The Influence of Surface Alumina and Silica on the Photocatalytic Degradation of Organic Pollutants

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction and Scope
2. Materials and Their Characterization
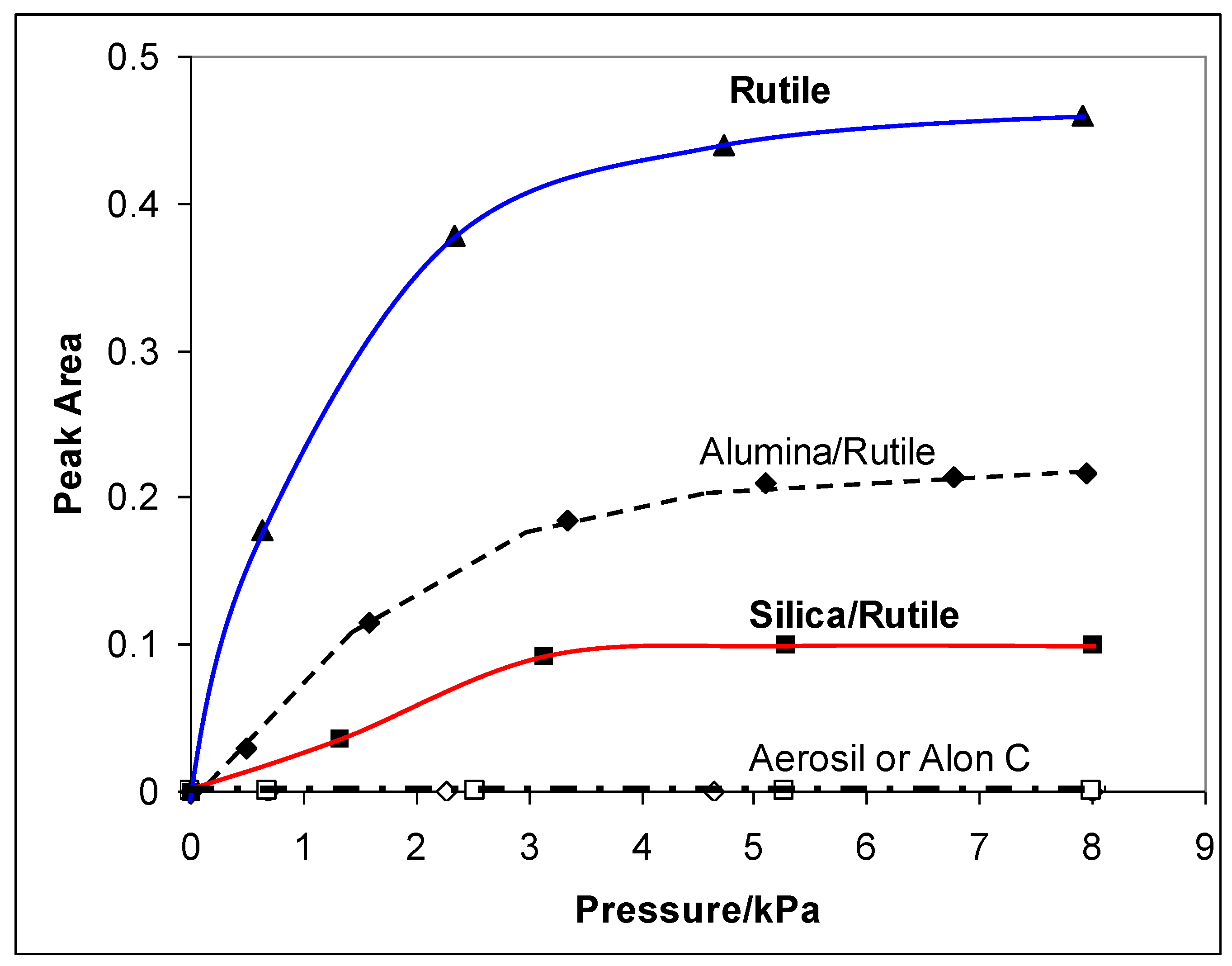
2.1. Preparation of Alumina and Silica Coatings on High Area Rutile
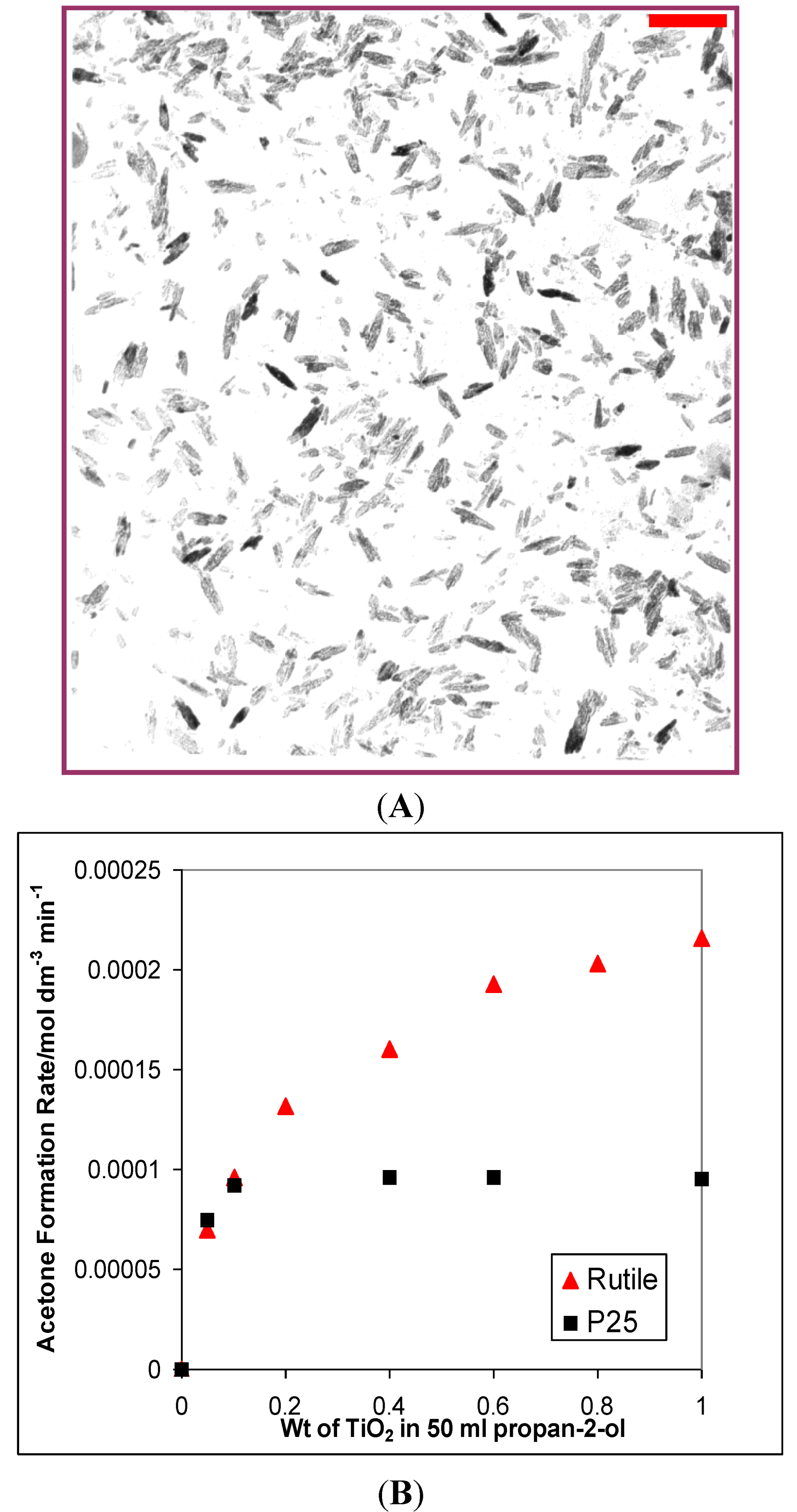
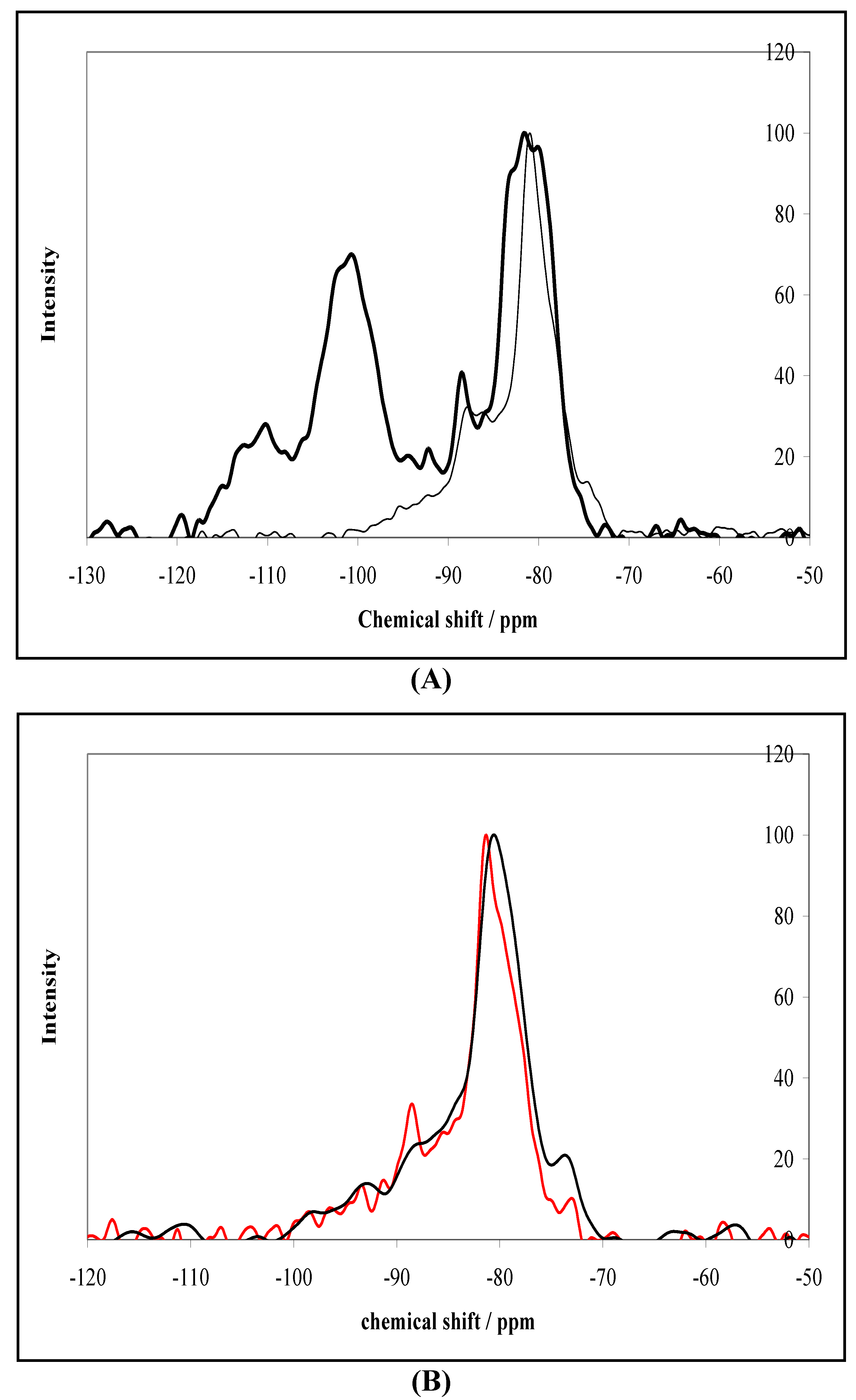
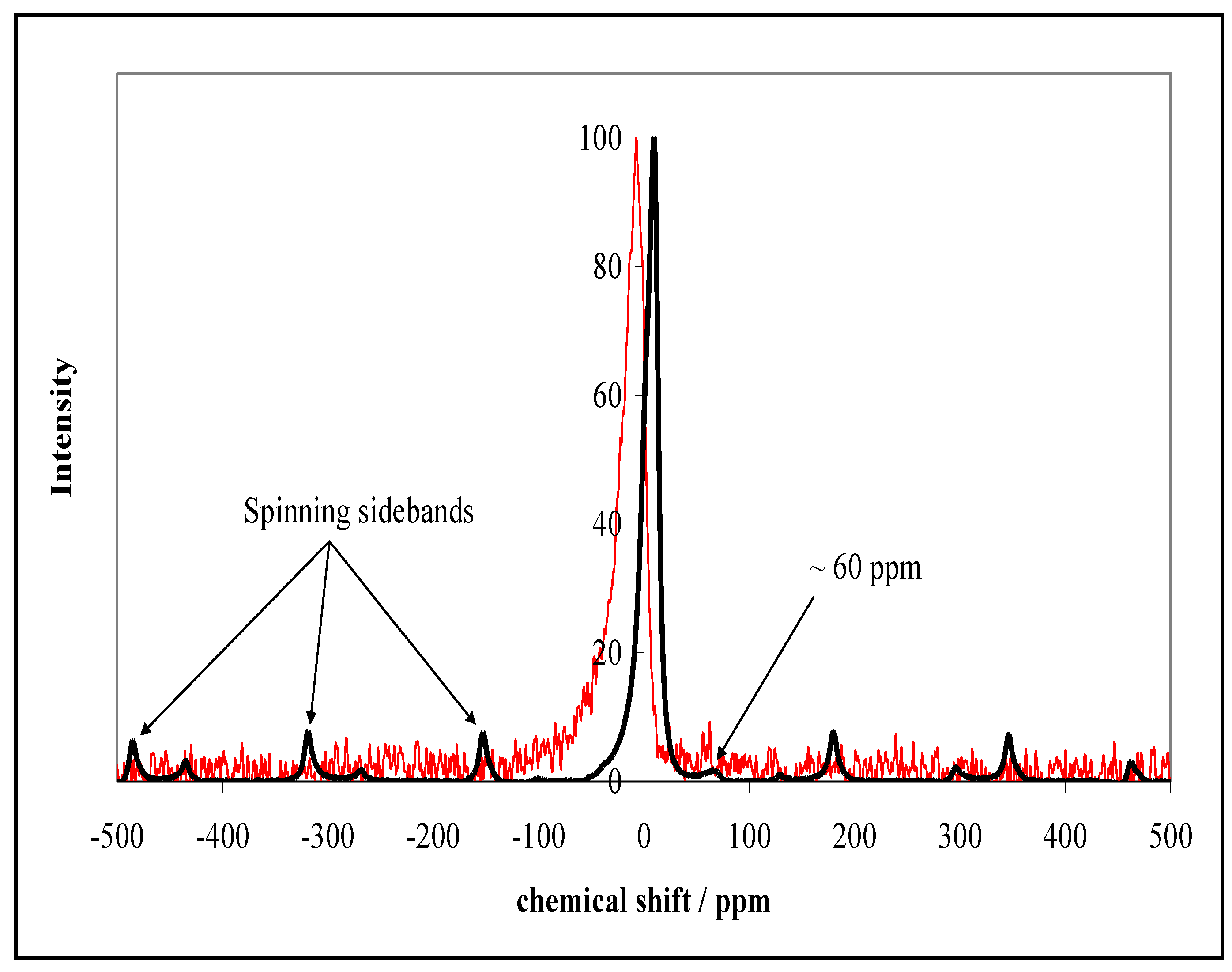
2.2. NMR Characterization of Silica and Alumina Coated 123 m2 g−1 Rutile


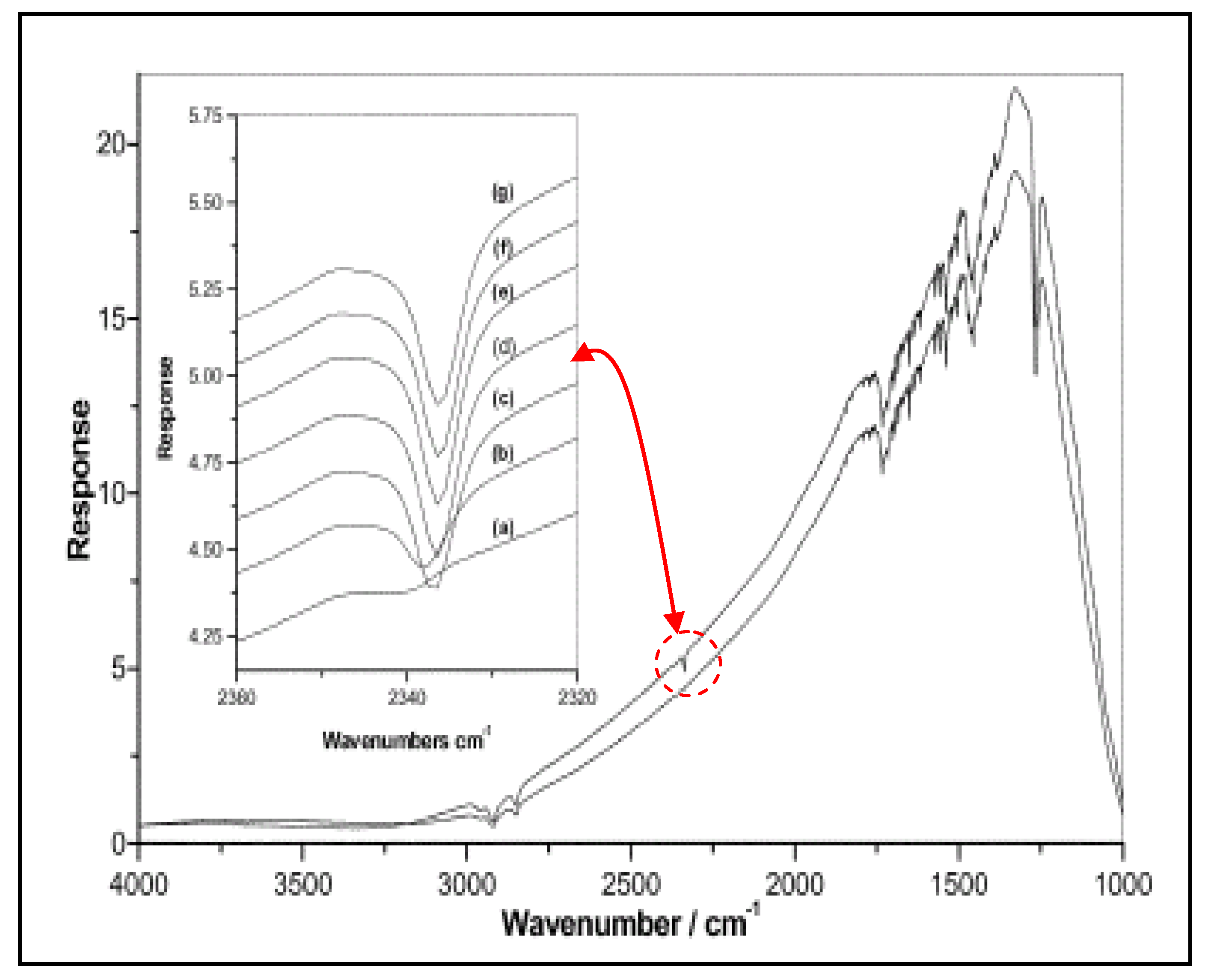
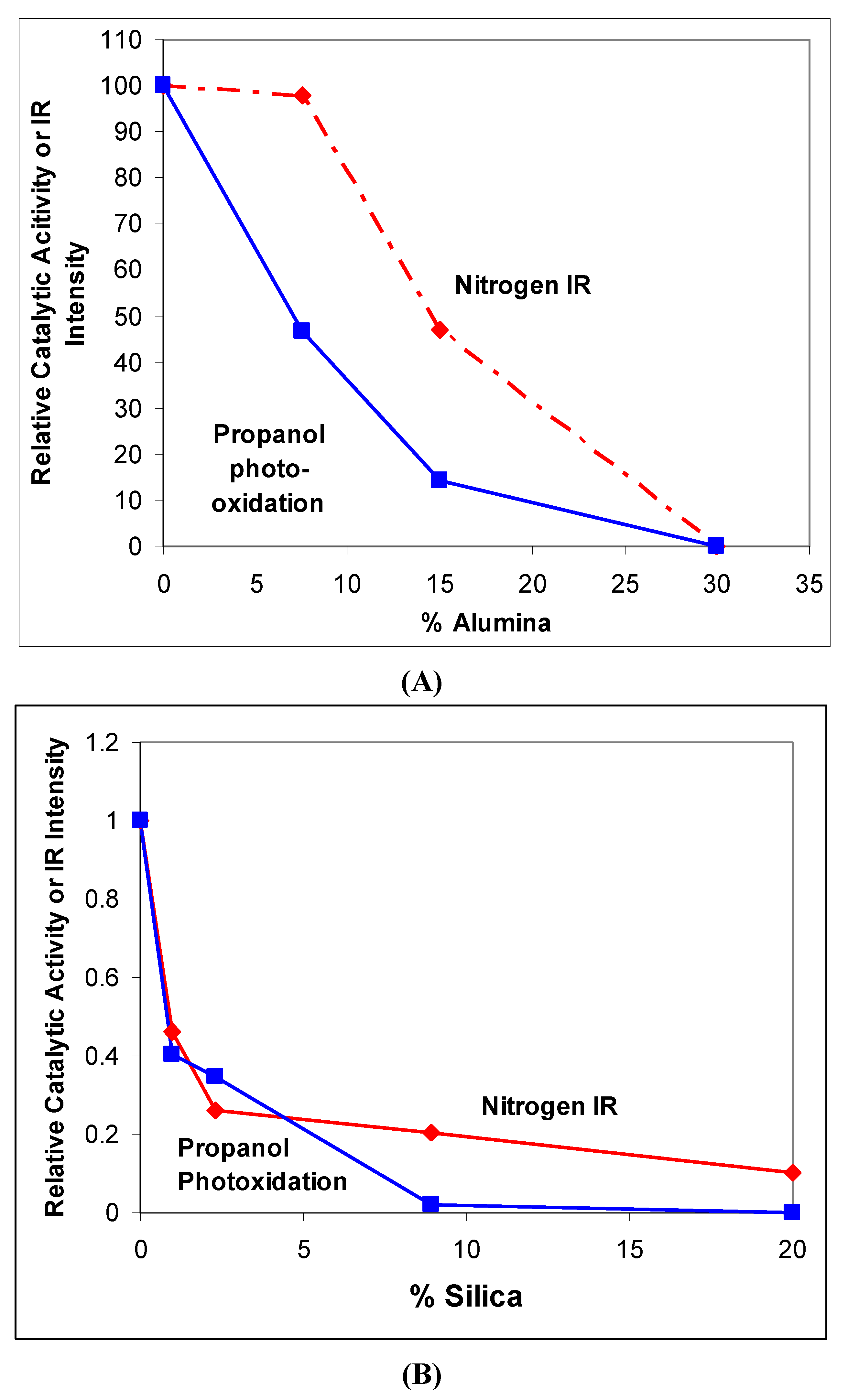
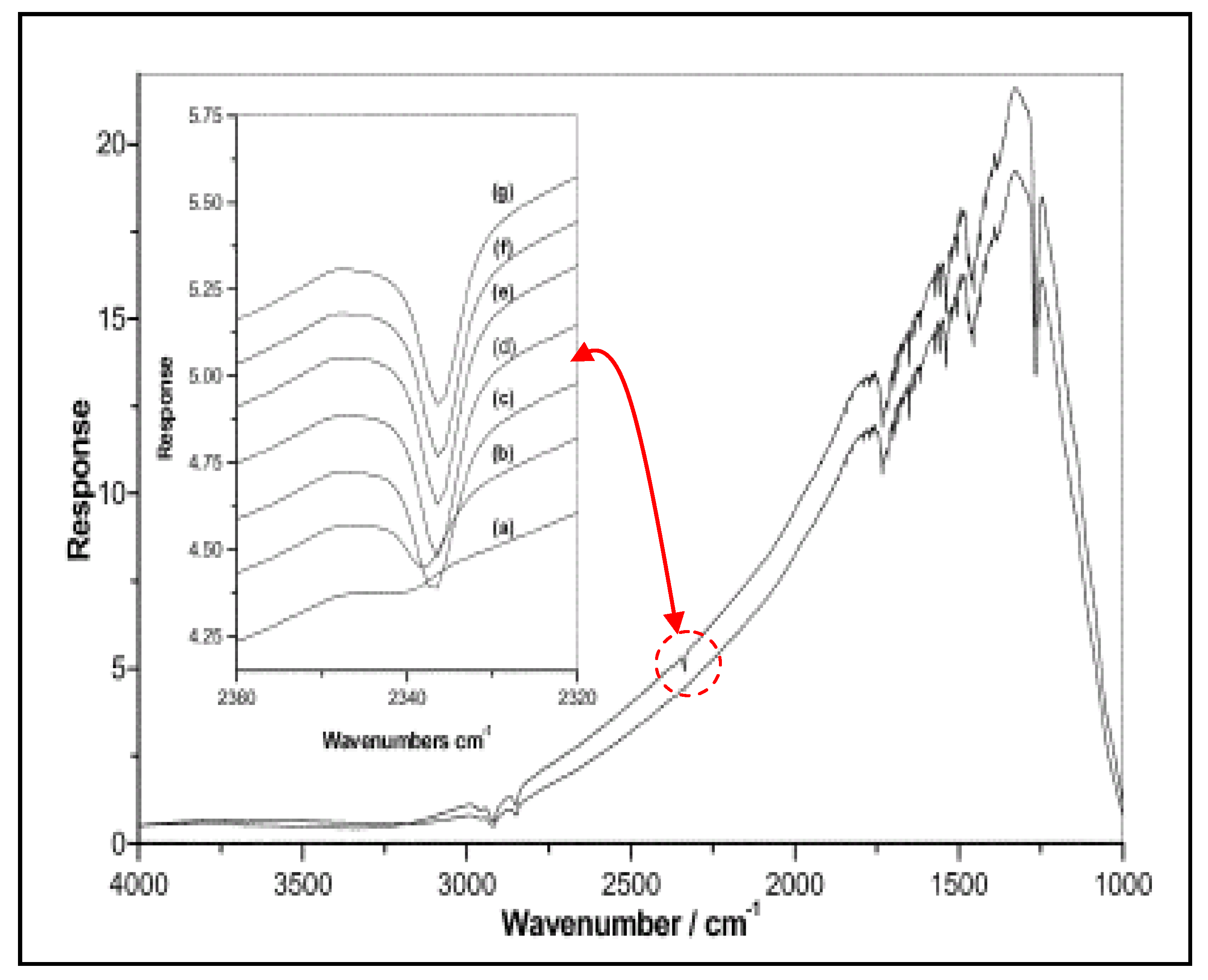
2.3. Characterization by IR Spectroscopy of Adsorbed Nitrogen
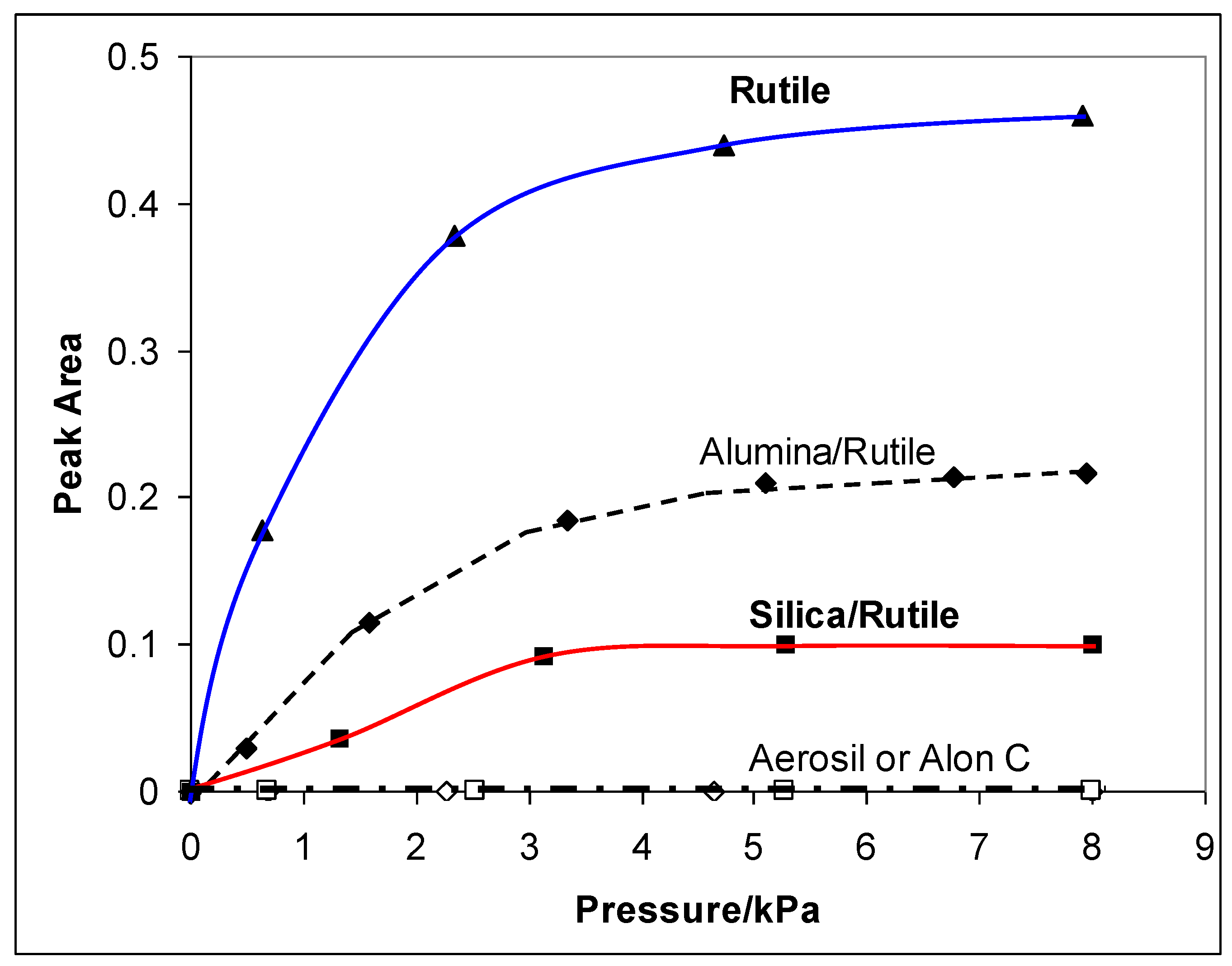
3. Photocatalytic Oxidation
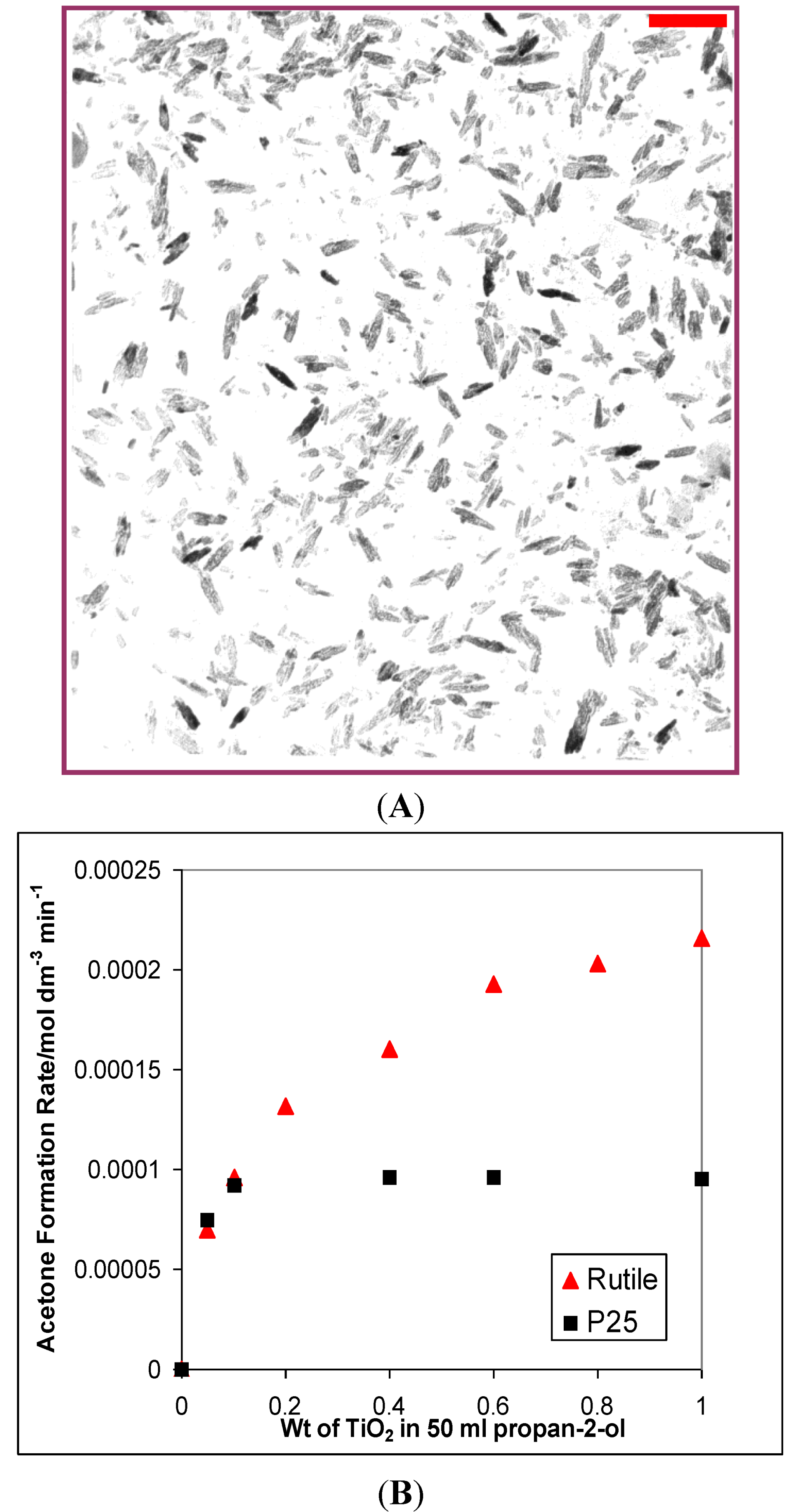
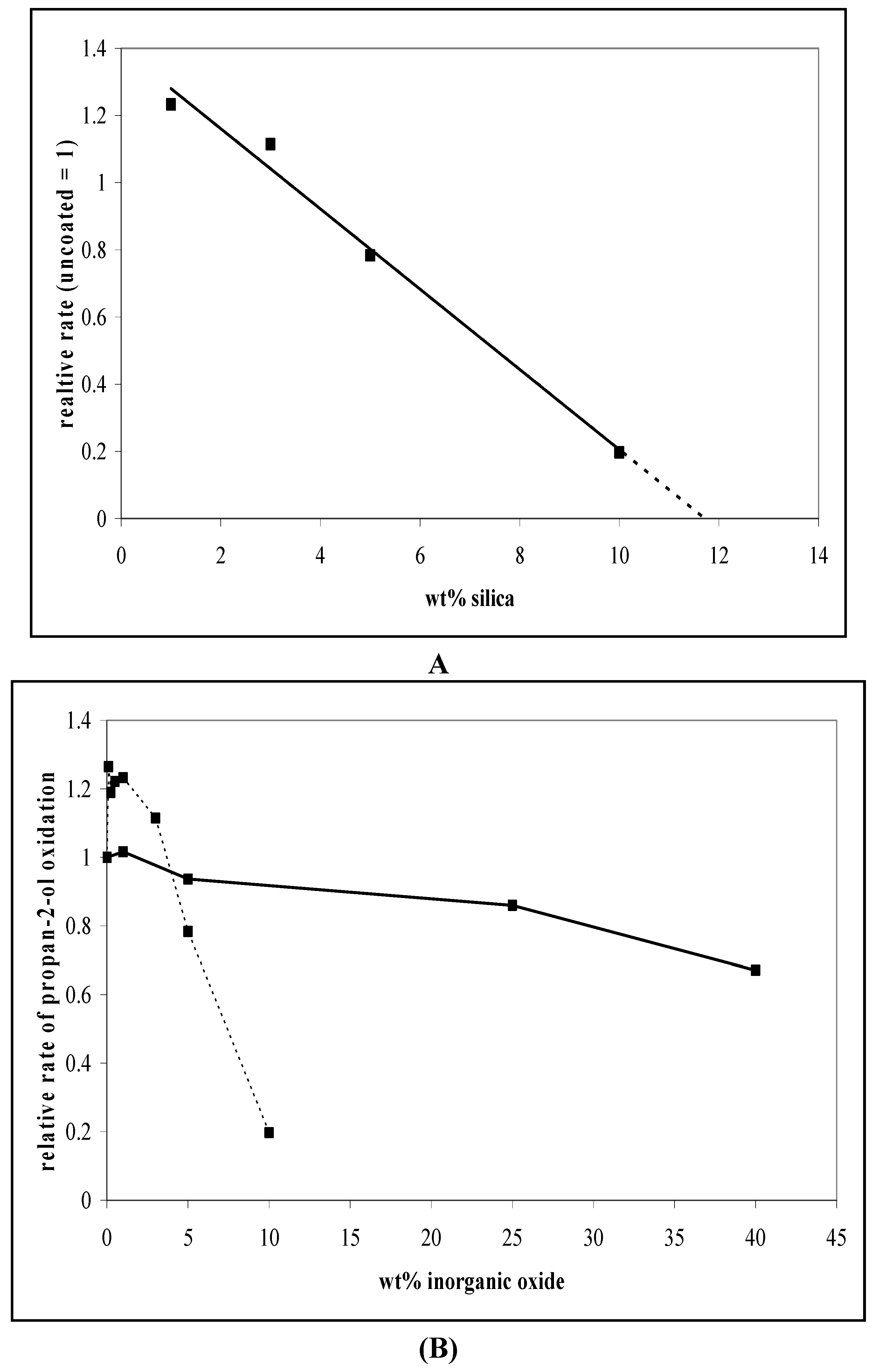
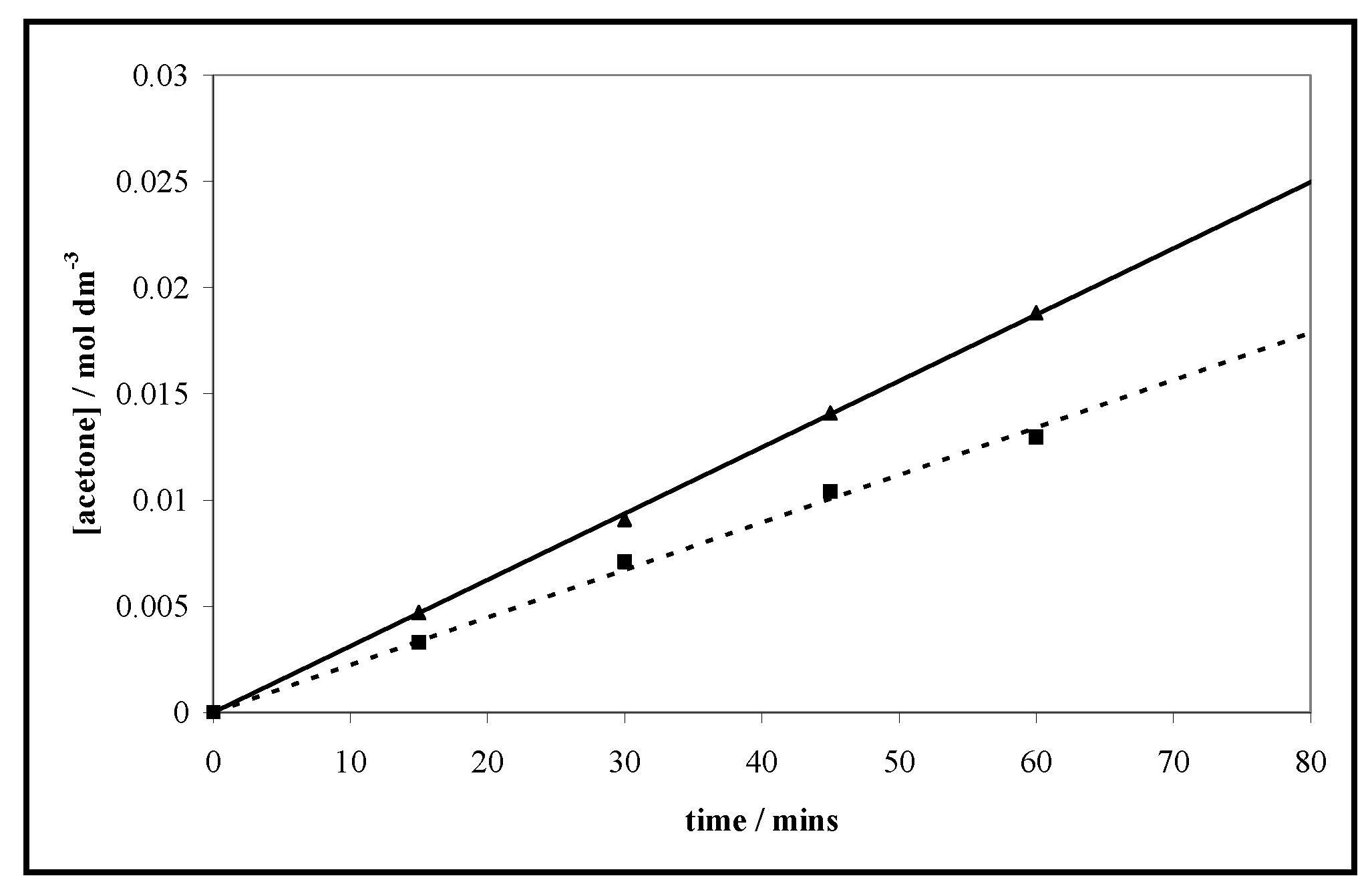
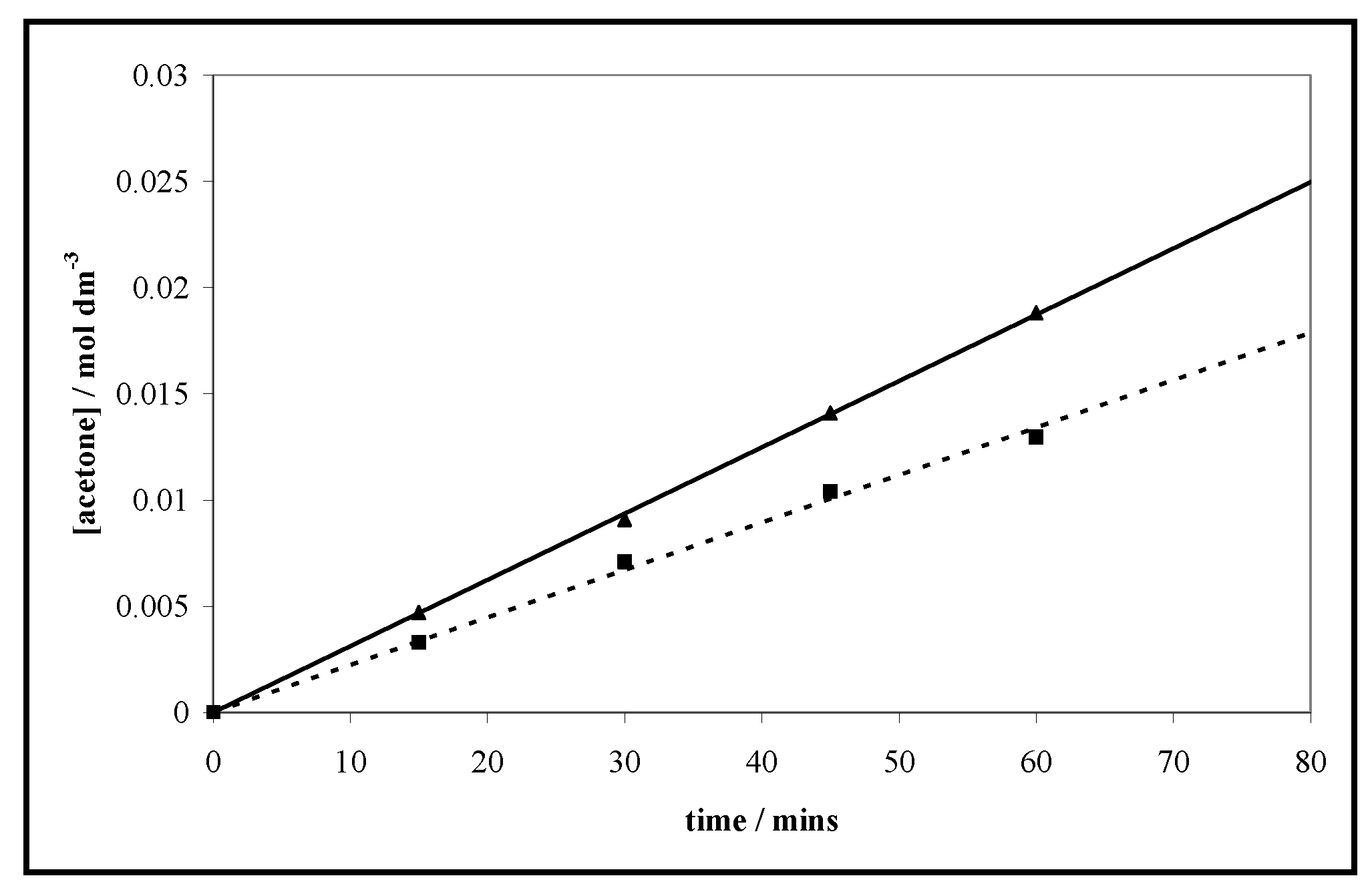
3.1. Photocatalytic Oxidation of Propan-2-ol

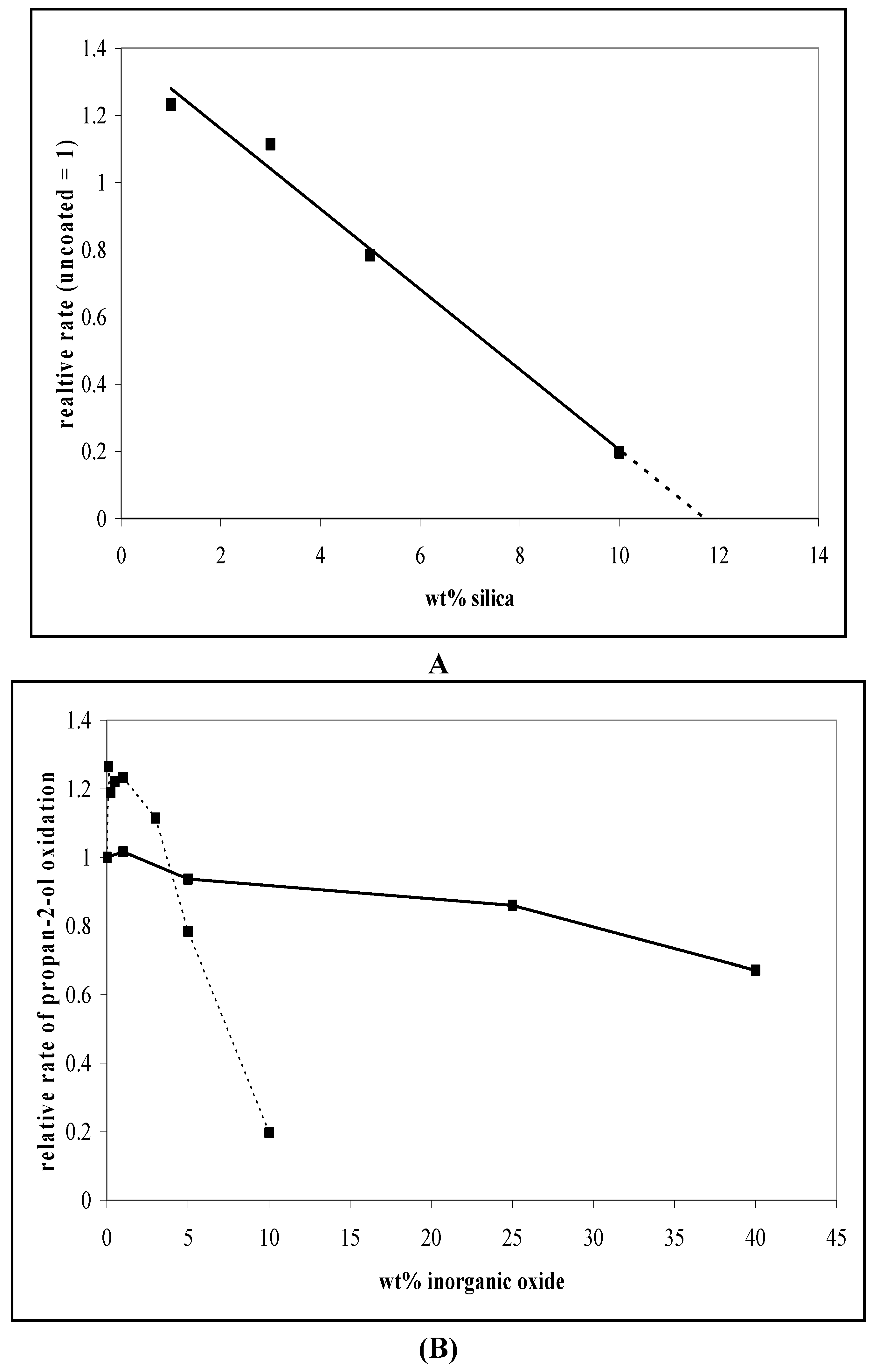
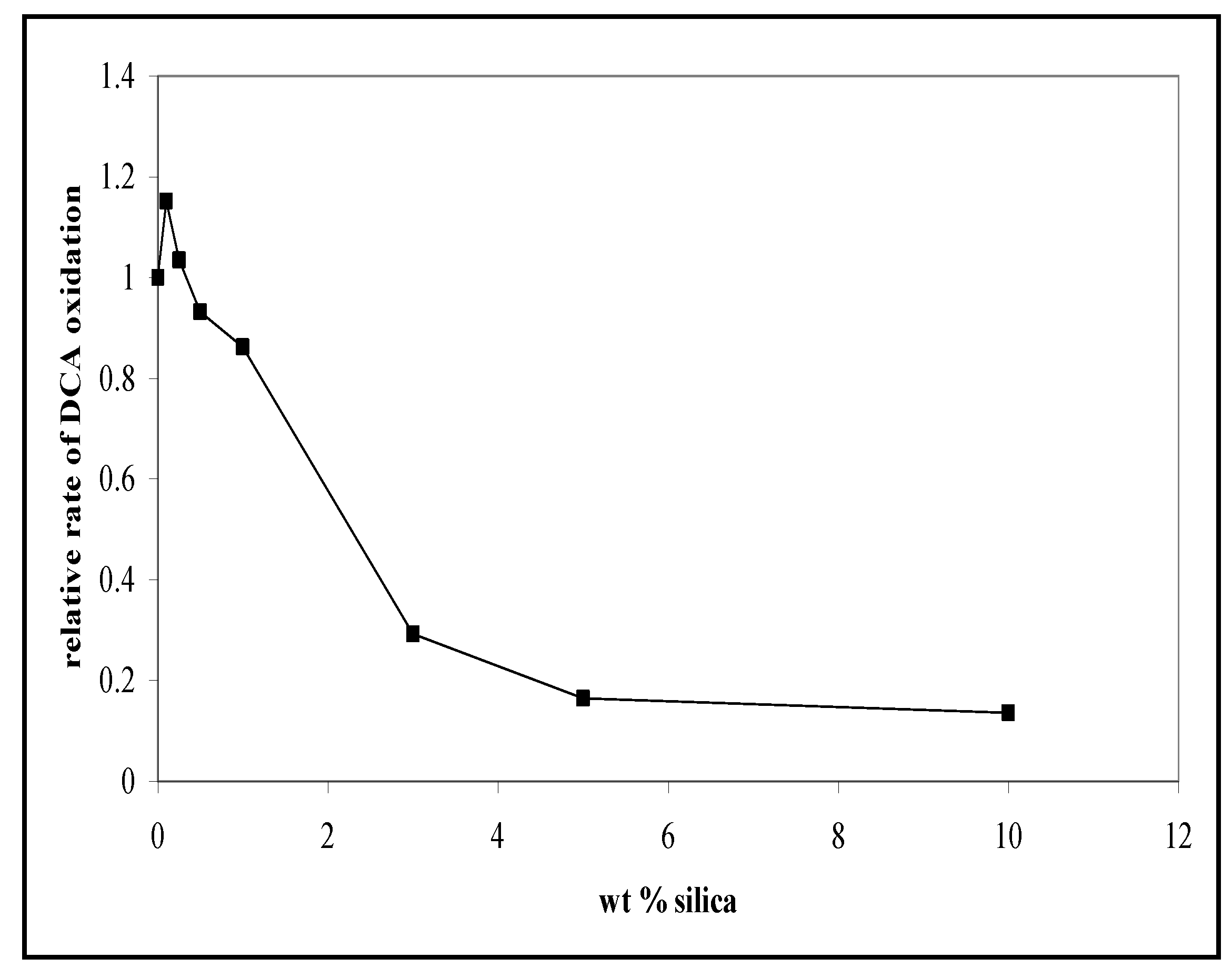
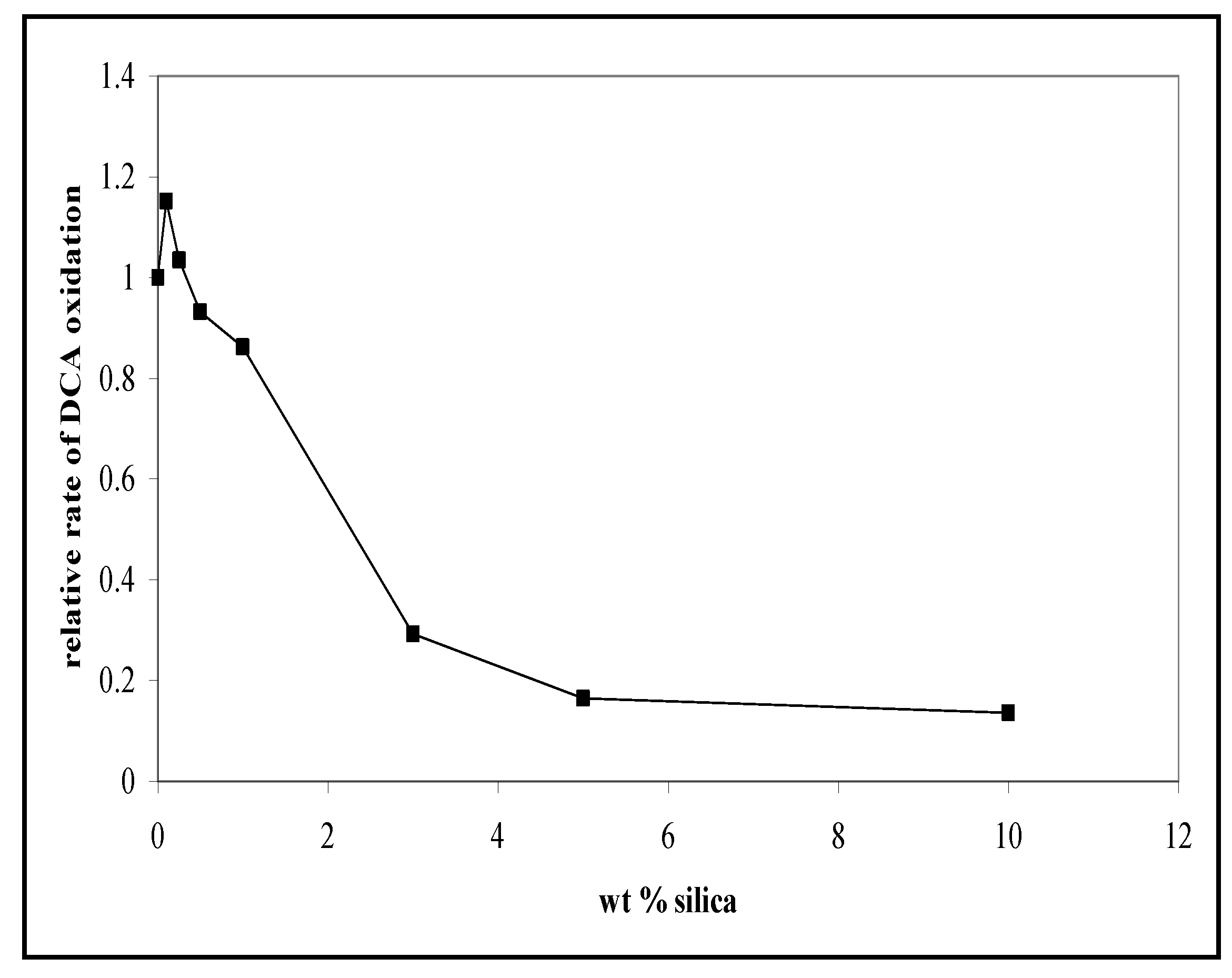
3.2. The Photocatalytic Oxidation of DCA
4. Photocatalytic Reduction of DPPH and Photogreying
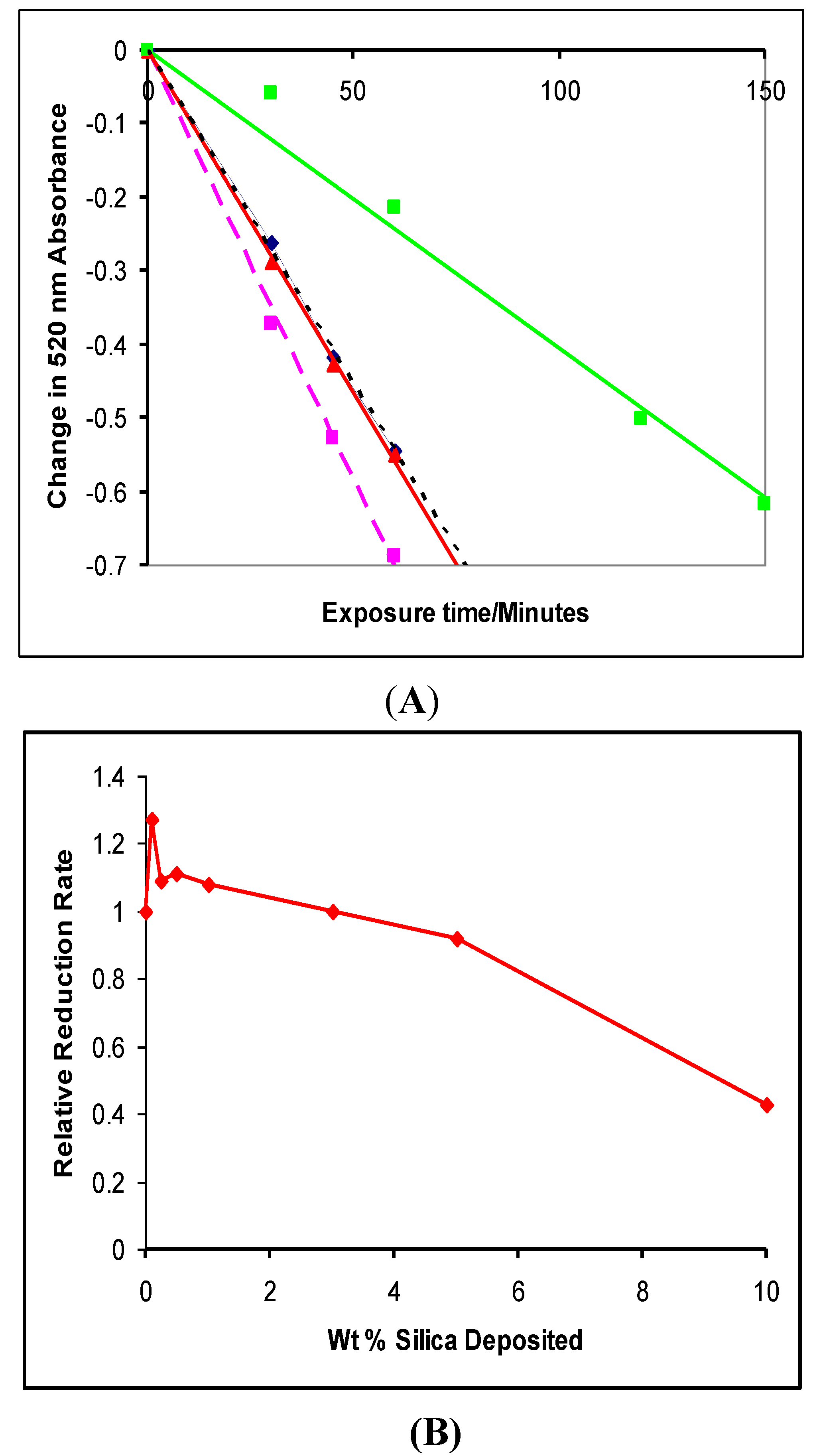
4.1. Photocatalytic Reduction of DPPH
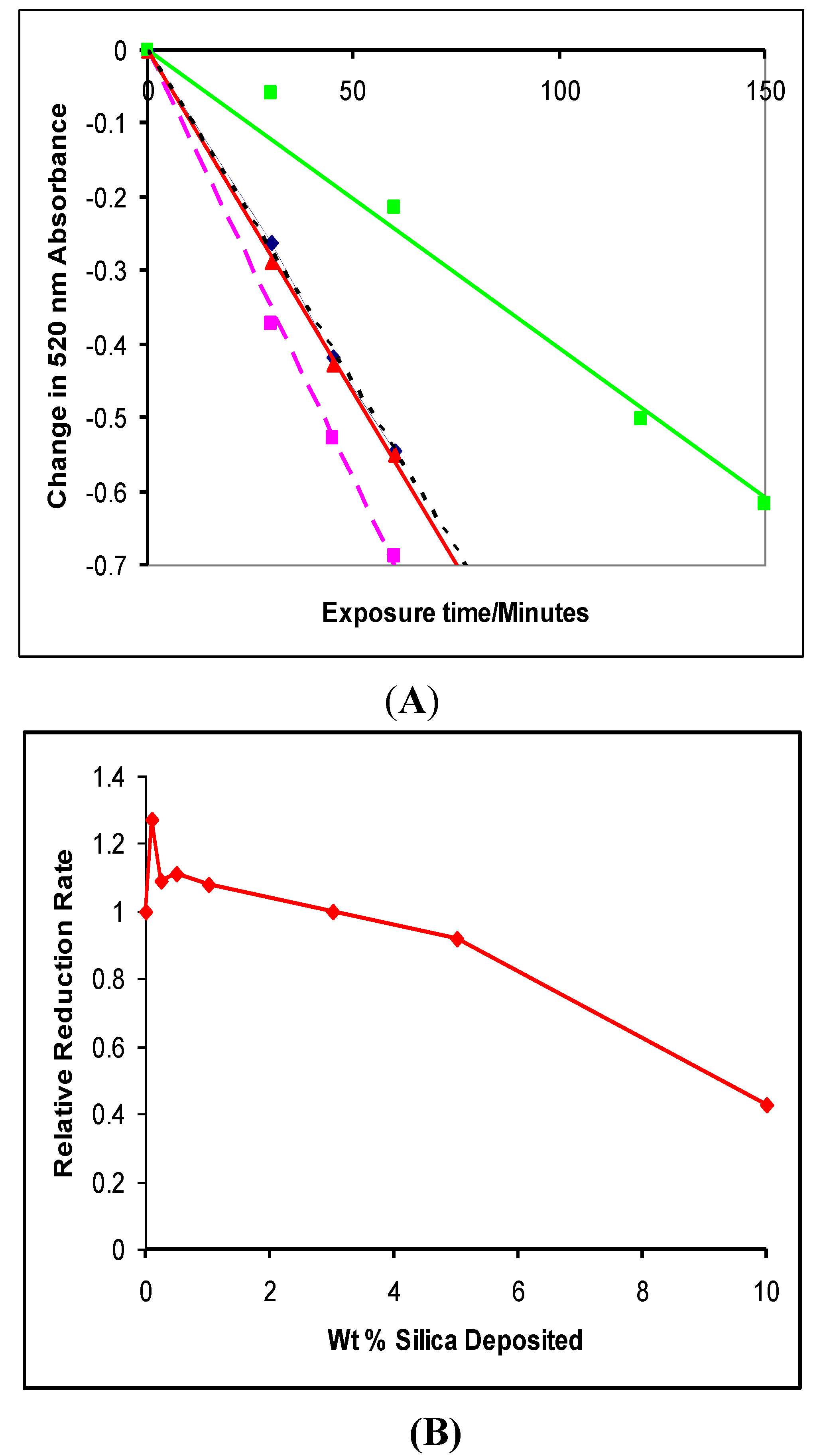
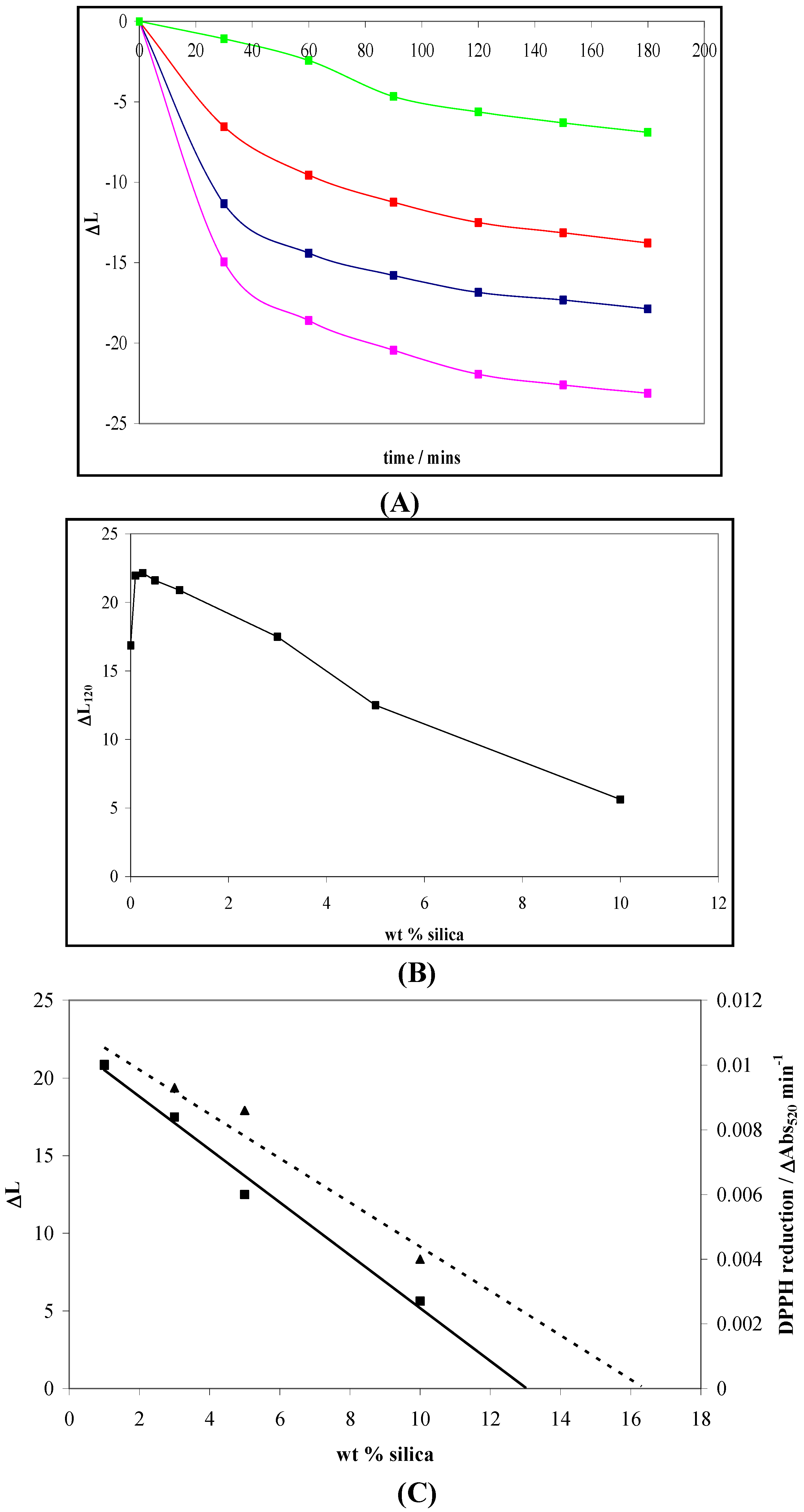
4.2. Photogreying
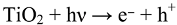
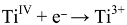
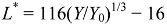
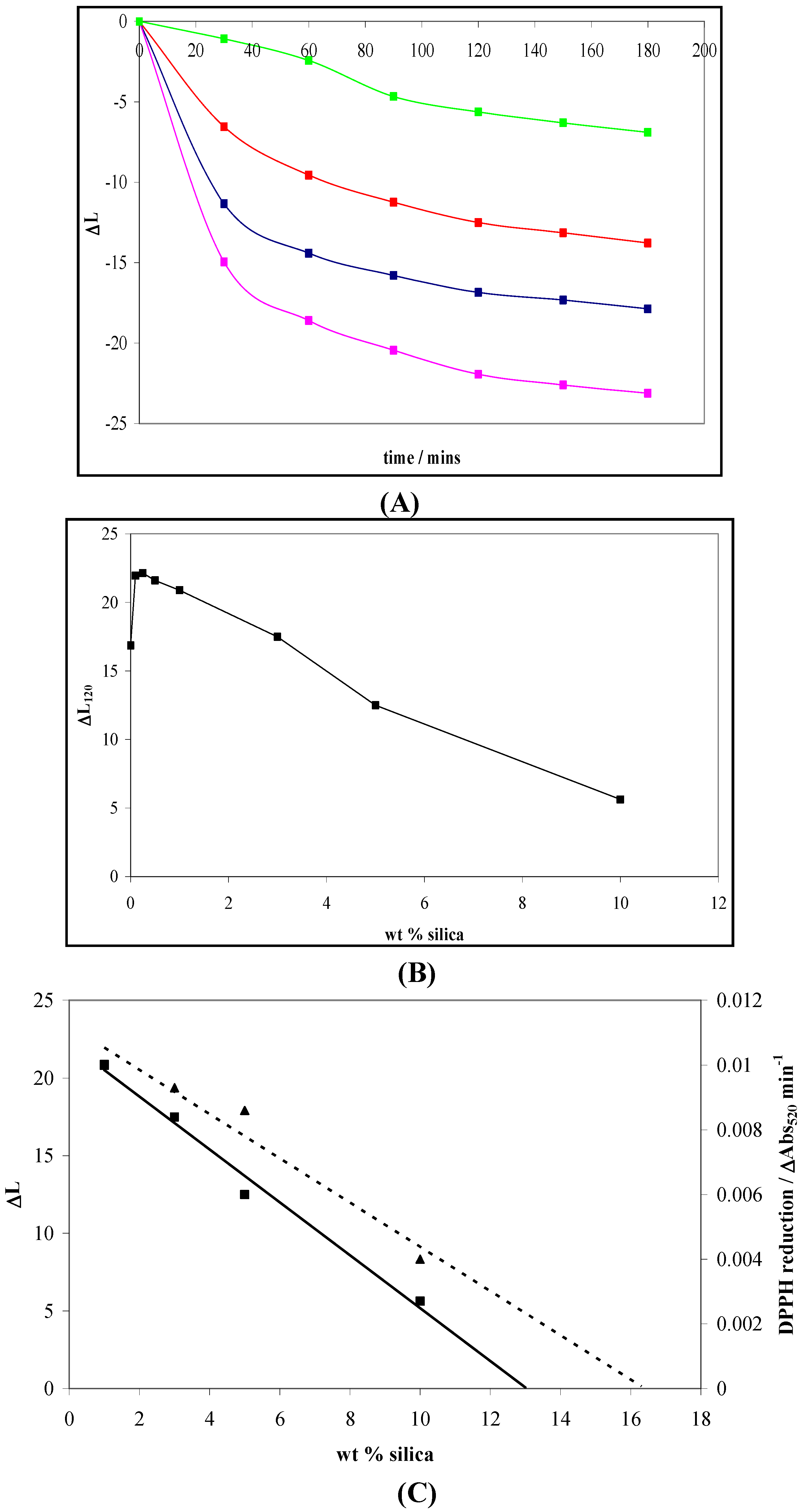
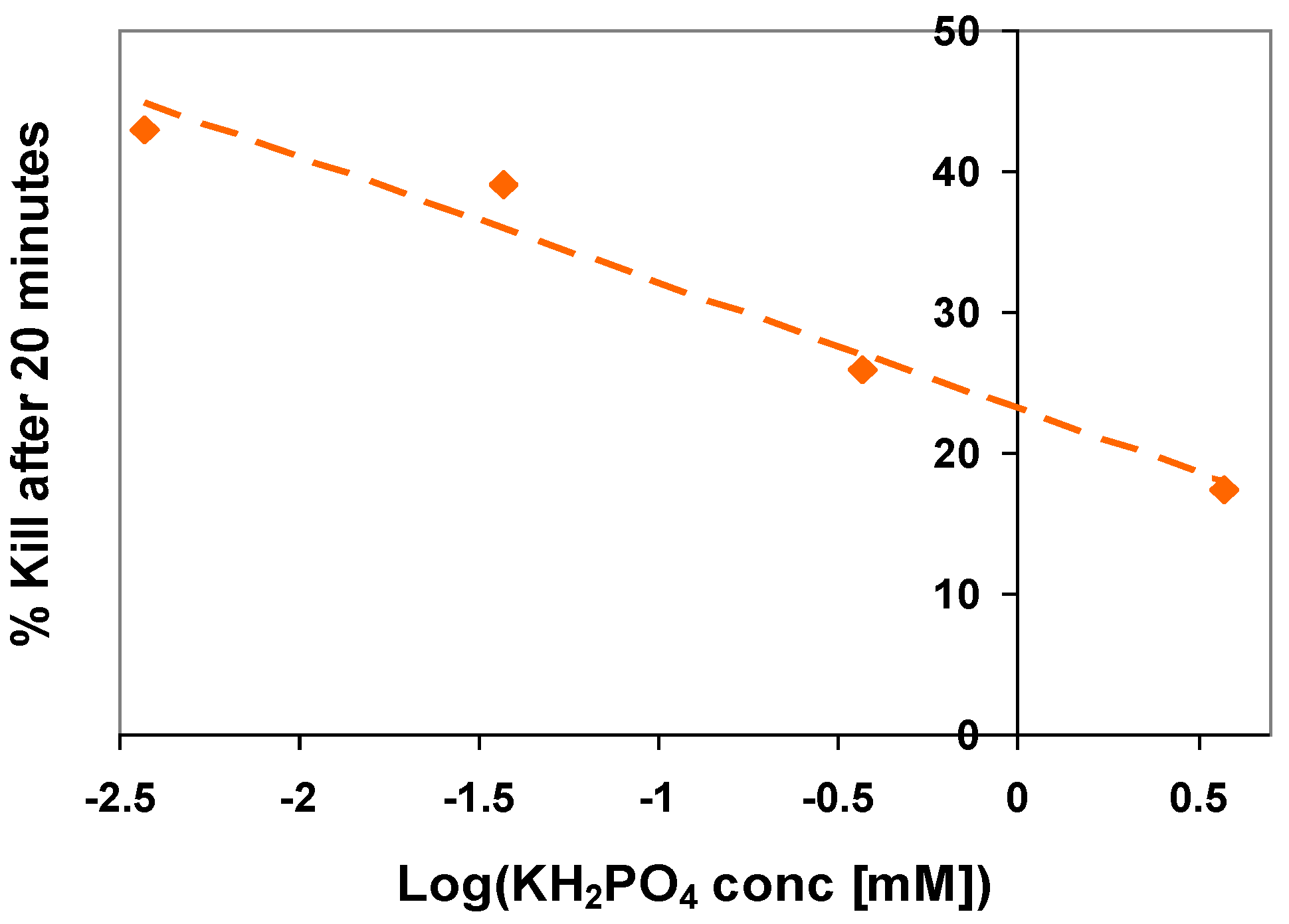
5. The Influence of Phosphate on Photocatalytic Disinfection
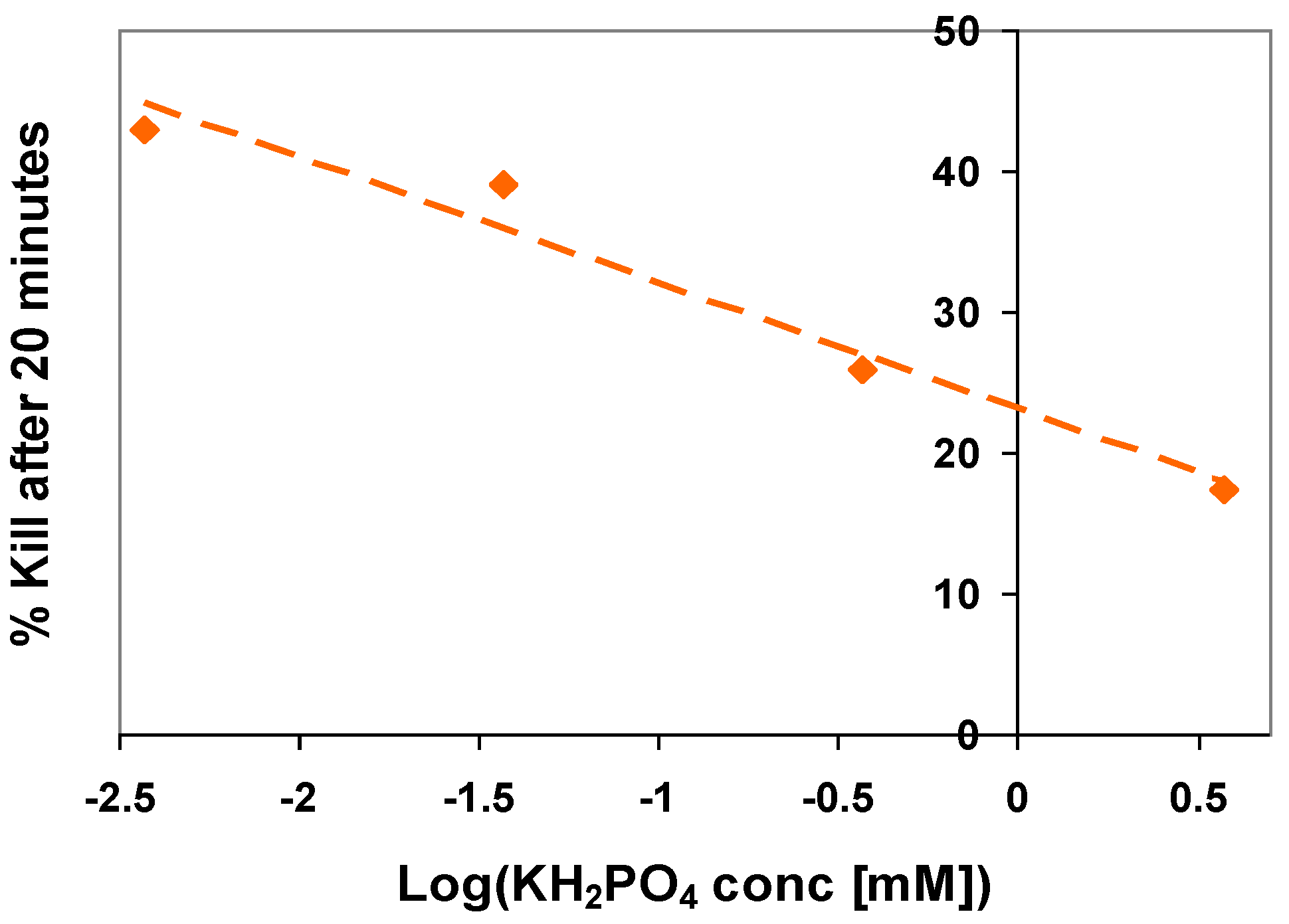
6. An Overview of the Effect of Surface Inorganics on Photoactivity
6.1. The Effectiveness of the Different Coating Procedures
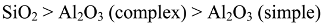

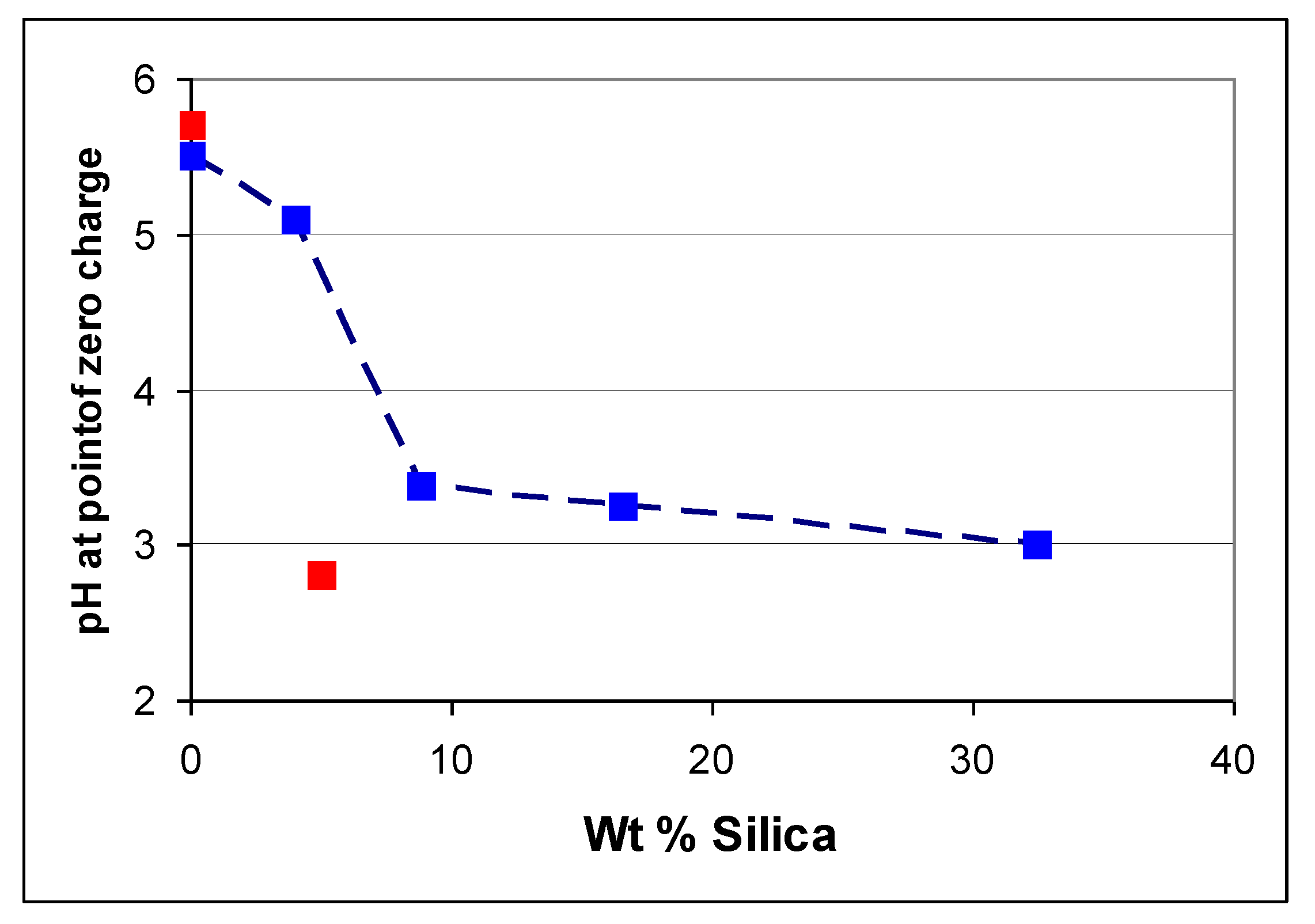
6.2. Dependence of Photocatalysis on Coating Induced Changes in Adsorption
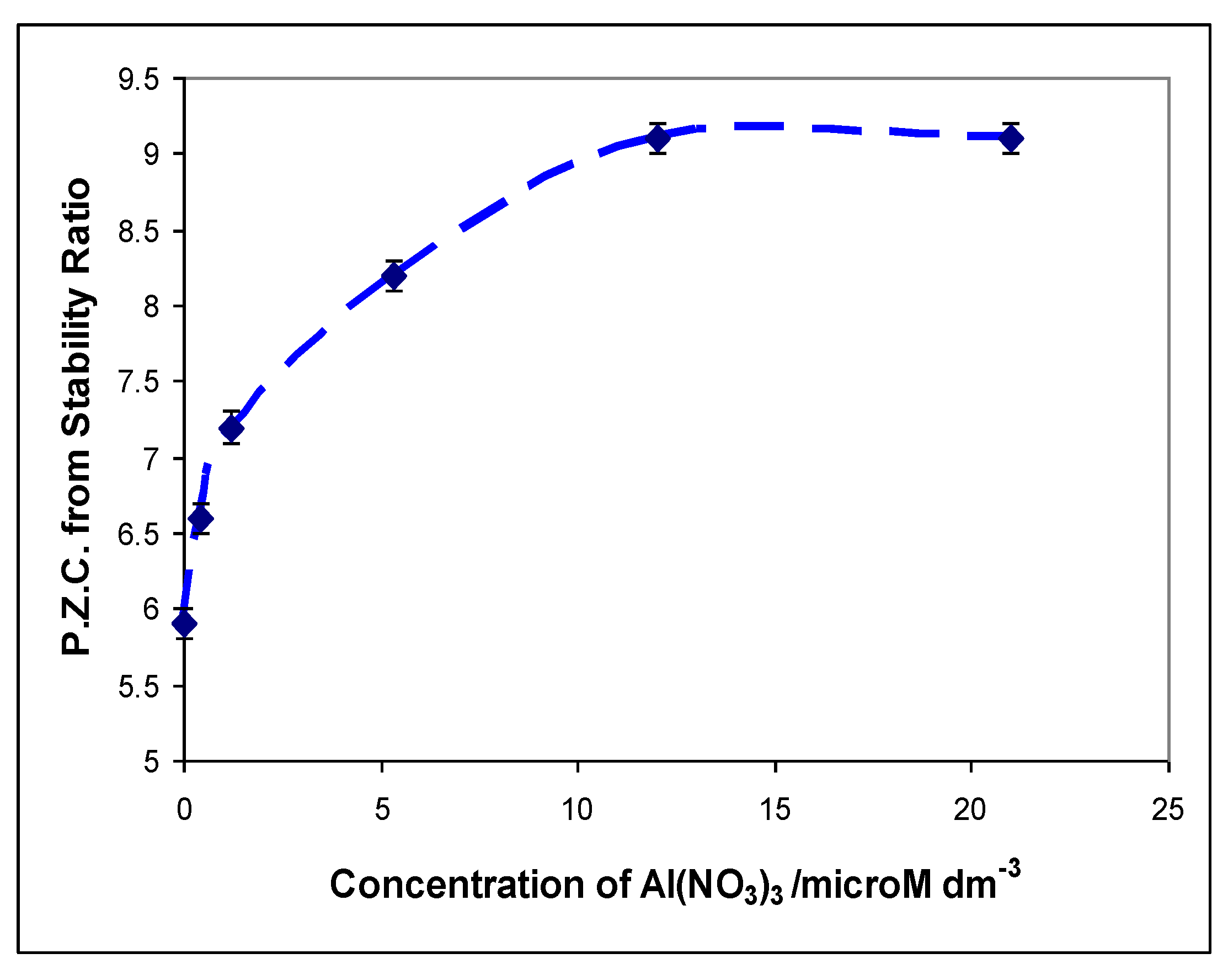
 . and p.z.c.’s measured by Furlong et al. [50], but “correcting” the silica levels to allow for the lower surface area of the rutile used by them (20 m2 g−1)
. and p.z.c.’s measured by Furlong et al. [50], but “correcting” the silica levels to allow for the lower surface area of the rutile used by them (20 m2 g−1)  .
. and p.z.c.’s measured by Furlong et al. [50], but “correcting” the silica levels to allow for the lower surface area of the rutile used by them (20 m2 g−1) .
.
. and p.z.c.’s measured by Furlong et al. [50], but “correcting” the silica levels to allow for the lower surface area of the rutile used by them (20 m2 g−1) .
6.3. Possible Perturbation of the Photocatalytic Results Caused by Dispersion Changes
7. The Implications of These Results for Practical Treatment of Water

8. Summary and Conclusions
Acknowledgments
Conflict of Interest
References
- Arslan-Aloton, I. Advanced Oxidation of Textile Industry Dyes. In Advanced Oxidation Processes for Water and Wastewater Treatment; Parsons, S., Ed.; IWA Publishing: London, UK, 2004; pp. 302–328. [Google Scholar]
- Karkmaz, M.; Puzenat, E.; Guillard, C.; Herrmann, J.M. Photocatalytic degradation of the alimentary azo dye amaranth. Mineralization of the azo group to nitrogen. Appl. Catal. B 2004, 51, 183–194. [Google Scholar] [CrossRef]
- Muruganandham, M.; Swaminathan, M. Solar driven decolourisation of reactive yellow 14 by advanced oxidation processes in heterogeneous and homogeneous media. Dyes Pigments 2007, 72, 137–143. [Google Scholar] [CrossRef]
- Upadhyay, S.; Liss, P.S.; Jickells, T.D. Sorption Model for Dissolved Aluminium in Freshwaters. Aquat. Geochem. 2002, 8, 255–275. [Google Scholar] [CrossRef]
- Mackin, J.E.; Aller, R.C. Processes affecting the behavior of dissolved aluminium in estuarine waters. Mar. Chem. 1984, 14, 213–232. [Google Scholar] [CrossRef]
- Hydes, D.J.; Liss, P.S. Behavior of Dissolved Aluminum in Estuarine and Coastal Waters. Estuar. Coast. Mar. Sci. 1977, 5, 755–776. [Google Scholar] [CrossRef]
- Tipping, E. Modelling A1 competition for heavy metal binding by dissolved organic matter in soil and surface waters of acid and neutral pH. Geoderma 2005, 127, 293–304. [Google Scholar] [CrossRef]
- Wiese, G.R.; Healy, T.W. Coagulation and electrokinetic behavior of TiO2 and Al2O3 colloidal dispersions. J. Colloid Interf. Sci. 1975, 51, 427–433. [Google Scholar] [CrossRef]
- Wiese, G.R.; Healy, T.W. Adsorption of Al(III) at the TiO2-H2O interface. J. Colloid Interf. Sci. 1975, 51, 434–442. [Google Scholar] [CrossRef]
- Healy, T.W.; Wiese, G.R.; Yates, D.E.; Kavanagh, B.V. Heterocoagulation in mixed oxide colloidal dispersions. J. Colloid Interf. Sci. 1973, 42, 647–649. [Google Scholar] [CrossRef]
- Wiese, G.R.; Healy, T.W. Heterocoagulation in mixed TiO2-Al2O3 dispersions. J. Colloid Interf. Sci. 1975, 52, 458–467. [Google Scholar] [CrossRef]
- Harrison, J.A.; Frings, P.J.; Beusen, A.H.W.; Conley, D.J.; McCrackin, M.L. Global importance, patterns, and controls of dissolved silica retention in lakes and reservoirs. Global Biogeochem. Cycle 2012, 26. [Google Scholar] [CrossRef]
- Mackin, J.E.; Aller, R.C. The effects of clay mineral reactions on dissolved Al distributions in sediments and waters of the Amazon continental-shelf. Cont. Shelf Res. 1986, 6, 245–262. [Google Scholar] [CrossRef]
- Egerton, T.A.; Everall, N.J.; Tooley, I.R. Characterization of TiO2 Nanoparticles Surface Modified with Aluminum Stearate. Langmuir 2005, 21, 3172–3178. [Google Scholar] [CrossRef]
- Egerton, T.A.; Tooley, I.R. The surface characterisation of coated titanium dioxide by FTIR spectroscopy of adsorbed nitrogen. J. Mater. Chem. 2002, 12. [Google Scholar] [CrossRef]
- Howard, P.B.; Parfitt, G.D. Precipitation of silica-alumina on titanium-dioxide surfaces. Croat. Chem. Acta 1977, 50, 15–30. [Google Scholar]
- Iler, R.K. Product comprising a skin of dense, hydrated amorphous silica bound upon a core of another solid material and process of making same. US Patent No. 2,885,366, 5 May 1959. [Google Scholar]
- Egerton, T.A. The modification of fine powders by Inorganic Coatings. Kona 1998, 16, 46–59. [Google Scholar]
- Gesenhues, U. Coprecipitation of hydrous alumina and silica with TiO2 pigment as substrate. J. Coll. Inter. Sci. 1994, 168, 428–436. [Google Scholar] [CrossRef]
- Exley, C.; Birchall, J.D. Hydroxy aluminosilicate formation in solutions of low total aluminium concentration. Polyhedron 1992, 11, 1901–1907. [Google Scholar] [CrossRef]
- Birchall, J.D.; Chappell, J.S. Aluminum, water chemistry, and Alzheimers-Disease. Lancet 1989, 1, 953–953. [Google Scholar] [CrossRef]
- Marsac, R.; Davranche, M.; Gruau, G.; Dia, A.; Bouhnik-Le Coz, M. Aluminium competitive effect on rare earth elements binding to humic acid. Geochim. Cosmochim. Acta 2012, 89, 1–9. [Google Scholar] [CrossRef]
- Cabaniss, S.E. Forward Modeling of Metal Complexation by NOM: II. Prediction of Binding Site Properties. Environ. Sci. Technol. 2011, 45, 3202–3209. [Google Scholar] [CrossRef]
- Sen Kavurmaci, S.; Bekbolet, M. The Role of Oxidative Treatment on the Trivalent Cation Complexation Properties of Natural Organic Matter. J. Adv. Oxid. Technol. 2010, 13, 212–220. [Google Scholar]
- Engelhardt, G.; Michel, D. High Resolution Solid State NMR of Silicates and Zeolites; Wiley: New York, NY, USA, 1987. [Google Scholar]
- Egerton, T.A.; Mattinson, J.A. Comparison of photooxidation and photoreduction reactions on TiO2 nanoparticles. J. Photochem. Photobiol. A 2007, 186, 115–120. [Google Scholar] [CrossRef]
- Day, R.E.; Egerton, T.A. Surface Studies of TiO, Pigment with Especial Reference to the Role of Coatings. Colloid Surface 1987, 23, 137–155. [Google Scholar] [CrossRef]
- Mattinson, J.A. Effect of Inorganic Surface Treatment on TiO2 Photoactivity. Ph.D. Thesis, Newcastle University, Newcastle-upon-Tyne, UK, November 2008. [Google Scholar]
- Kraeutler, B.; Bard, A.J. Heterogeneous photocatalytic decomposition of saturated carboxylic-acids on tio2 powder-decarboxylative route to alkanes. J. Am. Chem. Soc. 1978, 100, 5985–5992. [Google Scholar] [CrossRef]
- Bahnemann, D.W.; Kholuiskaya, S.N.; Dillert, R.; Kulak, A.I.; Kokorin, A.I. Photodestruction of dichloroacetic acid catalyzed by nano-sized TiO2 particles. Appl. Catal. B 2002, 36, 161–169. [Google Scholar] [CrossRef]
- Enriquez, R.; Pichat, P. Different net effect of TiO2 sintering temperature on the photocatalytic removal rates of 4-chlorophenol, 4-chlorobenzoic acid and dichloroacetic acid in water. J. Environ. Sci. Health A 2006, 41, 955–966. [Google Scholar] [CrossRef]
- McGarvey, D.J.; Lyth, P.L.; Guest, P.J.; Dransfield, G.; Truscott, T.G. Photoactivity tests of TiO2-based inorganic sunscreens - Part 1: Non-aqueous dispersions. J. Photochem. Photobiol. B. 2000, 59, 151–174. [Google Scholar]
- Egerton, T.A.; Kessell, L.M.; Tooley, I.R.; Wang, L.W. Photogreying of TiO2 nanoparticles. J. Nanopart. Res. 2007, 9, 251–260. [Google Scholar] [CrossRef]
- Howe, R.F.; Gratzel, M. EPR observation of trapped electrons in colloidal TiO2. J. Phys. Chem. 1985, 89, 4495–4499. [Google Scholar] [CrossRef]
- Nakaoka, Y.; Nosaka, Y.Y. ESR investigation into the effects heat treatment and crystal structure of radical produced over irradiated TiO2 powder. J. Photochem. Photobiol. A 1997, 110, 299–305. [Google Scholar] [CrossRef]
- Attwood, A.L.; Murphy, D.M.; Edwards, J.L. An EPR study of thermally and photochemically generated oxygen radicals on hydrated and dehydrated titania surfaces. Res. Chem. Intermediat. 2003, 29, 449–465. [Google Scholar] [CrossRef]
- Deskins, N.A.; Dupuis, M. Electron transfer via polaron hopping in bulk TiO2: A density functional theory characterization. Phys. Rev. B 2007, 75, 195212–195217. [Google Scholar] [CrossRef]
- MacDonald, I.R.; Howe, R.F.; Zhang, X.; Zhou, W. In situ EPR studies of electron trapping in a nanocrystalline rutile. J. Photochem. Photobiol. A 2010, 216, 238–243. [Google Scholar] [CrossRef]
- Flaig-Baumann, R.; Hermann, M.; Boehm, H.P. Chemistry of titanium dioxide surface .3. Reactions of basic hydroxyl on surface. Z. Anorg. Allg. Chem. 1970, 372, 296–297. [Google Scholar] [CrossRef]
- Criado, J.; Real, C. Mechanism of the inhibiting effect of phosphate on the anatase-rutile transformation induced by thermal and mechanical treatment of TiO2. J. Chem. Soc. Faraday Trans. 1983, 79, 2765–2771. [Google Scholar] [CrossRef]
- Okazaki, S.; Aoki, T.; Tani, K. The adsorption of basic alpha-amino-acids in an aqueous-solution by titanium (iv) oxide. Bull. Chem. Soc. Jpn. 1981, 54, 1595–1599. [Google Scholar] [CrossRef]
- Abdullah, M.; Low, G.K.C.; Matthews, R.W. Effects of common inorganic anions on rates of photocatalytic oxidation of organic-carbon over illuminated titanium-dioxide. J. Phys. Chem. 1990, 94, 6820–6825. [Google Scholar] [CrossRef]
- Christensen, P.A.; Curtis, T.P.; Egerton, T.A.; Kosa, S.A.M.; Tinlin, J.R. Photoelectrocatalytic and photocatalytic disinfection of E. coli suspensions by titanium dioxide. Appl. Catal. B 2003, 41, 371–386. [Google Scholar] [CrossRef]
- Egerton, T.A.; Parfitt, G.D.; Kang, Y.; Wightman, J.P. XPS analysis of uncoated and silica-coated titanium-dioxide powders. Colloid Surface 1983, 7, 311–323. [Google Scholar] [CrossRef]
- Mizushima, K.; Tanaka, M.; Asai, A.; Iida, S.; Goodenough, J.B. Impurity levels of iron-group ions in TiO2. J. Phys. Chem. Solids 1979, 40, 1129–1140. [Google Scholar] [CrossRef]
- Martin, S.T.; Morrison, C.L.; Hoffmann, M.R. Photochemical Mechanism of Size-Quantized Vanadium-Doped TiO2 Particles. J. Phys. Chem. 1994, 98, 13695–13704. [Google Scholar] [CrossRef]
- Palmisano, L.; Augugliaro, V.; Sclafani, A.; Schiavello, M. Activity of chromium-ion-doped titania for the dinitrogen photoreduction to ammonia and for the phenol photodegradation. J. Phys. Chem. 1988, 92, 6710–6713. [Google Scholar] [CrossRef]
- Egerton, T.A.; Kosa, S.A.M.; Christensen, P.A. Photoelectrocatalytic disinfection of E. coli suspensions by iron doped TiO2. Phys. Chem. Chem. Phys. 2006, 8, 398–406. [Google Scholar] [CrossRef]
- Furlong, D.N.; Parfit, G.D. Electrokinetics of titanium dioxide. J. Colloid. Interf. Sci. 1978, 65, 548–554. [Google Scholar] [CrossRef]
- Furlong, D.N.; Sing, K.S.W.; Parfitt, G.D.P. Precipitation of silica on titanium dioxide surfaces. 1. Prepartion of coated surfaces and examination by electrophoresis. J. Colloid Interf. Sci. 1979, 69, 409–419. [Google Scholar] [CrossRef]
- Parfitt, G.D.; Ramsbotham, J. Study of surface properties of coated titanium dioxide pigments by electrophoresis. J. Oil Colour Chem. Ass. 1971, 54, 356–358. [Google Scholar]
- Losoi, T. Surface studies of titanium dioxide pigments. J. Coating Technol. 1989, 61, 57–63. [Google Scholar]
- Egerton, T.A.; Harrison, R.W.; Hill, S.E.; Mattinson, J.A.; Purnama, H. Effects of particle dispersion on the measurement of semi-conductor photocatalytic activity. J. Photochem. Photobiol. A 2010, 216, 268–274. [Google Scholar] [CrossRef]
- Egerton, T.A.; Tooley, I.R. Effect of changes in TiO2 dispersion on its measured photocatalytic activity. J. Phys. Chem. B 2004, 108, 5066–5072. [Google Scholar] [CrossRef]
- Egerton, T.A.; Tooley, I.R. UV absorption and scattering properties of inorganic-based sunscreens. Int. J. Cosmetic Sci. 2012, 34, 117–122. [Google Scholar] [CrossRef]
- Egerton, T.A.; King, C.J. The influence of light intensity on photoactivity in TiO2 pigmented systems. J. Oil. Colour Chem. Assoc. 1979, 62, 386–391. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Egerton, T.A. The Influence of Surface Alumina and Silica on the Photocatalytic Degradation of Organic Pollutants. Catalysts 2013, 3, 338-362. https://doi.org/10.3390/catal3010338
Egerton TA. The Influence of Surface Alumina and Silica on the Photocatalytic Degradation of Organic Pollutants. Catalysts. 2013; 3(1):338-362. https://doi.org/10.3390/catal3010338
Chicago/Turabian StyleEgerton, Terry A. 2013. "The Influence of Surface Alumina and Silica on the Photocatalytic Degradation of Organic Pollutants" Catalysts 3, no. 1: 338-362. https://doi.org/10.3390/catal3010338