Gold Nanoparticle-Biological Molecule Interactions and Catalysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Why Use GNPs in Biology?
- Templating: This refers to templating of GNPs by biological molecules. Here the ability of biological molecules to assemble into ordered arrays is used to provide a scaffold upon which GNPs can be assembled. This provides ordered, potentially programmable patterns of GNPs, which may be useful for biosensors and/or catalysis.
- Catalytic Effects: Here the catalytic effects of GNPs are considered. This is further subdivided into catalysis by the GNP itself, where the gold atoms on the GNP surface are the catalysts, and catalysis by the ligands, in which the molecules attached to the surface of the GNP are responsible for the catalytic effect and the GNP plays an important scaffolding role. In both cases catalysis relevant to biological molecules is considered particularly where the GNPs carry out catalytic function usually performed by a biological molecule and/or perform catalysis on a biological molecule.
3. Templating
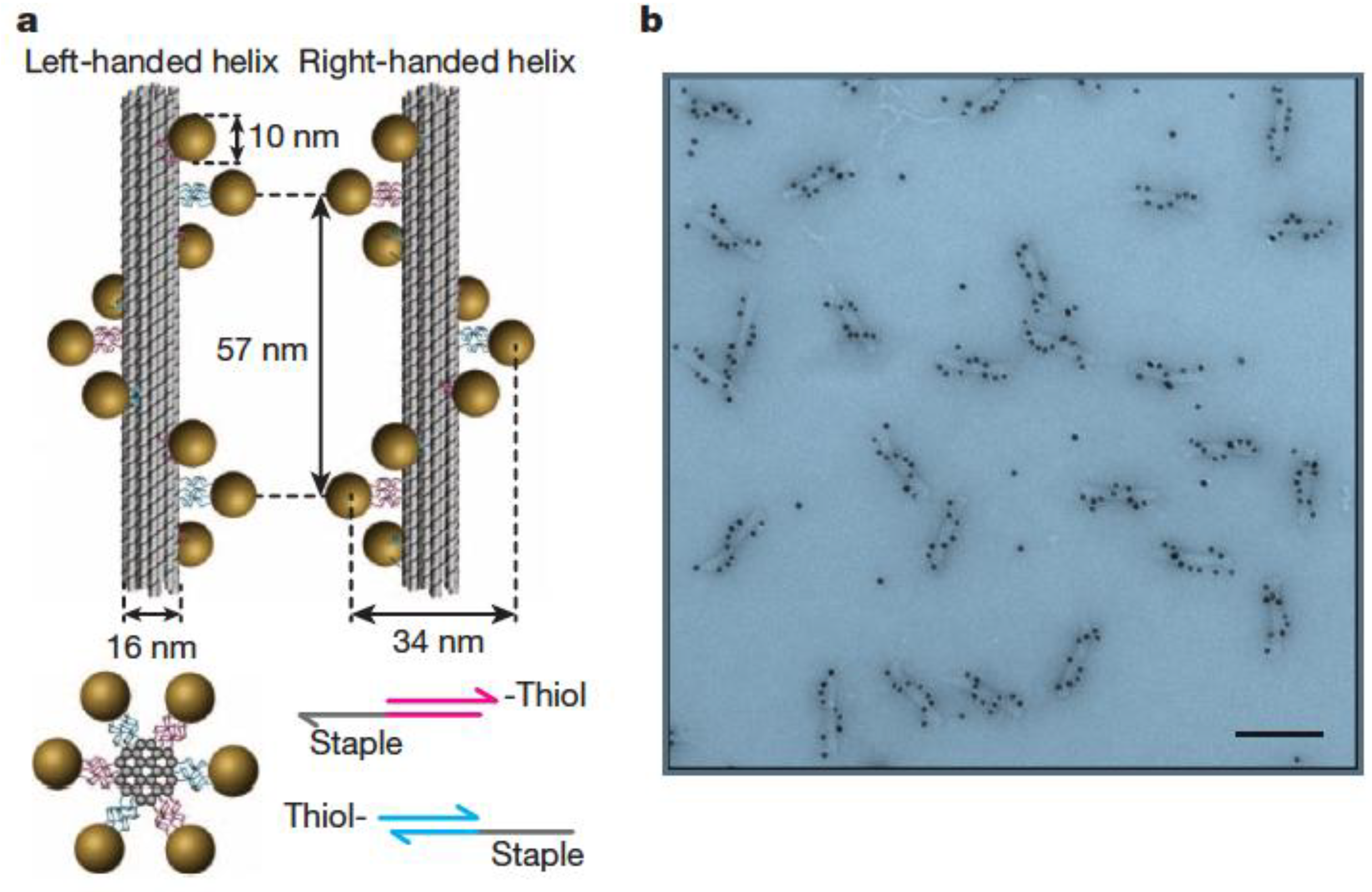
3.1. Templating by DNA


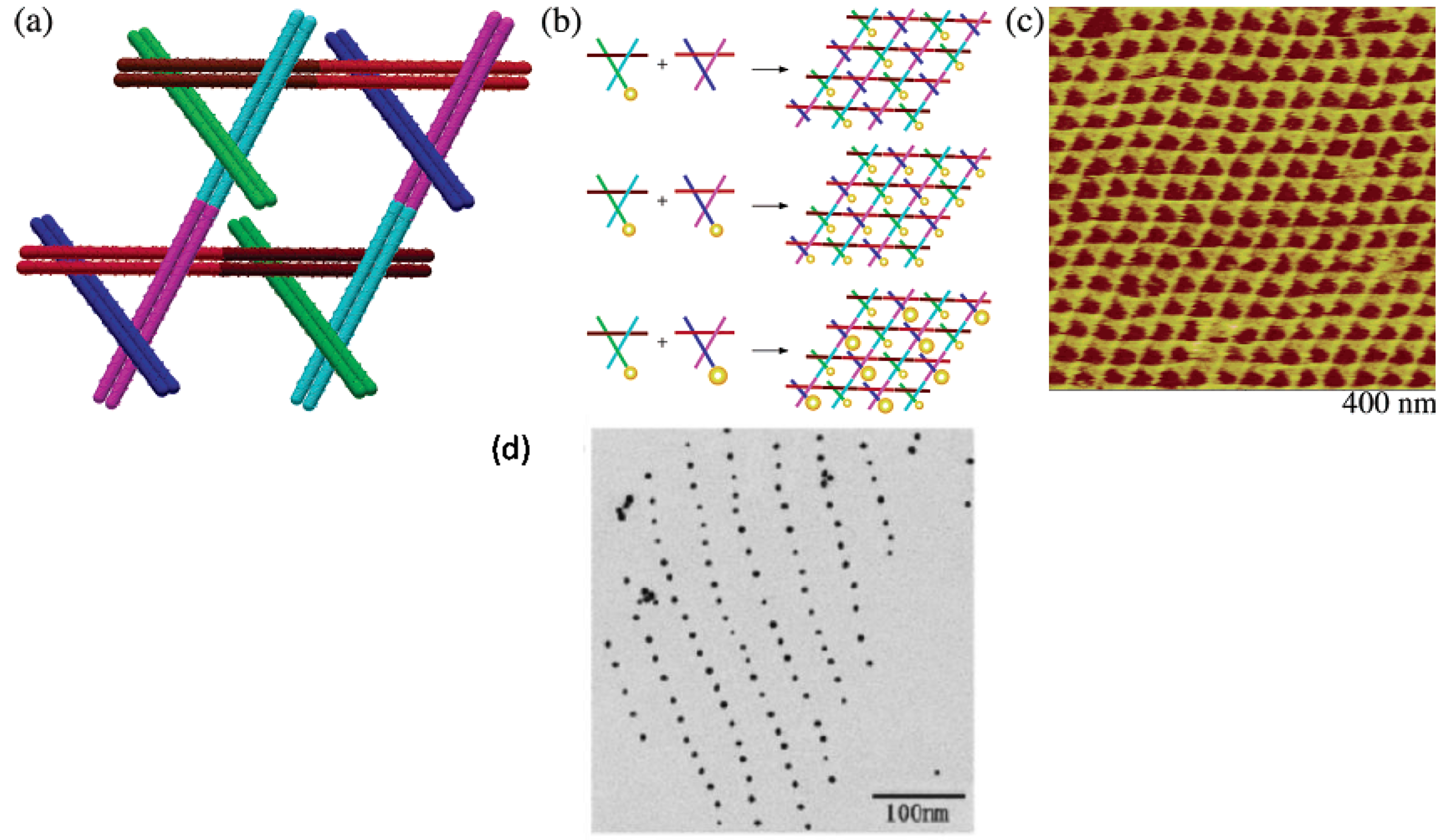
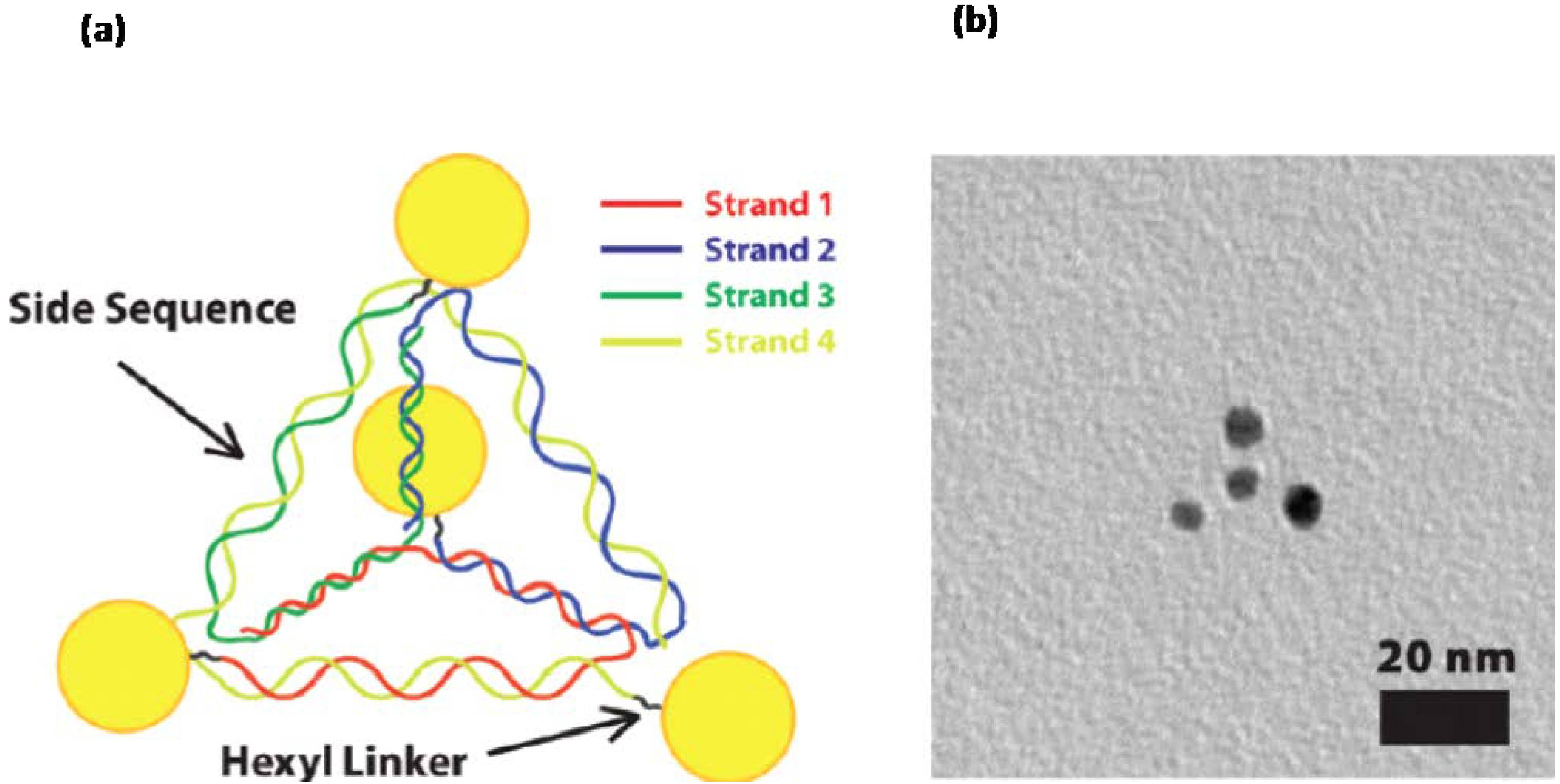
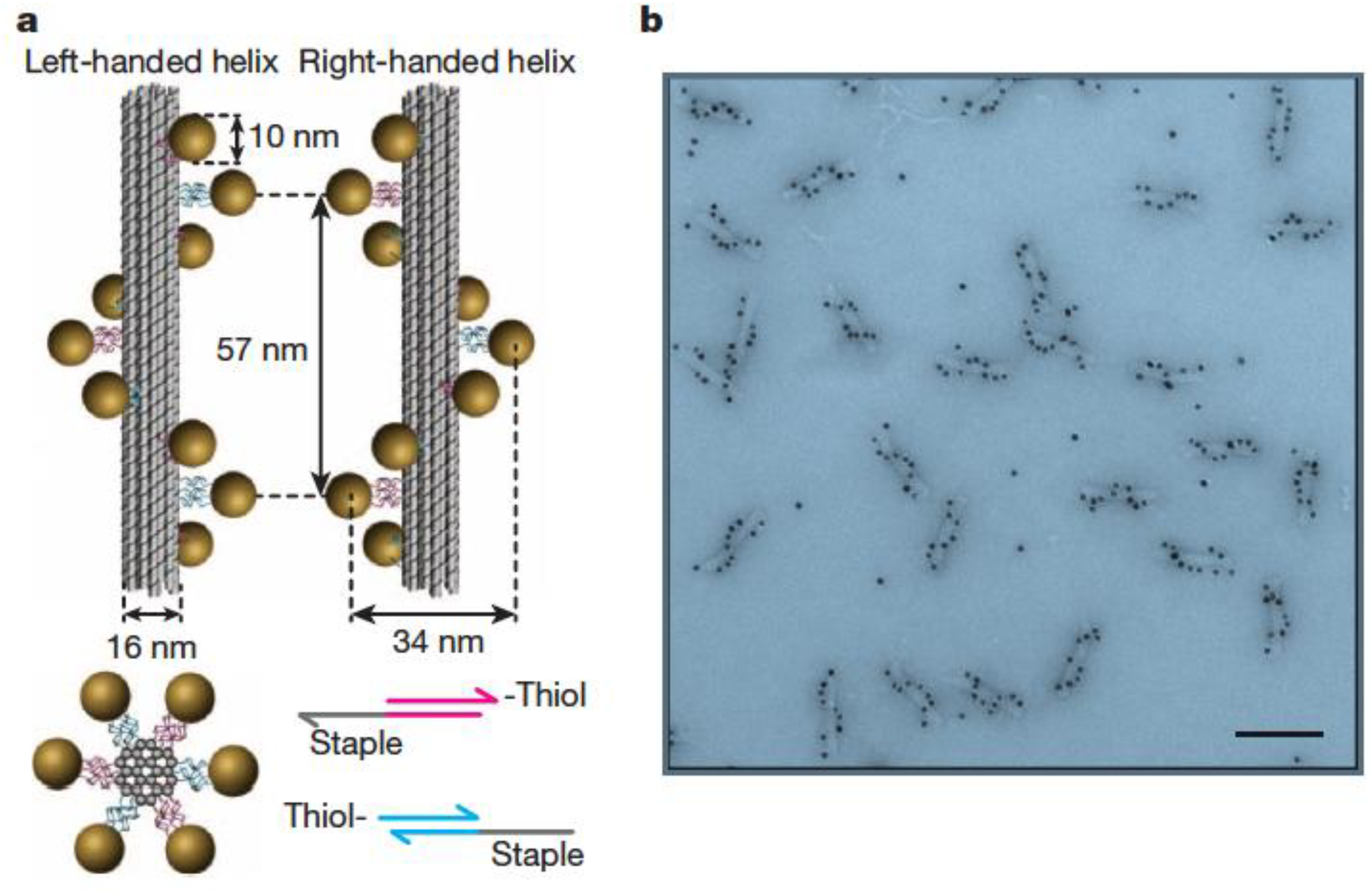
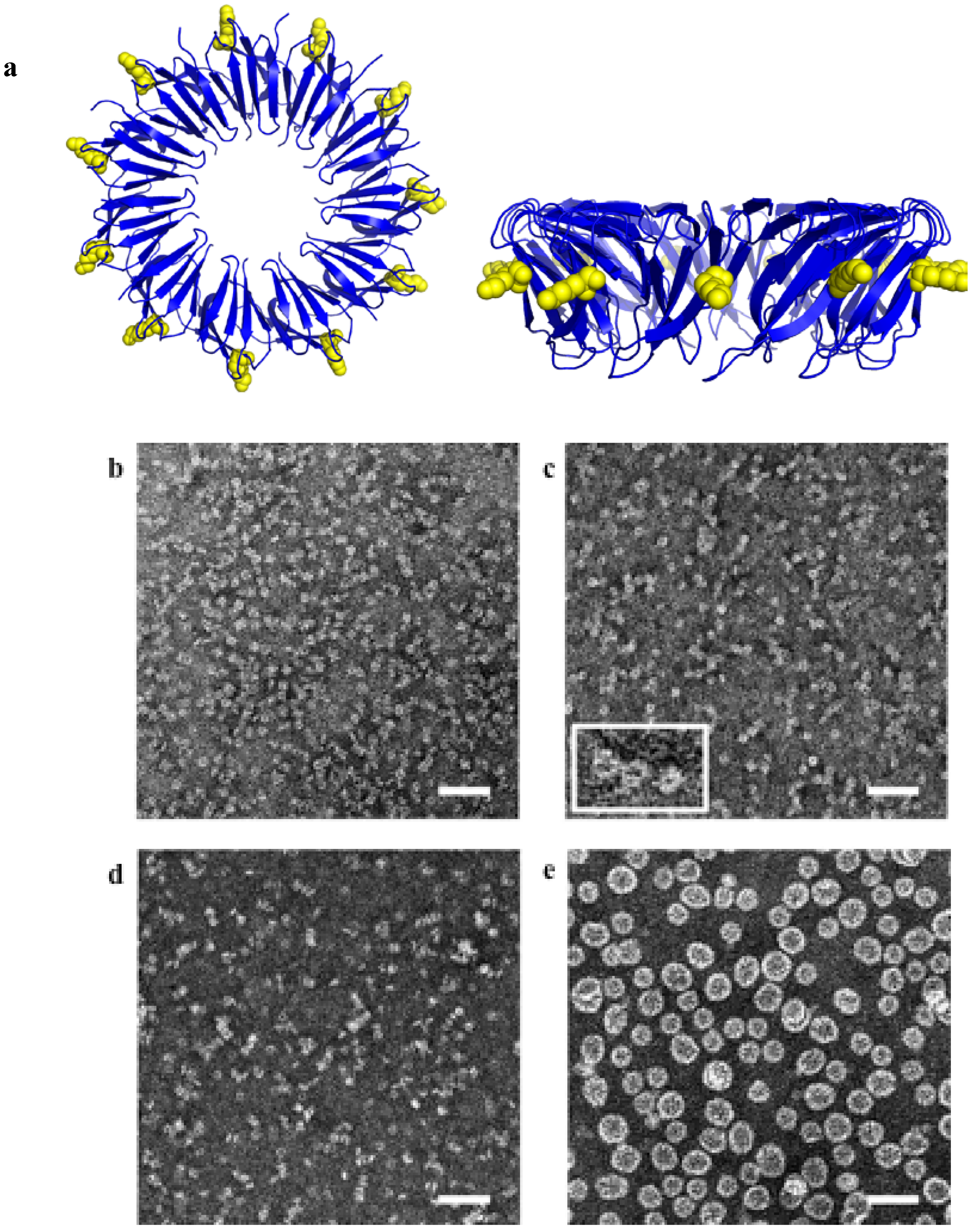
3.2. Templating by Protein


4. Catalytic Effects of GNPs on Biological Molecules
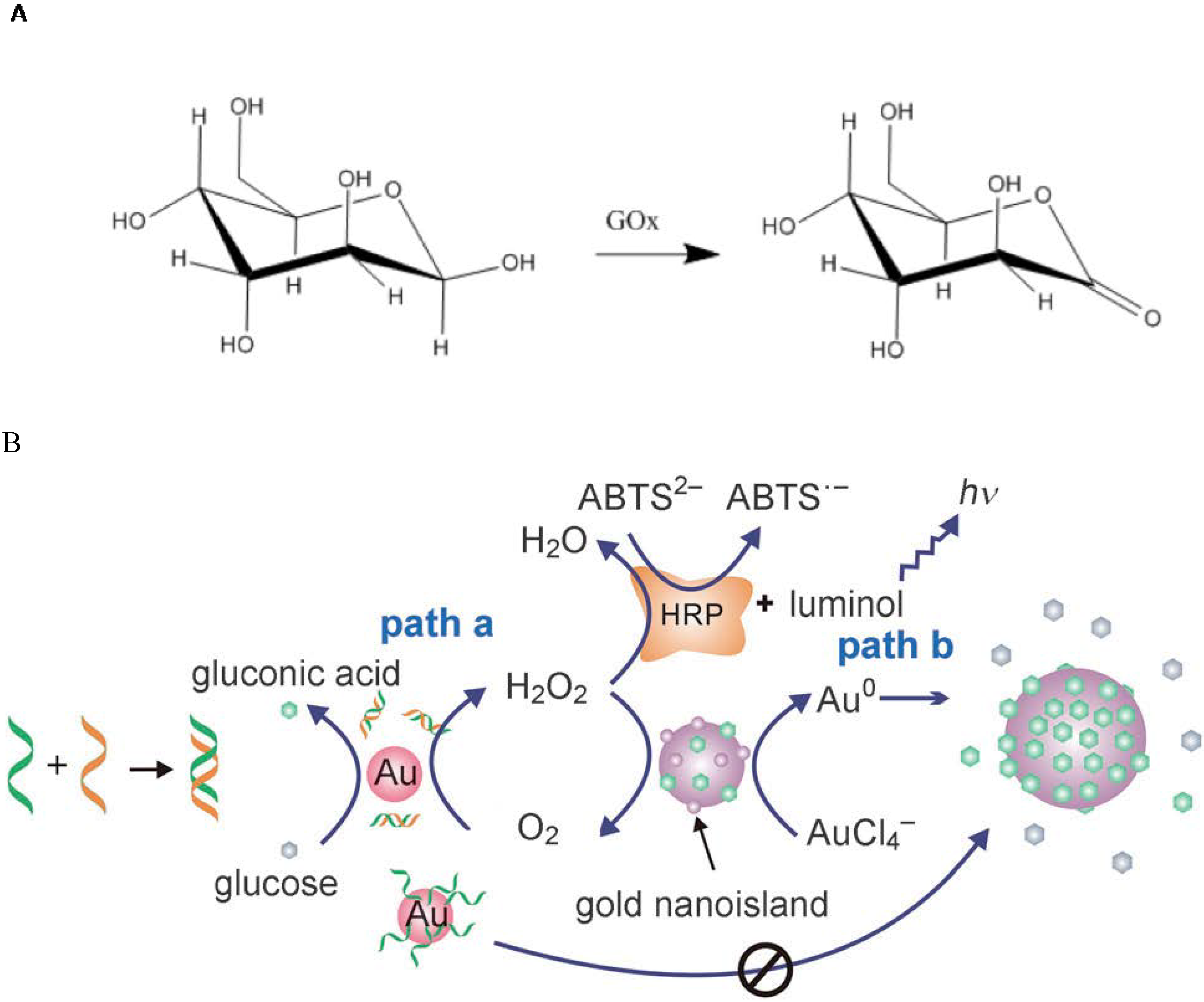
4.1. Catalysis by GNP Itself
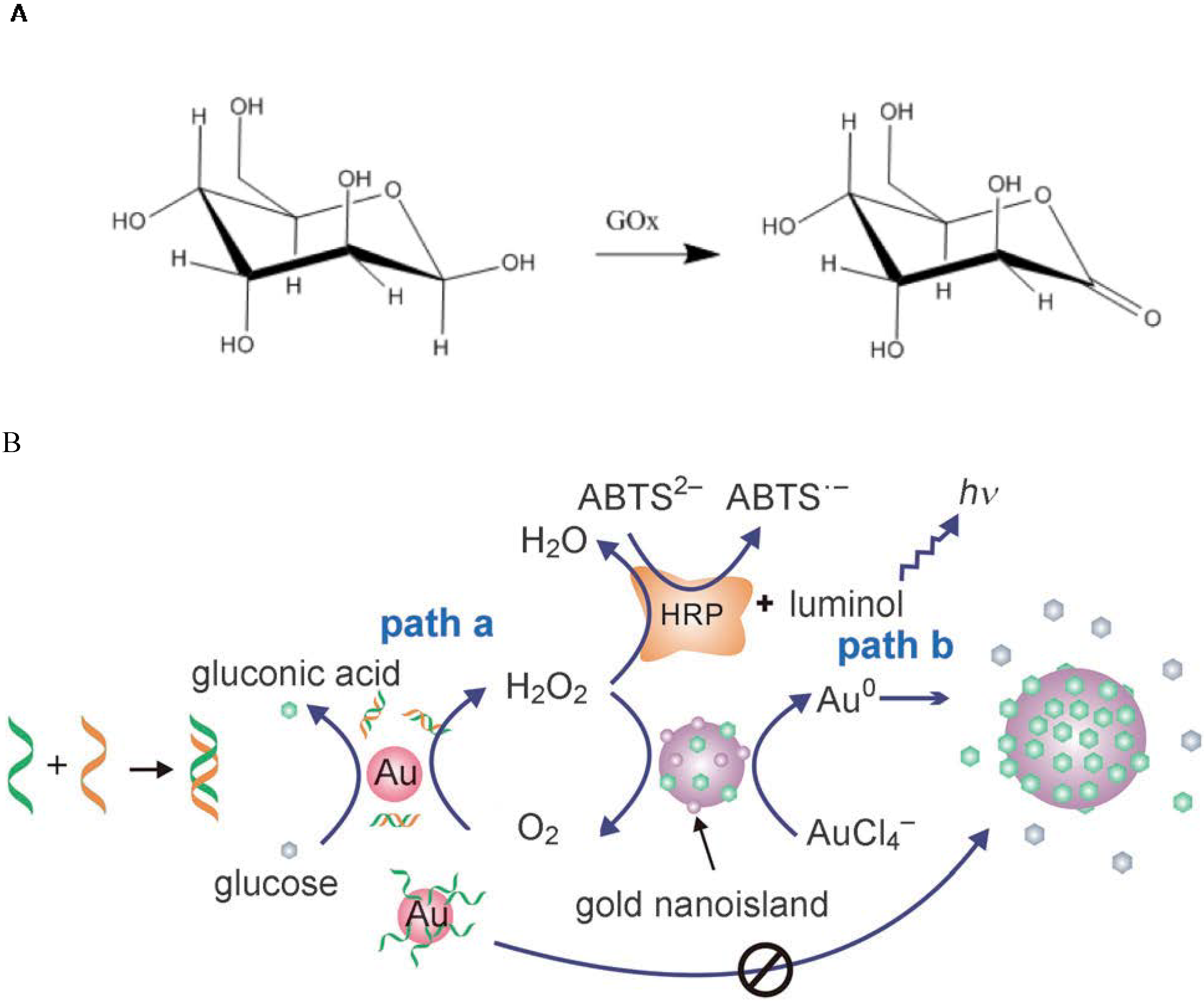
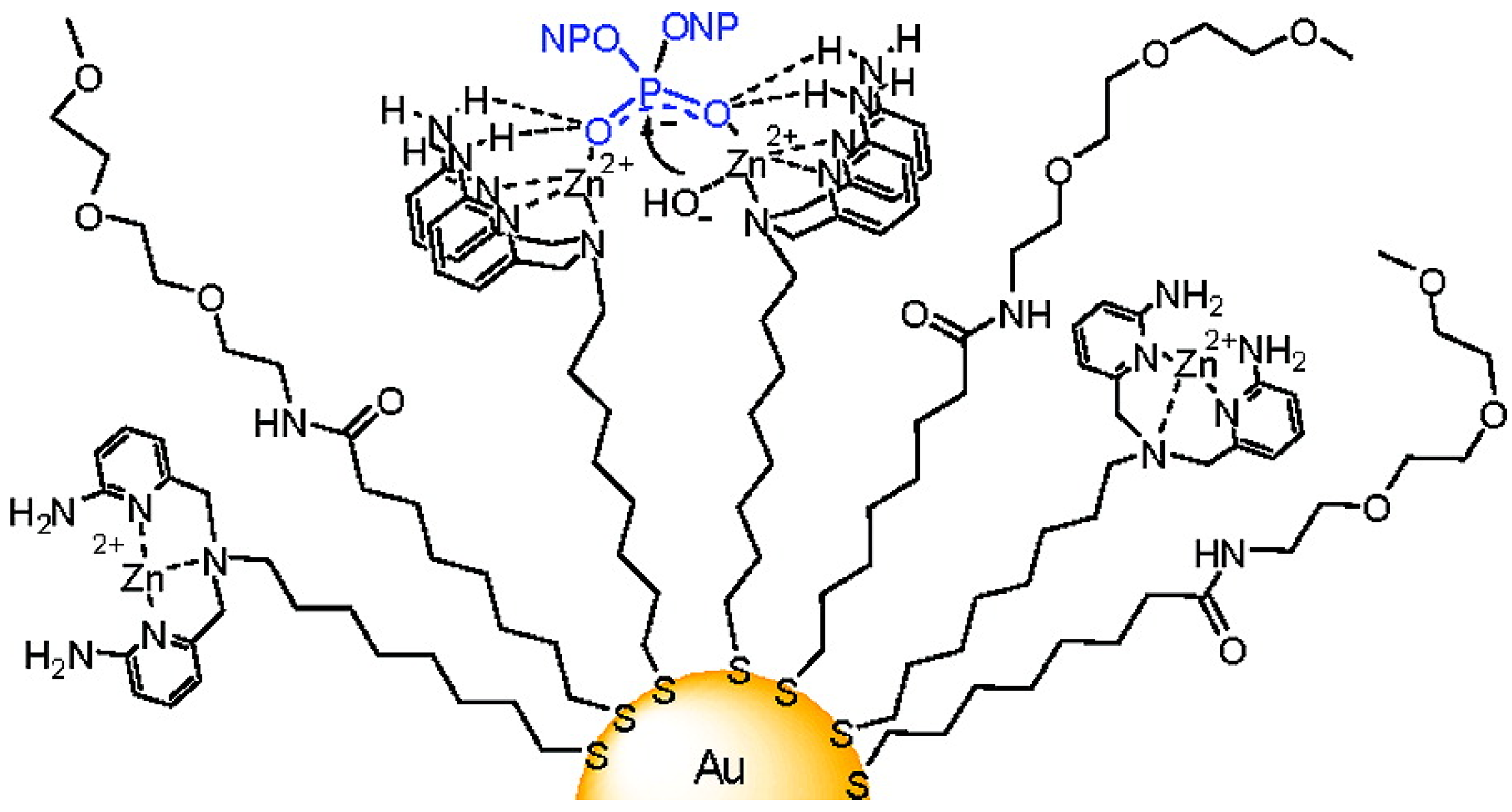
4.2. Biological Catalysis by the Ligand
5. Conclusions and Future Potential
Acknowledgments
Conflicts of Interest
References
- Faraday, M. The Bakerian lecture: experimental relations of gold (and other metals) to light. Philos. Trans. R. Soc. London 1857, 147, 145–181. [Google Scholar] [CrossRef]
- Edwards, P.P.; Thomas, J.M. Gold in a metallic divided state—From Faraday to present-day nanoscience. Angew. Chem. Int. Ed. 2007, 46, 5480–5486. [Google Scholar] [CrossRef]
- Tweney, R.D. Discovering discovery: how Faraday found the first metallic colloid. Perspect. Sci. 2006, 14, 97–121. [Google Scholar] [CrossRef]
- Haruta, M.; Kobayashi, T.; Sano, H.; Yamada, N. Novel gold catalysts for the oxidation of carbon monoxide at a temperature far below 0 °C. Chem. Lett. 1987, 16, 405–408. [Google Scholar]
- Turkevich, J.; Stevenson, P.C.; Hillier, J. A study of the nucleation and growth processes in the synthesis of colloidal gold. Discuss. Faraday Soc. 1951, 11, 55–75. [Google Scholar] [CrossRef]
- Enustun, B.V.; Turkevich, J. Coagulation of colloidal gold. J. Am. Chem. Soc. 1963, 85, 3317–3328. [Google Scholar] [CrossRef]
- Turkevich, J. Colloidal gold. Part I. Gold Bull. 1985, 18, 86–91. [Google Scholar] [CrossRef]
- Brust, M.; Walker, M.; Bethell, D.; Schiffrin, D.J.; Whyman, R. Synthesis of thiol-derivatised gold nanoparticles in a two-phase Liquid-Liquid system. J. Chem. Soc. Chem. Commun. 1994, 801–802. [Google Scholar]
- Grzelczak, M.; Perez-Juste, J.; Mulvaney, P.; Liz-Marzan, L.M. Shape control in gold nanoparticle synthesis. Chem. Soc. Rev. 2008, 37, 1783–1791. [Google Scholar] [CrossRef]
- Pan, Y.; Neuss, S.; Leifert, A.; Fischler, M.; Wen, F.; Simon, U.; Schmid, G.; Brandau, W.; Jahnen-Dechent, W. Size-dependent cytotoxicity of gold nanoparticles. Small 2007, 3, 1941–1949. [Google Scholar] [CrossRef]
- Pan, Y.; Leifert, A.; Ruau, D.; Neuss, S.; Bornemann, J.; Schmid, G.; Brandau, W.; Simon, U.; Jahnen-Dechent, W. Gold nanoparticles of diameter 1.4 nm trigger necrosis by oxidative stress and mitochondrial damage. Small 2009, 5, 2067–2076. [Google Scholar] [CrossRef]
- Hoa, X.D.; Kirk, A.G.; Tabrizian, M. Towards integrated and sensitive surface plasmon resonance biosensors: A review of recent progress. Biosens. Bioelectron. 2007, 23, 151–160. [Google Scholar] [CrossRef]
- Liedberg, B.; Nylander, C.; Lunström, I. Surface plasmon resonance for gas detection and biosensing. Sens. Actuators 1983, 4, 299–304. [Google Scholar] [CrossRef]
- Murphy, C.J.; Gole, A.M.; Stone, J.W.; Sisco, P.N.; Alkilany, A.M.; Goldsmith, E.C.; Baxter, S.C. Gold nanoparticles in biology: beyond toxicity to cellular imaging. Acc. Chem. Res. 2008, 41, 1721–1730. [Google Scholar] [CrossRef]
- Kelly, K.L.; Coronado, E.; Zhao, L.L.; Schatz, G.C. The Optical Properties of metal nanoparticles: The influence of size, shape, and dielectric environment. J. Phys. Chem. B 2002, 107, 668–677. [Google Scholar]
- Dusemund, B.; Hoffmann, A.; Salzmann, T.; Kreibig, U.; Schmid, G. Cluster matter: The transition of optical elastic scattering to regular reflection. Z. Phys. D 1991, 20, 305–308. [Google Scholar]
- Katz, E.; Willner, I. Integrated nanoparticle-biomolecule hybrid systems: Synthesis, properties, and applications. Angew. Chem. Int. Ed. 2004, 43, 6042–6108. [Google Scholar] [CrossRef]
- Saha, K.; Agasti, S.S.; Kim, C.; Li, X.; Rotello, V.M. Gold nanoparticles in chemical and biological sensing. Chem. Rev. 2012, 112, 2739–2779. [Google Scholar] [CrossRef]
- Stockman, M.I. Nanoplasmonics: Past, present, and glimpse into future. Opt. Express. 2011, 19, 22029–22106. [Google Scholar] [CrossRef]
- Tanaka, R.; Yuhi, T.; Nagatani, N.; Endo, T.; Kerman, K.; Takamura, Y.; Tamiya, E. A novel enhancement assay for immunochromatographic test strips using gold nanoparticles. Anal. Bioanal. Chem. 2006, 385, 1414–1420. [Google Scholar] [CrossRef]
- Cole, J.R.; Mirin, N.A.; Knight, M.W.; Goodrich, G.P.; Halas, N.J. Photothermal efficiencies of nanoshells and nanorods for clinical therapeutic applications. J. Phys. Chem. C 2009, 113, 12090–12094. [Google Scholar] [CrossRef]
- Jain, P.K.; Lee, K.S.; El-Sayed, I.H.; El-Sayed, M.A. Calculated absorption and scattering properties of gold nanoparticles of different size, shape, and composition: applications in biological imaging and biomedicine. J. Phys. Chem. B 2006, 110, 7238–7248. [Google Scholar] [CrossRef]
- Loo, C.; Lin, A.; Hirsch, L.; Lee, M.H.; Barton, J.; Halas, N.; West, J.; Drezek, R. Nanoshell-enabled photonics-based imaging and therapy of cancer. Technol. Cancer Res. Treat. 2004, 3, 33–40. [Google Scholar]
- O'Neal, D.P.; Hirsch, L.R.; Halas, N.J.; Payne, J.D.; West, J.L. Photo-thermal tumor ablation in mice using near infrared-absorbing nanoparticles. Cancer Lett. 2004, 209, 171–176. [Google Scholar] [CrossRef]
- Hirsch, L.R.; Stafford, R.J.; Bankson, J.A.; Sershen, S.R.; Rivera, B.; Price, R.E.; Hazle, J.D.; Halas, N.J.; West, J.L. Nanoshell-mediated near-infrared thermal therapy of tumors under magnetic resonance guidance. Proc. Natl. Acad. Sci. USA 2003, 100, 13549–13554. [Google Scholar] [CrossRef]
- Cardinal, J.; Klune, J.R.; Chory, E.; Jeyabalan, G.; Kanzius, J.S.; Nalesnik, M.; Geller, D.A. Noninvasive radiofrequency ablation of cancer targeted by gold nanoparticles. Surgery 2008, 144, 125–132. [Google Scholar] [CrossRef]
- Pissuwan, D.; Valenzuela, S.M.; Cortie, M.B. Therapeutic possibilities of plasmonically heated gold nanoparticles. Trends Biotechnol. 2006, 24, 62–67. [Google Scholar] [CrossRef]
- Haynes, C.L.; McFarland, A.D.; Duyne, R.P.V. Surface-enhanced raman spectroscopy. Anal. Chem. 2005, 77, 338A–346A. [Google Scholar]
- Watson, J.D.; Crick, F.H. Molecular structure of nucleic acids; A structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef]
- Alivisatos, A.; Johnsson, K.; Peng, X.; Wilson, T.; Loweth, C.; Bruchez, M.; Schultz, P. Organization of ‘nanocrystal molecules’ using DNA. Nature 1996, 382, 609–611. [Google Scholar] [CrossRef]
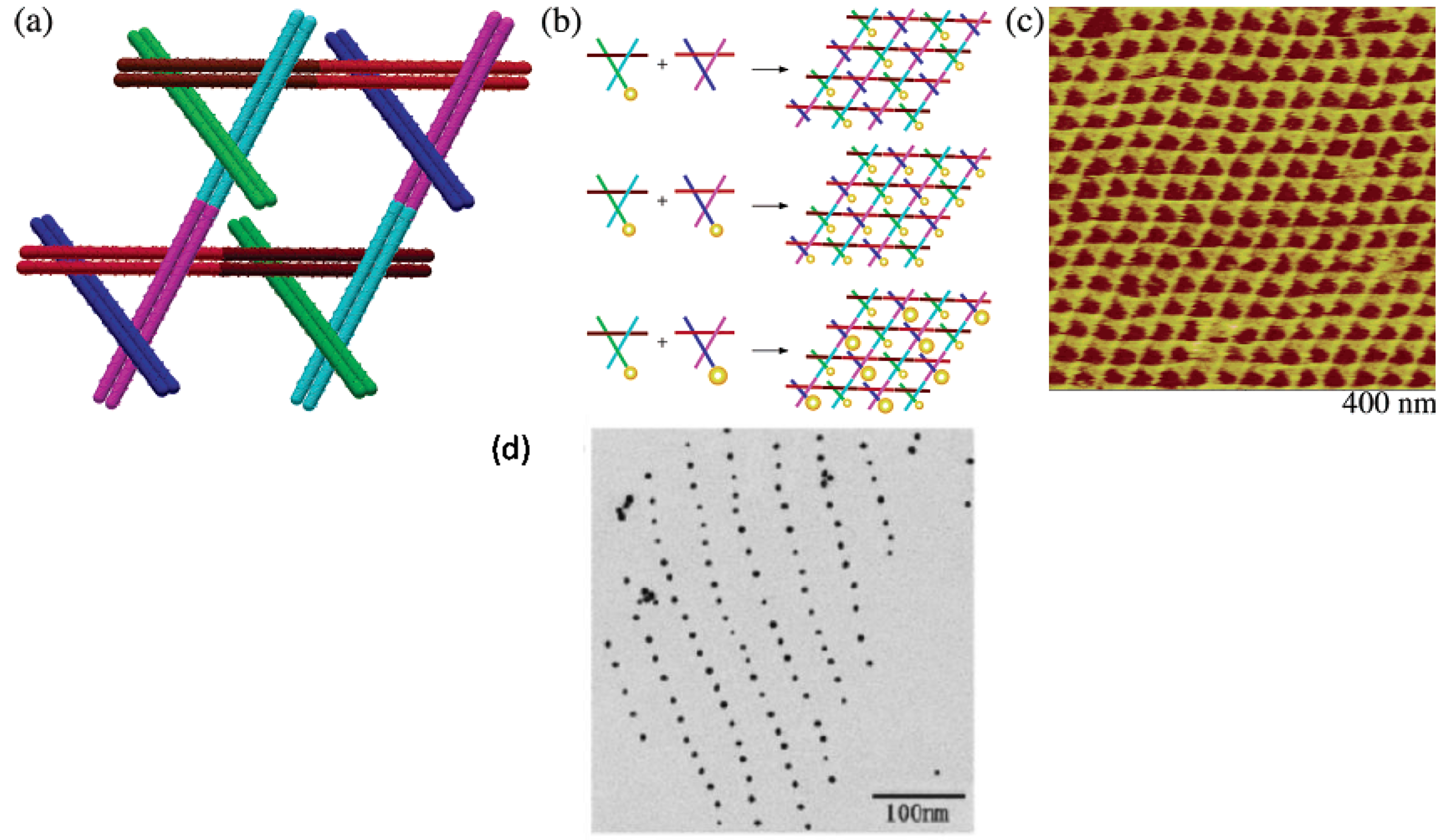
- Pinto, Y.Y.; Le, J.D.; Seeman, N.C.; Musier-Forsyth, K.; Taton, T.A.; Kiehl, R.A. Sequence-encoded self-assembly of multiple-nanocomponent arrays by 2D DNA scaffolding. Nano Lett. 2005, 5, 2399–2402. [Google Scholar] [CrossRef]
- Zheng, J.; Constantinou, P.E.; Micheel, C.; Alivisatos, A.P.; Kiehl, R.A.; Seeman, N.C. Two-dimensional nanoparticle arrays show the organizational power of robust DNA motifs. Nano Lett. 2006, 6, 1502–1504. [Google Scholar] [CrossRef]
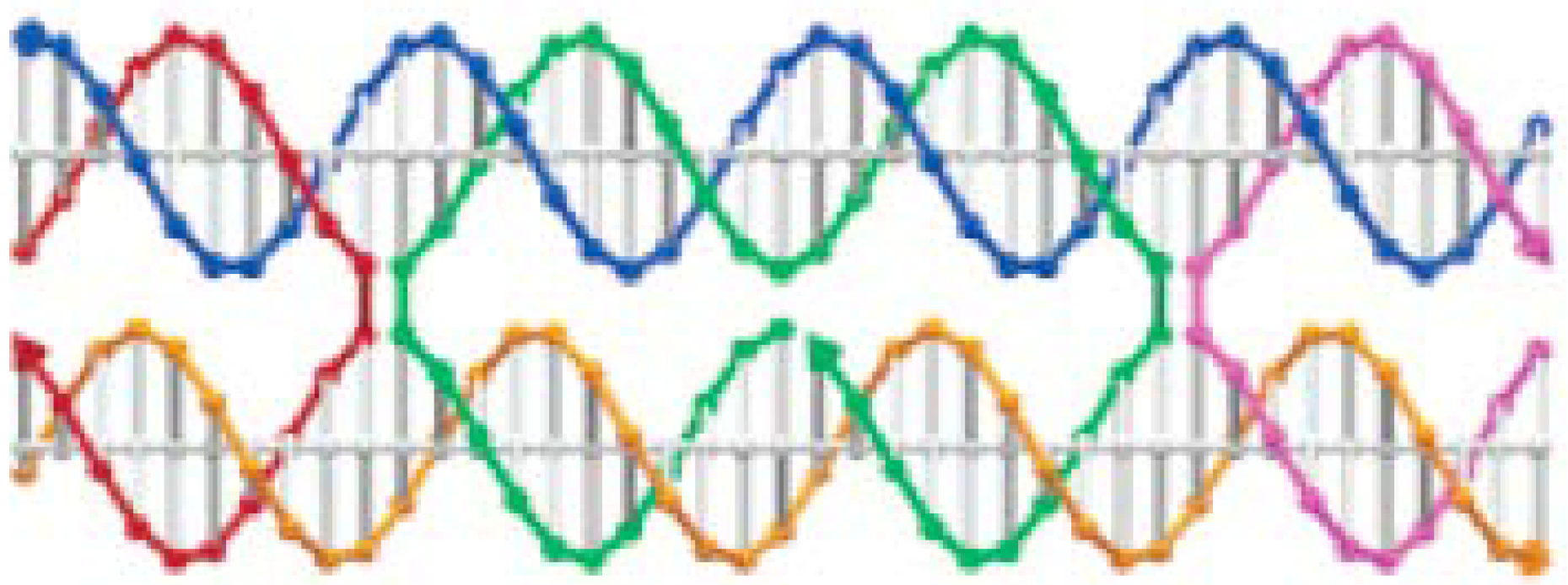
- Sharma, J.; Chhabra, R.; Liu, Y.; Ke, Y.; Yan, H. DNA-templated self-assembly of two-dimensional and periodical gold nanoparticle arrays. Angew. Chem. Int. Ed. 2006, 45, 730–735. [Google Scholar] [CrossRef]
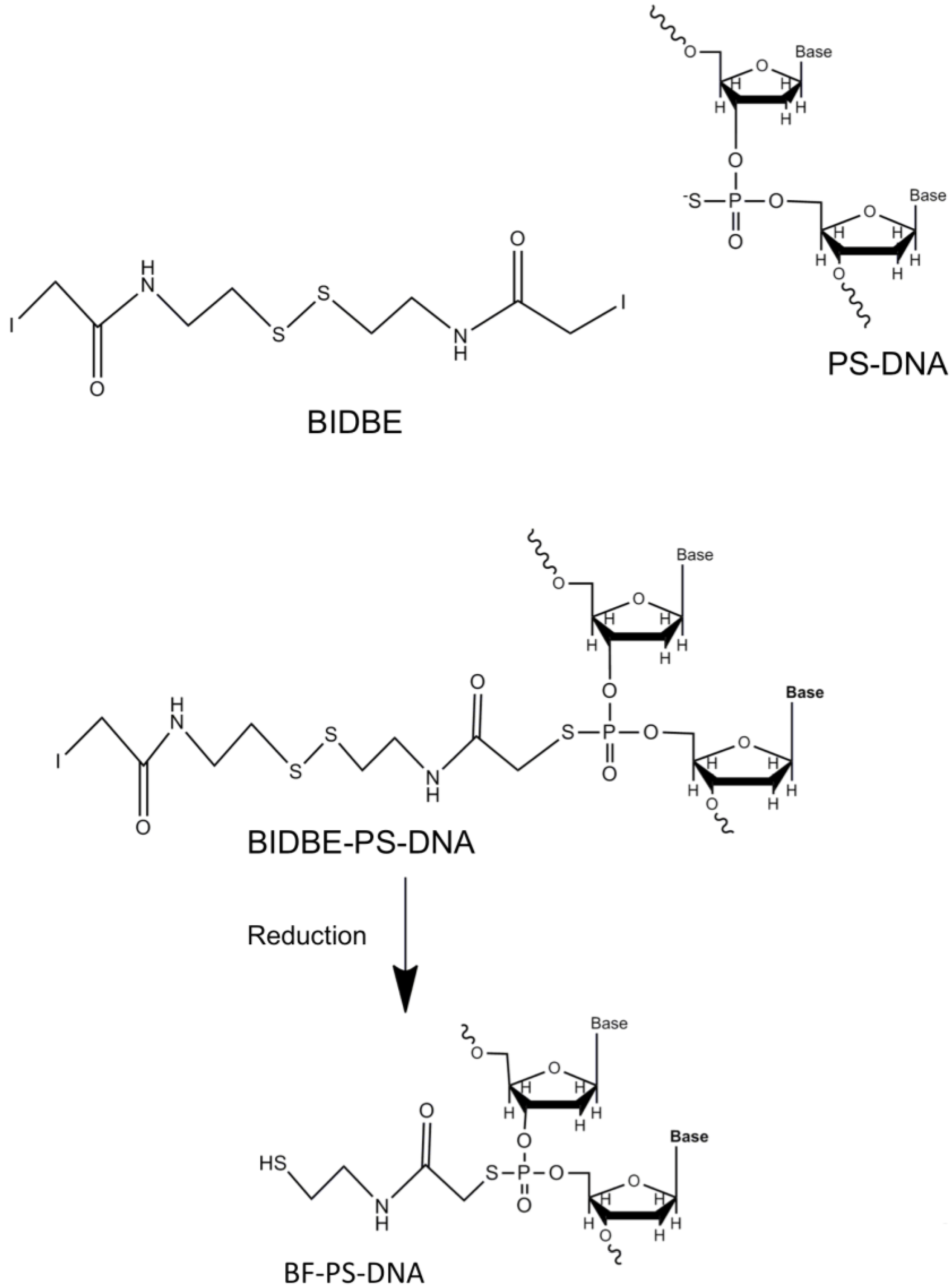
- Lee, J.H.; Wernette, D.P.; Yigit, M.V.; Liu, J.; Wang, Z.; Lu, Y. Site-specific control of distances between gold nanoparticles using phosphorothioate anchors on DNA and a short bifunctional molecular fastener. Angew. Chem. Int. Ed. 2007, 46, 9006–9010. [Google Scholar] [CrossRef]
- Ludueña, R.F.; Roach, M.C.; Trcka, P.P.; Weintraub, S. N,N-Bis(α-iodoacetyl)-2,2′-dithiobis(ethylamine), a reversible crosslinking reagent for protein sulfhydryl groups. Anal. Biochem. 1981, 117, 76–80. [Google Scholar] [CrossRef]
- Ozbay, E. Plasmonics: Merging photonics and electronics at nanoscale dimensions. Science 2006, 311, 189–193. [Google Scholar] [CrossRef]
- Liu, Z.; Searson, P.C. Single nanoporous gold nanowire sensors. J. Phys. Chem. B 2006, 110, 4318–4322. [Google Scholar] [CrossRef]
- Chirea, M.; Freitas, A.; Vasile, B.S.; Ghitulica, C.; Pereira, C.M.; Silva, F. Gold nanowire networks: Synthesis, characterization, and catalytic activity. Langmuir 2011, 27, 3906–3913. [Google Scholar] [CrossRef]
- Bai, X.; Gao, Y.; Liu, H.-g.; Zheng, L. Synthesis of amphiphilic ionic liquids terminated gold nanorods and their superior catalytic activity for the reduction of nitro compounds. J. Phys. Chem. C 2009, 113, 17730–17736. [Google Scholar] [CrossRef]
- Richter, J. Metallization of DNA. Physica E 2003, 16, 157–173. [Google Scholar] [CrossRef]
- Ongaro, A.; Griffin, F.; Beecher, P.; Nagle, L.; Iacopino, D.; Quinn, A.; Redmond, G.; Fitzmaurice, D. DNA-Templated Assembly of Conducting Gold Nanowires between Gold Electrodes on a Silicon Oxide Substrate. Chem. Mater. 2005, 17, 1959–1964. [Google Scholar] [CrossRef]
- Kim, H.J.; Roh, Y.; Hong, B. Selective Formation of a Latticed Nanostructure with the Precise Alignment of DNA-Templated Gold Nanowires. Langmuir 2010, 26, 18315–18319. [Google Scholar] [CrossRef]
- Harnack, O.; Ford, W.E.; Yasuda, A.; Wessels, J.M. Tris(hydroxymethyl)phosphine-Capped Gold Particles Templated by DNA as Nanowire Precursors. Nano Lett. 2002, 2, 919–923. [Google Scholar] [CrossRef]
- Yonezawa, T.; Onoue, S.-y.; Kimizuka, N. Metal Coating of DNA Molecules by Cationic, Metastable Gold Nanoparticles. Chem. Lett. 2002, 31, 1172–1173. [Google Scholar] [CrossRef]
- Patolsky, F.; Weizmann, Y.; Lioubashevski, O.; Willner, I. Au-Nanoparticle Nanowires Based on DNA and Polylysine Templates. Angew. Chem. Int. Ed. 2002, 41, 2323–2327. [Google Scholar] [CrossRef]
- Haruta, M. Catalysis of Gold Nanoparticles Deposited on Metal Oxides. CATTECH 2002, 6, 102–115. [Google Scholar] [CrossRef]
- Fu, T.J.; Seeman, N.C. DNA double-crossover molecules. Biochemistry 1993, 32, 3211–3220. [Google Scholar] [CrossRef]
- Ding, B.; Sha, R.; Seeman, N.C. Pseudohexagonal 2D DNA Crystals from Double Crossover Cohesion. J. Am. Chem. Soc. 2004, 126, 10230–10231. [Google Scholar] [CrossRef]
- Le, J.D.; Pinto, Y.; Seeman, N.C.; Musier-Forsyth, K.; Taton, T.A.; Kiehl, R.A. DNA-Templated Self-Assembly of Metallic Nanocomponent Arrays on a Surface. Nano Lett. 2004, 4, 2343–2347. [Google Scholar] [CrossRef]
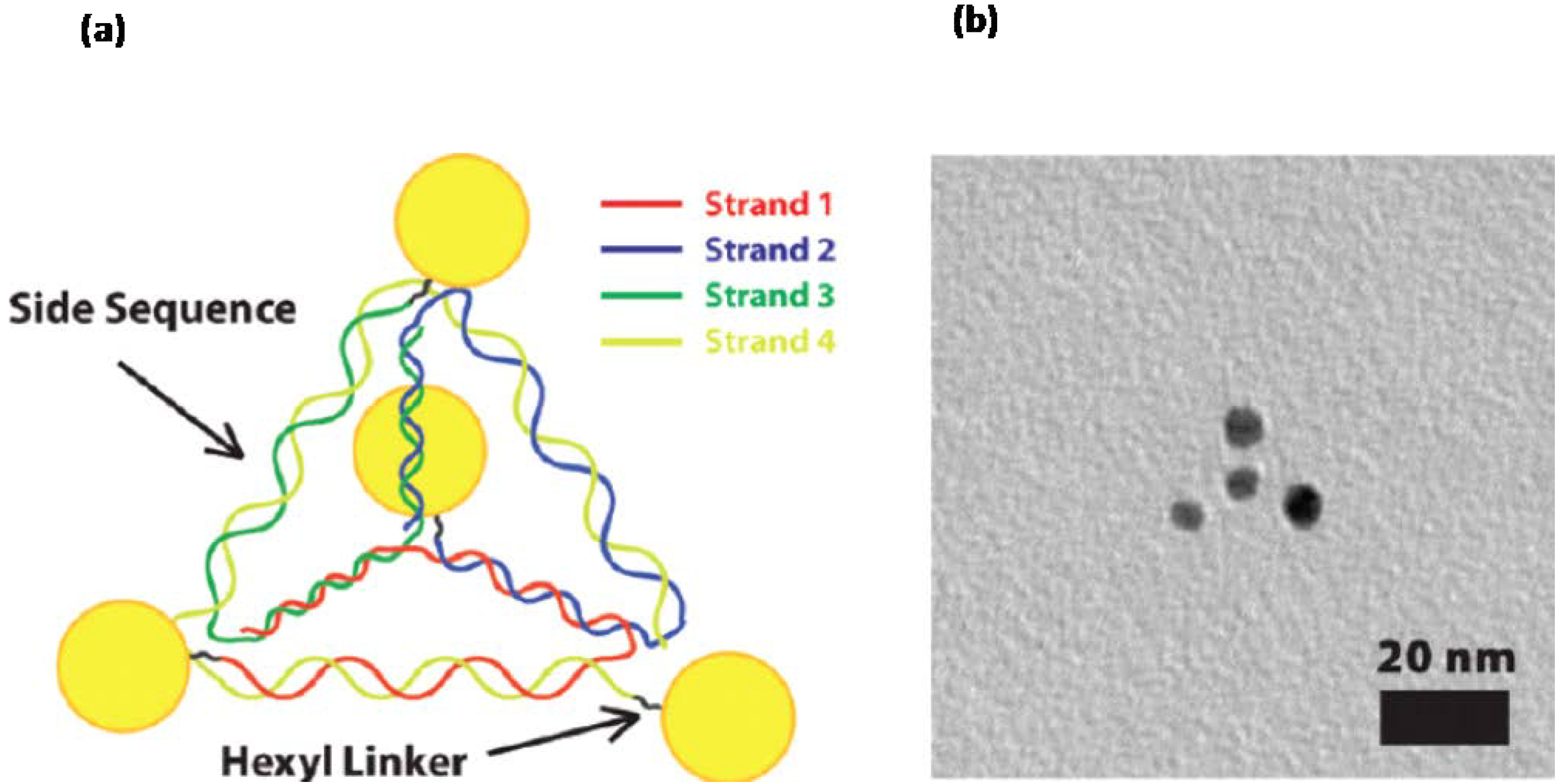
- Mastroianni, A.J.; Claridge, S.A.; Alivisatos, A.P. Pyramidal and Chiral Groupings of Gold Nanocrystals Assembled Using DNA Scaffolds. J. Am. Chem. Soc. 2009, 131, 8455–8459. [Google Scholar] [CrossRef]
- Chen, J.H.; Seeman, N.C. Synthesis from DNA of a molecule with the connectivity of a cube. Nature 1991, 350, 631–633. [Google Scholar] [CrossRef]
- Erben, C.M.; Goodman, R.P.; Turberfield, A.J. A Self-Assembled DNA Bipyramid. J. Am. Chem. Soc. 2007, 129, 6992–6993. [Google Scholar] [CrossRef]
- Zhang, Y.; Seeman, N.C. Construction of a DNA-Truncated Octahedron. J. Am. Chem. Soc. 1994, 116, 1661–1669. [Google Scholar] [CrossRef]
- Rothemund, P.W. Folding DNA to create nanoscale shapes and patterns. Nature 2006, 440, 297–302. [Google Scholar] [CrossRef]
- Andersen, E.S.; Dong, M.; Nielsen, M.M.; Jahn, K.; Subramani, R.; Mamdouh, W.; Golas, M.M.; Sander, B.; Stark, H.; Oliveira, C.L.; et al. Self-assembly of a nanoscale DNA box with a controllable lid. Nature 2009, 459, 73–76. [Google Scholar] [CrossRef]
- Kuzuya, A.; Komiyama, M. Design and construction of a box-shaped 3D-DNA origami. Chem. Commun. 2009, 4182–4184. [Google Scholar] [CrossRef]
- Kuzyk, A.; Schreiber, R.; Fan, Z.; Pardatscher, G.; Roller, E.-M.; Hogele, A.; Simmel, F.C.; Govorov, A.O.; Liedl, T. DNA-based self-assembly of chiral plasmonic nanostructures with tailored optical response. Nature 2012, 483, 311–314. [Google Scholar] [CrossRef]
- Yang, C.; Manocchi, A.K.; Lee, B.; Yi, H. Viral templated palladium nanocatalysts for dichromate reduction. Appl. Catal. B 2010, 93, 282–291. [Google Scholar] [CrossRef]
- Nam, Y.S.; Magyar, A.P.; Lee, D.; Kim, J.-W.; Yun, D.S.; Park, H.; Pollom, T.S.; Weitz, D.A.; Belcher, A.M. Biologically templated photocatalytic nanostructures for sustained light-driven water oxidation. Nat. Nanotechnol. 2010, 5, 340–344. [Google Scholar] [CrossRef]
- Scheibel, T.; Parthasarathy, R.; Sawicki, G.; Lin, X.-M.; Jaeger, H.; Lindquist, S.L. Conducting nanowires built by controlled self-assembly of amyloid fibers and selective metal deposition. Proc. Natl. Acad. Sci. USA 2003, 100, 4527–4532. [Google Scholar]
- Glover, J.R.; Kowal, A.S.; Schirmer, E.C.; Patino, M.M.; Liu, J.J.; Lindquist, S. Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell 1997, 89, 811–819. [Google Scholar]
- Steinmetz, N.F.; Evans, D.J. Utilisation of plant viruses in bionanotechnology. Org. Biomol. Chem. 2007, 5, 2891–2902. [Google Scholar] [CrossRef]
- Lin, T.; Chen, Z.; Usha, R.; Stauffacher, C.V.; Dai, J.B.; Schmidt, T.; Johnson, J.E. The refined crystal structure of cowpea mosaic virus at 2.8 A resolution. Virology 1999, 265, 20–34. [Google Scholar] [CrossRef]
- Steinmetz, N.F.; Lomonossoff, G.P.; Evans, D.J. Cowpea Mosaic Virus for Material Fabrication: Addressable Carboxylate Groups on a Programmable Nanoscaffold. Langmuir 2006, 22, 3488–3490. [Google Scholar] [CrossRef]
- Steinmetz, N.F.; Lomonossoff, G.P.; Evans, D.J. Decoration of Cowpea Mosaic Virus with Multiple, Redox-Active, Organometallic Complexes. Small 2006, 2, 530–533. [Google Scholar] [CrossRef]
- Blum, A.S.; Soto, C.M.; Wilson, C.D.; Cole, J.D.; Kim, M.; Gnade, B.; Chatterji, A.; Ochoa, W.F.; Lin, T.; Johnson, J.E.; et al. Cowpea Mosaic Virus as a Scaffold for 3-D Patterning of Gold Nanoparticles. Nano Lett. 2004, 4, 867–870. [Google Scholar] [CrossRef]
- Zafeiratos, S.; Piccinin, S.; Teschner, D. Alloys in catalysis: Phase separation and surface segregation phenomena in response to the reactive environment. Catal. Sci. Technol. 2012, 2, 1787–1801. [Google Scholar] [CrossRef]
- Evans, D.J. The bionanoscience of plant viruses: templates and synthons for new materials. J. Mater. Chem. 2008, 18, 3746–3754. [Google Scholar] [CrossRef]
- Shenton, W.; Douglas, T.; Young, M.; Stubbs, G.; Mann, S. Inorganic-Organic Nanotube Composites from Template Mineralization of Tobacco Mosaic Virus. Adv. Mater. 1999, 11, 253–256. [Google Scholar] [CrossRef]
- Knez, M.; Kadri, A.; Wege, C.; Gösele, U.; Jeske, H.; Nielsch, K. Atomic Layer Deposition on Biological Macromolecules: Metal Oxide Coating of Tobacco Mosaic Virus and Ferritin. Nano Lett. 2006, 6, 1172–1177. [Google Scholar] [CrossRef]
- Dujardin, E.; Peet, C.; Stubbs, G.; Culver, J.N.; Mann, S. Organization of Metallic Nanoparticles Using Tobacco Mosaic Virus Templates. Nano Lett. 2003, 3, 413–417. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Royston, E.; Culver, J.N.; Harris, M.T. Improved metal cluster deposition on a genetically engineered tobacco mosaic virus template. Nanotechnology 2005, 16, S435. [Google Scholar] [CrossRef]
- Lim, J.-S.; Kim, S.-M.; Lee, S.-Y.; Stach, E.A.; Culver, J.N.; Harris, M.T. Formation of Au/Pd Alloy Nanoparticles on TMV. J. Nanomater. 2010, 2010. [Google Scholar] [CrossRef]
- Khan, A.A.; Fox, E.K.; Gorzny, M.L.; Nikulina, E.; Brougham, D.F.; Wege, C.; Bittner, A.M. pH control of the electrostatic binding of gold and iron oxide nanoparticles to tobacco mosaic virus. Langmuir 2013, 29, 2094–2098. [Google Scholar] [CrossRef]
- Guli, M.; Lambert, E.M.; Li, M.; Mann, S. Template-directed synthesis of nanoplasmonic arrays by intracrystalline metalization of cross-linked lysozyme crystals. Angew. Chem. Int. Ed. 2010, 49, 520–523. [Google Scholar] [CrossRef]
- Wei, H.; Wang, Z.; Zhang, J.; House, S.; Gao, Y.G.; Yang, L.; Robinson, H.; Tan, L.H.; Xing, H.; Hou, C.; et al. Time-dependent, protein-directed growth of gold nanoparticles within a single crystal of lysozyme. Nat. Nanotechnol. 2011, 6, 93–97. [Google Scholar]
- Wei, H.; Lu, Y. Catalysis of gold nanoparticles within lysozyme single crystals. Chem. Asian J. 2012, 7, 680–683. [Google Scholar] [CrossRef]
- McMillan, R.A.; Paavola, C.D.; Howard, J.; Chan, S.L.; Zaluzec, N.J.; Trent, J.D. Ordered nanoparticle arrays formed on engineered chaperonin protein templates. Nat. Mater. 2002, 1, 247–252. [Google Scholar] [CrossRef]
- Trent, J.D.; Kagawa, H.K.; Yaoi, T.; Olle, E.; Zaluzec, N.J. Chaperonin filaments: the archaeal cytoskeleton? Proc. Natl. Acad. Sci. USA 1997, 94, 5383–5388. [Google Scholar] [CrossRef]
- Ellis, M.J.; Knapp, S.; Koeck, P.J.B.; Fakoor-Biniaz, Z.; Ladenstein, R.; Hebert, H. Two-Dimensional Crystallization of the Chaperonin TF55 from the Hyperthermophilic ArchaeonSulfolobus solfataricus. J. Struct. Biol. 1998, 123, 30–36. [Google Scholar] [CrossRef]
- Bond, G.C.; Louis, C.; Thompson, D.T. Catalysis by Gold; Imperial College Press: London, UK, 2006; Volume 6. [Google Scholar]
- Hashmi, A.S.; Hutchings, G.J. Gold catalysis. Angew. Chem. Int. Ed. 2006, 45, 7896–7936. [Google Scholar] [CrossRef]
- Della Pina, C.; Falletta, E.; Rossi, M. Update on selective oxidation using gold. Chem. Soc. Rev. 2012, 41, 350–369. [Google Scholar] [CrossRef]
- Mills, G.; Gordon, M.S.; Metiu, H. Oxygen adsorption on Au clusters and a rough Au(111) surface: The role of surface flatness, electron confinement, excess electrons, and band gap. J. Chem. Phys. 2003, 118, 4198–4205. [Google Scholar] [CrossRef]
- Bond, G.C.; Thompson, D.T. Catalysis by Gold. Cataly. Rev. 1999, 41, 319–388. [Google Scholar] [CrossRef]
- Wales, D.J. Structure, Dynamics, and Thermodynamics of Clusters: Tales from Topographic Potential Surface. Science 1996, 271, 925–929. [Google Scholar]
- Boyen, H.G.; Kastle, G.; Weigl, F.; Koslowski, B.; Dietrich, C.; Ziemann, P.; Spatz, J.P.; Riethmuller, S.; Hartmann, C.; Moller, M.; et al. Oxidation-resistant gold-55 clusters. Science 2002, 297, 1533–1536. [Google Scholar] [CrossRef]
- Bond, G.C.; Thompson, D.T. Gold-catalysed oxidation of carbon monoxide. Gold Bull. 2000, 33, 41–50. [Google Scholar] [CrossRef]
- Benkovic, S.J.; Hammes-Schiffer, S. A perspective on enzyme catalysis. Science 2003, 301, 1196–1202. [Google Scholar] [CrossRef]
- Comotti, M.; Della Pina, C.; Matarrese, R.; Rossi, M. The catalytic activity of “naked” gold particles. Angew. Chem. Int. Ed. 2004, 43, 5812–5815. [Google Scholar] [CrossRef]
- Boronat, M.; Corma, A. Oxygen activation on gold nanoparticles: Separating the influence of particle size, particle shape and support interaction. Dalton Trans. 2010, 39, 8538–8546. [Google Scholar] [CrossRef]
- Kotov, N.A. Inorganic Nanoparticles as Protein Mimics. Science 2010, 330, 188–189. [Google Scholar] [CrossRef]
- Witt, S.; Wohlfahrt, G.; Schomburg, D.; Hecht, H.J.; Kalisz, H.M. Conserved arginine-516 of Penicillium amagasakiense glucose oxidase is essential for the efficient binding of beta-D-glucose. Biochem. J. 2000, 347, 553–559. [Google Scholar] [CrossRef]
- Wild, S.; Roglic, G.; Green, A.; Sicree, R.; King, H. Global Prevalence of Diabetes: Estimates for the year 2000 and projections for 2030. Diabetes Care 2004, 27, 1047–1053. [Google Scholar] [CrossRef]
- Zheng, X.; Liu, Q.; Jing, C.; Li, Y.; Li, D.; Luo, W.; Wen, Y.; He, Y.; Huang, Q.; Long, Y.T.; et al. Catalytic gold nanoparticles for nanoplasmonic detection of DNA hybridization. Angew. Chem. Int. Ed. 2011, 50, 11994–11998. [Google Scholar] [CrossRef]
- Newman, J.D.; Turner, A.P.F. Home blood glucose biosensors: A commercial perspective. Biosens. Bioelectron. 2005, 20, 2435–2453. [Google Scholar] [CrossRef] [Green Version]
- Beltrame, P.; Comotti, M.; Della Pina, C.; Rossi, M. Aerobic oxidation of glucose: II. Catalysis by colloidal gold. Appl. Catal. A 2006, 297, 1–7. [Google Scholar] [CrossRef]
- Biella, S.; Prati, L.; Rossi, M. Selective Oxidation of d-Glucose on Gold Catalyst. J. Catal. 2002, 206, 242–247. [Google Scholar] [CrossRef]
- Luo, W.; Zhu, C.; Su, S.; Li, D.; He, Y.; Huang, Q.; Fan, C. Self-Catalyzed, Self-Limiting Growth of Glucose Oxidase-Mimicking Gold Nanoparticles. ACS Nano 2010, 4, 7451–7458. [Google Scholar] [CrossRef]
- Li, H.; Rothberg, L. Colorimetric detection of DNA sequences based on electrostatic interactions with unmodified gold nanoparticles. Proc. Natl. Acad. Sci. USA 2004, 101, 14036–14039. [Google Scholar] [CrossRef]
- Li, H.; Rothberg, L.J. Label-Free Colorimetric Detection of Specific Sequences in Genomic DNA Amplified by the Polymerase Chain Reaction. J. Am. Chem. Soc. 2004, 126, 10958–10961. [Google Scholar] [CrossRef]
- Cedervall, T.; Lynch, I.; Lindman, S.; Berggard, T.; Thulin, E.; Nilsson, H.; Dawson, K.; Linse, S. Understanding the nanoparticle-protein corona using methods to quntify exchange rates and affinities of proteins for nanoparticles. Proc. Natl. Acad. Sci. USA 2007, 104, 2050–2055. [Google Scholar]
- Lacerda, S.H.D.P.; Park, J.J.; Meuse, C.; Pristinski, D.; Becker, M.L.; Karim, A.; Douglas, J.F. Interaction of Gold Nanoparticles with Common Human Blood Proteins. ACS Nano 2009, 4, 365–379. [Google Scholar]
- Rocker, C.; Potzl, M.; Zhang, F.; Parak, W.J.; Nienhaus, G.U. A quantitative fluorescence study of protein monolayer formation on colloidal nanoparticles. Nat. Nanotechnol. 2009, 4, 577–580. [Google Scholar] [CrossRef]
- Babitzke, P.; Gollnick, P. Posttranscription initiation control of tryptophan metabolism in Bacillus subtilis by the trp RNA-binding attenuation protein (TRAP), anti-TRAP, and RNA structure. J. Bacteriol. 2001, 183, 5795–5802. [Google Scholar] [CrossRef]
- Chen, X.; Antson, A.A.; Yang, M.; Li, P.; Baumann, C.; Dodson, E.J.; Dodson, G.G.; Gollnick, P. Regulatory features of the trp operon and the crystal structure of the trp RNA-binding attenuation protein from Bacillus stearothermophilus. J. Mol. Biol. 1999, 289, 1003–1016. [Google Scholar] [CrossRef]
- Antson, A.A.; Otridge, J.; Brzozowski, A.M.; Dodson, E.J.; Dodson, G.G.; Wilson, K.S.; Smith, T.M.; Yang, M.; Kurecki, T.; Gollnick, P. The structure of trp RNA-binding attenuation protein. Nature 1995, 374, 693–700. [Google Scholar] [CrossRef]
- Heddle, J.G.; Okajima, T.; Scott, D.J.; Akashi, S.; Park, S.Y.; Tame, J.R. Dynamic allostery in the ring protein TRAP. J. Mol. Biol. 2007, 371, 154–167. [Google Scholar] [CrossRef]
- Heddle, J.G.; Yokoyama, T.; Yamashita, I.; Park, S.Y.; Tame, J.R. Rounding up: Engineering 12-membered rings from the cyclic 11-mer TRAP. Structure 2006, 14, 925–933. [Google Scholar] [CrossRef]
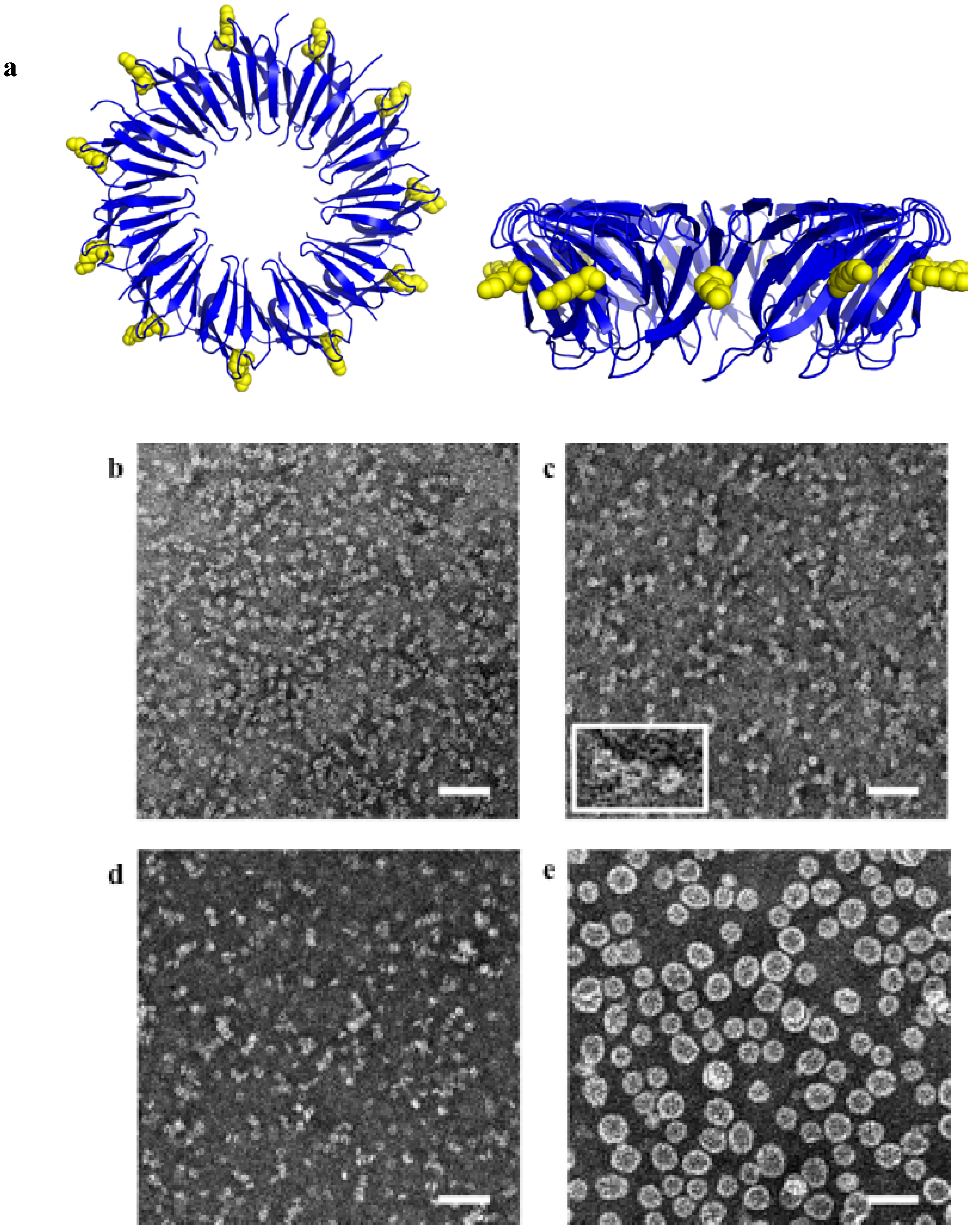
- Heddle, J.G.; Fujiwara, I.; Yamadaki, H.; Yoshii, S.; Nishio, K.; Addy, C.; Yamashita, I.; Tame, J.R. Using the ring-shaped protein TRAP to capture and confine gold nanodots on a surface. Small 2007, 3, 1950–1956. [Google Scholar] [CrossRef]
- Miranda, F.F.; Iwasaki, K.; Akashi, S.; Sumitomo, K.; Kobayashi, M.; Yamashita, I.; Tame, J.R.H.; Heddle, J.G. A Self-Assembled Protein Nanotube with High Aspect Ratio. Small 2009, 5, 2077–2084. [Google Scholar] [CrossRef]
- Malay, A.D.; Heddle, J.G.; Tomita, S.; Iwasaki, K.; Miyazaki, N.; Sumitomo, K.; Yanagi, H.; Yamashita, I.; Uraoka, Y. Gold Nanoparticle-Induced Formation of Artificial Protein Capsids. Nano Lett. 2012, 12, 2056–2059. [Google Scholar] [CrossRef]
- Wittstock, A.; Zielasek, V.; Biener, J.; Friend, C.M.; Baumer, M. Nanoporous gold catalysts for selective gas-phase oxidative coupling of methanol at low temperature. Science 2010, 327, 319–322. [Google Scholar] [CrossRef]
- Hakkinen, H. The gold-sulfur interface at the nanoscale. Nat. Chem. 2012, 4, 443–455. [Google Scholar] [CrossRef]
- Zhang, D.; Neumann, O.; Wang, H.; Yuwono, V.M.; Barhoumi, A.; Perham, M.; Hartgerink, J.D.; Wittung-Stafshede, P.; Halas, N.J. Gold Nanoparticles Can Induce the Formation of Protein-based Aggregates at Physiological pH. Nano Lett. 2009, 9, 666–671. [Google Scholar] [CrossRef]
- Aubin-Tam, M.E.; Hamad-Schifferli, K. Structure and function of nanoparticle-protein conjugates. Biomed. Mater. 2008, 3, 034001. [Google Scholar] [CrossRef]
- Treuel, L.; Malissek, M.; Gebauer, J.S.; Zellner, R. The Influence of Surface Composition of Nanoparticles on their Interactions with Serum Albumin. ChemPhysChem 2010, 11, 3093–3099. [Google Scholar] [CrossRef]
- Lynch, I.; Dawson, K.A. Protein-nanoparticle interactions. Nano Today 2008, 3, 40–47. [Google Scholar] [CrossRef]
- Mancin, F.; Prins, L.J.; Scrimin, P. Catalysis on gold-nanoparticle-passivating monolayers. Curr. Opin. Colloid Interface Sci. 2013, 18, 61–69. [Google Scholar] [CrossRef]
- Mikami, Y.; Dhakshinamoorthy, A.; Alvaro, M.; Garcia, H. Catalytic activity of unsupported gold nanoparticles. Cataly. Sci. Technol. 2013, 3, 58–69. [Google Scholar] [CrossRef]
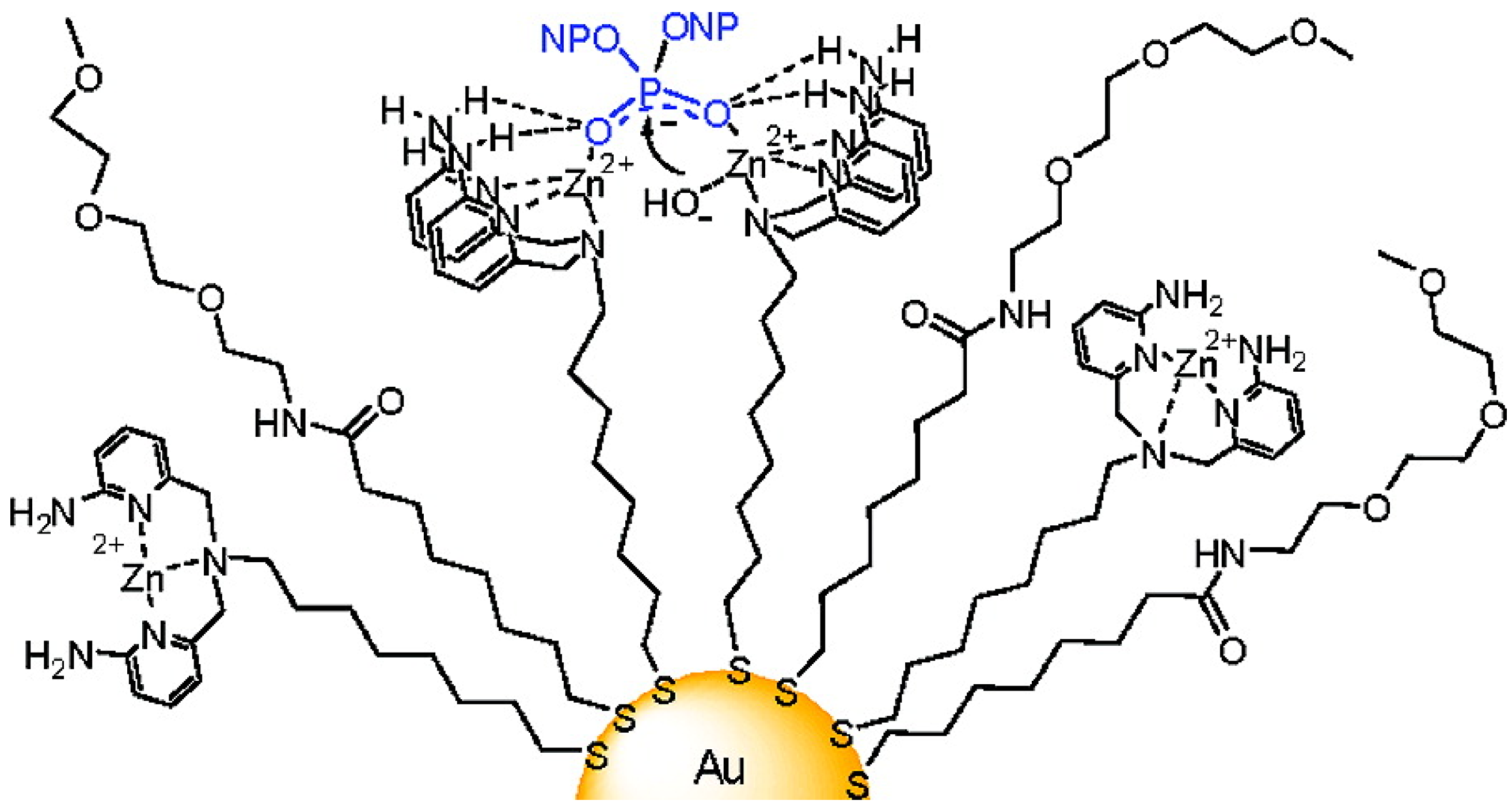
- Manea, F.; Houillon, F.B.; Pasquato, L.; Scrimin, P. Nanozymes: Gold-Nanoparticle-Based Transphosphorylation Catalysts. Angew. Chem. Int. Ed. 2004, 43, 6165–6169. [Google Scholar] [CrossRef]
- Pingoud, A.; Fuxreiter, M.; Pingoud, V.; Wende, W. Type II restriction endonucleases: Structure and mechanism. Cell. Mol. Life Sci. 2005, 62, 685–707. [Google Scholar] [CrossRef]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef]
- Zaupa, G.; Mora, C.; Bonomi, R.; Prins, L.J.; Scrimin, P. Catalytic self-assembled monolayers on Au nanoparticles: The source of catalysis of a transphosphorylation reaction. Chemistry A 2011, 17, 4879–4889. [Google Scholar]
- Pengo, P.; Polizzi, S.; Pasquato, L.; Scrimin, P. Carboxylate-imidazole cooperativity in dipeptide-functionalized gold nanoparticles with esterase-like activity. J. Am. Chem. Soc. 2005, 127, 1616–1617. [Google Scholar] [CrossRef]
- Northrop, D.B. Follow the Protons: A Low-Barrier Hydrogen Bond Unifies the Mechanisms of the Aspartic Proteases. Acc. Chem. Res. 2001, 34, 790–797. [Google Scholar] [CrossRef]
- Hsu, M.H.; Josephrajan, T.; Yeh, C.S.; Shieh, D.B.; Su, W.C.; Hwu, J.R. Novel arylhydrazone-conjugated gold nanoparticles with DNA-cleaving ability: The first DNA-nicking nanomaterial. Bioconj. Chem. 2007, 18, 1709–1712. [Google Scholar] [CrossRef]
- Hwu, J.R.; Chieh Lin, C.; Hsien Chuang, S.; Yung King, K.; Su, T.-R.; Tsay, S.-C. Aminyl and iminyl radicals from arylhydrazones in the photo-induced DNA cleavage. Bioorg. Med. Chem. 2004, 12, 2509–2515. [Google Scholar] [CrossRef]
- Bonomi, R.; Selvestrel, F.; Lombardo, V.; Sissi, C.; Polizzi, S.; Mancin, F.; Tonellato, U.; Scrimin, P. Phosphate diester and DNA hydrolysis by a multivalent, nanoparticle-based catalyst. J. Am. Chem. Soc. 2008, 130, 15744–15745. [Google Scholar] [CrossRef]
- Fillon, Y.; Verma, A.; Ghosh, P.; Ernenwein, D.; Rotello, V.M.; Chmielewski, J. Peptide ligation catalyzed by functionalized gold nanoparticles. J. Am. Chem. Soc. 2007, 129, 6676–6677. [Google Scholar] [CrossRef]
- Dawson, P.; Muir, T.; Clark-Lewis, I.; Kent, S. Synthesis of proteins by native chemical ligation. Science 1994, 266, 776–779. [Google Scholar]
- Yarus, M. Getting past the RNA world: The initial Darwinian ancestor. Cold Spring Harb. Perspect. Biol. 2011. [Google Scholar] [CrossRef]
- Silverman, S.K. Deoxyribozymes: DNA catalysts for bioorganic chemistry. Org. Biomol. Chem. 2004, 2, 2701–2706. [Google Scholar] [CrossRef]
- Fiammengo, R.; Jäschke, A. Nucleic acid enzymes. Curr. Opin. Biotech. 2005, 16, 614–621. [Google Scholar]
- Lee, J.H.; Wang, Z.; Liu, J.; Lu, Y. Highly sensitive and selective colorimetric sensors for uranyl (UO2(2+)): Development and comparison of labeled and label-free DNAzyme-gold nanoparticle systems. J. Am. Chem. Soc. 2008, 130, 14217–14226. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, J. Functional DNA nanotechnology: Emerging applications of DNAzymes and aptamers. Curr. Opin. Biotech. 2006, 17, 580–588. [Google Scholar] [CrossRef]
- Zhao, W.; Lam, J.C.; Chiuman, W.; Brook, M.A.; Li, Y. Enzymatic cleavage of nucleic acids on gold nanoparticles: A generic platform for facile colorimetric biosensors. Small 2008, 4, 810–816. [Google Scholar] [CrossRef]
- Santoro, S.W.; Joyce, G.F. A general purpose RNA-cleaving DNA enzyme. Proc. Natl. Acad. Sci. USA 1997, 94, 4262–4266. [Google Scholar] [CrossRef]
- Li, J.; Lu, Y. A Highly Sensitive and Selective Catalytic DNA Biosensor for Lead Ions. J. Am. Chem. Soc. 2000, 122, 10466–10467. [Google Scholar] [CrossRef]
- Liu, J.; Lu, Y. A Colorimetric Lead Biosensor Using DNAzyme-Directed Assembly of Gold Nanoparticles. J. Am. Chem. Soc. 2003, 125, 6642–6643. [Google Scholar] [CrossRef]
- Brennan, J.L.; Hatzakis, N.S.; Tshikhudo, T.R.; Dirvianskyte, N.; Razumas, V.; Patkar, S.; Vind, J.; Svendsen, A.; Nolte, R.J.; Rowan, A.E.; et al. Bionanoconjugation via click chemistry: The creation of functional hybrids of lipases and gold nanoparticles. Bioconj. Chem. 2006, 17, 1373–1375. [Google Scholar] [CrossRef]
- Prati, L.; Spontoni, P.; Gaiassi, A. From Renewable to Fine Chemicals Through Selective Oxidation: The Case of Glycerol. Top. Catal. 2009, 52, 288–296. [Google Scholar] [CrossRef]
- Villa, A.; Wang, D.; Su, D.S.; Prati, L. Gold Sols as Catalysts for Glycerol Oxidation: The Role of Stabilizer. ChemCatChem 2009, 1, 510–514. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Heddle, J.G. Gold Nanoparticle-Biological Molecule Interactions and Catalysis. Catalysts 2013, 3, 683-708. https://doi.org/10.3390/catal3030683
Heddle JG. Gold Nanoparticle-Biological Molecule Interactions and Catalysis. Catalysts. 2013; 3(3):683-708. https://doi.org/10.3390/catal3030683
Chicago/Turabian StyleHeddle, Jonathan G. 2013. "Gold Nanoparticle-Biological Molecule Interactions and Catalysis" Catalysts 3, no. 3: 683-708. https://doi.org/10.3390/catal3030683