Kinetic Isotope Effect of Prostaglandin H Synthase Exhibits Inverted Temperature Dependence


Abstract
:1. Introduction
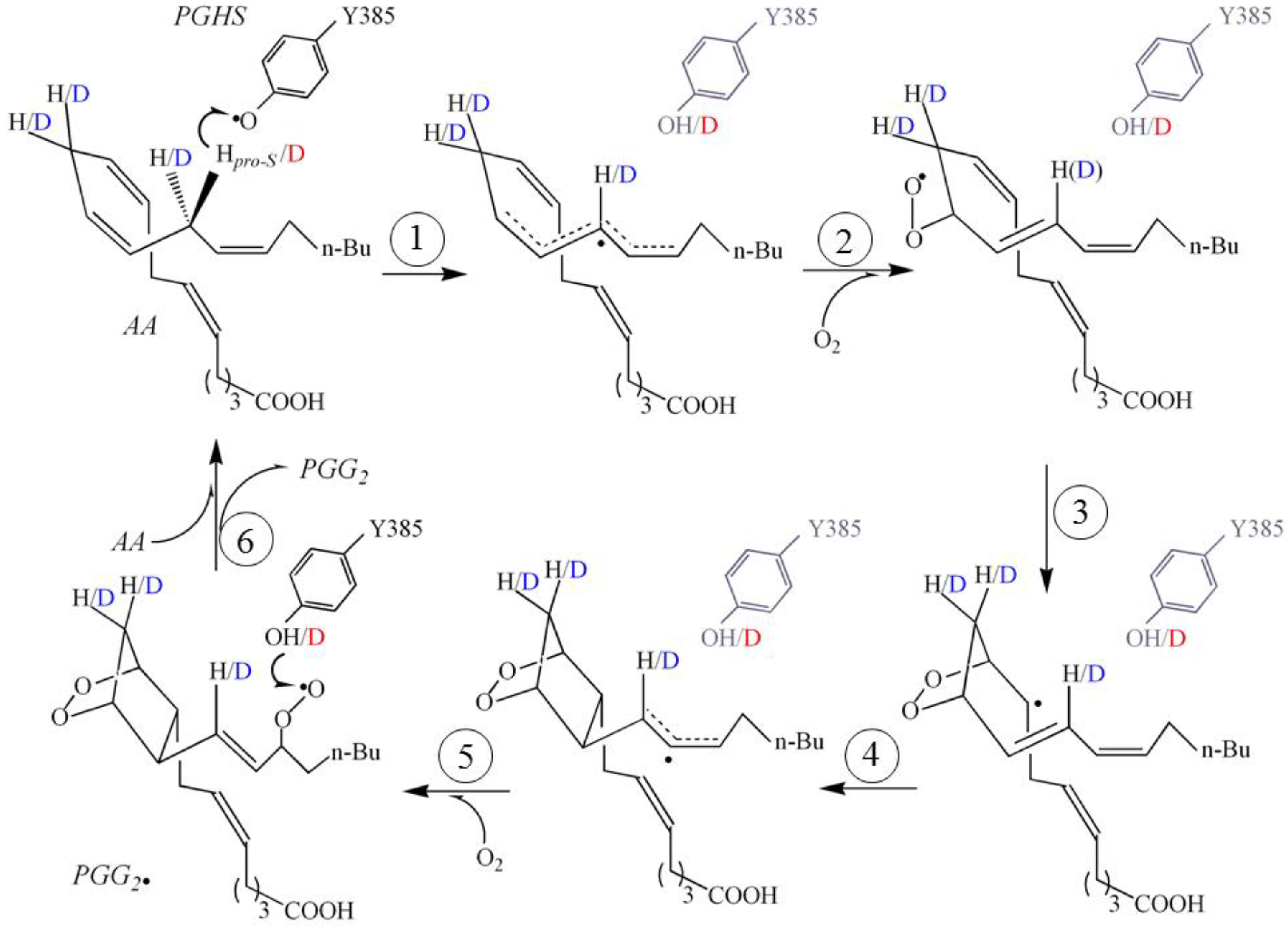
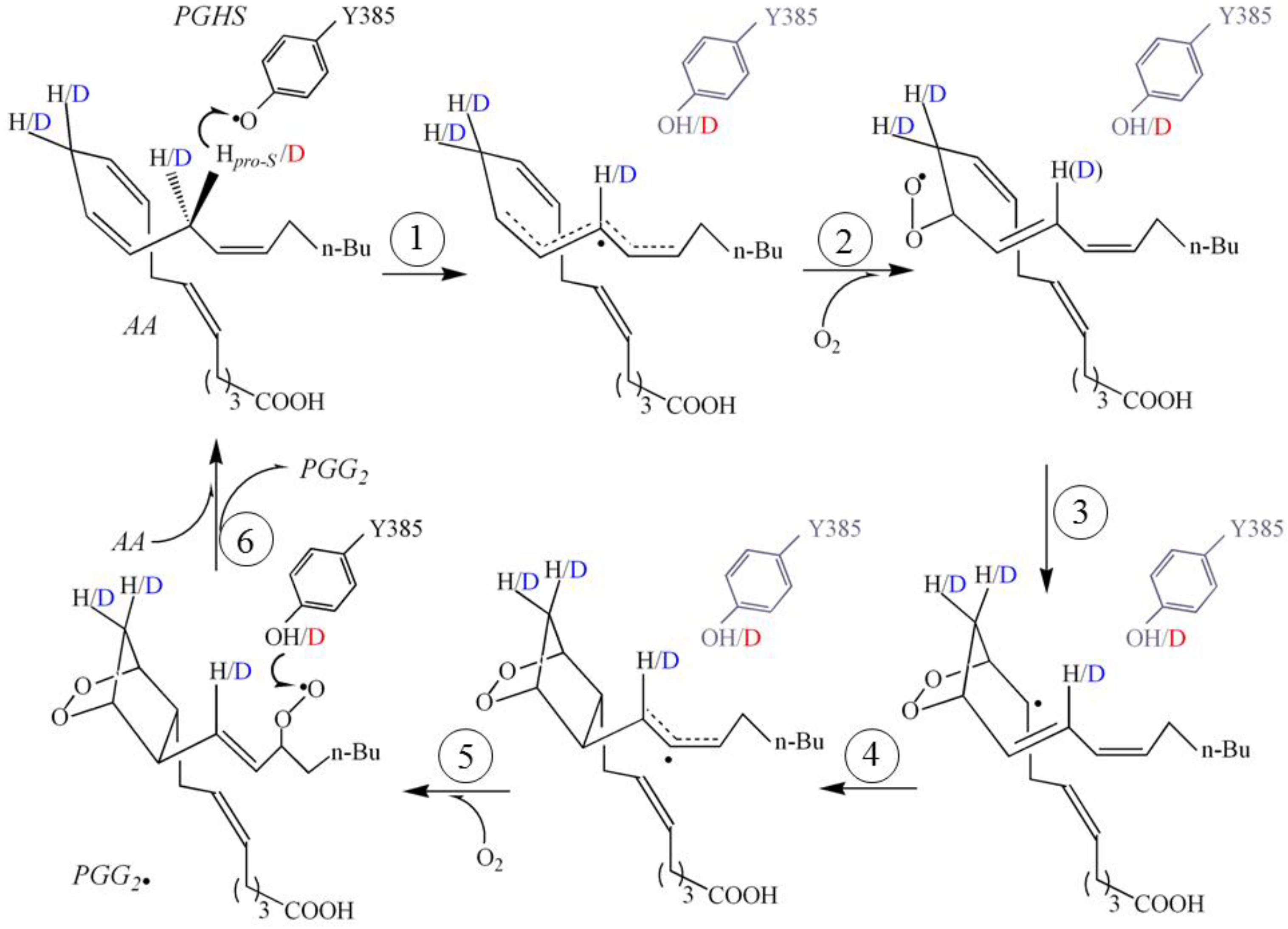
2. Results and Discussion

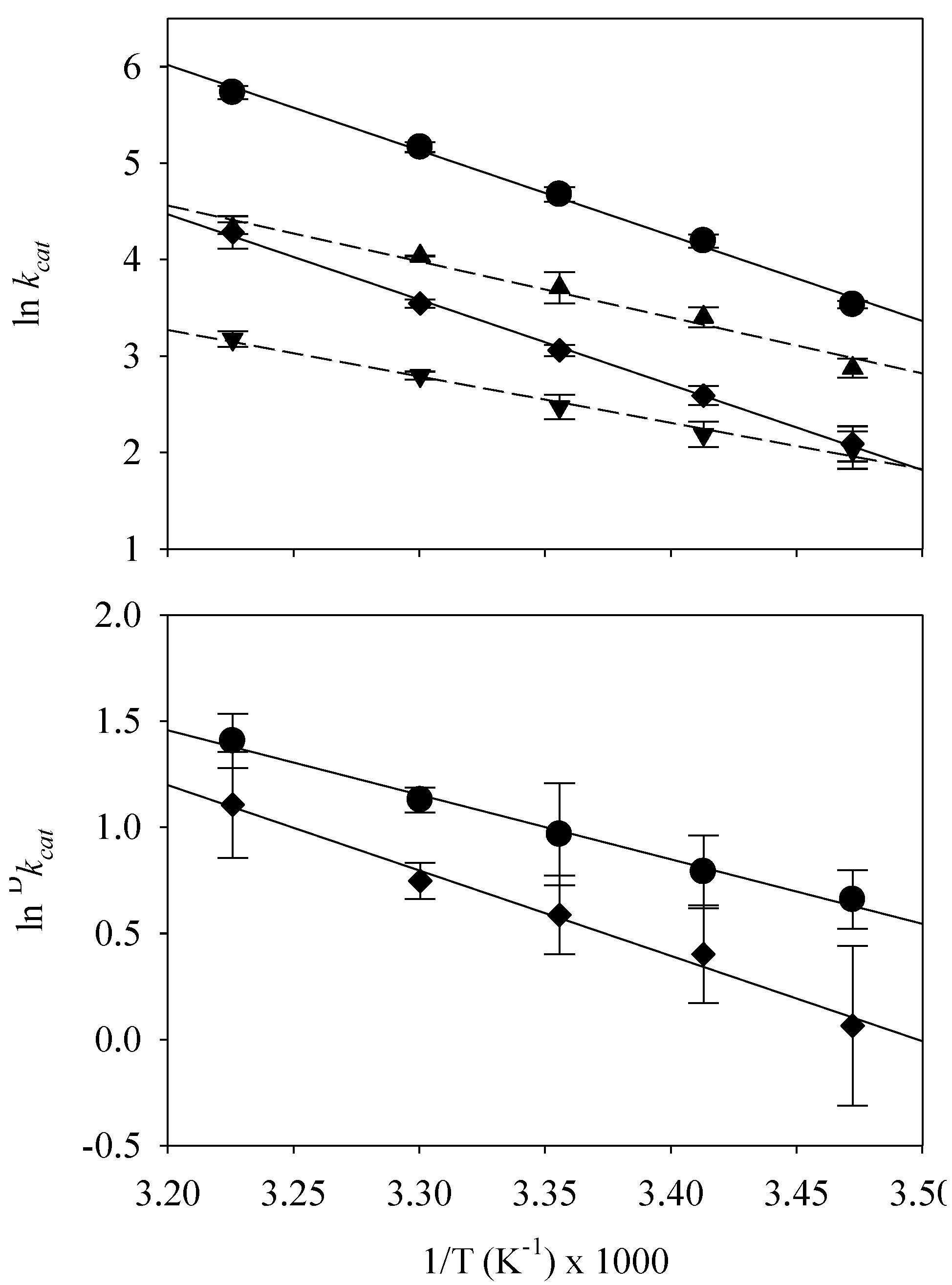
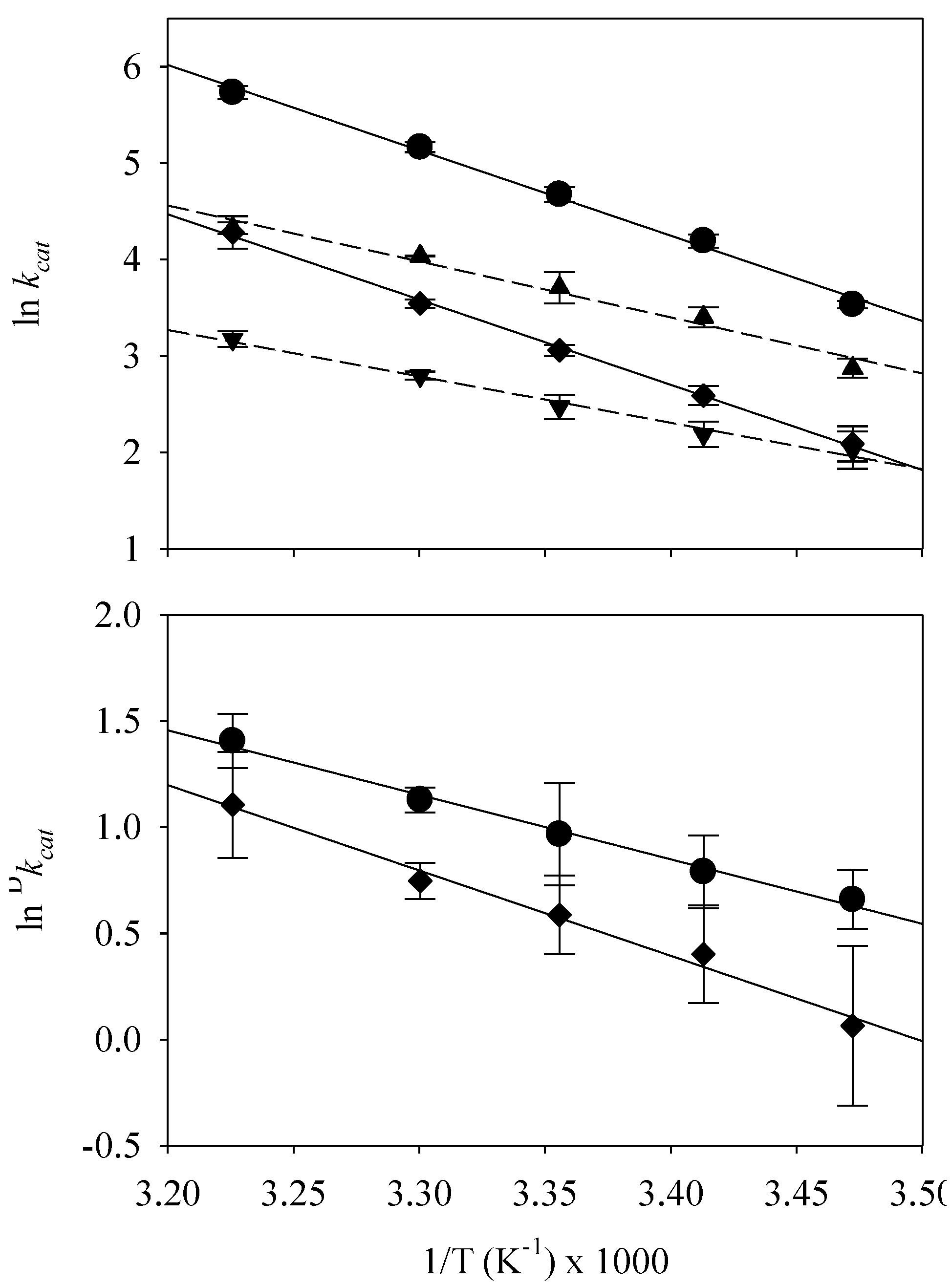{kind=link}
{kind=link}
| Temp. K | PGHS-1 | PGHS-2 | ||||
|---|---|---|---|---|---|---|
| kcat,AA, s−1 | kcat,d4-AA, s−1 | Dkcat | kcat,AA, s−1 | kcat,d4-AA, s−1 | Dkcat | |
| 288 | 34.2 ± 1.3 | 17.7 ± 1.8 | 1.9 ± 0.3 | 8.1 ± 1.8 | 7.6 ± 1.5 | 1.1 ± 0.4 |
| 293 | 66.2 ± 4.5 | 30.1 ± 3.1 | 2.2 ± 0.4 | 13.3 ± 3.1 | 8.9 ± 1.2 | 1.5 ± 0.3 |
| 298 | 107.3 ± 8.3 | 40.8 ± 6.7 | 2.6 ± 0.6 | 21.3 ± 6.7 | 11.9 ± 1.5 | 1.8 ± 0.3 |
| 303 | 175.1 ± 9.2 | 56.6 ± 0.3 | 3.1 ± 0.2 | 34.6 ± 0.3 | 16.4 ± 0.7 | 2.1 ± 0.2 |
| 310 | 308.6 ± 20.7 | 75.6 ± 4.6 | 4.1 ± 0.5 | 72.3 ± 4.6 | 23.9 ± 1.9 | 3.0 ± 0.8 |
| Ea,H (kJ·mol−1) | Ea,D (kJ·mol−1) | ΔEa (kJ·mol−1) | AH (s−1) | AD (s−1) | AH/AD | |
|---|---|---|---|---|---|---|
| PGHS-1/AA a | 74 | 48 | 25 | 7.9 × 1014 | 1.1 × 1010 | 7.9 × 104 |
| PGHS-2/AA a | 73 | 40 | 33 | 1.6 × 1014 | 1.3 × 108 | 1.2 × 106 |
| PGHS-2/LA b | 0.04 | 20 | ||||
| Soybean LOX/LA c | 8.8 | 3.8 | 9 × 103 | 18 |
3. Experimental Section
3.1. Materials
3.2. Measurement of Non-Competitive Kinetic Isotope Effect
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Smith, W.L.; Garavito, R.M.; DeWitt, D.L. Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and -2. J. Biol. Chem. 1996, 271, 33157–33160. [Google Scholar]
- Samuelsson, B.; Goldyne, M.; Granstrom, E.; Hamberg, M.; Hammarstrom, S.; Malmsten, C. Prostaglandins and thromboxanes. Annu. Rev. Biochem. 1978, 47, 997–1029. [Google Scholar] [CrossRef]
- Shimokawa, T.; Kulmacz, R.J.; Dewitt, D.L.; Smith, W.L. Tyrosine-385 of prostaglandin endoperoxide synthase is required for cyclooxygenase catalysis. J. Biol. Chem. 1990, 265, 20073–20076. [Google Scholar]
- Tsai, A.-L.; Kulmacz, R.J. Tyrosyl radicals in prostaglandin H synthase-1 and-2. Prostaglandins Other Lipid Mediat. 2000, 62, 231–254. [Google Scholar] [CrossRef]
- Kiefer, J.R.; Pawlitz, J.L.; Moreland, K.T.; Stegeman, R.A.; Hood, W.F.; Gierse, J.K.; Stevens, A.M.; Goodwin, D.C.; Rowlinson, S.W.; Marnett, L.J.; et al. Structural insights into the stereochemistry of the cyclooxygenase reaction. Nature 2000, 405, 97–101. [Google Scholar] [CrossRef]
- Tsai, A.-L.; Kulmacz, R.J. Prostaglandin H synthase: Resolved and unresolved mechanistic issues. Arch. Biochem. Biophys. 2010, 493, 103–124. [Google Scholar] [CrossRef]
- Rouzer, C.A.; Marnett, L.J. Mechanism of free radical oxygenation of polyunsaturated fatty acids by cyclooxygenases. Chem. Rev. 2003, 103, 2239–2304. [Google Scholar] [CrossRef]
- Van der Donk, W.A.; Tsai, A.-L.; Kulmacz, R.J. The cyclooxygenase reaction mechanism. Biochemistry 2002, 41, 15451–15458. [Google Scholar] [CrossRef]
- Hamberg, M.; Samuelsson, B. On the mechanism of the biosynthesis of prostaglandins E-1 and F-1-α. J. Biol. Chem. 1967, 242, 5336–5343. [Google Scholar]
- Wu, G.; Lu, J.M.; van der Donk, W.A.; Kulmacz, R.J.; Tsai, A.-L. Cyclooxygenase reaction mechanism of prostaglandin H synthase from deuterium kinetic isotope effects. J. Inorg. Biochem. 2011, 105, 382–390. [Google Scholar] [CrossRef]
- Mukherjee, A.; Brinkley, D.W.; Chang, K.M.; Roth, J.P. Molecular oxygen dependent steps in fatty acid oxidation by cyclooxygenase-1. Biochemistry 2007, 46, 3975–3989. [Google Scholar] [CrossRef]
- Danish, H.H.; Doncheva, I.S.; Roth, J.P. Hydrogen tunneling steps in cyclooxygenase-2 catalysis. J. Am. Chem. Soc. 2011, 133, 15846–15849. [Google Scholar] [CrossRef]
- Andreou, A.; Feussner, I. Lipoxygenases—Structure and reaction mechanism. Phytochemistry 2009, 70, 1504–1510. [Google Scholar] [CrossRef]
- Prigge, S.T.; Boyington, J.C.; Faig, M.; Doctor, K.S.; Gaffney, B.J.; Amzel, L.M. Structure and mechanism of lipoxygenases. Biochimie 1997, 79, 629–636. [Google Scholar] [CrossRef]
- Rickert, K.W.; Klinman, J.P. Nature of hydrogen transfer in soybean lipoxygenase 1: Separation of primary and secondary isotope effects. Biochemistry 1999, 38, 12218–12228. [Google Scholar] [CrossRef]
- Glickman, M.H.; Klinman, J.P. Nature of rate-limiting steps in the soybean lipoxygenase-1 reaction. Biochemistry 1995, 34, 14077–14092. [Google Scholar] [CrossRef]
- Glickman, M.H.; Klinman, J.P. Lipoxygenase reaction mechanism: Demonstration that hydrogen abstraction from substrate precedes dioxygen binding during catalytic turnover. Biochemistry 1996, 35, 12882–12892. [Google Scholar] [CrossRef]
- Knapp, M.J.; Rickert, K.; Klinman, J.P. Temperature-dependent isotope effects in soybean lipoxygenase-1: Correlating hydrogen tunneling with protein dynamics. J. Am. Chem. Soc. 2002, 124, 3865–3874. [Google Scholar] [CrossRef]
- Peng, S.; van der Donk, W.A. An unusual isotope effect on substrate inhibition in the oxidation of arachidonic acid by lipoxygenase. J. Am. Chem. Soc. 2003, 125, 8988–8989. [Google Scholar] [CrossRef]
- Jonsson, T.; Glickman, M.H.; Sun, S.J.; Klinman, J.P. Experimental evidence for extensive tunneling of hydrogen in the lipoxygenase reaction: Implications for enzyme catalysis. J. Am. Chem. Soc. 1996, 118, 10319–10320. [Google Scholar] [CrossRef]
- Pu, J.; Gao, J.; Truhlar, D.G. Multidimensional tunneling, recrossing, and the transmission coefficient for enzymatic reactions. Chem. Rev. 2006, 106, 3140–3169. [Google Scholar] [CrossRef]
- Ludlow, M.K.; Soudackov, A.V.; Hammes-Schiffer, S. Theoretical analysis of the unusual temperature dependence of the kinetic isotope effect in quinol oxidation. J. Am. Chem. Soc. 2009, 131, 7094–7102. [Google Scholar] [CrossRef]
- Koch, H.F.; Dahlberg, D.B.; McEntee, M.F.; Klecha, C.J. Use of kientic isotope effects in mechanism studies. Anomalous arrhenius parameters in the study of elimination reactions. J. Am. Chem. Soc. 1976, 98, 1060–1061. [Google Scholar] [CrossRef]
- Koch, H.F.; Koch, A.S. Proton-transfer reactions. 5. An observed primary kinetic isotope effect that increases with increasing temperature. J. Am. Chem. Soc. 1984, 106, 4536–4539. [Google Scholar] [CrossRef]
- Nagaoka, S.; Nishioku, Y.; Mukai, K. Tunneling effect in the regeneration reaction of vitamin E by ubiquinol. Chem. Phys. Lett. 1998, 287, 70–74. [Google Scholar] [CrossRef]
- Zhu, X.Q.; Li, X.T.; Han, S.H.; Mei, L.R. Conversion and origin of normal and abnormal temperature dependences of kinetic isotope effect in hydride transfer reactions. J. Org. Chem. 2012, 77, 4774–4783. [Google Scholar] [CrossRef]
- Cape, J.L.; Bowman, M.K.; Kramer, D.M. Reaction intermediates of quinol oxidation in a photoactivatable system that mimics electron transfer in the cytochrome bc1 complex. J. Am. Chem. Soc. 2005, 127, 4208–4215. [Google Scholar] [CrossRef]
- Wecksler, A.T.; Jacquot, C.; van der Donk, W.A.; Holman, T.R. Mechanistic investigations of human reticulocyte 15- and platelet 12-lipoxygenases with arachidonic acid. Biochemistry 2009, 48, 6259–6267. [Google Scholar] [CrossRef]
- Yellow Springs Instrument Co. Instructions for YSI Model 53 Biological Oxygen Monitor; Yellow Springs Instrument Co.: Yellow Springs, OH, USA, 1987. [Google Scholar]
- Blomberg, L.M.; Blomberg, M.R.A.; Siegbahn, P.E.M.; van der Donk, W.A.; Tsai, A.L. A quantum chemical study of the synthesis of prostaglandin G2 by the cyclooxygenase active site in prostaglandin endoperoixde H synthase 1. J. Phys. Chem. B 2003, 107, 3297–3308. [Google Scholar] [CrossRef]
- Malkowski, M.G.; Ginell, S.L.; Smith, W.L.; Garavito, R.M. The productive conformation of arachidonic acid bound to prostaglandin synthase. Science 2000, 289, 1933–1937. [Google Scholar] [CrossRef]
- Garavito, R.M.; Mulichak, A.M. The structure of mammalian cyclooxygenases. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 183–206. [Google Scholar] [CrossRef]
- Simon, H.; Palm, D. Isotope effects in organic chemistry and biochemistry. Angew. Chem. Int. Ed. 1966, 5, 920–933. [Google Scholar] [CrossRef]
- Laneuville, O.; Breuer, D.K.; Xu, N.; Huang, Z.H.; Gage, D.A.; Watson, J.T.; Lagarde, M.; DeWitt, D.L.; Smith, W.L. Fatty acid substrate specificities of human prostaglandin-endoperoxide H synthase-1 and -2. Formation of 12-hydroxy-(9Z, 13E/Z, 15Z)-octadecatrienoic acids from alpha-linolenic acid. J. Biol. Chem. 1995, 270, 19330–19336. [Google Scholar] [CrossRef]
- Liu, W.; Cao, D.; Oh, S.F.; Serhan, C.N.; Kulmacz, R.J. Divergent cyclooxygenase responses to fatty acid structure and peroxide level in fish and mammalian prostaglandin H synthases. FASEB J. 2006, 20, 1097–1108. [Google Scholar] [CrossRef]
- Vecchio, A.J.; Simmons, D.M.; Malkowski, M.G. Structural basis of fatty acid substrate binding to cyclooxygenase-2. J. Biol. Chem. 2010, 285, 22151–22163. [Google Scholar]
- Vecchio, A.J.; Orlando, B.J.; Nandagiri, R.; Malkowski, M.G. Investigating substrate promiscuity in cyclooxygenase-2: The role of Arg-120 and residues lining the hydrophobic groove. J. Biol. Chem. 2012, 287, 24619–24630. [Google Scholar]
- Janssen-Timmen, U.; Tomic, I.; Specht, E.; Beilecke, U.; Habenicht, A.J. The arachidonic acid cascade, eicosanoids, and signal transduction. Ann. N. Y. Acad. Sci. 1994, 733, 325–334. [Google Scholar] [CrossRef]
- Wensel, S.E. Arachidonic acid metabolites: Mediatiors of inflammation in asthma. Pharmacotherapy 1997, 17, 3S–12S. [Google Scholar]
- Peng, S.; Okeley, N.M.; Tsai, A.-L.; Wu, G.; Kulmacz, R.J.; van der Donk, W.A. Structural characterization of a pentadienyl radical intermediate formed during catalysis by prostaglandin H synthase-2. J. Am. Chem. Soc. 2001, 123, 3609–3610. [Google Scholar] [CrossRef]
- Peng, S.; Okeley, N.M.; Tsai, A.-L.; Wu, G.; Kulmacz, R.J.; van der Donk, W.A. Synthesis of isotopically labeled arachidonic acids to probe the reaction mechanism of prostaglandin H synthase. J. Am. Chem. Soc. 2002, 124, 10785–10796. [Google Scholar]
- Wu, G.; Wei, C.; Kulmacz, R.J.; Osawa, Y.; Tsai, A.-L. A mechanistic study of self-inactivation of the peroxidase activity in prostaglandin H synthase-1. J. Biol. Chem. 1999, 274, 9231–9237. [Google Scholar]
- Wu, G.; Tsai, A.-L.; Kulmacz, R.J. Cyclooxygenase competitive inhibitors alter tyrosyl radical dynamics in prostaglandin h synthase-2. Biochemistry 2009, 48, 11902–11911. [Google Scholar] [CrossRef]
- Kulmacz, R.J.; Tsai, A.-L.; Palmer, G. Heme spin states and peroxide-induced radical species in prostaglandin H synthase. J. Biol. Chem. 1987, 262, 10524–10531. [Google Scholar]
- Kulmacz, R.J.; Palmer, G.; Tsai, A.-L. Prostaglandin H synthase: Perturbation of the tyrosyl radical as a probe of anticyclooxygenase agents. Mol. Pharmacol. 1991, 40, 833–837. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wu, G.; Kulmacz, R.J.; Tsai, A.-L. Kinetic Isotope Effect of Prostaglandin H Synthase Exhibits Inverted Temperature Dependence. Catalysts 2014, 4, 174-185. https://doi.org/10.3390/catal4020174
Wu G, Kulmacz RJ, Tsai A-L. Kinetic Isotope Effect of Prostaglandin H Synthase Exhibits Inverted Temperature Dependence. Catalysts. 2014; 4(2):174-185. https://doi.org/10.3390/catal4020174
Chicago/Turabian StyleWu, Gang, Richard J. Kulmacz, and Ah-Lim Tsai. 2014. "Kinetic Isotope Effect of Prostaglandin H Synthase Exhibits Inverted Temperature Dependence" Catalysts 4, no. 2: 174-185. https://doi.org/10.3390/catal4020174
APA StyleWu, G., Kulmacz, R. J., & Tsai, A.-L. (2014). Kinetic Isotope Effect of Prostaglandin H Synthase Exhibits Inverted Temperature Dependence. Catalysts, 4(2), 174-185. https://doi.org/10.3390/catal4020174