Abstract
Hydrogen is the ideal fuel for the future because it is clean, energy efficient, and abundant in nature. While various technologies can be used to generate hydrogen, only some of them can be considered environmentally friendly. Recently, solar hydrogen generated via photocatalytic water splitting has attracted tremendous attention and has been extensively studied because of its great potential for low-cost and clean hydrogen production. This paper gives a comprehensive review of the development of photocatalytic water splitting for generating hydrogen, particularly under visible-light irradiation. The topics covered include an introduction of hydrogen production technologies, a review of photocatalytic water splitting over titania and non-titania based photocatalysts, a discussion of the types of photocatalytic water-splitting approaches, and a conclusion for the current challenges and future prospects of photocatalytic water splitting. Based on the literatures reported here, the development of highly stable visible-light-active photocatalytic materials, and the design of efficient, low-cost photoreactor systems are the key for the advancement of solar-hydrogen production via photocatalytic water splitting in the future.
1. Alternative Energies and Hydrogen
Human civilization is built by our energy system, which facilitates the development of technologies that provide us with a higher standard of living. Energy is an essential part of productivity and is as important as raw materials, capital, and labor. Today, most of the energy that we use comes from fossil fuels, which are not considered ideal due to the following reasons: First, the combustion of fossil fuels, such as coal and petroleum, will produce carbon dioxide (CO2), which is one of the major greenhouse gases that causes climate change [1]; Second, the amount of fossil fuel on the Earth is limited and will be depleted someday. Nature has stored solar energy in the form of mineral organic compounds or in fossil fuels such as coal, petroleum, and natural gas through millions of years of biological and non-biological processes [2]. However, the rate of global energy consumption has far exceeded the rate of energy storage, implying that fossil fuels will soon be exhausted [3]; Third, fossil fuels are generally controlled by certain nations in the world. As a result, a significant amount of time and money will be spent for the relocation and distribution of these fuels. To satisfy the energy demand, competition for resources among nations will continue to be seen. Therefore, it is imperative for us to search for a sustainable energy source that can be easily produced at low cost and that is friendly to the environment.
To replace or reduce the use of fossil fuels, several alternative energies have been developed. Alternative energies are renewable and have lower carbon emissions when compared to conventional energy sources. These energy sources include wind, hydropower, solar, geothermal, etc. The term “wind energy” describes the process by which the wind is used to generate mechanical power or electricity [4]. Although wind energy is a free, renewable resource, its major challenge is that wind cannot be stored, and not all wind can be harnessed to meet the timing of the demand for electricity.
Hydropower is energy that is generated by water and converted to electricity [5]. The most common method of using energy from water is a hydroelectric dam in which water passes through turbines, causing them to rotate, and then the energy is captured to run a generator. The major disadvantage of hydropower is that dams are extremely expensive to build, which means that they must operate for many decades to become profitable. Moreover, the building of large dams can often cause serious geological and ecological damage.
Solar energy is a free, inexhaustible resource from the sun that can be converted to electricity (photovoltaic power) or heat using devices such as solar cells [6] or concentrators [7]. Electricity produced from solar energy has advantages over wind power and hydropower, since the later two need turbines with moving parts that are both noisy and require much maintenance. However, the major disadvantage of solar energy is its intermittent nature. That is, the amount of sunlight a location receives varies greatly depending on the geographical location, time of day, season, and even clouds.
Geothermal energy is obtained by extracting heat from water or rocks deep underground [8]. Unlike wind and solar energy, geothermal energy is not intermittent, and hence it can be a reliable energy source for several years. However, heat typically is extracted from the rocks much more rapidly than it is restored from the environment. Therefore, geothermal plants have limited lifetimes and require periodic drilling of new holes for continued operation, which in turn increases the cost of electricity. In sum, the availability of energy from renewable sources is unstable with variability in location and time, and the energy may not be available to the end-users when it is needed. Therefore, it is necessary to identify a medium or container in which to store the energy.
Hydrogen is an ideal energy storage medium or carrier because of the following reasons; First, it is the most abundant element and it exists in both water and biomass; Second, it has a high energy yield (122 kJ/g) compared to other fuels such as gasoline (40 kJ/g); Third, it is environmentally friendly because its end use will not produce pollutants, greenhouse gases, nor any harmful effect on the environment. Last, but not least, hydrogen can be stored in gaseous, liquid or metal hydride form and can be distributed over large distances through pipelines or via tankers.
1.1. Some Concerns about Hydrogen
Despite the above advantages, application of hydrogen technologies may have some limitations. In order to serve as a practical fuel for transportation, hydrogen must be compressed to minimize its storage volume because of its low energy density. Hydrogen with low volumetric energy is generally stored as a compressed gas or liquid, meaning that an advanced compression process is needed. However, such processes will require energy and expansive equipment, which adds costs to the use of hydrogen. The storage of hydrogen in metal hydride form is another alternative to compression. However, metal hydrides are often expensive, heavy, and have a limited lifetime, making the process costly and less practical [9]. Considering the application of hydrogen in road transportation, present efforts are based on two directions. One is to make hydrogen-combustion vehicles, and the other is to make hydrogen fuel-cell vehicles. The advantages of hydrogen vehicles include a reduction in the emission of carbon dioxide and other smog-producing pollutants, as well as a great reduction in the release of nitrogen oxides (NOx). Unfortunately, the introduction of hydrogen vehicles into the commercial market has faced the challenges of inadequate hydrogen fueling infrastructure and high production cost in comparison to other petroleum-based vehicles. In fact, these problems are interconnected in the sense that customers will not purchase hydrogen vehicles unless adequate fueling is available, that manufacturers will not produce vehicles that people will not buy, and that fuel providers will not install hydrogen stations for vehicles that do not exist.
1.2. Hydrogen Production
Currently, most of the world’s hydrogen is produced by a process called “steam reforming” [10]. In this process, methane is widely used as fuel since it has the highest hydrogen-to-carbon ratio among hydrocarbons; hence, the by-products generated are minimized. In general, the steam methane reforming (SMR) process consists of two steps. First is the reformation process in which methane mixed with steam is passed over a catalyst bed at high temperature (700-900 °C) and high pressure (1.5-3 MPa) to form a mixture of hydrogen and carbon monoxide (CO) as shown in Equation 1. The second step is the shift reaction in which CO from the first step reacts with additional steam to give CO2 and more hydrogen (Equation 2).
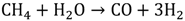
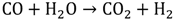
Another process used for hydrogen generation that involves fossil fuels is coal gasification [11]. In this process, the coal undergoes partial oxidation at high temperature and pressure (~5 MPa) with the help of oxygen and steam to produce a mixture of hydrogen, CO, CO2, methane and other compounds. At temperatures above 1000 ℃ and pressures of 1 bar, mostly hydrogen and CO remain. The process can be represented by the following reactions (3 and 4).
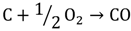
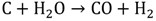
Biomass, such as crops, plants, and animal wastes, can also be used to produce hydrogen via thermochemical and biological processes. Pyrolysis [12] and gasification are feasible thermochemical routes for hydrogen production, whereas biophotolysis, biological gas shift reaction, and fermentation are promising biological processes that are under development [13]. In the pyrolysis process, biomass is heated rapidly to a high temperature in the absence of oxygen to produce hydrogen, methane, CO, CO2, carbon, and other compounds, depending on the nature of the biomass. The temperature used for pyrolysis ranges from 400 to 600 °C, and pressure ranges from 0.1 to 0.5 MPa.
There is a general perception that hydrogen is a clean fuel, but this may not be necessarily correct. If hydrogen is produced from natural gas, coal, or biomass, it will use a lot of energy, not to mention the substantial amount of CO2 that will be generated as a by-product. Therefore, the best way of producing hydrogen is to utilize an alternative energy, such as hydropower, wind energy, and solar energy, to carry out the water-splitting reaction. Among these alternative energies, solar energy is the most promising approach since region-related limitations are less rigorous as compared to wind energy and hydropower.
1.3. Hydrogen Production by Solar Energy
Hydrogen production via solar water splitting generally can be categorized into 3 types: (1) thermochemical water splitting; (2) photobiological water splitting, and (3) photocatalytic water splitting. The principle of thermochemical water splitting is to use concentrators to collect the heat from sunlight, which typically can reach around 2000 °C, and to utilize the collected heat to perform the water-splitting reaction under the presence of a catalyst such as ZnO [14]. The reactions are shown in Equations 5 and 6. Even though this technique appears to be unsophisticated, heat management/control and the search for appropriate heat-resisting materials has become the greatest challenge. Furthermore, large-scale solar concentrator systems are essential to achieve the high temperature requirement; therefore, such a technique is often costly.
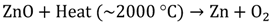
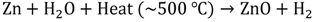
Photobiological water splitting [15] basically can be divided into two groups based on the microorganisms selected, products generated, and reaction mechanisms involved. Hydrogen production by photosynthetic oxygenic cyanobacteria or green algae under light irradiation and anaerobic condition is referred to as water biophotolysis, while hydrogen production by photosynthetic anoxygenic bacteria under light irradiation and anaerobic condition is referred to as organic biophotolysis.
Although organic biophotolysis is capable of decomposing organic wastes to give a high hydrogen yield, the reactions will generate CO2 as the by-product, which has made the technology less environmentally friendly as compared with water biophotolysis. In water biophotolysis, on the other hand, water is transformed into hydrogen and oxygen in the presence of light by cyanobacteria or green algae with the help of a special enzyme such as hydrogenase or nitrogenase, as illustrated in Equations 7 and 8 [16,17].
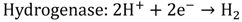
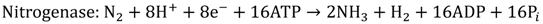
Despite water biophotolysis being a “cleaner” way to produce hydrogen as comparing with organic biophotolysis, it still has many problems waiting to be solved, including low hydrogen yield, the poisoning effect of enzymes under the existence of oxygen (generated simultaneously during biophotolysis), and the difficulty in designing and scaling up the bioreactor for the process.
Photocatalytic water splitting is another promising technology to produce “clean” hydrogen. Compared with thermochemical and photobiological water-splitting techniques, it has the following advantages: (1) reasonable solar-to-hydrogen efficiency; (2) low process cost; (3) the ability to achieve separate hydrogen and oxygen evolution during reaction; and (4) small reactor systems suitable for household applications, thus providing for a huge market potential. The following is an overview of hydrogen generation by photocatalytic water splitting.
2. Photocatalytic Water Splitting
Photocatalysis is defined as the chemical reaction induced by photoirradiation in the presence of a catalyst, or more specifically, a photocatalyst. Such material will facilitate chemical reactions without being consumed or transformed. Photosynthesis by plants is a well-known example of photocatalysis in nature, where chlorophyll serves as the photocatalyst. The basic working principle of photocatalysis is simple. First, irradiation of light with energy greater than the bandgap of photocatalyst, separating the vacant conduction band (CB) and filled valence band (VB), excites an electron in VB into CB to result in the formation of an electron (e−)-hole (h+) pair. These e− and h+ reduce and oxidize respectively chemical species on the surface of photocatalyst, unless they recombine to give no net chemical reaction. The original structure (or chemical composition) of photocatalyst remains unchanged if an equal number of e− and h+ are consumed for chemical reaction and/or recombination.
Several terms have been adopted to describe the efficiency for converting solar energy, namely the Applied Bias Photon-to-Current Efficiency (ABPE), and Quantum Efficiency (QE). ABPE is usually used to characterize the photo-response efficiency of a photoelectrode material under an applied voltage. ABPE sometimes is referred to as the photo-conversion efficiency [18]. Due to the voltage applied, such terms cannot be used to represent the true photo-conversion efficiency for photocatalytic water splitting. The definition of photo-conversion efficiency is shown in Equation 9:
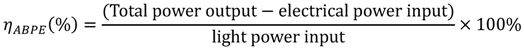
where E0rev is 1.23 V, the standard state-reversible potential, I0 is the power density of incident light, and Eapp is the applied potential.
As for quantum efficiency, it can represent the characteristic photon conversion of photoactive films. It is defined as the percentage of generated electrons and incident photons while the photoactive films are irradiated under a specific wavelength, as shown in Equation 10 [19].
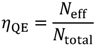
In the above equation, Neff is the number of effective generated electron-hole pairs under light irradiation, and Ntotal is the total number of incident photons. It is noted that ηQE neglects the energy loss of solar irradiance and the chemical conversion efficiency. Therefore, it is suitable to qualify the photoactive films but not to represent the water-splitting reaction conversion efficiency.
To describe the true hydrogen production efficiency of a water-splitting reaction under sunlight, a term called “solar-to-hydrogen” conversion efficiency (STH) [20] is often used. The definition of STH conversion efficiency is shown in Equation 11:
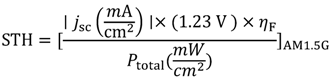
where, Ptotal represents the power density of incident simulative sunlight (AM1.5G) and the numerator is the product of photocurrent density (jsc) at zero bias (short-circuit photocurrent), the thermodynamic voltage required for water splitting (1.23 V), and the faradic efficiency (ηF). A different form of solar-to-hydrogen conversion efficiency which can also be used is shown in Equation 12:
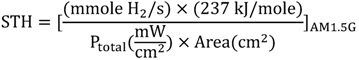
where the denominator is the total power of incident simulative sunlight (AM1.5G) multiplied by the area irradiated by the incident light, and the numerator is the product of the H2 evolution rate and the Gibbs free energy for generating one mole of H2 from water.
In a photocatalytic water splitting reaction, photocatalyst plays a crucial role. Until now, titania (TiO2) has been a widely used photocatalyst for photocatalytic water splitting because it is stable, non-corrosive, environmentally friendly, abundant, and cost-effective. More importantly, its energy levels are appropriate to initiate the water-splitting reaction [21]. In other words, the CB of TiO2 is more negative than the reduction energy level of water (EH+/H2 = 0 V), while the VB is more positive than the oxidation energy level of water (EO2/H2O = +1.23 V), as shown in Figure 1.
Despite the many advantages of TiO2, its photocatalytic water-splitting efficiency under solar energy is still quite low, mainly due to the following reasons; First, the photo-generated electrons in the CB of TiO2 may recombine with the VB holes quickly to release energy in the form of unproductive heat or photons; Second, the decomposition of water into hydrogen and oxygen is a chemical reaction with large positive Gibbs free energy (∆G = 237 kJ/mol), thus the backward reaction (recombination of hydrogen and oxygen into water) easily proceeds; Third, the bandgap of TiO2 is about 3.2 eV, and therefore, only UV light can be utilized to activate the photocatalyst. Since UV light only accounts for approximately 4% of solar energy, while visible light contributes about 50%, the inability to utilize visible light limits the efficiency of TiO2 in solar photocatalytic hydrogen production.
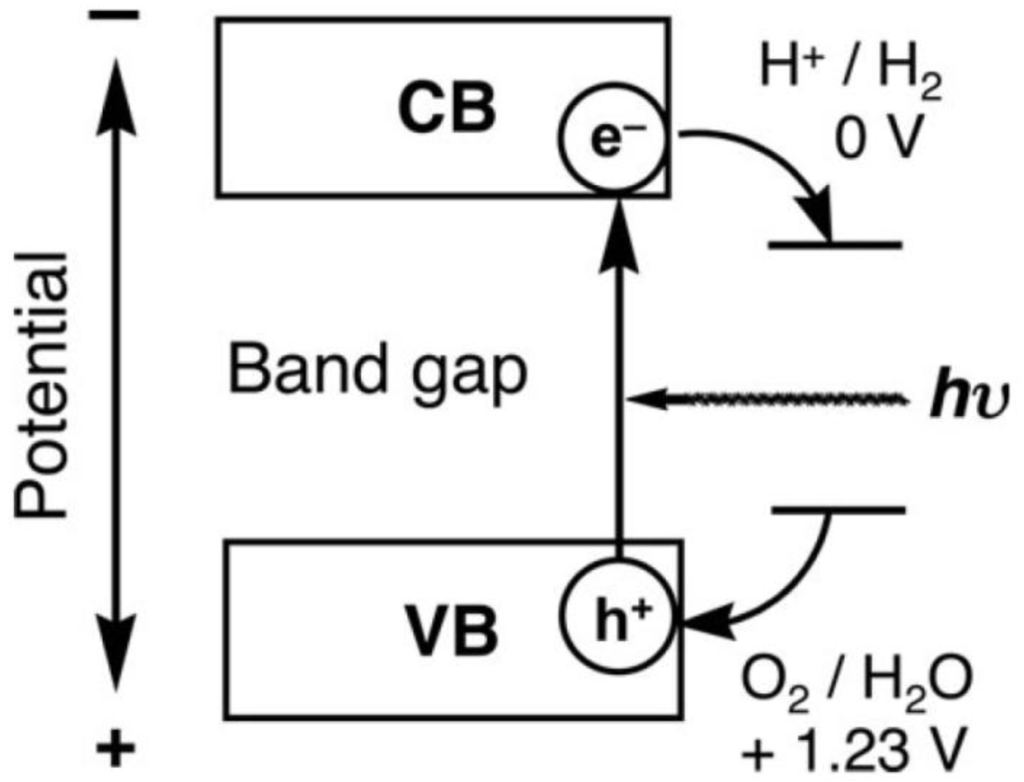
Figure 1.
Mechanism of photocatalytic water splitting reproduced (“adapted” or “in part”) from ref [21] with permission of The Royal Society of Chemistry.
2.1. How to Improve the Photoactivity of TiO2
In order to solve the problems mentioned above and to make solar photocatalytic water splitting of TiO2 feasible, continuous efforts have been made to promote the photocatalytic activity of TiO2 and enhance its visible-light response. The techniques that have been investigated in the past include the addition of sacrificial agent/carbonate salts, metal loading, dye sensitization, ion (cation, and anion) doping, etc. Some of them have been proven to be useful for improving the photocatalytic activity of TiO2.
Due to rapid recombination of photo-generated CB electrons and VB holes, it is difficult to achieve water splitting for hydrogen production using TiO2 photocatalyst in pure water. Adding electron donors or sacrificial reagents to react with the photo-generated VB holes is an effective measure to enhance the electron-hole separation, resulting in higher quantum efficiency. However, the drawback of this technique is the need to continuously add electron donors in order to sustain the reaction since they will be consumed during photocatalytic reaction. Li et al. [22] reported enhanced photocatalytic hydrogen production when organic pollutants acting as electron donors, such as oxalic acid, formic acid, and formaldehyde, were added into the reaction system. Decomposition of the organic pollutants was reported to be consistent with hydrogen production. Besides the use of sacrificial agents, the addition of carbonate salts was found to improve photocatalytic hydrogen production by suppressing its backward reaction to form water. Sayama et al. [23,24] reported that adding carbonate salts to Pt-loaded TiO2 suspensions led to highly efficient stoichiometric photocatalytic decomposition of water into H2 and O2. It was found that Pt-loaded TiO2 photocatalyst during reaction was covered with several types of carbonate species. These carbonate species can effectively suppress the back reaction of water splitting to form water and alleviate the photoabsorption of oxygen on the TiO2.
Usually, loading of metals that act as co-catalysts on the surface of photocatalyst, such as Pt, Pd, or Rh, is essential for enhancing its performance. Loading of metals, including Pt, Au, Pd, Rh, Ni, Cu, and Ag, have been reported to be very effective for improving TiO2’s activity in photocatalysis. As the Fermi levels of these metals are lower than the CB of TiO2 [25], photo-excited electrons can be transferred from the CB of TiO2 to metal particles deposited on its surface, while photo-generated VB holes remain on the photocatalyst. Accumulated electrons on metal particles can then be used to carry out a reduction reaction, while holes on the photocatalyst can be used to carry out the oxidation reaction. Therefore, metals with suitable work-function can help prevent electron-hole recombination, leading to higher photocatalytic activity of TiO2. Bamwenda et al. [26] prepared Au and Pt loaded TiO2 photocatalysts by deposition precipitation, impregnation, photodeposition, and colloidal mixing methods for hydrogen production. It was found that synthesis methods and metal loadings affect H2 production significantly. Gold and platinum precursors calcined in air at 300 °C were found to have the highest activity towards H2 generation, followed by a decline in activity with increasing calcinations temperature. The maximum H2 yield observed for Pt-TiO2 and Au-TiO2 corresponded to metal loadings of 0.3-1 and 1-2 wt.%, respectively. The roles of Au and Pt on TiO2 include the trapping of photogenerated electrons, the reduction of protons, and the formation/desorption of hydrogen. Murdoch and co-workers studied the effect of Au loading and particle size on photocatalytic hydrogen production over Au/TiO2 nanoparticles [27]. It was concluded that the increase in the hydrogen production rate is simply due to the greater availability of Au particles at the interface with TiO2, trapping electrons to reduce hydrogen ions into hydrogen molecules. Anpo and Takeuchi [28] employed ESR signals to investigate electron transfer from TiO2 to Pt particles. It was found that Ti3+ signals increased with irradiation time, and the loading of Pt reduced the amount of Ti3+. This observation indicates the occurrence of electron transfer from TiO2 to Pt particles.
Besides the role of electron traps to improve the photo electron-hole separation, loading of metal, such as Au or Ag, may also promote the activity of photocatalyst by the surface plasmon resonance (SPR) effect. SPR is defined as the collective motions of the conduction electrons induced by light irradiation, which is associated with a considerable enhancement of the electric near-field [29]. The resonance wavelength strongly depends on the size and shape of the nanoparticles, the inter-particle distance, and the dielectric property of the surrounding medium [30,31]. As for Au loaded TiO2, electrons from the valence band of photocatalyst are excited to the conduction band by UV light irradiation, and then transferred to the gold particles on TiO2. The SPR effect induced by appropriate visible-light irradiation (~560 nm for Au) can then boost the energy intensity of the trapped electrons, resulting in enhancement of photocatalytic activity [32]. Kowalska and co-workers have examined a series of Au/TiO2 samples prepared by photodeposition for the degradation of carboxylic acids. It was observed that the position of the surface plasmon band varies from 520 to 570 nm depending on the average particle size of the TiO2 support [33]. It was also observed that gold deposition significantly enhanced the activity of photocatalyst due to the surface plasmon resonance effect. Silva et al. reported the investigation of Au/TiO2 nanoparticles for the generation of hydrogen and oxygen from water [34]. They demonstrated that gold nanoparticles exhibit a dual role as light harvesters, injecting electrons into TiO2’s conduction band, and also as catalytic sites for gas generation, depending on the excitation light source used. For instance, when using excitation wavelengths corresponding to gold plasmon band, gold nanoparticles will absorb photons and inject electrons to the conduction band of TiO2 to perform water reduction.
Dye sensitization is a widely used technique to utilize visible light for energy conversion. Some dyes having redox property and visible-light sensitivity can be used in solar cells as well as photocatalytic systems [35]. Under illumination by visible light, the excited dyes can inject electrons to the CB of photocatalyst to initiate the catalytic reactions as illustrated in Figure 2. Higher photoactivity can be obtained by efficient absorption of visible light and efficient transfer of electrons from excited dyes to the CB of TiO2. The CB electrons can then be transferred to the metal particle or co-catalyst (such as Pt) loaded on the surface to initiate the reduction reaction. In order to regenerate dyes, redox systems or sacrificial agents, such as I3−/I− pair and EDTA, can be added to the solution to sustain the reaction cycle. The benefits of adopting dye-sensitized photocatalyst systems include inhibiting charge recombination by improving electron-hole separation, increasing the spectrum response range of photocatalyst (i.e., excitation of wide bandgap photocatalyst by visible light), and changing the selectivity or yield of a particular product [36]. Based on the literature reported in the past, inorganic sensitizers, organic dyes, and coordination metal complexes are very effective photosensitizers that have been studied [37,38]. Among them, photosensitization by organic dyes is the most widely studied method because it is known that the organic dyes have prominent visible light absorption properties and that their structures can be changed by easy and low-cost approaches [39]. In past years, many organic dyes, such as eosin Y, riboflavin, cyanine, cresyl violet, hemicyanine, and merocyanine [40,41], have been tested as photosensitizers. However, the stability of pure organic dyes is a notable problem that should be solved immediately before dye sensitization can be applied for practical uses.
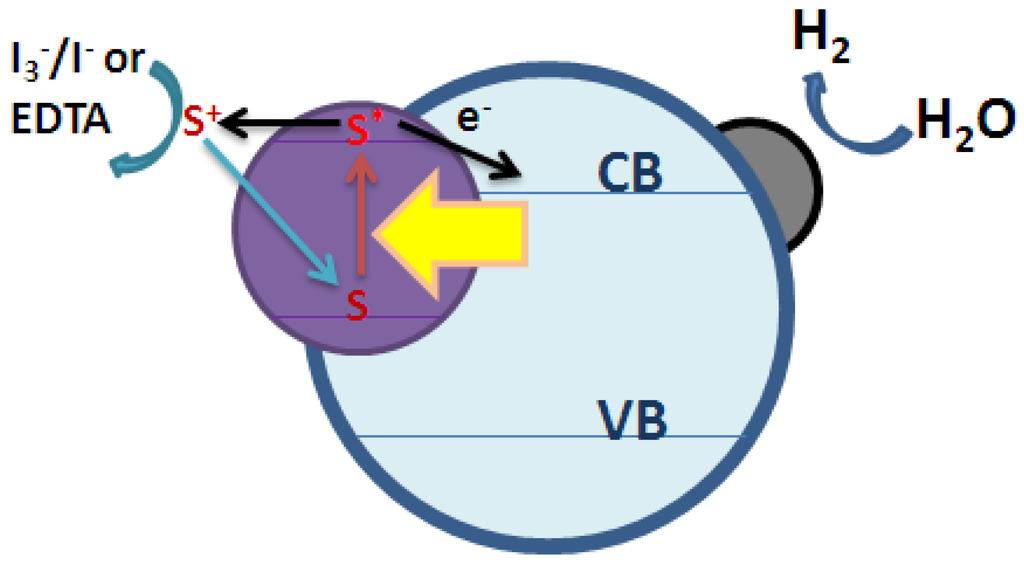
Figure 2.
Mechanism of dye-sensitized photocatalytic hydrogen production under visible light irradiation.
Another common practice for modifying the bandgap of photocatalyst is the so-called metal ion doping practice in which a small percentage of metal ion(s) is incorporated into the crystal lattice of photocatalyst. Transitional metal ion doping and rare-earth metal ion doping have been extensively investigated for enhancing photocatalytic activities of photocatalyst under visible light. Choi et al. [42] carried out a systematic investigation to study the photoactivity of 21 metal ions doped into TiO2. It was found that doping of metal ions could expand the photo-response of TiO2 into visible-light spectrum. As metal ions are incorporated into the TiO2 lattice, impurity energy levels in the bandgap of TiO2 are formed. For photocatalytic reactions, carrier transferring is as important as carrier trapping. Only when the trapped electron and hole are transferred to the surface can photocatalytic reactions occur. Therefore, metal ions should be doped near the surface of photocatalysts for better charge transferring. In the case of deep doping, metal ions are likely to behave as recombination centers, which is unfavorable for the photocatalytic reactions. Among the 21 metal ions studied, Fe, Mo, Ru, Os, Re, V, and Rh ions can increase visible light-induced photocatalytic activity, while dopants of Co and Al ions cause detrimental effects. The different effects of metal ions result from their abilities to trap and transfer electrons/holes [43].
The use of anion doping to improve photocatalytic activity under visible light is a new method with few investigations reported in the literature. Doping of anions (N, F, C, S etc.) in TiO2 crystalline could shift its photo-response into visible-light spectrum. Unlike metal ions (cations), anions are less likely to form recombination centers and, therefore, are more effective at enhancing photocatalytic activity. Asahi et al. [44] determined the substitutional doping contents of C, N, F, P, and S for O in anatase TiO2. It was found that mixing of p states of N with 2p of O could shift the VB edge upwards to narrow down the bandgap of TiO2. Although doping of S had resulted in a similar effect of bandgap narrowing, the ionic radius of S was reported to be too large to be incorporated into the lattice of TiO2. It was reported by Umebayashi et al. [45] that S-doped TiO2 could be prepared by oxidation annealing of TiS2. Annealed at 600 °C, TiS2 was partly changed to anatase TiO2. The residual S atoms in the anatase TiO2 formed S-doped TiO2 by Ti-S bonds. It was found that when TiO2 was doped with S, the mixing of S 3p states with the VB of TiO2 increased the width of VB, resulting in bandgap narrowing. Since the bandgap narrowing was caused by VB upward shifting, the CB of the photocatalyst remained unchanged. Therefore, the S-doped TiO2 should be able to reduce protons for hydrogen production under visible light. On the other hand, the upward shift of VB may reduce the oxidation ability under visible light. Also, N-doped TiO2 have been extensively investigated. The reported methods to dope N are heating of titanium hydroxide and urea, reactive DC magnetron sputtering, nitriding of anatase TiO2 with alkylammonium salts, and treating TiO2 powder in NH3/Ar gas flow at 550 °C [46,47,48]. Similar to S-doping, N-doping also caused a VB upward shift resulting in a narrowed bandgap. To achieve a highly efficient photocatalytic water-splitting reaction, the coupling of different approaches may sometimes be necessary.
2.2. High-Efficient Photocatalytic System for Water Splitting
Even though modified TiO2 has shown improved photocatalytic activity towards water-splitting reaction, its performance is still far below the requirement for commercialization because of the intrinsic limitation of TiO2. As a result, researchers have started to develop other potential photocatalysts to improve the efficiency of water-splitting reaction. Most of the high-efficiency photocatalysts synthesized for H2 production via photocatalytic water splitting are composed of two or more components that are more complicated than TiO2. An example is the NiO-SrTiO3 photocatalyst prepared by Domen and his group [49]. Nickel oxide (NiO) as a co-catalyst was first loaded on the surface of SrTiO3, which then underwent reduction and oxidation by hydrogen and oxygen, respectively, to form a core (Ni)-shell (NiO) structure, as shown in Figure 3. The co-catalyst with a core-shell structure is believed to facilitate the transport of electrons toward the surface of the photocatalyst, hence improving the photoactivity.
Kudo et al. [50] also prepared NiO-Ni loaded Sr2Ta2O7 and Sr2Nb2O7 photocatalysts for water splitting reaction. However, the loading of NiO-Ni only improved the activity of Sr2Nb2O7. A possible reason is that the transfer electrons from the CB of Sr2Ta2O7 to that of NiO, the active site for hydrogen generation, is likely, whereas the transfer of electrons from the CB of Sr2Nb2O7 to that of NiO is difficult because of their similar CB energy levels, as shown in Figure 4. In addition to that, photocatalytic activities of various tantalates for water decomposition were also investigated by Kudo et al. [51]. In the alkali and alkaline earth tantalates, LiTaO3, NaTaO3, KTaO3, MgTa2O6, and BaTa2O6 showed photocatalytic activities for water decomposition without co-catalysts. In the transition metal tantalates, on the other hand, NiTa2O6 produced both H2 and O2 without co-catalysts.
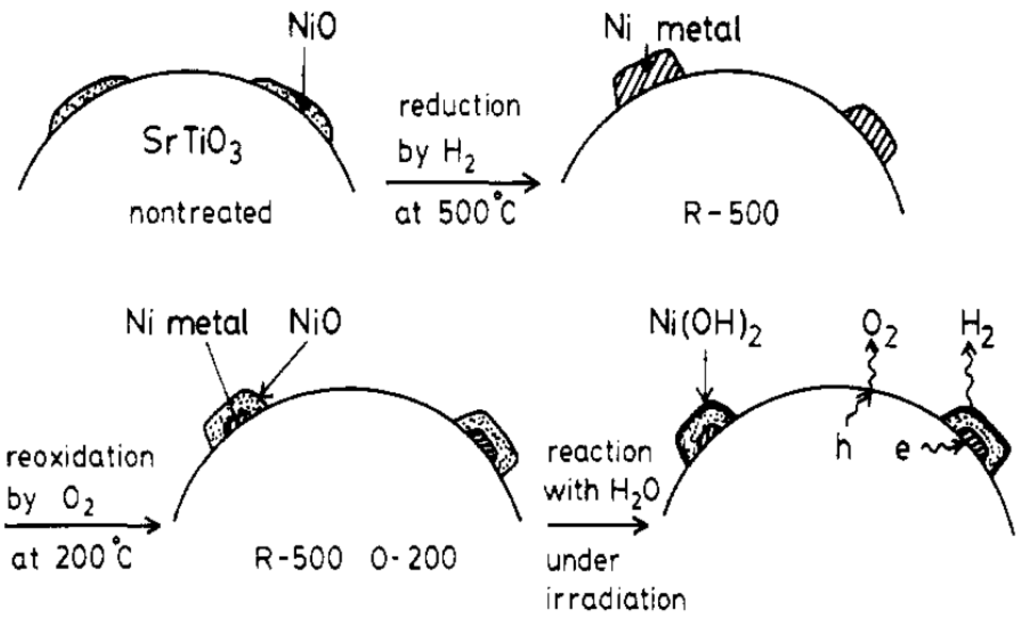
Figure 3.
NiO-SrTiO3 photocatalyst after reduction and oxidation treatments reprinted (adapted) with permission from ref [49]. Copyright (1986) American Chemical Society.
From the above results, it is noteworthy that not only the nature of the photocatalyst is crucial in determining its activity, but the loaded co-catalyst is as well. Sato et al. compared the activity of CaIn2O4 and RuO2-loaded CaIn2O4 and concluded that the performance of CaIn2O4 will be enhanced by loading RuO2. However, excess RuO2 loaded on CaIn2O4 will cause an adverse effect because of the aggregation of RuO2, which lowers the active surface area of the photocatalyst [52].
Sayama et al. [53] reported the preparation of new layered compounds, A4TaxNb6−xO17 (A = K or Rb, x = 2, 3, 4 and 6), which had two different kinds of interlayer spaces. It was found that these compounds with intercalated nickel metal particles showed a remarkable photocatalytic activity for water splitting. In the case of Ni- K4TaxNb6-xO17, the rate of H2 and O2 evolutions decreased with the increase of Ta substitution, even though the UV absorption shifted to a longer wavelength, suggesting that the extended absorption to a longer wavelength did not contribute to the photocatalytic water splitting. It was also found that A4TaxNb6−xO17 itself without any modification could decompose water, which confirmed that a structure consisting of two different kinds of interlayer spaces is essential for water splitting.
Coupling of non-oxide photocatalyst with oxide or other non-oxide photocatalysts (the so-called composite photocatalyst) is another approach to increase the photocatalytic activity by achieving efficient charge separation and by expanding the absorption spectrum of the photocatalyst at the same time. Examples of composite photocatalyst include CdS-TiO2, CdS-ZnO, and CdS-AgI. Taking CdS-TiO2 as an example [54], the electrons generated on the CB of visible-light active CdS can be transferred to the CB of TiO2, while holes remain on the VB of CdS as shown in Figure 5. The difference in the energy level of the two photocatalysts plays an important role in achieving such charge separation. Despite the improved activity of composite photocatalysts, most of the narrowed bandgap non-oxide photocatalysts involved may encounter photocorrosion problems in aqueous solution because of their material nature [55], which greatly confines their application in photocatalytic water splitting.
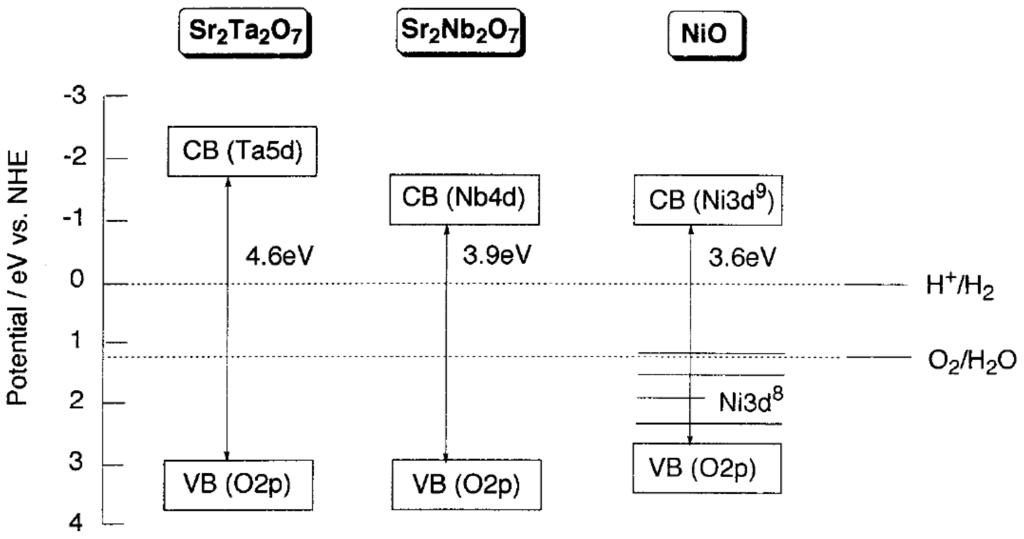
Figure 4.
Band structures of Sr2M2O7 (M = Nb and Ta) photocatalysts and NiO co-catalyst Reprinted (adapted) with permission from ref [50]. Copyright (2000) American Chemical Society.
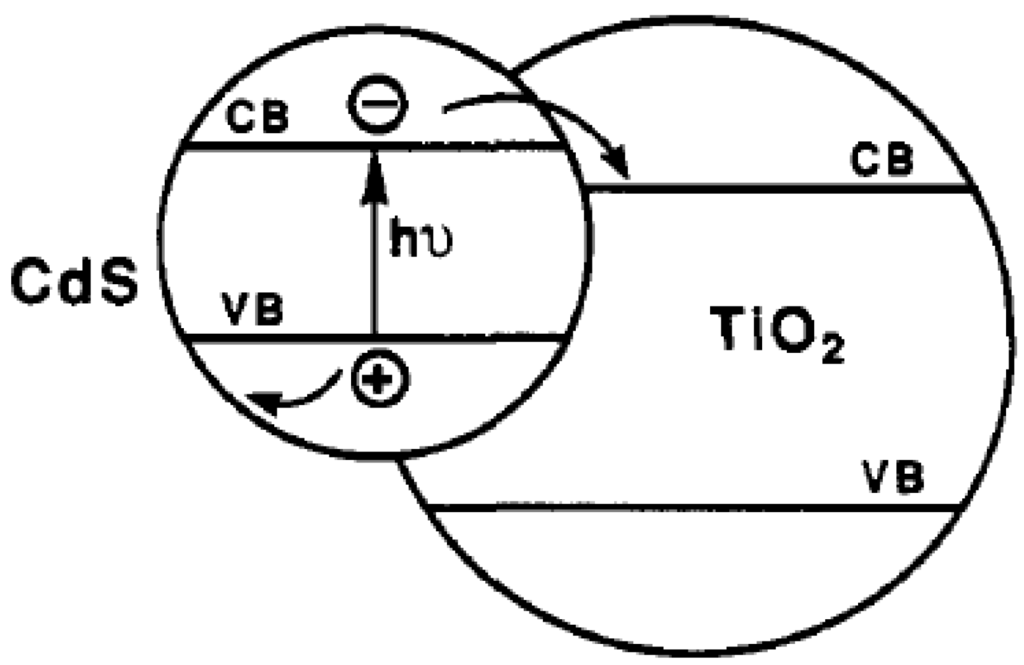
Figure 5.
Schematic diagram of the energy levels of CdS-TiO2 composite photocatalyst Reprinted (adapted) with permission from ref [55]. Copyright (1990) American Chemical Society.
To overcome the photocorrosion problem of composite photocatalyst while maintaining its high efficiency, a photocatalytic system called Z-scheme, which mimics the Z-scheme mechanism in the natural photosynthesis of green plants, has been developed [56] to generate H2 and O2 simultaneously. The Z-scheme is a dual-photocatalyst system that basically consists of a H2-photocatalyst and an O2-photocatalyst to perform water reduction and oxidation, respectively. In addition, a reversible redox mediator, such as Fe2+/Fe3+, is essential to regenerate the photocatalyst so that un-reacted electrons and holes in O2-photocatalyst and H2-photocatalyst, respectively, can be removed to allow sustained photoreaction. The detailed mechanism of a Z-scheme system is shown in Figure 6. There are some limitations to the redox mediators used for Z-scheme process. First, the redox potential of mediator must be appropriate for the photocatalysts selected. For instance, the oxidation potential of the reducing agent (i.e., Fe2+) must be higher than the valence band of H2-photocatalyst, and the reduction potential of oxidizing agent (i.e., Fe3+) must be lower than the conduction band of O2-photocatalyst. Second, some redox mediators are only chemically stable under specific pH condition; for example, Fe3+ will form precipitate under basic conditions. Last but not least, some redox mediators, such as Fe2+/Fe3+ and I-/IO3-, are light absorbers that will compete with photocatalysts in light absorption.
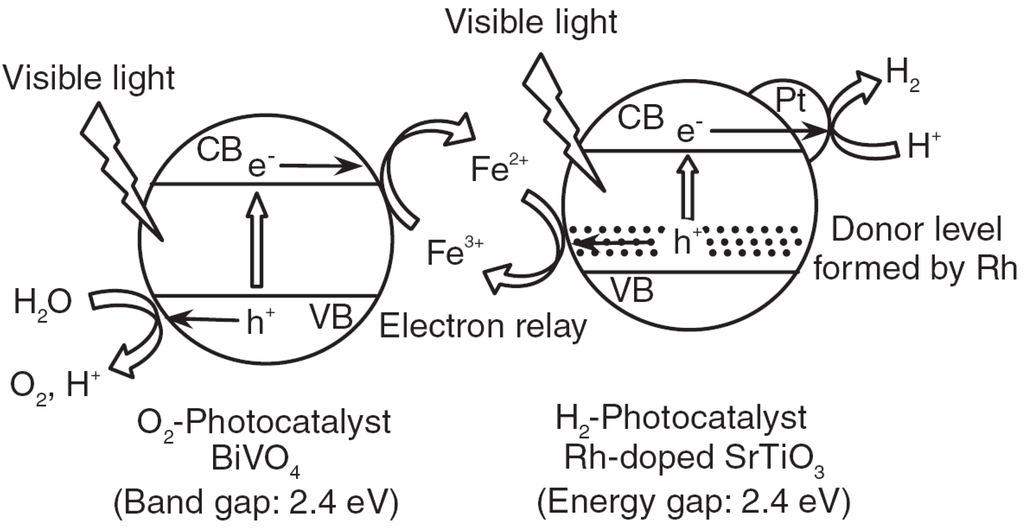
Figure 6.
Mechanism of Z-scheme system for water-splitting reaction reprinted from ref [56], Copyright (2008), with permission from Elsevier.
Fujihara and coworkers [57] reported the photocatalytic water splitting using photocatalyst-coated platinum electrodes and a cation-exchange membrane in a two-compartment reactor, where H2 was evolved on Pt/TiO2 (anatase) photocatalyst suspended in a Br2/Br- redox mediator solution and O2 was evolved on Pt/TiO2 (rutile) photocatalyst in a Fe3+/Fe2+ redox mediator solution. Abe et al. reported the Z-scheme photocatalytic system consisted of Pt/TaON as H2-photocatalyst, Pt/WO3 as O2-photocatalyst, and I−/IO3− as the redox mediator to perform water splitting under visible-light irradiation [45]. Sayama et al. also used I-/IO3- as the redox mediator to carry out water splitting under visible light, while Pt/SrTiO3:Cr/Ta and Pt/WO3 were selected as H2-photocatalyst and O2-photocatalyst, respectively [58]. Higashi et al. reported the Z-scheme system for water-splitting reaction using ATaO2N (A = Ca, Sr, Ba) as H2-photocatalyst and WO3 as O2-photocatalyst in the IO3-/I-solution [59]. ATaO2N was prepared by calcining A2Ta2O7 in NH3 for 20 h. Among these prepared H2-photocatalysts, BaTaO2N showed the largest absorption spectrum, which extended over the wavelength of 600 nm. The system using Pt/BaTaO2N and Pt/WO3 as the H2-photocatalyst and O2-photocatalyst, respectively, in the solution of 5 mM NaI has demonstrated hydrogen yield of 95 μmol in a total reaction time of 50 h. Kato et al. used Pt/SrTiO3:Rh as the H2-photocatalyst and a variety of O2-photocatalysts, such as BiVO4, Bi2MoO6, and WO3 in 2 mM Fe3+ solution to conduct the water-splitting reaction under visible-light irradiation [60]. The total amount of hydrogen produced for BiVO4 was 1800 μmol in 120 h, and that for WO3 was 1240 μmol in 158 h.
2.3. Types of Photocatalytic Water-Splitting Reaction
In general, the literature on hydrogen production via photocatalytic water splitting can be classified into 2 types: (1) photochemical-cell reaction; and (2) photoelectrochemical-cell reaction. In a photochemical cell, powder photocatalyst as suspended particles in solution is used to perform the water-splitting reaction. Most of the photocatalytic water-splitting reactions that we have introduced so far are examples of photochemical-cell reaction. In a photoelectrochemical cell, on the other hand, photocatalyst is deposited as a thin film on a substrate to form a photo-anode (or photoelectrode) for carrying out the water-splitting reaction in solution. An external circuit is required to direct the photo-generated electrons from photo-anode to a cathode where hydrogen is evolved. An example of photocatalytic water splitting performed in a photoelectrochemical cell (PEC) was first demonstrated by Fujishima and Honda in 1972 [61]. Figure 7 is a schematic diagram of the photoelectrochemical cell (PEC) used to carry out the reaction. The mechanism basically involves 4 major steps: (1) generation of electron-hole pairs upon light irradiation on the photo-anode; (2) oxidation of water by photo-generated holes on the photo-anode surface to give O2 and H+; (3) transfer of photo-generated electrons through an external circuit to the cathode; and (4) reduction of H+ by photo-generated electrons on the cathode surface to give H2.
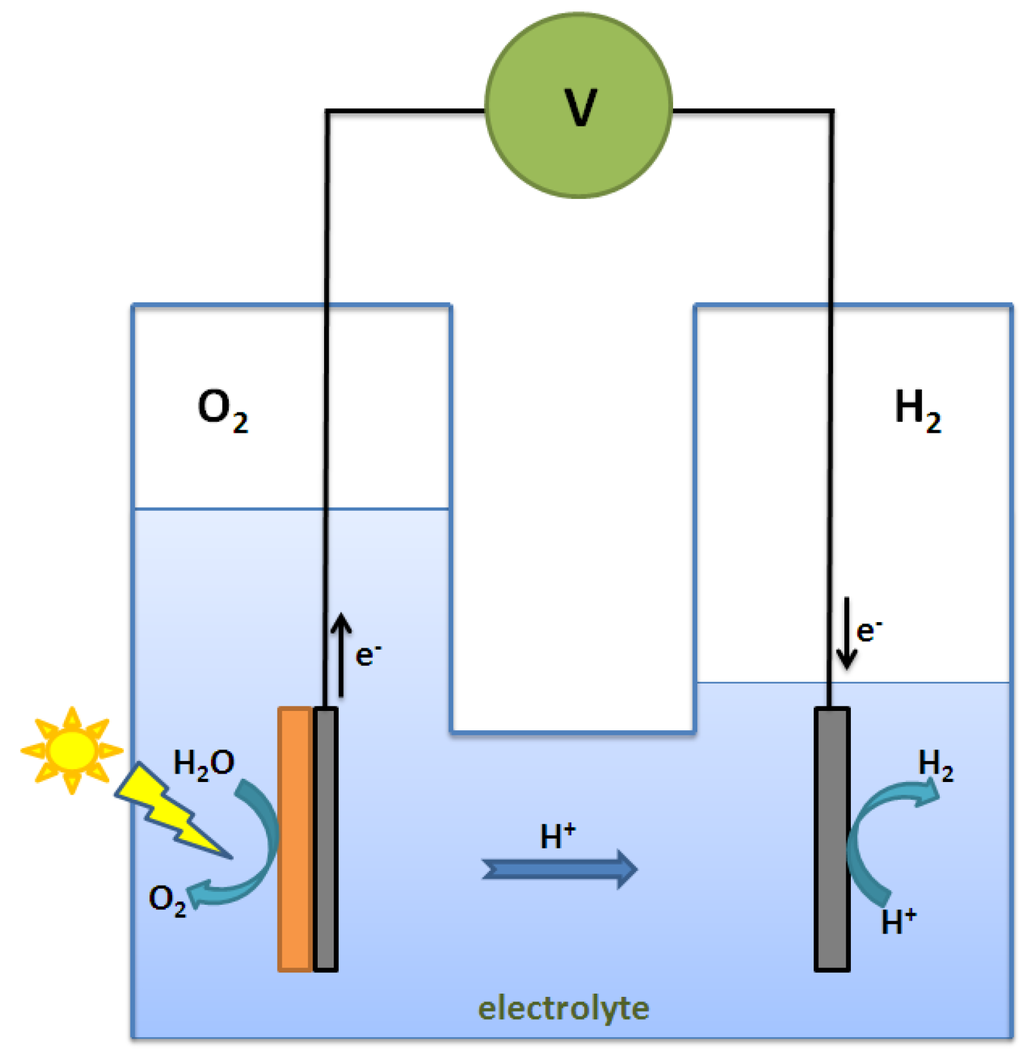
Figure 7.
Schematic diagram of a photoelectrochemical cell.
Usually, photochemical cells have the advantage of a simple process because additional film deposition or coating equipment is not required. Another advantage is that suspended photocatalyst tends to have a larger surface area per unit weight available for photocatalytic reaction, which means more active sites for photocatalytic reaction. The advantage of photoelectrochemical cell is that an internal bias can be easily achieved by the photo-anode with a combination of different materials. The bias formed will facilitate electron-hole separation and result in higher photocatalytic activity. Other than the internal bias, an external bias can also be applied between the electrodes for further enhancement. Under standard conditions, water can be reversibly electrolyzed at a potential of 1.23 V. If, however, the maximum open circuit photopotential of a water-splitting system falls short of 1.23 V, an external bias can be provided to increase the reduction potential energy of the electrodes, making the transfer of electrons energetically feasible. Wrighton et al. [62] reported the use of an n-type semiconductor SrTiO3 electrode in a photoelectrochemical cell to convert H2O to H2 and O2. The results reported herein show for the first time that the decomposition of H2O can be driven photochemically without any external bias. However, when an external potential was applied, photocurrent measured, as well as the hydrogen yield obtained, increased significantly. Photoelectrode stability was confirmed by experiments carried out in oxygen-18 labeled H2O and by the lack of weight loss in the SrTiO3. Ki et al. [63] also investigated the photo-effects of undoped and Nb2O5-, Sb2O3-, and V2O5-doped SrTiO3 electrodes. Photoresponses in undoped SrTiO3 electrodes appeared at a wavelength of about 390 nm, and the quantum efficiency was about 3.5% at a wavelength of 340 nm for the applied voltage of 0.5 V vs. Ag/AgCl. Photocurrents of Nb2O5-, Sb2O3-, and V2O5-doped SrTiO3 electrodes decreased as the amount of dopant increased.
Since water oxidation (O2 evolution) and reduction (H2 evolution) in photoelectrochemical cell occurs at different sites (electrodes), simultaneous separation of evolved O2 and H2 is possible, which is the biggest advantage of photoelectrochemical cells. Instantaneous separation of the produced O2 and H2 not only avoids the backward reaction of water splitting to form water again but also saves on the cost for additional hydrogen separation before usage. Moreover, since the mixture of O2 and H2 is easily combustible, instantaneous separation makes the entire system safe for commercial operation and scale-up. An example that best demonstrates the merits of photoelectrochemical cells is the H-type reactor system proposed by Anpo et al. [64]. The reactor system consisted of an H-type reactor, a photoelectrode, and a Nafion or proton-exchange membrane. Water solution inside the reactor was separated by the photoelectrode and proton-exchange membrane into two compartments as shown in Figure 8. The photoelectrode was made up of a Ti foil substrate sandwiched by a visible light-active TiO2 photocatalyst anode and a Pt cathode, both of which were prepared by sputtering. The metal Ti foil provides the channel for electron transfer so that the external circuit can be eliminated and electrical resistance can be significantly reduced. Upon light irradiation, water oxidation occurred on TiO2 to give oxygen gas and protons, which then were transferred to the Pt side via proton exchange membrane, while reduction of hydrogen ion occurred on Pt to give hydrogen gas. As a result, separate evolution of H2 and O2 can be achieved.
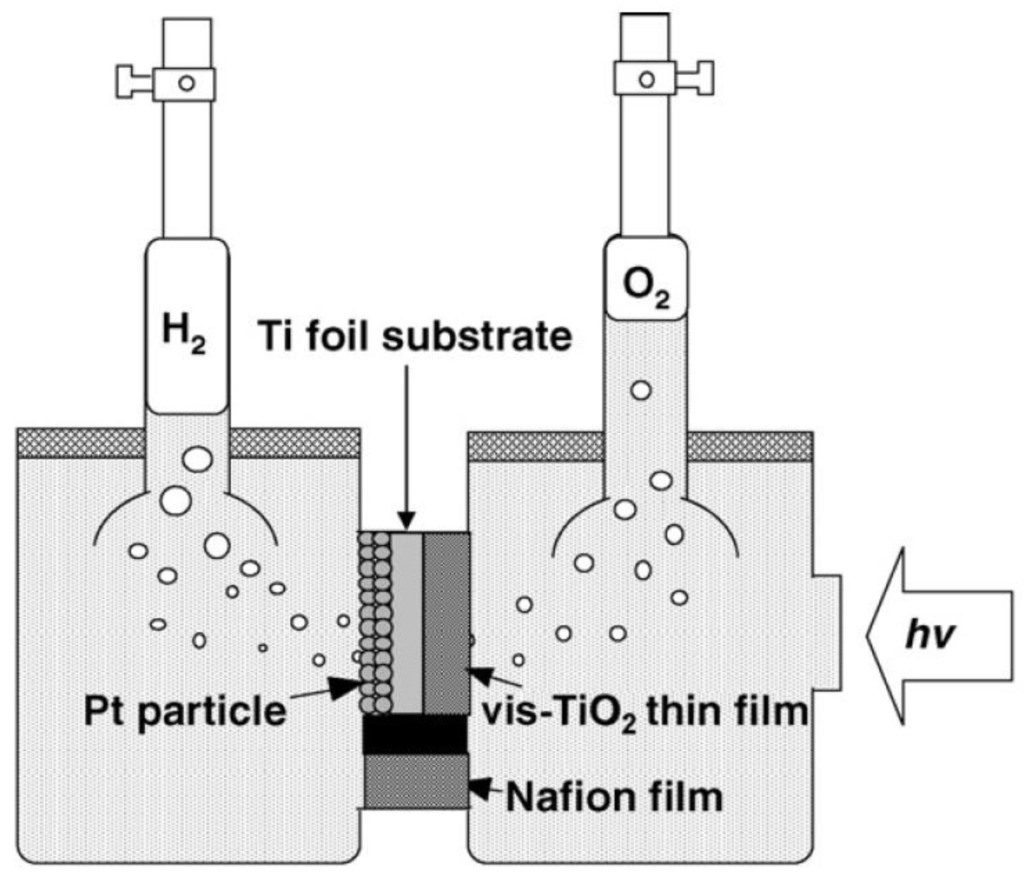
Figure 8.
H-type reaction system for photocatalytic water splitting Reprinted from ref [64], Copyright (2007), with permission from Elsevier.
In collaboration with Anpo’s group, Wu and his group adopted a similar system to carry out water-splitting reaction in which the visible-light TiO2 of the photoelectrode was prepared by a different physical deposition technique called “electron beam-induced deposition” [65]. Later, a novel dual-layer photoelectrode, which consisted of a layer of visible-light WO3, and a layer of visible-light TiO2 deposited on a Pt coated Ti foil was developed by Wu et al. [66]. The dual-layer photoelectrode has the advantages of improved light absorption efficiency in the visible region and better charge separation. The activity of the prepared dual-layer photoelectrode under both UV and visible-light irradiations were evaluated by conducting photovoltammetry and water-splitting reaction in an H-type reactor. The dual-layer photoelectrode showed enhanced photocurrent comparing with TiO2-only photoelectrode, which has been proved to result mainly from the improved charge separation of the dual-layer structure. Moreover, the H2 and O2 yields obtained from the water-splitting reactions were consistent with the photocurrent results, showing dual-layer photoelectrodes with the highest photoactivity. Besides the physical methods such as sputtering and electron beam-induced deposition, a chemical deposition method called “evaporation-induced self-assembling” (EISA) process was also adopted by Wu et al. to prepare mesoporous TiO2 thin films (MTTFs) for fabricating the photoelectrode [67]. In general, the efficiency of photocatalyst strongly depends on its surface area. Therefore, it is important to prepare porous TiO2 thin film with high surface area for carrying out water-splitting reaction. In this study, mesoporous TiO2 thin films with pillar and tube structures were synthesized and characterized (Figure 9). The difference in structure has been revealed to affect not only the translation efficiency of excited electrons, but also influence the interfacial barrier and concentration gradient of the reaction solution. Even though the yield of hydrogen produced in this study was lower than that reported by Anpo et al. [68], it has been demonstrated that mesoporous titania thin films can be successfully prepared by a simple and reliable chemical method instead of an expensive physical method.

Figure 9.
SEM image of (a) pillar-MTTFs, and (b) tube-MTTFs Reprinted from ref [67], Copyright (2012), with permission from Elsevier.
In addition to the H-type reactor system, Wu and his group has recently developed a novel twin-reactor system that combines the advantages of both Z-scheme and H-type reactor systems to carry out water-splitting reaction [69]. In this novel system, Pt/SrTiO3:Rh and WO3, used as H2-photocatalyst and O2-photocatalyst, respectively, were discretely placed into the compartments of a connected twin reactor separated by a modified ion-exchange membrane, as shown in Figure 10. This modified ion-exchange membrane not only allows the transport of protons, but also the exchange of the mediator ions (Fe2+/Fe3+) in solution. The major merit of this novel system is that separate hydrogen and oxygen evolution can be achieved while using only powder photocatalysts to perform the water-splitting reaction. In a subsequent study, Pt/SrTiO3:Rh and BiVO4 were used as the H2-photocatalyst and the O2-photocatalyst, respectively, to run water-splitting reaction in the novel twin-reactor system. The transport phenomenon of iron mediators through modified Nafion membrane was investigated in detail [70]. The apparent diffusivities of the mediator ions, Fe3+ and Fe2+, were derived quantitatively by colorimetric method. By comparing the rate of hydrogen generation with the rate of diffusion for the mediator ions, it was concluded that the resistance of the modified Nafion membrane in the novel twin reactor would not hinder photocatalytic water-splitting reaction. Furthermore, the H2-generating side of the novel twin-reactor system was found to be the rate-limiting step for the water-splitting reaction. It was also concluded in the study that by using the novel twin reactor system, the deactivation of Pt/SrTiO3:Rh often occurring in the conventional Z-scheme system can be successfully minimized by suppressing the formation of Fe(OH)3 on the photocatalyst surface.
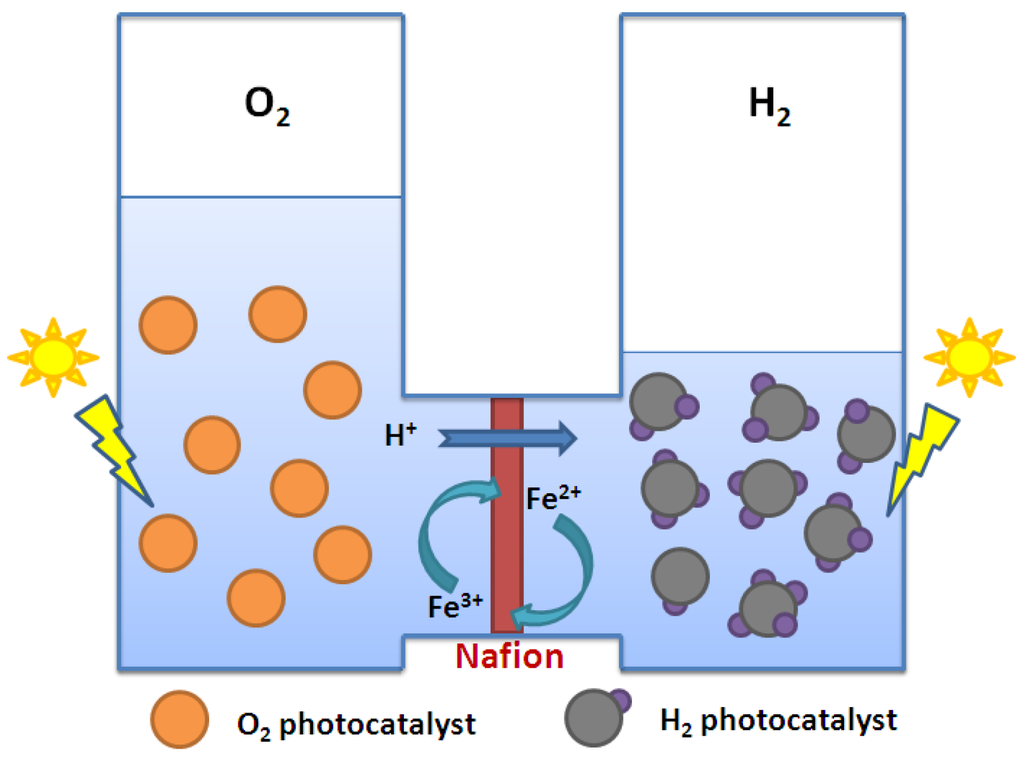
Figure 10.
Concept of a novel twin-reactor system.
Besides the photoelectrodes made from oxide-based material, semiconductor materials, or particularly, III-V semiconductors have also been used to prepare the high-efficient photoelectrode for water-splitting reaction. Prasad et al. [71] reported a photoanode that corresponds to the MX2 type layered material, WSe2. The WSe2 crystals were grown by the chemical vapor deposition technique with SeCI4 as the transporter. The photoelectrochemical efficiency (light-to-electricity conversion efficiency) of the as grown n-WSe2 crystals was approximately 17% after a photo-etching treatment was conducted on the crystal surface of WSe2. Licht et al. prepared the dual-junction semiconductor photoelectrode by a process called MOCVD (metal organic chemical vapor deposition). Such photoelectrode, comprised of AlGaAs and Si, can achieve higher conversion efficiency as compared with other single-junction semiconductor materials [72]. A monolithic photovoltaic-photoelectrochemical device for hydrogen production via water splitting was proposed by Khaselev et al. [73]. This photoelectrochemical cell, which was voltage-biased with an integrated photovoltaic device, could split water directly upon illumination (Figure 11). The hydrogen production efficiency of this system, based on the short-circuit current, was around 12.4%. Peharz et al. combined III-V solar cells and polymer electrolyte in an optical concentrator to achieve a photo-conversion efficiency of 18% [74]. Khaselev et al. prepared GaInP2/GaAs photoelectrode with photo-conversion efficiency of 12.4% under AM1.5 light irradiation [75]. Miller et al. proposed a photoelectrode (Figure 12), which is made up of a multi-junction solar cell and various photocatalysts, such as Fe2O3, WO3, TiO2 [76]. The hybrid planar photoelectrode was designed to absorb most of the sunlight to give better photocatalytic performance. The stability of photoelectrode was also improved by using encapsulant, which is transparent and anticorrosive to the electrolyte solution. A cell containing AlGaAs/Si RuO2/Ptblack was prepared by Licht et al. [77] for water splitting reaction. Under visible-light illumination, the bipolar configured Al0.15Ga0.85As (Eg = 1.6 eV) and Si (Eg = 1.1 eV) semiconductors generate open circuit and maximum power photopotentials of 1.30 and 1.57 V, respectively, well-suited to the water electrolysis thermodynamic potential of 1.23 V. The cell was then combined with an effective water electrolysis catalyst, RuO2, and achieved a photo-conversion efficiency of 18.3%.
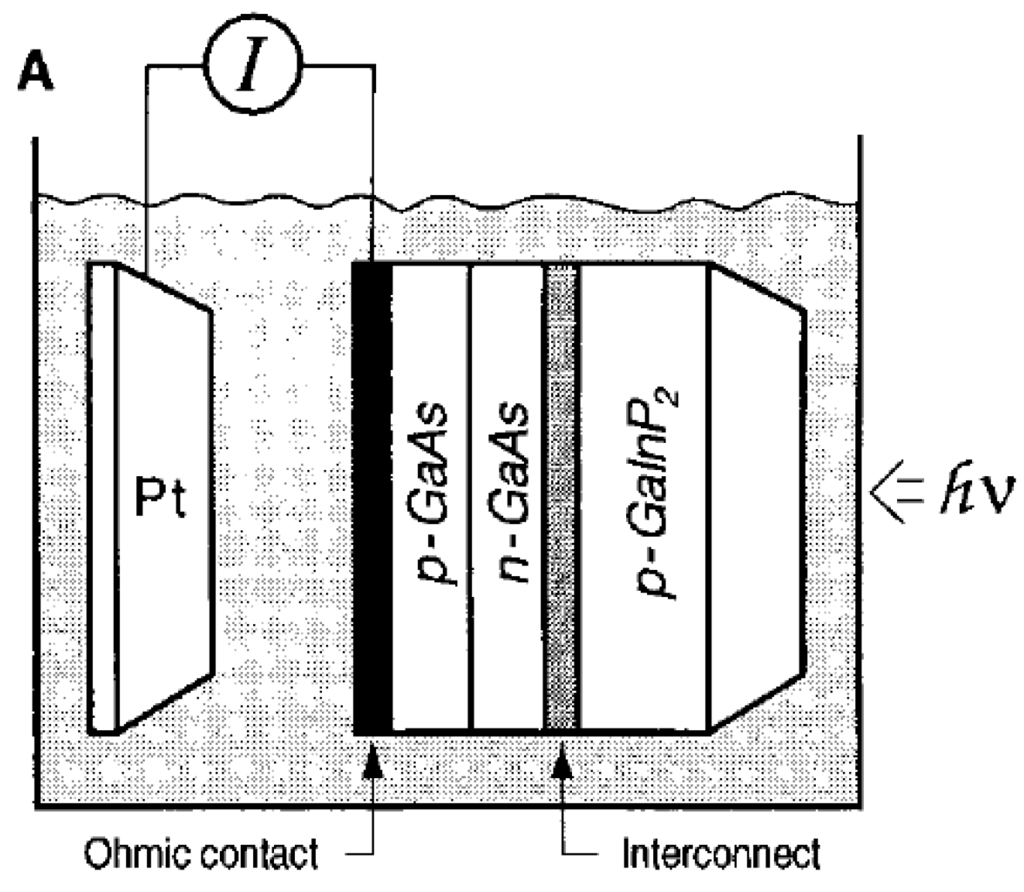
Figure 11.
Schematic of the monolithic PEC/PV device Reproduced with permission from ref [73]; published by (The American Association for the Advancement of Science), (1998).
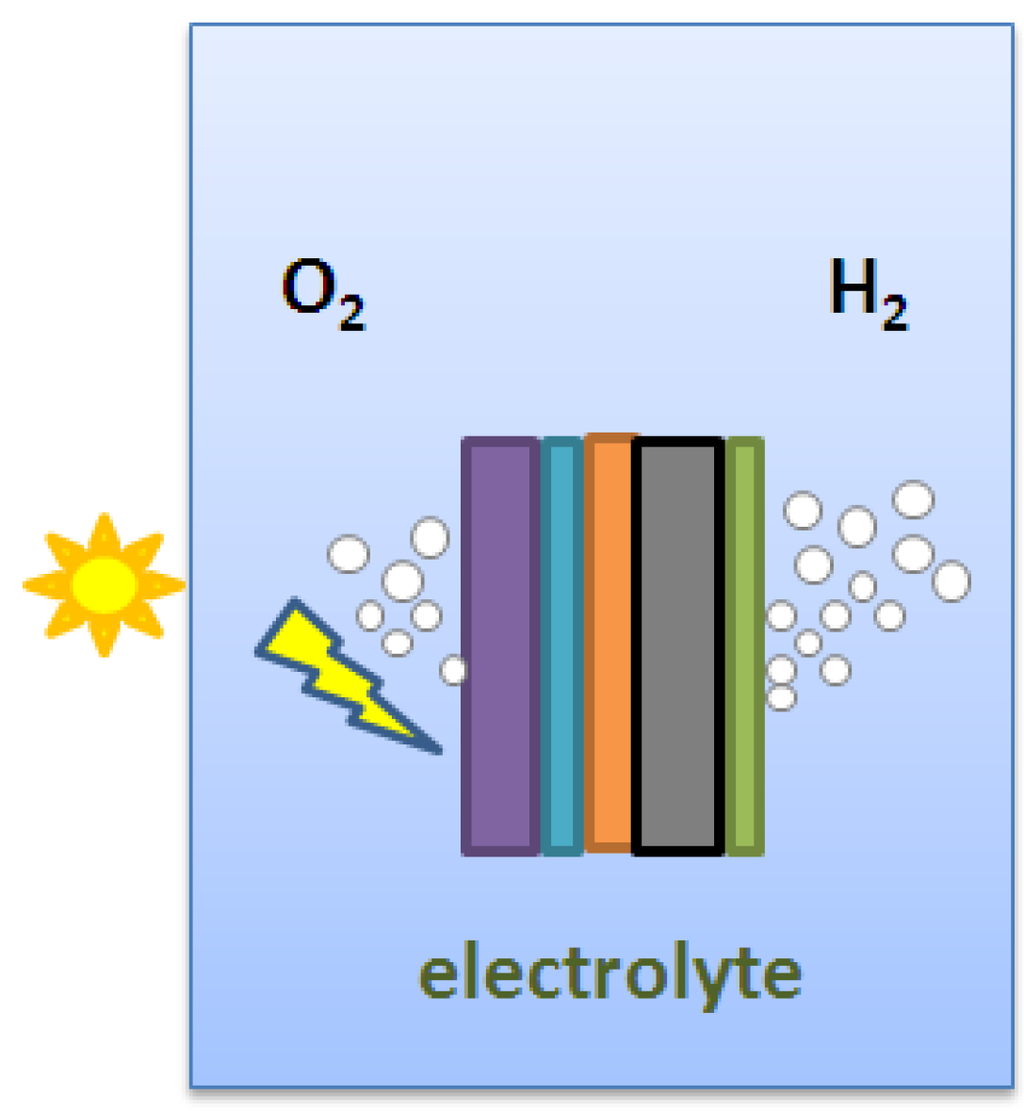
Figure 12.
Structure of the hybrid planar photoelectrode (photoactive semiconductor/ITO/solid-state multi-junction/ stainless steel/HER catalyst) for water splitting.
Although many studies have reported the fabrication of high-efficiency multi-junction solar cells for water-splitting reaction, most of the hydrogen yields obtained for these devices were based on the photocurrent generated under light irradiation. So far, none of them has presented the hydrogen yield obtained from the actual water-splitting reaction. Recently, Wu et al. applied a multi-junction solar cell (MJSC) that contains InGaP, GaAs, and Ge sub-cells to the H-type reactor system for carrying out water-splitting reaction [78]. Despite the observed corrosion phenomenon of the Ge bottom cell after photoreaction, a huge hydrogen yield of 440 μmol was observed under 8 h of visible-light irradiation (AM 1.5). Furthermore, by replacing the electrolyte solution of H2SO4/NaOH with Na2SO4, the corrosion phenomenon of the bottom cell was effectively suppressed.
2.4. Summary of Photocatalytic Water Splitting
In this section, a summary of the results reported from previous literature is presented. Table 1 and Table 2 show the results of several water-splitting reactions carried out in photochemical and photoelectrochemical cell, respectively, over various photocatalytic materials. The conditions at which the reactions were conducted are listed in the Table as well.

Table 1.
Water splitting by photochemical-cell reaction over various photocatalysts.
| Photocatalyst | Weight | Reaction solution | Light source | Rate of evolution (μmol h−1) | Reference | |
|---|---|---|---|---|---|---|
| H2 | O2 | |||||
| Pt/TiO2 | 0.3 g | 2.17M Na2CO3 | 400 W Hg lamp | 568 | 287 | [23] |
| ZrO2 | 1 g | distilled water | 400 W Hg lamp | 72 | 36 | [24] |
| ZrO2 | 1 g | 1.09M Na2CO3 | 400 W Hg lamp | 142 | 75 | [24] |
| Pt/ZrO2 | 1 g | 0.94M NaHCO3 | 400 W Hg lamp | 120 | 61 | [24] |
| Ru2O/ZrO2 | 1 g | distilled water | 400 W Hg lamp | 11 | 5 | [24] |
| Cu/ZrO2 | 1 g | distilled water | 400 W Hg lamp | 14 | 6 | [24] |
| NiO/Sr2Nb2O7 | 1 g | distilled water | 400 W Hg lamp | 110 | 36 | [50] |
| NiO/Sr2Ta2O7 | 1 g | distilled water | 400 W Hg lamp | 1000 | 480 | [50] |
| (Tetra)BaTa2O6 | 1 g | distilled water | 400 W Hg lamp | 21 | 10 | [51] |
| (Ortho)BaTa2O6 | 1 g | distilled water | 400 W Hg lamp | 33 | 15 | [51] |
| (Ortho)BaTa2O6 | 1 g | 0.0005 M Ba(OH)2 | 400 W Hg lamp | 126 | 59 | [51] |
| (Ortho)BaTa2O6 | 1 g | 0.001M KOH | 400 W Hg lamp | 24 | 11 | [51] |
| (Ortho)BaTa2O6 | 1 g | 0.0005 M BaCl2 | 400 W Hg lamp | 15 | 6 | [51] |
| NiO/BaTa2O6 | 1g | distilled water | 400 W Hg lamp | 629 | 303 | [51] |
| Ni/Rb4Nb6O17 | 1 g | distilled water | 400 W Hg lamp | 936 | 451 | [53] |
| Ni/K4Nb6O17 | 1 g | distilled water | 400 W Hg lamp | 403 | 197 | [53] |
| Pt/TiO2TiO2 | 12 mg | 2 M KBr6.5 mM FeCl2 | 500 W Hg | 2.8 | 1.3 | [57] |
| Pt-TaONPt-WO3 | 0.2 g | 5 mM NaI | 300 W Xe lamp with filters: λ > 420 nm | 24 | 12 | [45] |
| Pt/BaTaO2NPt/WO3 | 0.1 g | 5 mM NaI | 300 W Xe lamp with filters: λ > 420 nm | 6.6 | 3.1 | [59] |
| Pt/SrTiO3:Rh, BiVO4 | 0.1 g | 2 mM FeCl3 | 300W Xe with filter: λ > 420 nm | 15 | 7.2 | [60] |
| Pt/SrTiO3:Rh, Bi2MoO6 | 0.1 g | 2 mM FeCl3 | 300 W Xe with filter: λ > 420 nm | 19 | 8.9 | [60] |
| Pt/SrTiO3:Rh, WO3 | 0.1 g | 2 mM FeCl3 | 300 W Xe with filter: λ > 420 nm | 7.8 | 4.0 | [60] |
| K4Nb6O17 | 1 g | H2O | 450 W Hg lamp | 8 | 1 | [64] |
| NiO/ K4Nb6O17 | 1 g | H2O | 450 W Hg lamp | 77 | 37 | [64] |
| Pt/SrTiO3:RhWO3 | 0.3 | 2 mM FeCl2/FeCl3 | 500 W halogen lamp | 1.6 | 0.8 | [69] |
| Pt/SrTiO3:RhBiVO4 | 0.4 | 5mM FeCl2/FeCl3 | 300 W Xe lamp | 0.8 | 0.4 | [70] |
Table 2.
Water splitting by photoelectrochemical-cell reaction over various photoelectrodes.
| Photoelectrode | Surface area (cm2) | Electrolyte | Light source | Efficiency/H2 yield | Applied bias (V) | Reference |
|---|---|---|---|---|---|---|
| TiO2 | 1 | Fe3+ solution | 500 W Xenon lamp | QE = 10% | N/A | [61] |
| SrTiO3 | 0.25 | 9.5 M NaOH | Argon ion laser (351 nm) | QE = 11% | N/A | [62] |
| SrTiO3 | 1.539 | 1 M NaOH | 150 W halogen lamp (340 nm) | QE = 3.5% | 0.5 | [63] |
| TiO2 | 2 | 0.5 M H2SO4/1 M NaOH | UV light with intensity of 25 mW/cm2 | 60 μmol in 8 h | N/A | [65] |
| Vis-WO3/vis-TiO2 | 2 | 0.025 M H2SO4/0.05 M NaOH | UV light with intensity of 2.5 mW/cm2 | 39 μmol in 8 h | N/A | [66] |
| Vis-WO3/vis-TiO2 | 2 | 0.025 M H2SO4/0.05 M NaOH | AM 1. 5 | 6 μmol in 8 h | N/A | [66] |
| Pillar TiO2 | 2 | 0.5 M H2SO4/1M NaOH | UV light with intensity of 25 mW/cm2 | 37 μmol in 8 h | N/A | [67] |
| WSe2 | 0.0125 | 1 M KI+ 0.05 M I2 | 60 mW/cm2 tungsten lamp | ABPE = 17.1% | N/A | [71] |
| p-GaAs/ n-GaAs/p-GaInP2 | 0.2 | 3 M H2SO4 | 150 W tungsten-halogen lamp | ABPE = 12.4% | 0.3 | [73] |
| GaInP2/GaAs | 0.5 | 2 M KOH | 75 W Xe lamp | ABPE = 16.5% | N/A | [75] |
| Triple a-Si | 0.3 | 2 M KOH | 75 W Xe lamp | ABPE = 7.8% | N/A | [75] |
| AlGaAs/Si | 0.22 | 1 M HClO4 | 50 W tungsten-halogen lamp | ABPE = 18.3% | N/A | [77] |
| InGaP/GaAs/Ge | 2 | 0.5 M H2SO4/1 M NaOH | AM 1.5 | 440 μmol in 8 h | N/A | [78] |
| CM n-TiO2 | 0.2 | 5 M KOH | 150 W Xe lamp | ABPE = 8.35% | 0.3 | [18] |
| n-TiO2 | 0.2 | 5 M KOH | 150 W Xe lamp | ABPE = 1.08% | 0.6 | [18] |
3. Current Challenges and Future Prospects of Photocatalytic Water Splitting
Over the past few decades, several semiconductor materials and photocatalytic systems have been developed for the water-splitting reaction under UV and visible-light irradiation. It has been observed that photo-generated charge separation, prevention of water-splitting backward reaction, and utilization of a large fraction of the incident energy are the essential requirements for achieving high photo-conversion efficiency. Enhanced hydrogen production has been shown by the addition of hole scavengers or sacrificial agents that irreversibly react with the VB holes to inhibit charge recombination. However, to attain sustainable hydrogen production, sacrificial agents must be continuously added. Moreover, the design of novel photocatalytic reactor systems to achieve separate H2 and O2 evolution, such as the H-type reactor and the novel Z-scheme, have also shown enhanced hydrogen production by preventing the backward reaction of water splitting. Various syntheses procedures, such as loading and/or doping of metal or metal oxide particles on the photocatalyst, and the preparation of dye-sensitized or composite photocatalysts have been successfully employed to improve the performance of photocatalytic water splitting. These methods are effective in terms of tuning the bandgap of material for harnessing a greater portion of visible light, as well as preventing charge recombination. In addition, numerous non-oxide semiconductor materials have shown improved performances for photocatalytic water splitting. However, the stability of the materials remains a major challenge for their application.
In summary, photocatalytic water splitting is a cross-discipline technology that requires the involvement of experts from different fields (i.e., chemists, electrical engineers, material scientists, and physicists). A joint effort is needed to explore potential semiconductor materials and reactor systems that will generate the highest solar-to-hydrogen efficiency. The development of new technologies requires collaboration with a strong theoretical background for a better understanding of the hydrogen production mechanism in order to come up with a low-cost and environmentally friendly water-splitting process for hydrogen production.
References
- Solomon, S.; Plattner, G.K.; Knutti, R.; Friedlingstein, P. Irreversible climate change due to carbon dioxide emissions. Proc. Natl. Acad. Sc. USA 2009, 106, 1704–1709. [Google Scholar]
- Primio, R.D.; Horsfield, B.; Guzman-Vega, M.A. Determining the temperature of petroleum formation from the kinetic properties of petroleum asphaltenes. Nature 2000, 406, 173–176. [Google Scholar] [CrossRef]
- Chiari, L.; Zecca, A. Constraints of fossil fuels depletion on global warming projections. Energy Policy 2011, 39, 5026–5034. [Google Scholar] [CrossRef]
- Dincer, F. The analysis on wind energy electricity generation status, potential and policies in the world. Renew. Sustain. Energy Rev. 2011, 15, 5135–5142. [Google Scholar] [CrossRef]
- Yuksel, I. Hydropower for sustainable water and energy development. Renew. Sustain. Energy Rev. 2010, 14, 462–469. [Google Scholar] [CrossRef]
- Parida, B.; Iniyan, S.; Goic, R. A review of solar photovoltaic technologies. Renew. Sustain. Energy Rev. 2011, 15, 1625–1636. [Google Scholar] [CrossRef]
- Xie, W.T.; Dai, Y.J.; Wang, R.Z.; Sumathy, K. Concentrated solar energy applications using Fresnel lenses: A review. Renew. Sustain. Energy Rev. 2011, 15, 2588–2606. [Google Scholar] [CrossRef]
- Barbier, E. Geothermal energy technology and current status: an overview. Renew. Sustain. Energy Rev. 2002, 6, 3–65. [Google Scholar] [CrossRef]
- Midilli, A.; Ay, M.; Dincer, I.; Rosen, M.A. On hydrogen and hydrogen energy strategies I: Current status and needs. Renew. Sustain. Energy Rev. 2005, 9, 255–271. [Google Scholar] [CrossRef]
- Hou, K.H.; Hughes, R. The kinetics of methane steam reforming over a Ni/alpha-Al2O catalyst. Chem. Eng. J. 2001, 82, 311–328. [Google Scholar] [CrossRef]
- Nowotny, J.; Sorrell, C.C.; Sheppard, L.R.; Bak, T. Solar-hydrogen: Environmentally safe fuel for the future. Int. J. Hydrog. Energy 2005, 30, 521–544. [Google Scholar] [CrossRef]
- Czernik, S.; Evans, R.; French, R. Hydrogen from biomass-production by steam reforming of biomass pyrolysis oil. Catal. Today 2007 129, 265–268.
- Ni, M.; Leung, D.Y.C.; Leung, M.K.H.; Sumathy, K. An overview of hydrogen production from biomass. Fuel Process. Tech. 2006, 87, 461–472. [Google Scholar] [CrossRef]
- Steinfeld, A. Solar hydrogen production via a two-step water-splitting thermochemical cycle based on Zn/ZnO redox reactions. Int. J. Hydrog. Energy 2002, 27, 611–619. [Google Scholar] [CrossRef]
- Akkerman, I.; Janssen, M.; Rocha, J.; Wijffels, R.H. Photobiological hydrogen production: photochemical efficiency and bioreactor design. Int. J. Hydrog. Energy 2002, 27, 1195–1208. [Google Scholar] [CrossRef]
- Das, D.; Veziroglu, T.N. Advances in biological hydrogen production processes. Int. J. Hydrog. Energy 2008, 33, 6046–6057. [Google Scholar] [CrossRef]
- Guan, Y.F.; Deng, M.C.; Yu, X.J.; Zhang, W. Two-stage photo-biological production of hydrogen by marine green alga Platymonas subcordiformis. Biochem. Eng. J. 2004, 19, 69–73. [Google Scholar] [CrossRef]
- Khan, S.U.M.; Al-Shahry, M.; Ingler, W.B. Efficient photochemical water splitting by a chemically modified n-TiO2. Science 2002, 297, 2243–2245. [Google Scholar] [CrossRef]
- Bak, T.; Nowotny, J.; Rekas, M.; Sorrell, C.C. Photo-electrochemical hydrogen generation from water using solar energy. Materials-related aspects. Int. J. Hydrog. Energy 2002, 27, 991–1022. [Google Scholar] [CrossRef]
- Chen, Z.B.; Jaramillo, T.F.; Deutsch, T.G.; Kleiman-Shwarsctein, A.; Forman, A.J.; Gaillard, N.; Garland, R.; Takanabe, K.; Heske, C.; Sunkara, M.; et al. Accelerating materials development for photoelectrochemical hydrogen production: Standards for methods, definitions, and reporting protocols. J. Mater. Res. 2010, 25, 3–16. [Google Scholar] [CrossRef]
- Kudo, A.; Miseki, Y. Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 2009, 38, 253–278. [Google Scholar] [CrossRef]
- Li, Y.X.; Lu, G.X.; Li, S.B. Photocatalytic production of hydrogen in single component and mixture systems of electron donors and monitoring adsorption of donors by in situ infrared spectroscopy. Chemosphere 2003, 52, 843–850. [Google Scholar] [CrossRef]
- Sayama, K.; Arakawa, H. Effect of carbonate salt addition on the photocatalytic decomposition of liquid water over Pt-TiO2 catalyst. J. Chem. Soc.-Faraday Trans. 1997, 93, 1647–1654. [Google Scholar] [CrossRef]
- Sayama, K.; Arakawa, H. Photocatalytic decomposition of water and photocatalytic reduction of carbon-dioxide over ZrO2 catalyst. J.Phys. Chem. 1993, 97, 531–533. [Google Scholar] [CrossRef]
- Subramanian, V.; Wolf, E.E.; Kamat, P.V. Catalysis with TiO2/gold nanocomposites. Effect of metal particle size on the Fermi level equilibration. J. Am. Chem. Soc. 2004, 126, 4943–4950. [Google Scholar]
- Bamwenda, G.R.; Tsubota, S.; Nakamura, T.; Haruta, M. Photoassisted hydrogen production from a water-ethanol solution: a comparison of activities of Au-TiO2 and Pt-TiO2. J. Photochem. Photobiol. A 1995, 89, 177–189. [Google Scholar] [CrossRef]
- Murdoch, M.; Waterhouse, G.I.N.; Nadeem, M.A.; Metson, J.B.; Keane, M.A.; Howe, R.F.; Llorca, J.; Idriss, H. The effect of gold loading and particle size on photocatalytic hydrogen production from ethanol over Au/TiO2 nanoparticles. Nature Chem. 2011, 3, 489–492. [Google Scholar]
- Anpo, M.; Takeuchi, M. The design and development of highly reactive titanium oxide photocatalysts operating under visible light irradiation. J. Catal. 2003, 216, 505–516. [Google Scholar] [CrossRef]
- Merlen, A.; Gadenne, V.; Romann, J.; Chevallier, V.; Patrone, L.; Valmalette, J.C. Surface enhanced Raman spectroscopy of organic molecules deposited on gold sputtered substrates. Nanotechnology 2009, 20. [Google Scholar] [CrossRef]
- Primo, A.; Corma, A.; Garcia, H. Titania supported gold nanoparticles as photocatalyst. Phys. Chem. Chem. Phys. 2011, 13, 886–910. [Google Scholar]
- Primo, A.; Marino, T.; Corma, A.; Molinari, R.; Garcia, H. Efficient Visible-Light Photocatalytic Water Splitting by Minute Amounts of Gold Supported on Nanoparticulate CeO2 Obtained by a Biopolymer Templating Method. J. Am. Chem. Soc. 2012, 133, 6930–6933. [Google Scholar]
- Awazu, K.; Fujimaki, M.; Rockstuhl, C.; Tominaga, J.; Murakami, H.; Ohki, Y.; Yoshida, N.; Watanabe, T. A plasmonic photocatalyst consisting of sliver nanoparticles embedded in titanium dioxide. J. Am. Chem. Soc. 2008, 130, 1676–1680. [Google Scholar]
- Kowalska, E.; Abe, R.; Ohtani, B. Visible light-induced photocatalytic reaction of gold-modified titanium(IV) oxide particles: Action spectrum analysis. Chem. Commun. 2009. [Google Scholar] [CrossRef]
- Silva, C.G.; Juarez, R.; Marino, T.; Molinari, R.; Garcia, H. Influence of Excitation Wavelength (UV or Visible Light) on the Photocatalytic Activity of Titania Containing Gold Nanoparticles for the Generation of Hydrogen or Oxygen from Water. J. Am. Chem. Soc. 2011, 133, 595–602. [Google Scholar] [CrossRef]
- Gurunathan, K.; Maruthamuthu, P.; Sastri, M.V.C. Photocatalytic hydrogen production by dye-sensitized Pt/SnO2 AND Pt/SnO2/RuO2 in aqueous methyl viologen solution. Int. J. Hyd. Energy 1997, 22, 57–62. [Google Scholar] [CrossRef]
- Ni, M.; Leung, M.K.H.; Leung, D.Y.C.; Sumathy, K. A review and recent developments in photocatalytic water-splitting using TiO2 for hydrogen production. Renew. Sustain. Energy Rev. 2007, 11, 401–425. [Google Scholar] [CrossRef]
- Jing, D.; Guo, L. WS2 sensitized mesoporous TiO2 for efficient photocatalytic hydrogen production from water under visible light irradiation. Catal. Commun. 2007, 8, 795–799. [Google Scholar] [CrossRef]
- Sauve, G.; Cass, M.E.; Coia, G.; Doig, S.J.; Lauermann, I.; Pomykal, K.E.; Lewis, N.S. Dye sensitization of nanocrystalline titanium dioxide with osmium and ruthenium polypyridyl complexes. J. Phys. Chem. B 2000, 104, 6821–6836. [Google Scholar] [CrossRef]
- Chen, Y.S.; Li, C.; Zeng, Z.H.; Wang, W.B.; Wang, X.S.; Zhang, B.W. Efficient electron injection due to a special adsorbing group’s combination of carboxyl and hydroxyl: dye-sensitized solar cells based on new hemicyanine dyes. J. Mater. Chem. 2005, 15, 1654–1661. [Google Scholar] [CrossRef]
- Chen, C.P.; Qi, X.Y.; Zhou, B.M. Photosensitization of colloidal TiO2 with a cyanine dye. J. Photochem. Photobiol. 1997, 109, 155–158. [Google Scholar] [CrossRef]
- Chu, W.; Chan, K.H.; Jafvert, C.T.; Chan, Y.S. Removal of phenylurea herbicide monuron via riboflavin-mediated photo sensitization. Chemosphere 2007, 69, 177–183. [Google Scholar] [CrossRef]
- Choi, W.; Termin, A.; Hoffmann, M.R. The Role of Metal Ion Dopants in Quantum-Sized TiO2: Correlation between Photoreactivity and Charge Carrier Recombination Dynamics. J. Phys. Chem. 1994, 98, 13669–13679. [Google Scholar] [CrossRef]
- Litter, M.I. Heterogeneous photocatalysis: Transition metal ions in photocatalytic systems. Appl. Cataly. B 1999, 23, 89–114. [Google Scholar] [CrossRef]
- Asahi, R.; Morikawa, T.; Ohwaki, T.; Aoki, K.; Taga, Y. Visible-light photocatalysis in nitrogen-doped titanium oxides. Science 2001, 293, 269–271. [Google Scholar] [CrossRef]
- Abe, R.; Takata, T.; Sugihara, H.; Domen, K. Photocatalytic overall water splitting under visible light by TaON and WO3 with an IO3−/I− shuttle redox mediator. Chem. Commun. 2005, 38, 29–3831. [Google Scholar]
- Kobayakawa, K.; Murakami, Y.; Sato, Y. Visible-light active N-doped TiO2 prepared by heating of titanium hydroxide and urea. J. Photochem. Photobiol. A 2005, 170, 177–179. [Google Scholar] [CrossRef]
- Mrowetz, M.; Balcerski, W.; Colussi, A.J.; Hoffmann, M.R. Oxidative power of nitrogen-doped TiO2 photocatalysts under visible illumination. J. Phys. Chem. B 2004, 108, 17269–17273. [Google Scholar] [CrossRef]
- Torres, G.R.; Lindgren, T.; Lu, J.; Granqvist, C.-G.; Lindquist, S.-E. Photoelectrochemical Study of Nitrogen-Doped Titanium Dioxide for Water Oxidation. J. Phys. Chem. B 2004, 108, 5995–6003. [Google Scholar]
- Domen, K.; Kudo, A.; Onishi, T.; Kosugi, N.; Kuroda, H. Photocatalytic decomposition of water into hydrogen and oxygen over nickel(II) oxide-strontium titanate (SrTiO3) powder. 1. Structure of the catalysts. J. Phys. Chem. 1986, 90, 292–295. [Google Scholar]
- Kudo, A.; Kato, H.; Nakagawa, S. Water Splitting into H2 and O2 on New Sr2M2O7 (M = Nb and Ta) Photocatalysts with Layered Perovskite Structures: Factors Affecting the Photocatalytic Activity. J. Phys. Chem. B 1999, 104, 571–575. [Google Scholar]
- Kato, H.; Kudo, A. New tantalate photocatalysts for water decomposition into H2 and O2. Chem. Phys. Lett. 1998, 295, 487–492. [Google Scholar] [CrossRef]
- Sato, J.; Saito, N.; Nishiyama, H.; Inoue, Y. Photocatalytic Activity for Water Decomposition of Indates with Octahedrally Coordinated d10 Configuration. I. Influences of Preparation Conditions on Activity. J. Phys. Chem. B 2003, 107, 7965–7969. [Google Scholar] [CrossRef]
- Sayama, K.; Arakawa, H.; Domen, K. Photocatalytic water splitting on nickel intercalated A(4)Ta(x)Nb(6−x)O(17) (A = K, Rb). Cataly. Today 1996, 28, 175–182. [Google Scholar]
- Yamada, S.; Nosaka, A.Y.; Nosaka, Y. Fabrication of US photoelectrodes coated with titania nanosheets for water splitting with visible light. J. Electroanal. Chem. 2005, 585, 105–112. [Google Scholar] [CrossRef]
- Gopidas, K.R.; Bohorquez, M.; Kamat, P.V. Photophysical and photochemical aspects of coupled semiconductors: Charge-transfer processes in colloidal cadmium sulfide-titania and cadmium sulfide-silver(I) iodide systems. J. Phy. Chem. 1990, 94, 6435–6440. [Google Scholar] [CrossRef]
- Sasaki, Y.; Iwase, A.; Kato, H.; Kudo, A. The effect of co-catalyst for Z-scheme photocatalysis systems with an Fe3+/Fe2+ electron mediator on overall water splitting under visible light irradiation. J. Catal. 2008, 259, 133–137. [Google Scholar]
- Fujihara, K.; Ohno, T.; Matsumura, M. Splitting of water by electrochemical combination of two photocatalytic reactions on TiO2 particles. J. Chem. Soci. Faraday Trans. 1998, 94, 3705–3709. [Google Scholar] [CrossRef]
- Sayama, K.; Mukasa, K.; Abe, R.; Abe, Y.; Arakawa, H. A new photocatalytic water splitting system under visible light irradiation mimicking a Z-scheme mechanism in photosynthesis. J. Photochem. Photobiol. 2002, 148, 71–77. [Google Scholar] [CrossRef]
- Higashi, M.; Abe, R.; Takata, T.; Domen, K. Photocatalytic Overall Water Splitting under Visible Light Using ATaO2N (A = Ca, Sr, Ba) and WO3 in a IO3−/I− Shuttle Redox Mediated System. Chem.Mater. 2009, 21, 1543–1549. [Google Scholar] [CrossRef]
- Kato, H.; Hori, M.; Konta, R.; Shimodaira, Y.; Kudo, A. Construction of Z-scheme Type Heterogeneous Photocatalysis Systems for Water Splitting into H2 and O2 under Visible Light Irradiation. Chem. Lett. 2004, 33, 1348–1349. [Google Scholar] [CrossRef]
- Fujishima, A.; Honda, K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef]
- Wrighton, M.S.; Ellis, A.B.; Wolczanski, P.T.; Morse, D.L.; Abrahamson, H.B.; Ginley, D.S. Strontium-titanate photoelectrodes-efficient photoassisted electrolysis of water at zero applied potential. J. Am. Chem. Soc. 1976, 98, 2774–2779. [Google Scholar] [CrossRef]
- Ki, H.Y.; Tae, H.K. Photoeffects in undoped and doped SrTiO3 ceramic electrodes. J. Solid State Chem. 1987, 67, 359–363. [Google Scholar] [CrossRef]
- Matsuoka, M.; Kitano, M.; Takeuchi, M.; Tsujimaru, K.; Anpo, M.; Thomas, J.M. Photocatalysis for new energy production: Recent advances in photocatalytic water splitting reactions for hydrogen production. Catal. Today 2007, 122, 51–61. [Google Scholar]
- Huang, C.W.; Liao, C.H.; Wu, J.C.S.; Liu, Y.C.; Chang, C.L.; Wu, C.H.; Anpo, M.; Matsuoka, M.; Takeuchi, M. Hydrogen generation from photocatalytic water splitting over TiO2 thin film prepared by electron beam-induced deposition. Int. J. Hydrog. Energy 2010, 35, 12005–12010. [Google Scholar]
- Liao, C.-H.; Huang, C.-W.; Wu, J.C.S. Novel dual-layer photoelectrode prepared by RF magnetron sputtering for photocatalytic water splitting. Int. J. Hydrog. Energy 2012, 37, 11632–11639. [Google Scholar] [CrossRef]
- Liao, Y.-T.; Huang, C.-W.; Liao, C.-H.; Wu, J.C.S.; Wu, K.C.W. Synthesis of mesoporous titania thin films (MTTFs) with two different structures as photocatalysts for generating hydrogen from water splitting. Appl. Energy 2012. [Google Scholar] [CrossRef]
- Matsuoka, M.; Kitano, M.; Fukumoto, S.; Iyatani, K.; Takeuchi, M.; Anpo, M. The effect of the hydrothermal treatment with aqueous NaOH solution on the photocatalytic and photoelectrochemical properties of visible light-responsive TiO2 thin films. Catal. Today 2008, 132, 159–164. [Google Scholar]
- Lo, C.-C.; Huang, C.-W.; Liao, C.-H.; Wu, J.C.S. Novel twin reactor for separate evolution of hydrogen and oxygen in photocatalytic water splitting. Int. J. Hydrog. Energy 2010, 35, 1523–1529. [Google Scholar]
- Yu, S.C.; Huang, C.W.; Liao, C.H.; Wu, J.C.S.; Chang, S.T.; Chen, K.H. A novel membrane reactor for separating hydrogen and oxygen in photocatalytic water splitting. J. Membr. Sci. 2011, 382, 291–299. [Google Scholar] [CrossRef]
- Prasad, G.; Chandra Babu, K.S.; Srivastava, O.N. Structural and photoelectrochemical studies of In2O3-TiO2 and WSe2 photoelectrodes for photoelectrochemical production of hydrogen. Int. J. Hydrog. Energy 1989, 14, 537–544. [Google Scholar] [CrossRef]
- Licht, S.; Wang, B.; Mukerji, S.; Soga, T.; Umeno, M.; Tributsch, H. Over 18% solar energy conversion to generation of hydrogen fuel; theory and experiment for efficient solar water splitting. Int. J. Hydrog. Energy 2001, 26, 653–659. [Google Scholar] [CrossRef]
- Khaselev, O.; Turner, J.A. A monolithic photovoltaic-photoelectrochemical device for hydrogen production via water splitting. Science 1998, 280, 425–427. [Google Scholar] [CrossRef]
- Peharz, G.; Dimroth, F.; Wittstadt, U. Solar hydrogen production by water splitting with a conversion efficiency of 18%. Int. J. Hydrog. Energy 2007, 32, 3248–3252. [Google Scholar] [CrossRef]
- Khaselev, O.; Bansal, A.; Turner, J.A. High-efficiency integrated multijunction photovoltaic/electrolysis systems for hydrogen production. Int. J. Hydrog. Energy 2001, 26, 127–132. [Google Scholar] [CrossRef]
- Miller, E.L.; Rocheleau, R.E.; Khan, S. A hybrid multijunction photoelectrode for hydrogen production fabricated with amorphous silicon/germanium and iron oxide thin films. Int. J. Hydrog. Energy 2004, 29, 907–914. [Google Scholar] [CrossRef]
- Licht, S.; Wang, B.; Mukerji, S.; Soga, T.; Umeno, M.; Tributsch, H. Efficient solar water splitting, exemplified by RuO2-catalyzed AlGaAs/Si photoelectrolysis. J. Phys. Chem. B 2000, 104, 8920–8924. [Google Scholar] [CrossRef]
- Huang, C.-W.; Liao, C.-H.; Wu, C.-H.; Wu, J.C.S. Photocatalytic water splitting to produce hydrogen using multi-junction solar cell with different deposited thin films. Sol. Energy Mater. Sol. Cells 2012. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).