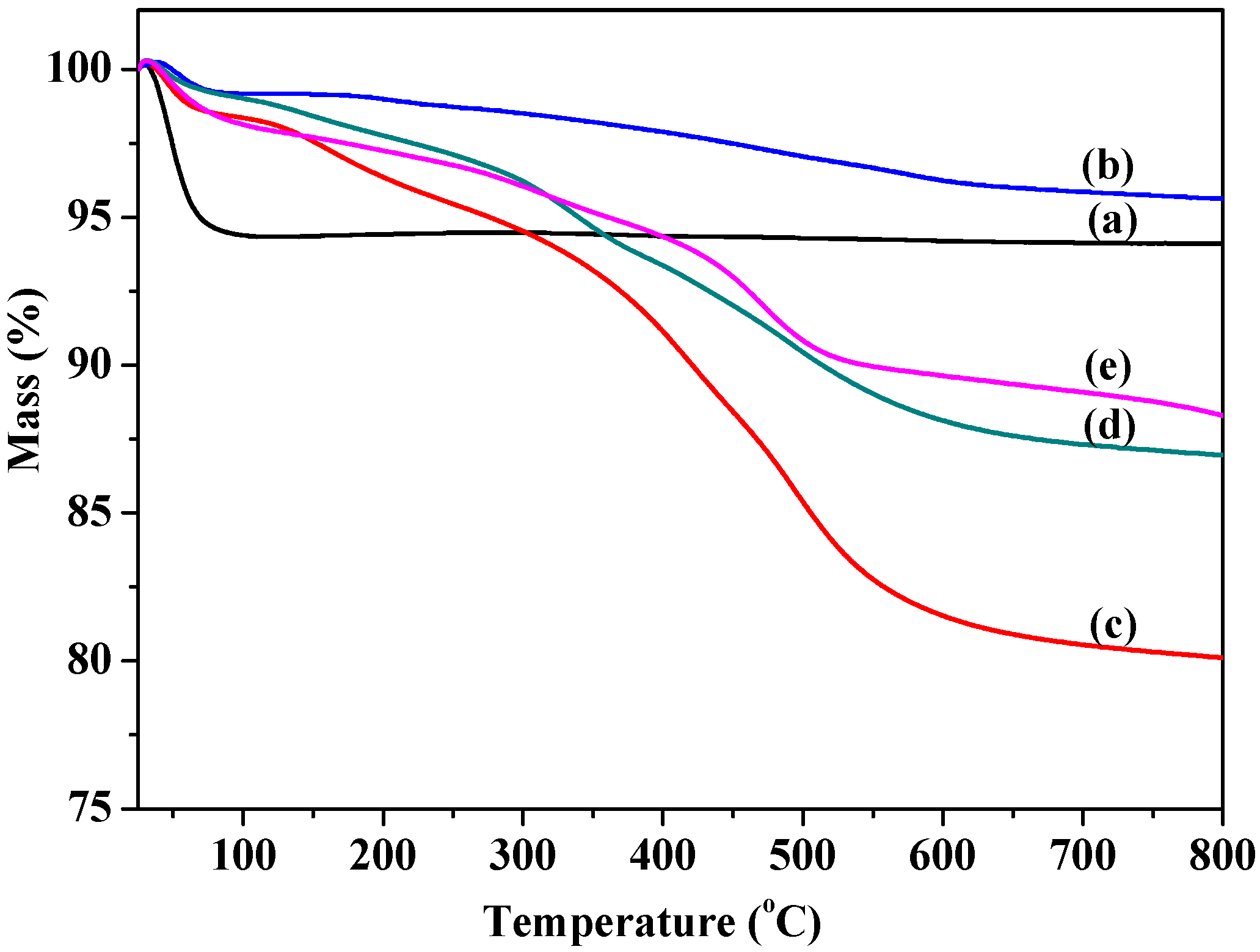
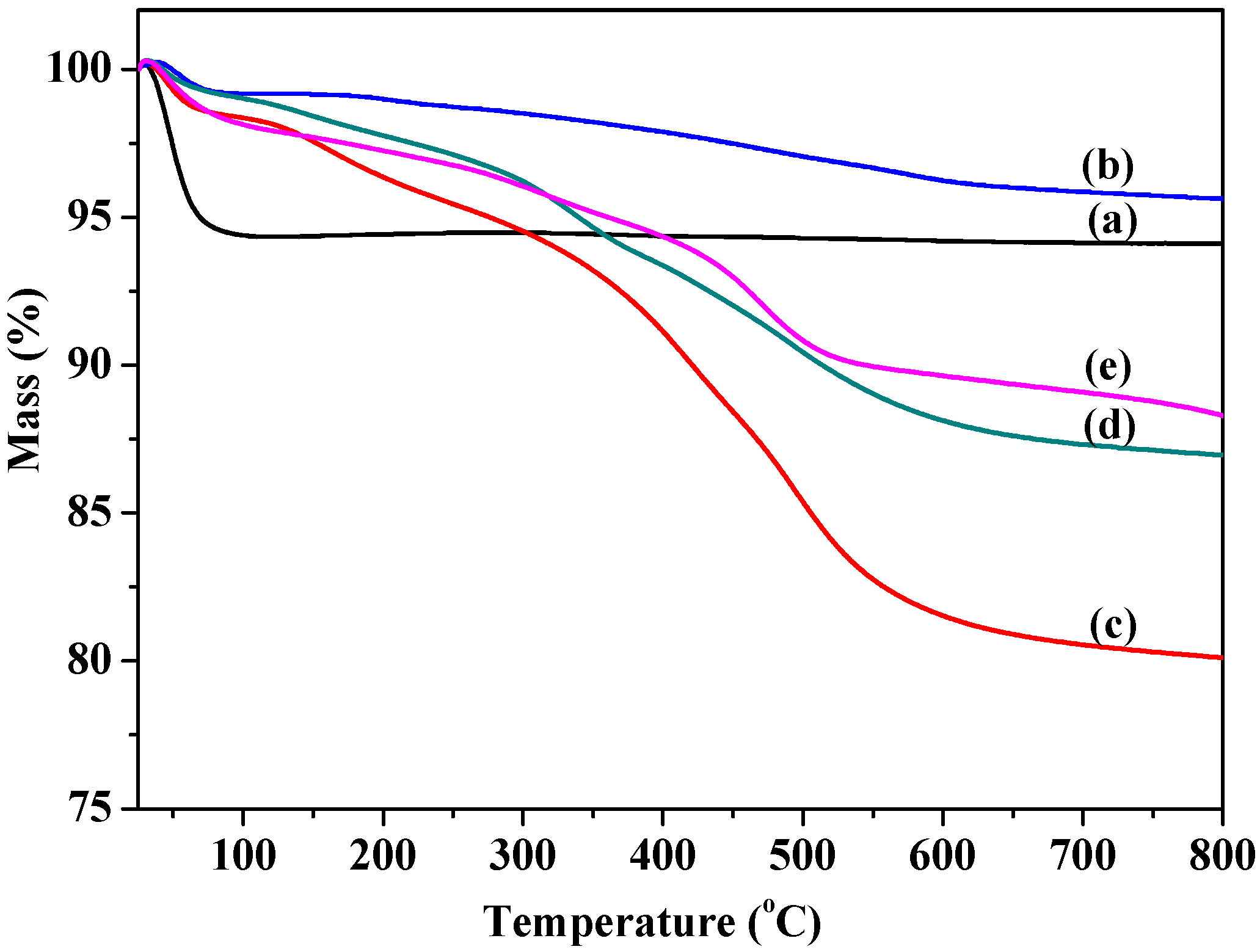
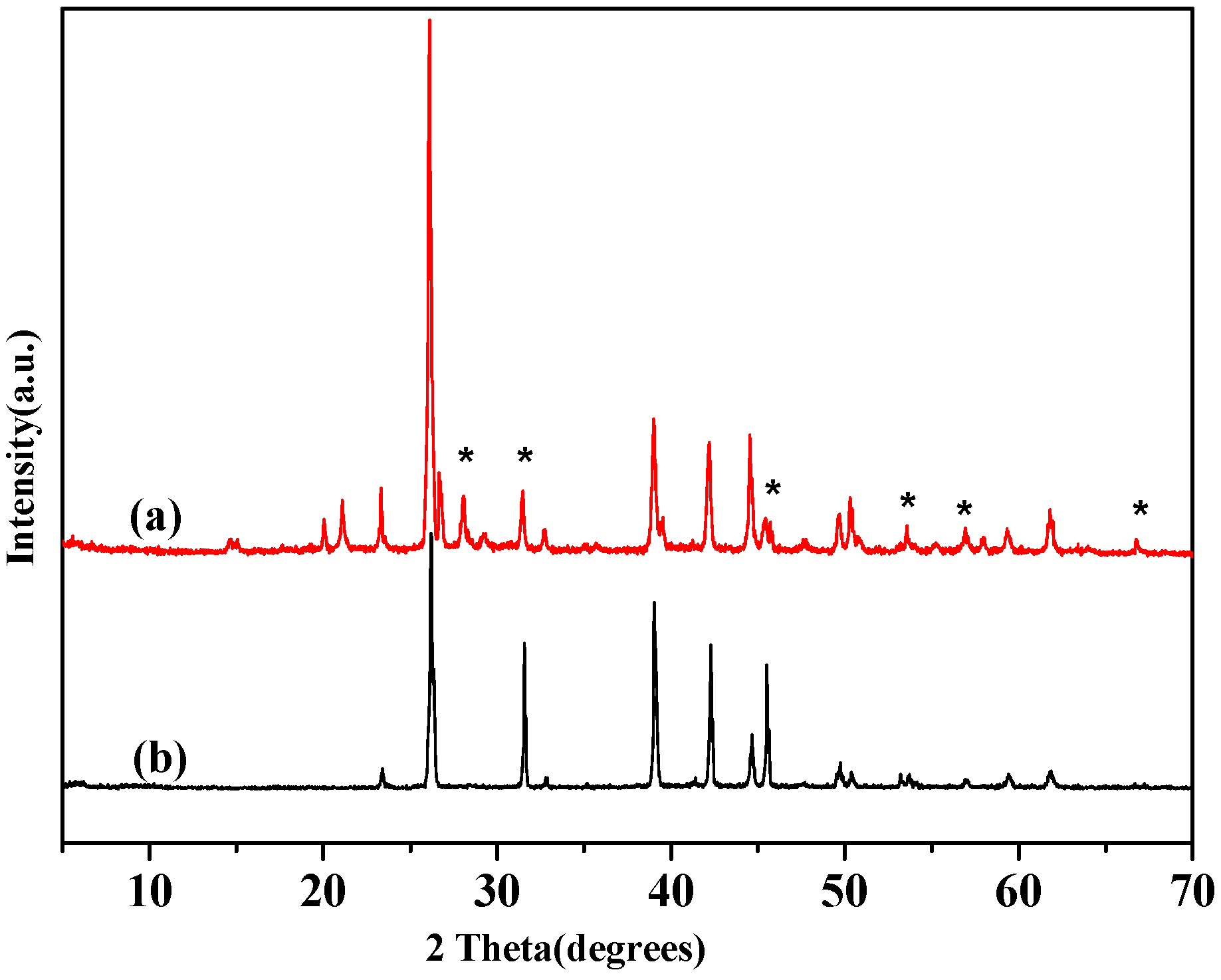
2.1.2. Thermal Stability and Structure
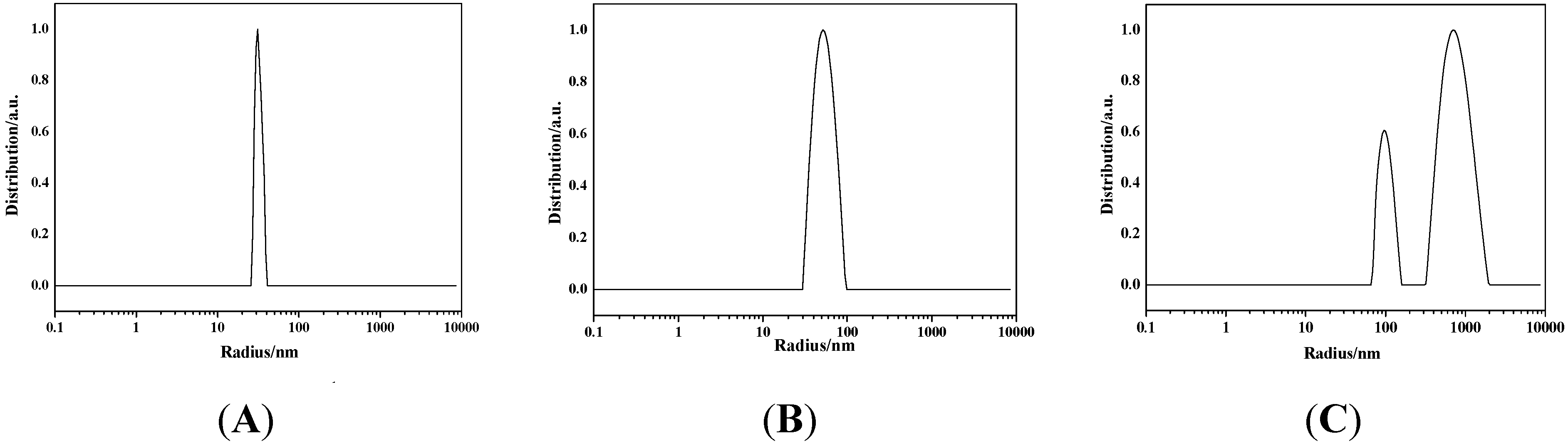
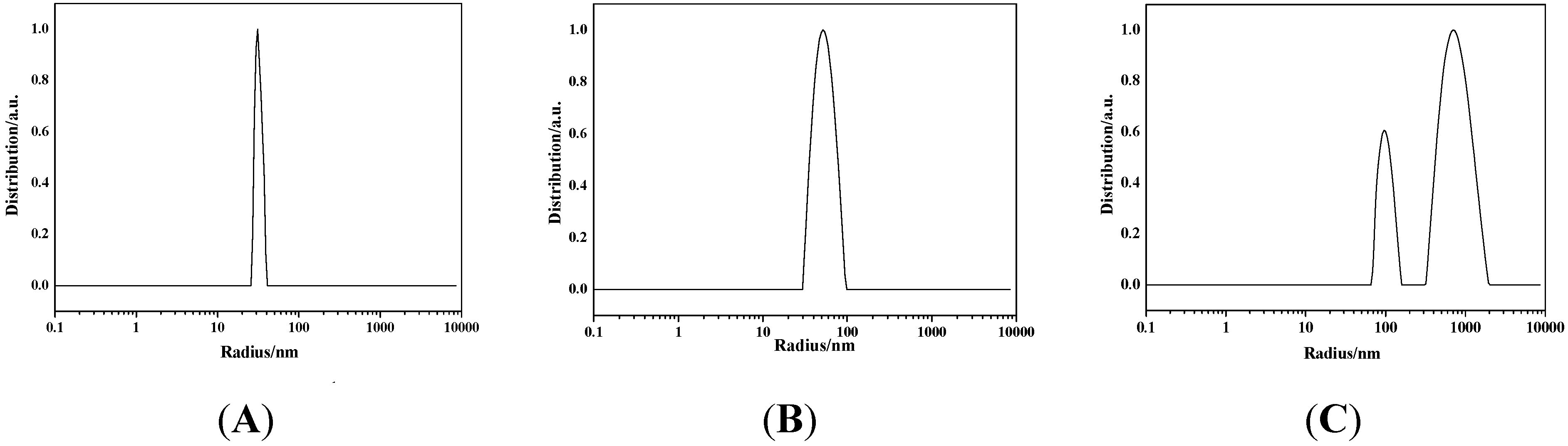
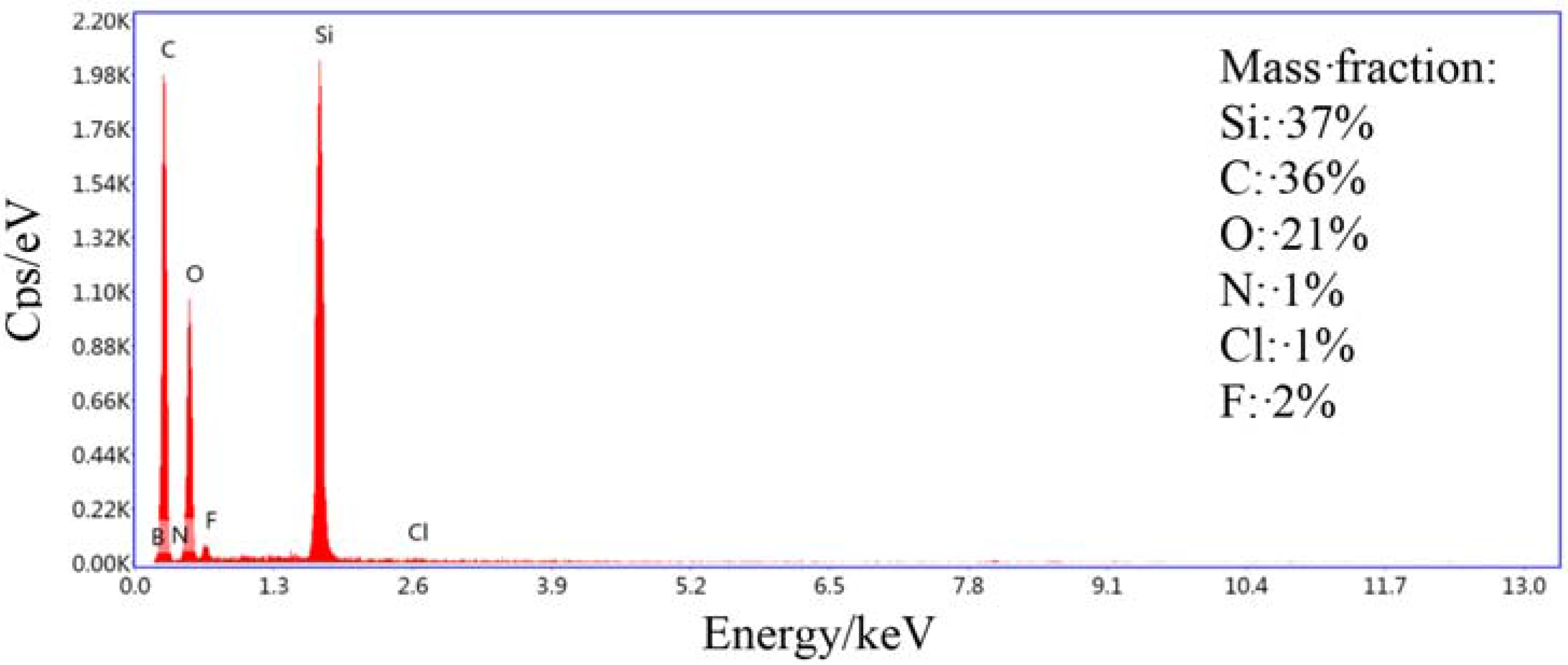
2.1.3. SEM, EDS, TEM, and DLS Characterization

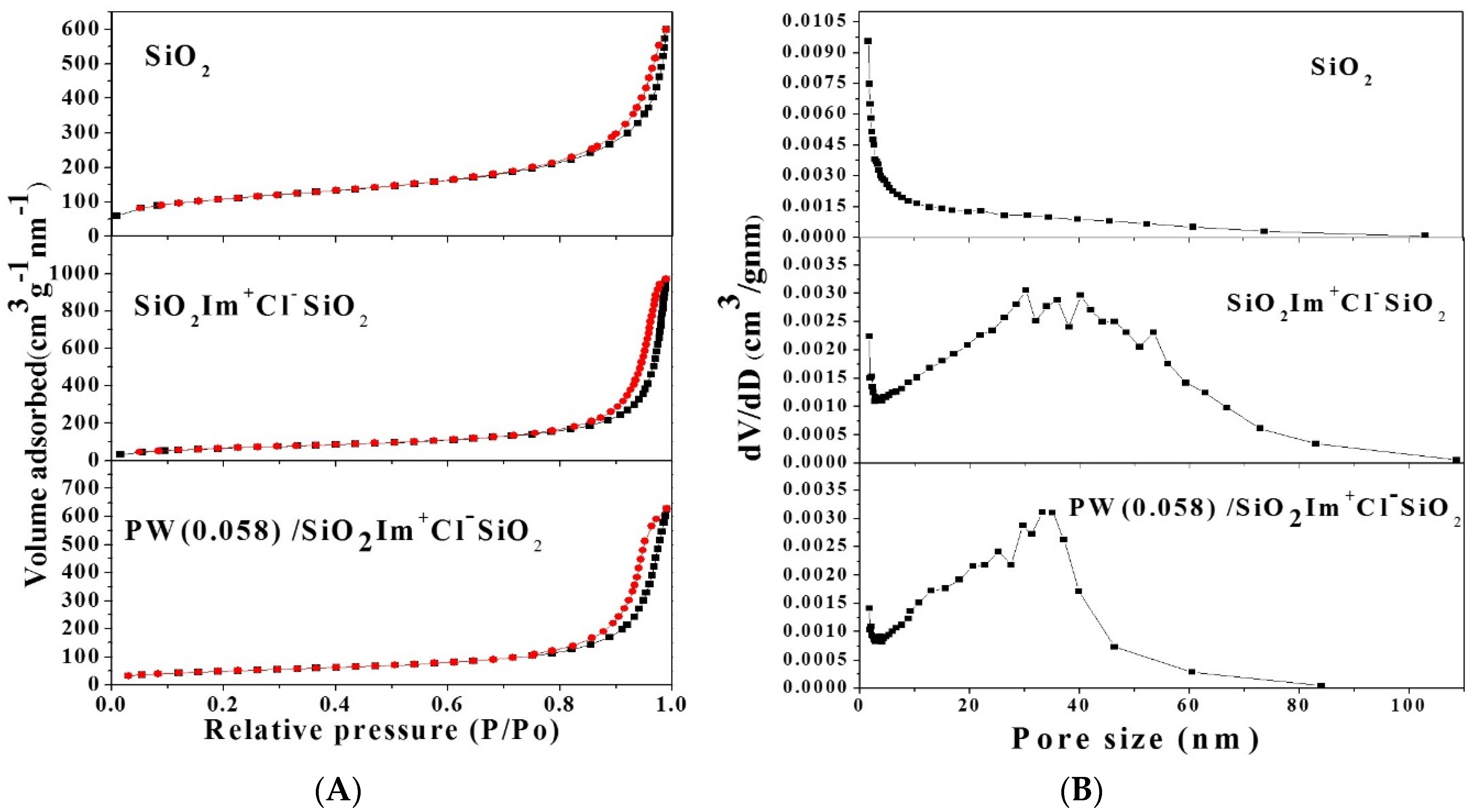
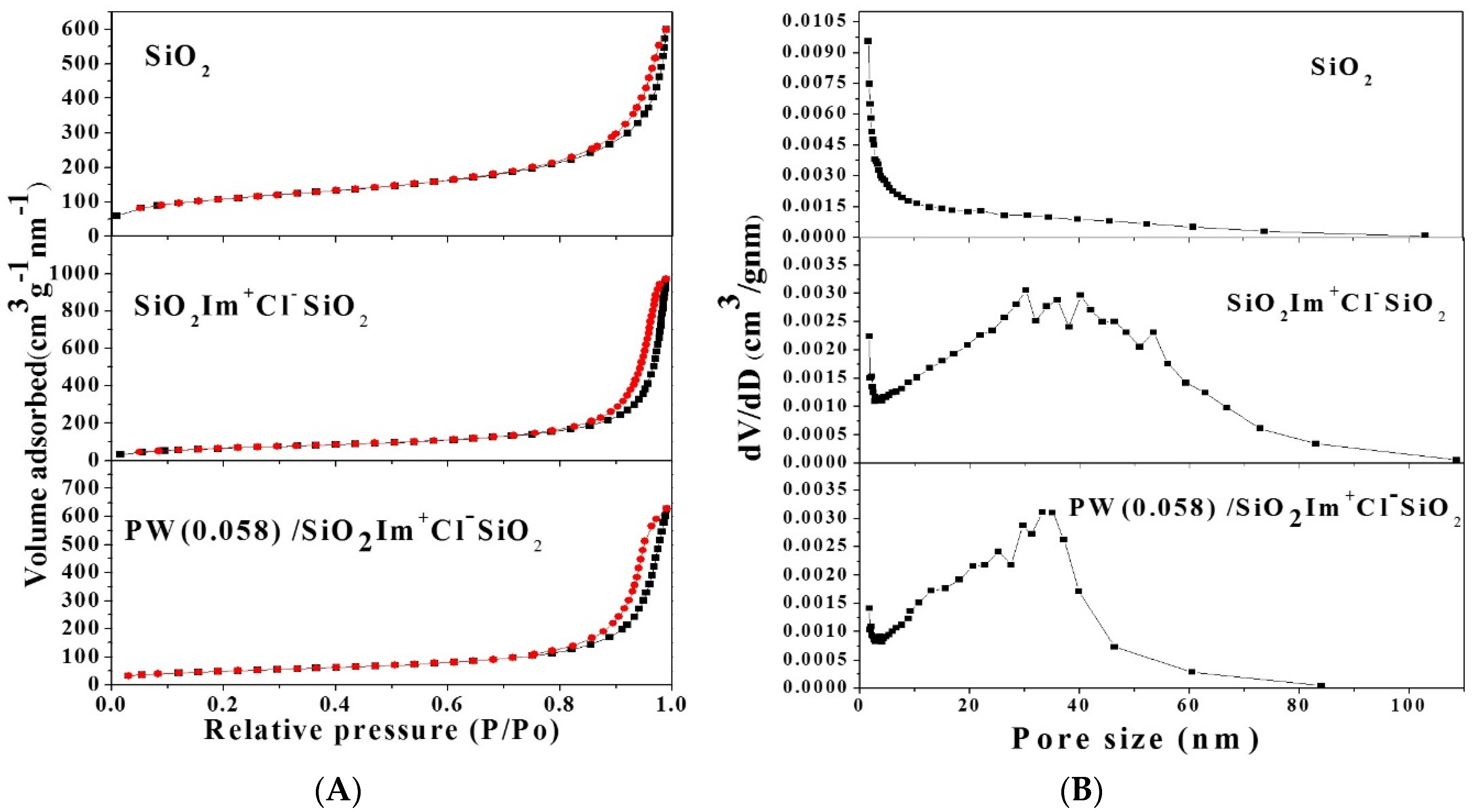
2.1.4. Nitrogen Sorption

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Compound | SBET (m2·g−1) | Vp (cm3·g−1) | Average Pore Size (nm) |
|---|---|---|---|---|
| 1 | SiO2 | 381 | 0.93 | 11.2 |
| 2 | SiO2 Im+Cl− SiO2 | 242 | 1.50 | 24.1 |
| 3 | PW(0.035)/SiO2Im+Cl−SiO2 | 182 | 1.24 | 24.0 |
| 4 | PW(0.058)/SiO2Im+Cl−SiO2 | 176 | 1.07 | 24.0 |
| 5 | PW(0.074)/SiO2Im+Cl−SiO2 | 168 | 1.01 | 22.4 |
| 6 | PW(0.17)/SiO2Im+Cl−SiO2 | 159 | 0.93 | 20.3 |
2.1.5. More Evidence of the Synthesis of the Network


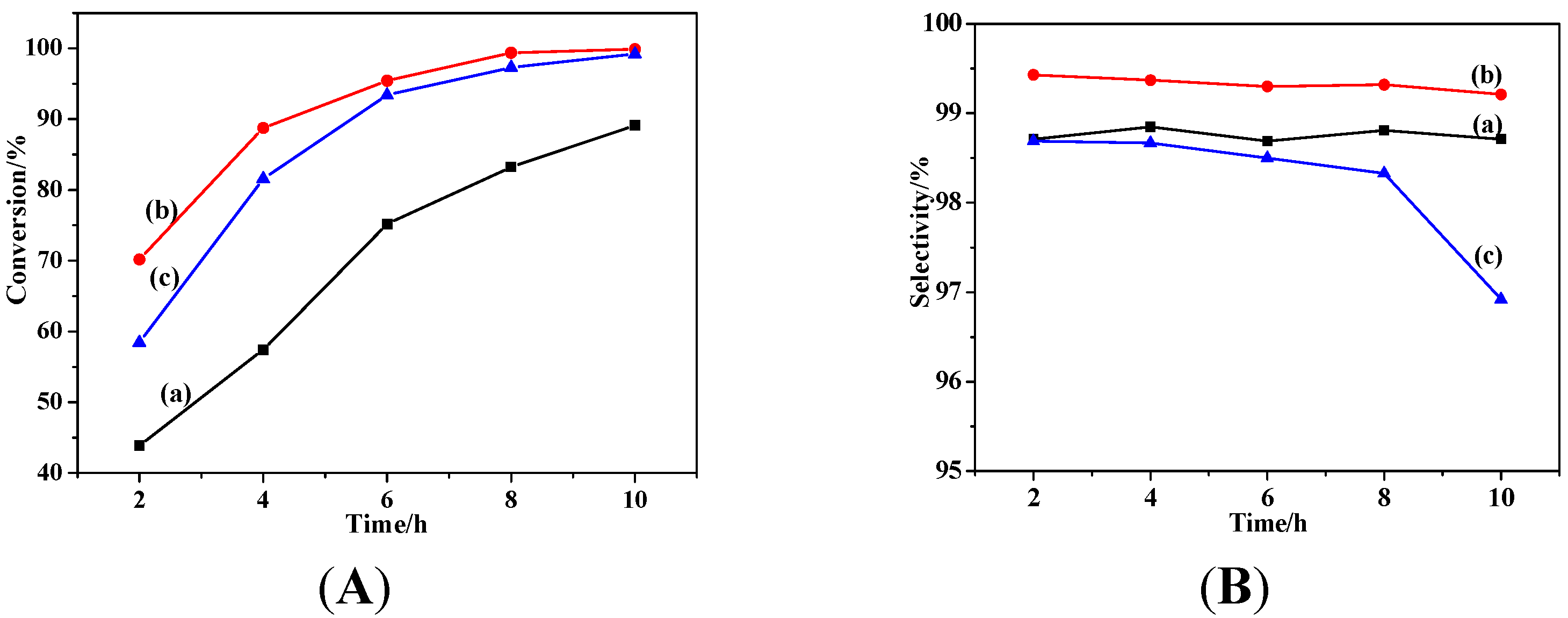
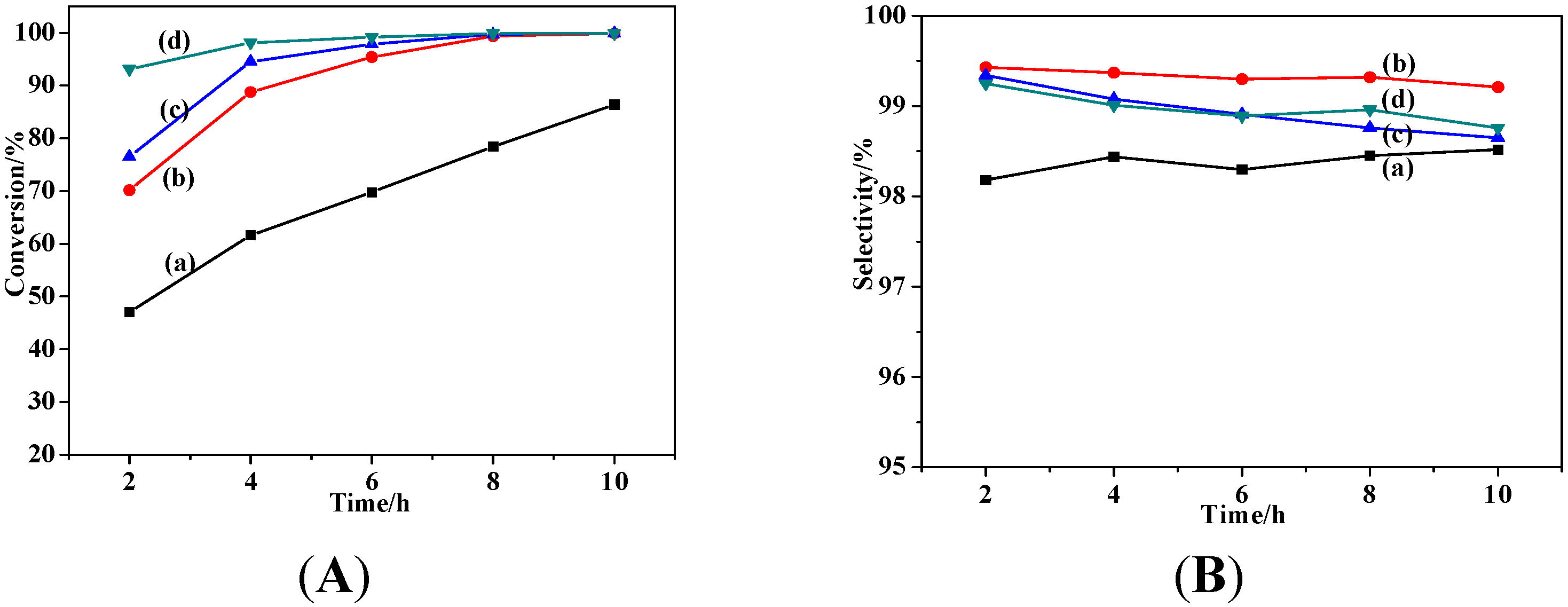
2.2. Catalytic Performances
| Catalysts | Conversion (%) | Selectivity (%) | ||||||
|---|---|---|---|---|---|---|---|---|
| 2 h | 4 h | 6 h | 8 h | 2 h | 4 h | 6 h | 8 h | |
| SiO2 | - | - | - | - | - | - | - | - |
| HPW | 4.9 | 8.6 | 11.2 | 14.2 | 70.3 | 67.9 | 61.8 | 43.2 |
| HPW/SiO2 | 0.8 | 1.6 | 4.4 | 6.6 | 36.8 | 34.5 | 35.8 | 32.4 |
| HPW/SiO2-IL | 30.5 | 52.7 | 72.4 | 79.8 | 98.7 | 98.1 | 97.4 | 97.0 |
| PW(0.035)/SiO2Im+Cl−SiO2 | 54.3 | 68.4 | 76.5 | 84.1 | 98.5 | 98.2 | 97.9 | 96.9 |
| PW(0.058)/SiO2Im+Cl−SiO2 | 77.2 | 88.8 | 94.4 | 99.4 | 99.4 | 99.4 | 99.3 | 99.2 |
| PW(0.074)/SiO2Im+Cl−SiO2 | 68.5 | 78.2 | 91.1 | 93.5 | 99.3 | 98.4 | 98.9 | 98.9 |
| PW(0.17)/SiO2Im+Cl−SiO2 | 45.3 | 63.6 | 70.2 | 77.6 | 98.7 | 98.1 | 98.7 | 98.2 |
| Run a | PW(0.058)/SiO2Im+Cl−SiO2 | PW(0.082)/SiO2-Im | ||
|---|---|---|---|---|
| Conversion (%) | Selectivity (%) | Conversion (%) | Selectivity (%) | |
| 1 | 94.4 | 99.3 | 96.0 | 98.2 |
| 2 | 88.7 | 97.5 | 27.1 | 87.3 |
| 3 | 82.1 | 95.1 | 25.8 | 81.6 |
| 4 | 75.2 | 94.6 | 18.6 | 78.3 |
2.2.1. Effects of Temperature and Time
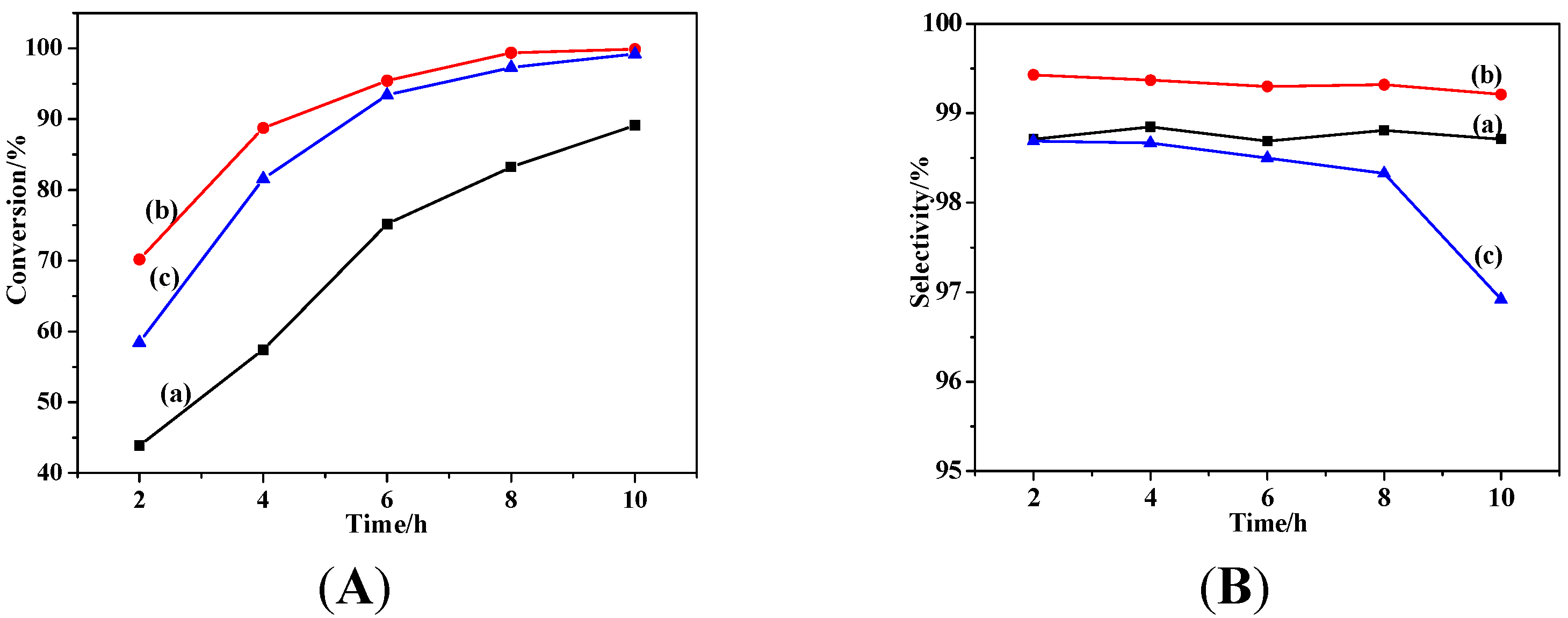
2.2.2. Effect of Catalyst Dosage
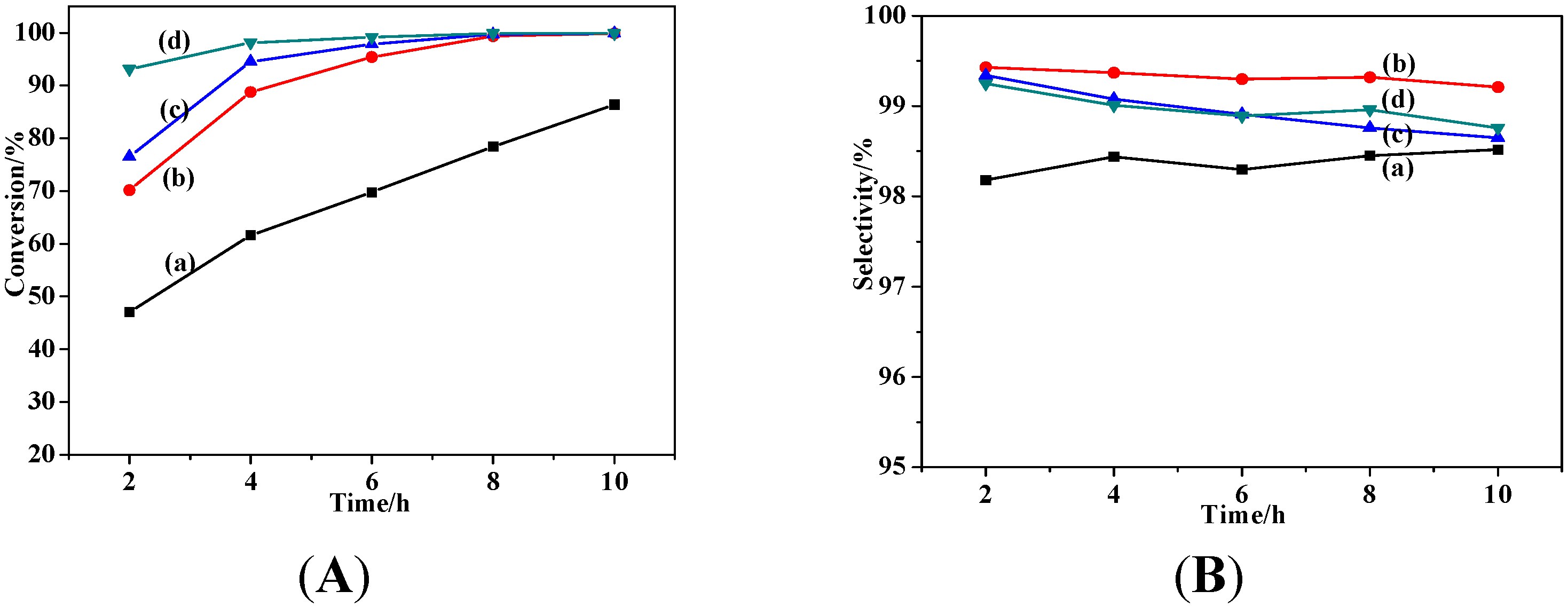
2.2.3. Effect of Solvent
| Entry | Solvent | Boiling Point (°C) | Conversion | Selectivity | ||
|---|---|---|---|---|---|---|
| 4 h | 8 h | 4 h | 8 h | |||
| 1 | Acetonitrile | 81.6 | 88.8 | 99.4 | 99.4 | 99.2 |
| 2 | Methanol | 64.5 | 92.9 | 96.8 | 99.1 | 96.6 |
| 3 | Ethanol | 78.2 | 52.0 | 77.1 | 98.8 | 98.8 |
| 4 | Chloroform | 61.2 | 98.9 | 100.0 | 98.6 | 96.6 |
| 5 | 1,2-Dichloroethane | 83.5 | 8.8 | 15.0 | 94.0 | 93.6 |
| 6 | Ethyl acetate | 78.3 | 4.5 | 5.5 | 85.8 | 88.2 |
3. Experimental Section
3.1. Materials and Methods
3.2. Catalyst Preparation
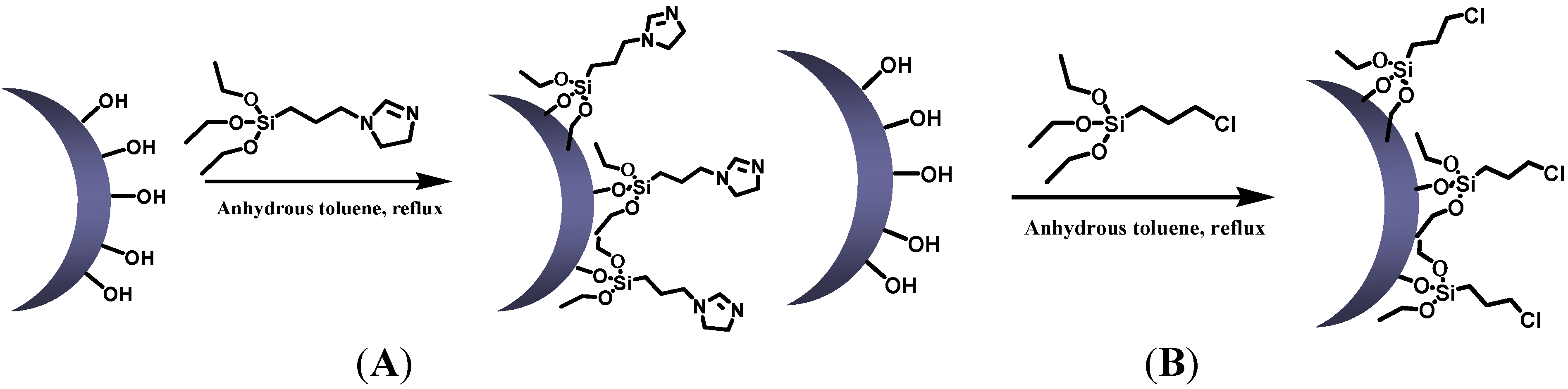
3.2.1. Synthesis of Silica Nanoparticles
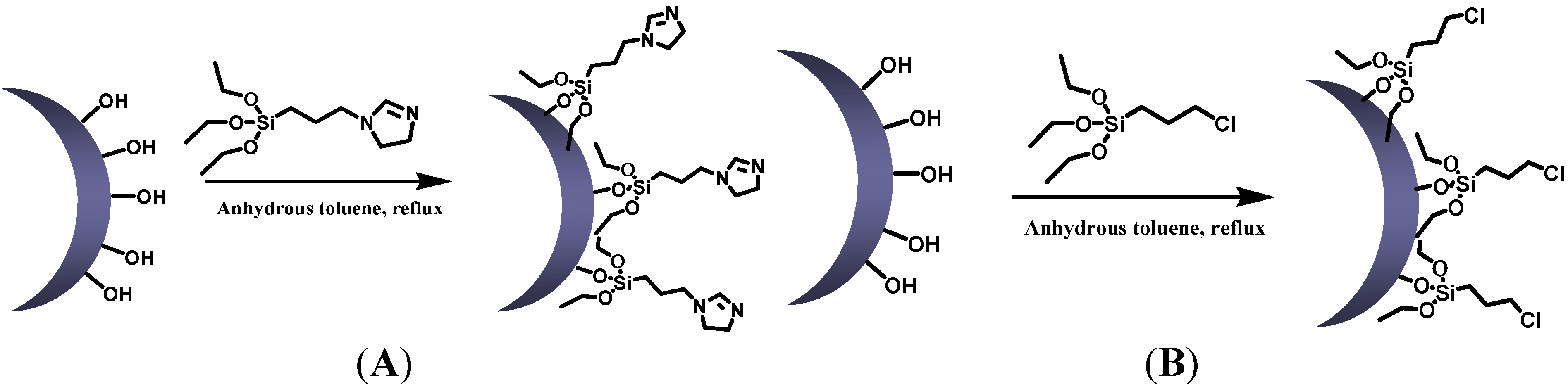
3.2.2. Synthesis of Modified Silica Nanoparticles
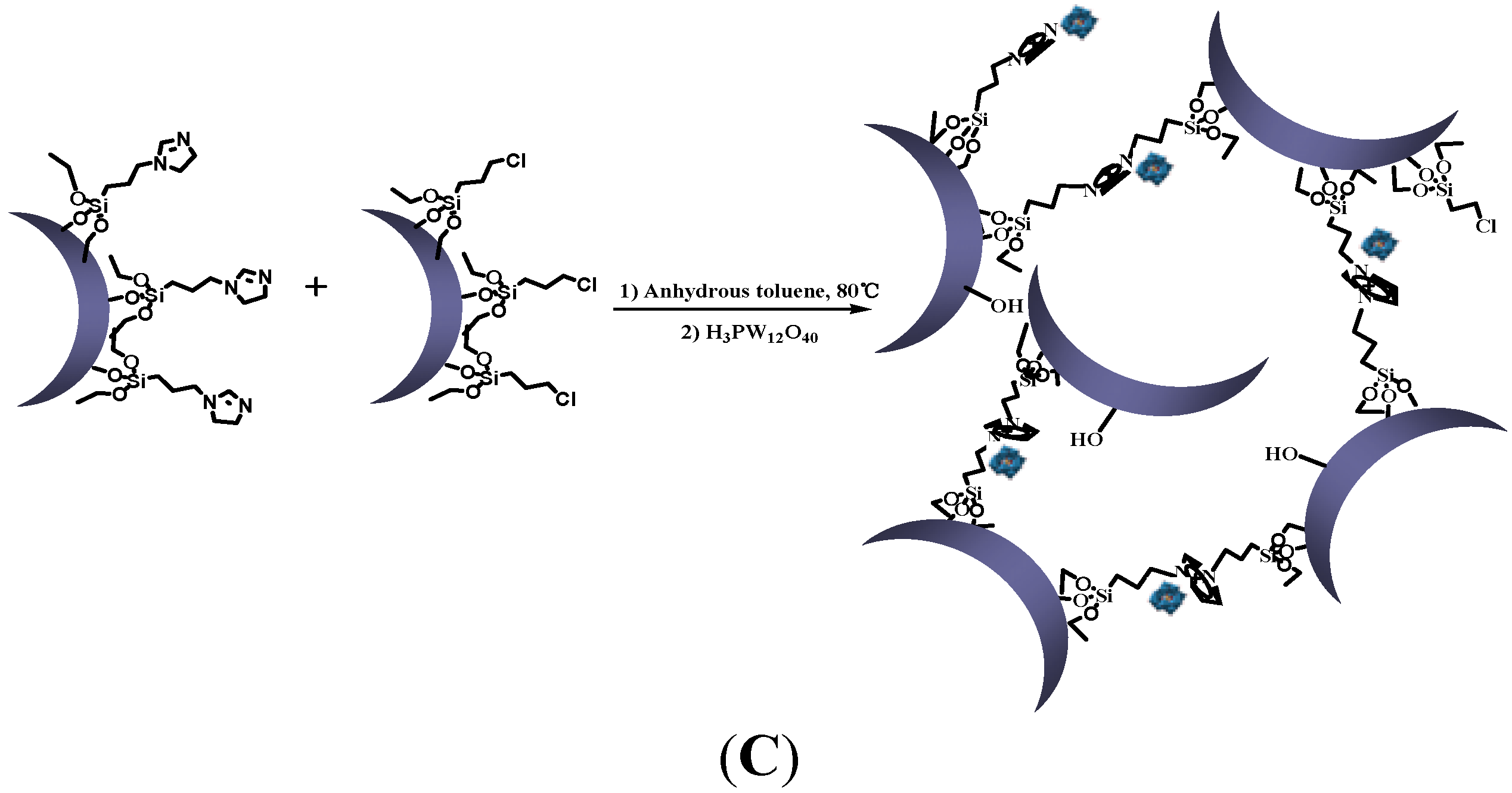
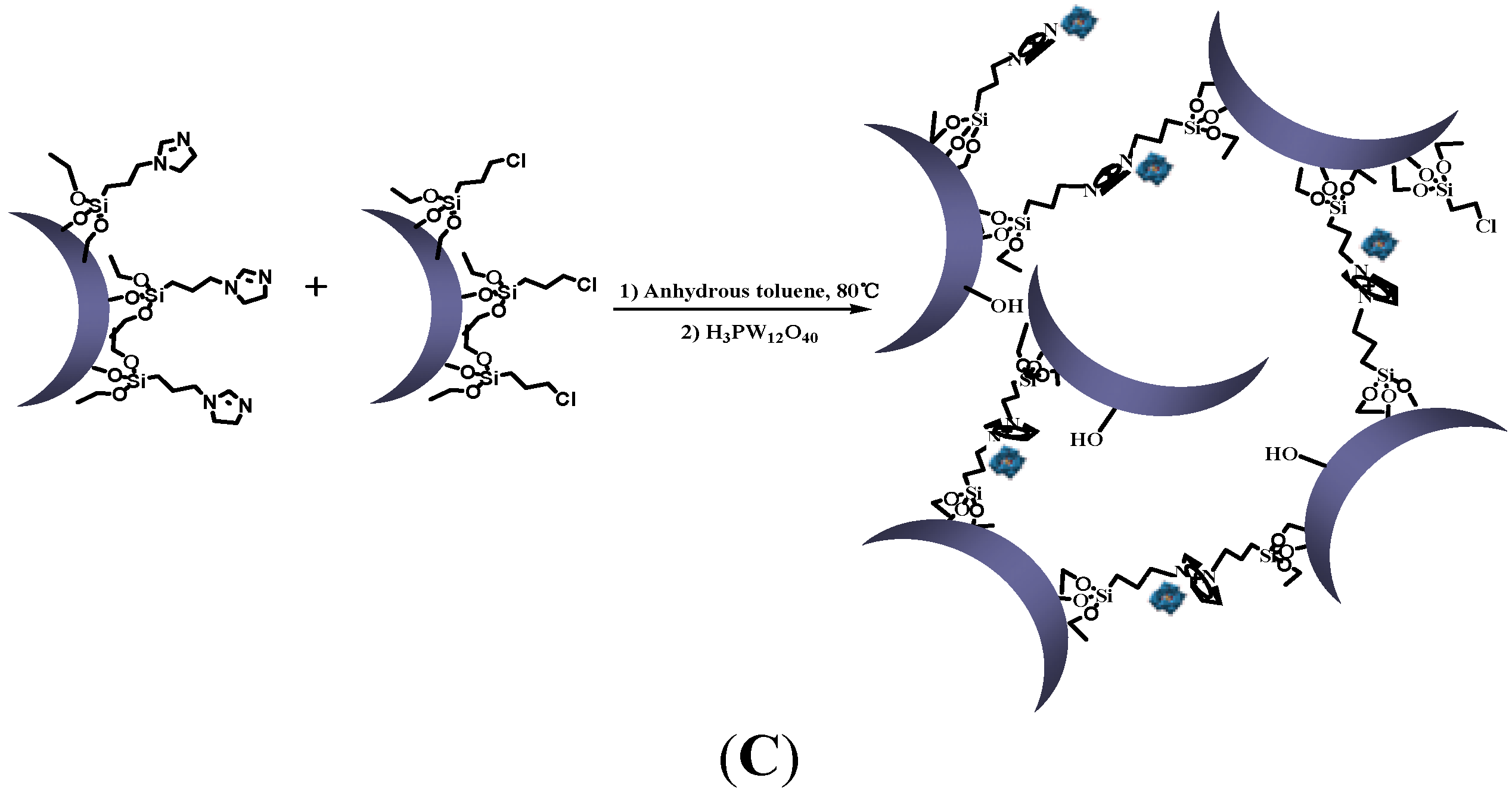
3.2.3. Synthesis of Silica Nanoparticle Network
3.2.4. Synthesis of Phosphotungstate-Loaded Silica Nanoparticle Network
3.2.5. Anion Exchange Experiment
3.3. Catalytic Performance
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhu, C.; Guo, S.; Zhai, Y.; Dong, S. Layer-by-Layer Self-Assembly for Constructing a Graphene/Platinum Nanoparticle Three-Dimensional Hybrid Nanostructure Using Ionic Liquid as a Linker. Langmuir 2010, 26, 7614–7618. [Google Scholar] [CrossRef] [PubMed]
- Corma, A.; Iborra, S.; Llabres i Xamena, F.X.; Monton, R.; Calvino, J.J.; Prestipino, C. Nanoparticles of Pd on Hybrid Polyoxometalate-Ionic Liquid Material: Synthesis, Characterization, and Catalytic Activity for Heck Reaction. J. Phys. Chem. C 2010, 114, 8828–8836. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, L.; Zhang, J.Z.; Li, J. Preparation of 1-Propyl-3-Methyl-Imidazolium Chloride Functionalized Organoclay for Protein Immobilization. Sci. Adv. Mater. 2009, 1, 55–62. [Google Scholar] [CrossRef]
- Karousis, N.; Economopoulos, S.P.; Sarantopoulou, E.; Tagmatarchis, N. Porphyrin counter anion in imidazolium-modified graphene-oxide. Carbon 2010, 48, 854–860. [Google Scholar] [CrossRef]
- Vangeli, O.C.; Romanos, G.E.; Beltsios, K.G.; Fokas, D.; Kouvelos, E.P.; Stefanopoulos, K.L.; Kanellopoulos, N.K. Grafting of Imidazolium Based Ionic Liquid on the Pore Surface of Nanoporous Materials-Study of Physicochemical and Thermodynamic Properties. J. Phys. Chem. B 2010, 114, 6480–6491. [Google Scholar] [CrossRef] [PubMed]
- Trilla, M.; Pleixats, R.; Man, M.W.C.; Bied, C. Organic-inorganic hybrid silica materials containing imidazolium and dihydroimidazolium salts as recyclable organocatalysts for Knoevenagel condensations. Green. Chem. 2009, 11, 1815–1820. [Google Scholar] [CrossRef]
- Tonle, I.K.; Letaief, S.; Ngameni, E.; Detellier, C. Nanohybrid materials from the grafting of imidazolium cations on the interlayer surfaces of kaolinite. Application as electrode modifier. J. Mater. Chem. 2009, 19, 5996–6003. [Google Scholar] [CrossRef]
- Crees, R.S.; Cole, M.L.; Hanton, L.R.; Sumby, C.J. Synthesis of a Zinc(II) Imidazolium Dicarboxylate Ligand Metal-Organic Framework (MOF): A Potential Precursor to MOF-Tethered N-Heterocyclic Carbene Compounds. Inorg. Chem. 2010, 49, 1712–1719. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Liu, Z.L.; Tay, S.W.; Fan, X.S.; Zhang, X.W.; He, C.B. An amphiphilic-like fluoroalkyl modified SiO2 nanoparticle@Nafion proton exchange membrane with excellent fuel cell performance. Chem. Commun. 2013, 49, 9639–9641. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jiang, P.P.; Shen, Y.R.; Zhang, W.J.; Li, X.T. Molybdenum(VI) complex with a tridentate Schiff base ligand immobilized on SBA-15 as effective catalysts in epoxidation of alkenes. Micropor. Mesopor. Mat. 2015, 206, 161–169. [Google Scholar] [CrossRef]
- Czakler, M.; Litschauer, M.; Fottinger, K.; Peterlik, H.; Neouze, M.A. Photoluminescence as Complementary Evidence for Short-Range Order in Ionic Silica Nanoparticle Networks. J. Phys. Chem. C 2010, 114, 21342–21347. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Yoshida, C.; Uchida, S.; Mizuno, N. Peroxotungstate Immobilized on Ionic Liquid-modified Silica as a Heterogeneous Epoxidation Catalyst with Hydrogen Peroxide. J. Am. Chem. Soc. 2005, 127, 530–531. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Shu, Y.; Wang, X.F.; Chen, X.W.; Wang, J.H. Encapsulation of silica nano-spheres with polymerized ionic liquid for selective isolation of acidic proteins. Anal. Bioanal. Chem. 2013, 405, 8799–8806. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, M.; Masteri-Farahani, M.; Ghorbani, M. Synthesis and characterization of heteropolytungstate-ionic liquid supported on the surface of silica coated magnetite nanoparticles. J. Magn. Magn. Mater. 2013, 327, 58–63. [Google Scholar] [CrossRef]
- Roeser, J.; Kronstein, M.; Litschauer, M.; Thomas, A.; Neouze, M.A. Ionic Nanoparticle Networks as Solid State Catalysts. Eur. J. Inorg. Chem. 2012, 32, 5305–5311. [Google Scholar] [CrossRef]
- Neouze, M.A.; Kronstein, M.; Tielens, F. Ionic nanoparticle networks: Development and perspectives in the landscape of ionic liquid based materials. Chem. Commun. 2014, 50, 10929–10936. [Google Scholar] [CrossRef] [PubMed]
- Neouze, M.A. Nanoparticle assemblies: Main synthesis pathways and brief overview on some important applications. J. Mater. Sci. 2013, 48, 7321–7349. [Google Scholar] [CrossRef]
- Herrmann, A.K.; Formanek, P.; Borchardt, L.; Klose, M.; Giebeler, L.; Eckert, J.; Kaskel, S.; Gaponik, N.; Eychmueller, A. Multimetallic Aerogels by Template-Free Self-Assembly of Au, Ag, Pt, and Pd Nanoparticles. Chem. Mater. 2014, 26, 1074–1083. [Google Scholar] [CrossRef]
- Lesnyak, V.; Voitekhovich, S.V.; Gaponik, P.N.; Gaponik, N.; Eychmueller, A. CdTe Nanocrystals Capped with a Tetrazolyl Analogue of Thioglycolic Acid: Aqueous Synthesis, Characterization, and Metal-Assisted Assembly. ACS Nano 2010, 4, 4090–4096. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.; Lesnyak, V.; Gaponik, N.; Eychmueller, A. Quantum-Dot-Based (Aero) gels: Control of the Optical Properties. J. Phys. Chem. Lett. 2012, 3, 2188–2193. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, K.; Yamaguchi, K.; Kamata, K. Epoxidation of olefins with hydrogen peroxide catalyzed by polyoxometalates. Coord. Chem. Rev. 2005, 249, 1944–1956. [Google Scholar] [CrossRef]
- Venturello, C.; D’Aloisio, R. Quaternary Ammonium Tetrakis(Diperoxotungsto) Phosphates(3-) As A New Class Of Catalysts For Efficient Alkene Epoxidation With Hydrogen-peroxide. J. Org. Chem. 1988, 53, 1553–1557. [Google Scholar] [CrossRef]
- Song, Y.; Tsunashima, R. Recent advances on polyoxometalate-based molecular and composite materials. Chem. Soc. Rev. 2012, 41, 7384–7402. [Google Scholar] [CrossRef] [PubMed]
- Karimi, Z.; Mahjoub, A.R.; Harati, S.M. Polyoxometalate-based hybrid mesostructured catalysts for green epoxidation of olefins. Inorg. Chim. Acta 2011, 376, 1–9. [Google Scholar] [CrossRef]
- Tan, R.; Liu, C.; Feng, N.D.; Xiao, J.; Zheng, W.G.; Zheng, A.M.; Yin, D.H. Phosphotungstic acid loaded on hydrophilic ionic liquid modified SBA-15 for selective oxidation of alcohols with aqueous H2O2. Micropor. Mesopor. Mat. 2012, 158, 77–87. [Google Scholar] [CrossRef]
- Kamata, K.; Yonehara, K.; Sumida, Y. Efficient epoxidation of olefins with ≥99% selectivity and use of hydrogen peroxide. Science 2003, 300, 964–966. [Google Scholar] [CrossRef] [PubMed]
- Litschauer, M.; Neouze, M.A. Nanoparticles connected through an ionic liquid-like network. J. Mater. Chem. 2008, 18, 640–646. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Jiang, P.; Wang, Z.; Huang, Y. Phosphotungstate-Based Ionic Silica Nanoparticles Network for Alkenes Epoxidation. Catalysts 2016, 6, 2. https://doi.org/10.3390/catal6010002
Li X, Jiang P, Wang Z, Huang Y. Phosphotungstate-Based Ionic Silica Nanoparticles Network for Alkenes Epoxidation. Catalysts. 2016; 6(1):2. https://doi.org/10.3390/catal6010002
Chicago/Turabian StyleLi, Xiaoting, Pingping Jiang, Zhuangqing Wang, and Yuandan Huang. 2016. "Phosphotungstate-Based Ionic Silica Nanoparticles Network for Alkenes Epoxidation" Catalysts 6, no. 1: 2. https://doi.org/10.3390/catal6010002
APA StyleLi, X., Jiang, P., Wang, Z., & Huang, Y. (2016). Phosphotungstate-Based Ionic Silica Nanoparticles Network for Alkenes Epoxidation. Catalysts, 6(1), 2. https://doi.org/10.3390/catal6010002