2.1. Results
A portion of the full cycling experiment carried out on Fe-doped MgH
2 is shown in
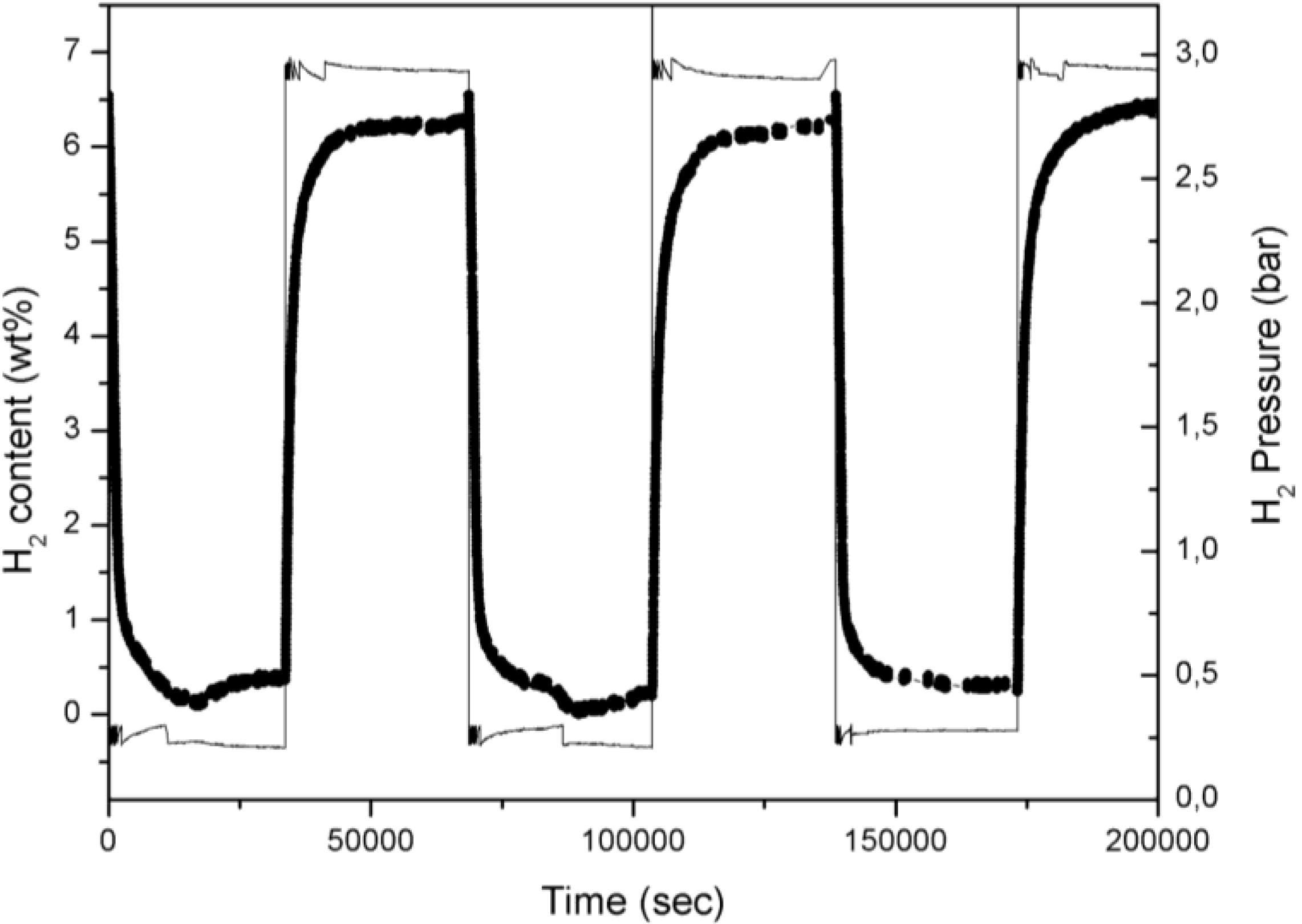
Figure 1 where a typical plot reporting the hydrogen content in Mg
versus time is reported.
Figure 1.
H2 desorption and absorption curve of three cycles for 5% Fe-doped MgH2 powder: scatter line; hydrogen pressure profile is also superimposed: full line.
Figure 1.
H2 desorption and absorption curve of three cycles for 5% Fe-doped MgH2 powder: scatter line; hydrogen pressure profile is also superimposed: full line.
In
Figure 1 the in-chamber H
2 pressure is also reported in order to illustrate the operating procedure. Changing periodically the H
2 pressure between 3.0 and 0.2 bars induces an alternate phase transformation between Mg and MgH
2. The temperature profile (data here not reported) evidences a practically constant temperature with negligible thermal fluctuation during the cycling time.
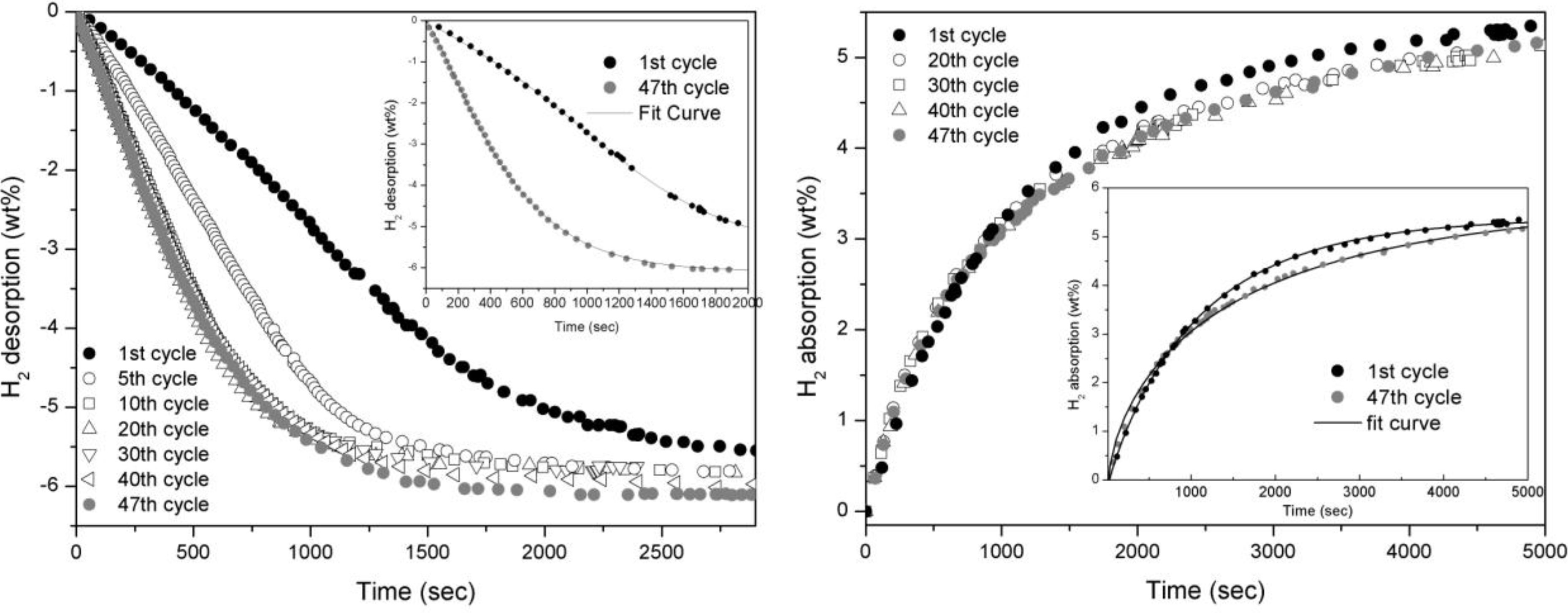
The absorption and desorption curves concerning doped MgH
2 are reported in
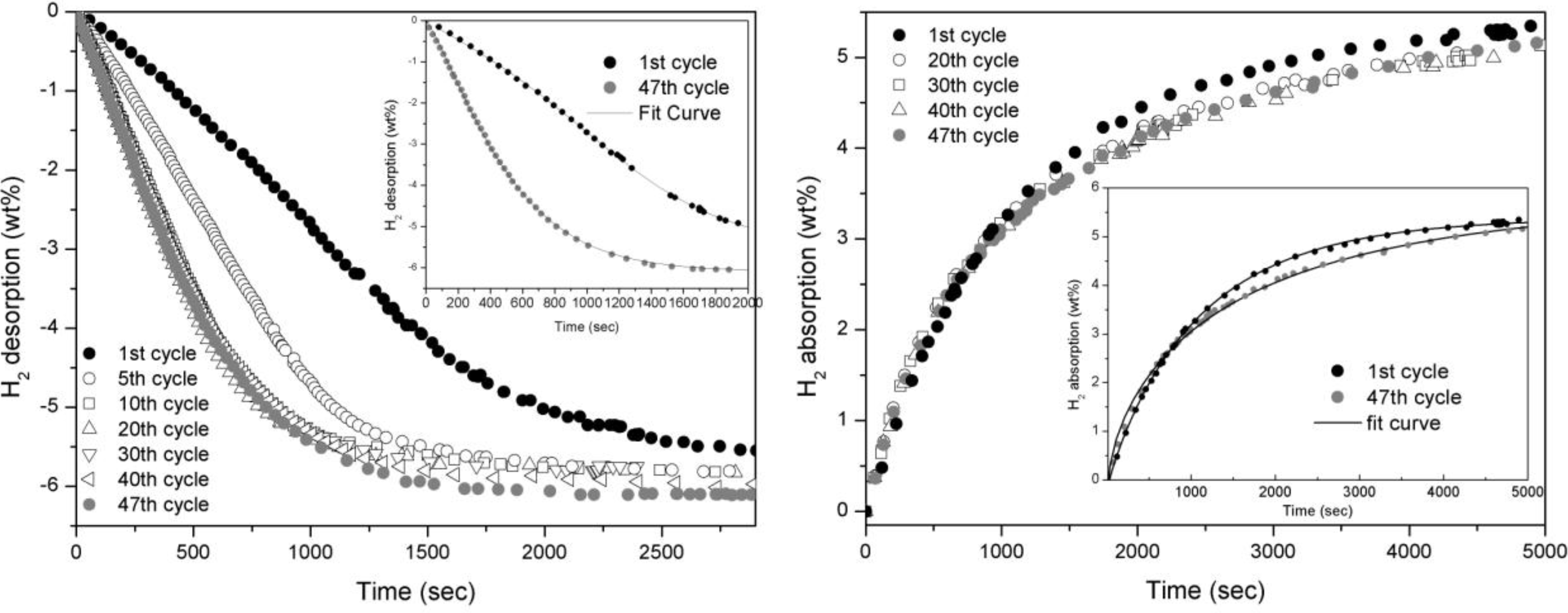
Figure 2. In desorption, kinetics undertake a small improvement in the firsts ten cycles and then maintain the same kinetic for the rest of cycling (
Figure 2a). In absorption, indeed, the performances are about constant all along the cycling apart from a slight worsening of the kinetic as it is shown in the inlet of
Figure 2 b.
Figure 2.
Desorption (left) and absorption (right) curves of MgH2 doped with 5 wt.% of Fe taken at 300°C and 0.2 bar for desorption and 10 bar for absorption.
Figure 2.
Desorption (left) and absorption (right) curves of MgH2 doped with 5 wt.% of Fe taken at 300°C and 0.2 bar for desorption and 10 bar for absorption.
The Johnson-Mehl-Avrami (JMA) model applied on these curves satisfactory fits the experimental data concerning all the hydrogen absorption and desorption curves. Avrami exponent n values shows a reduction by increasing the number of cycles, both for the absorption and the desorption reactions. In the former case we observe a reduction from ≈1 to ≈0.5 while in the latter from ≈1.7 to ≈1.2. These changes in the kinetic parameters indicate that repeated cycling induces major modifications of the sample structure, affecting the reaction path and the rate-limiting step.
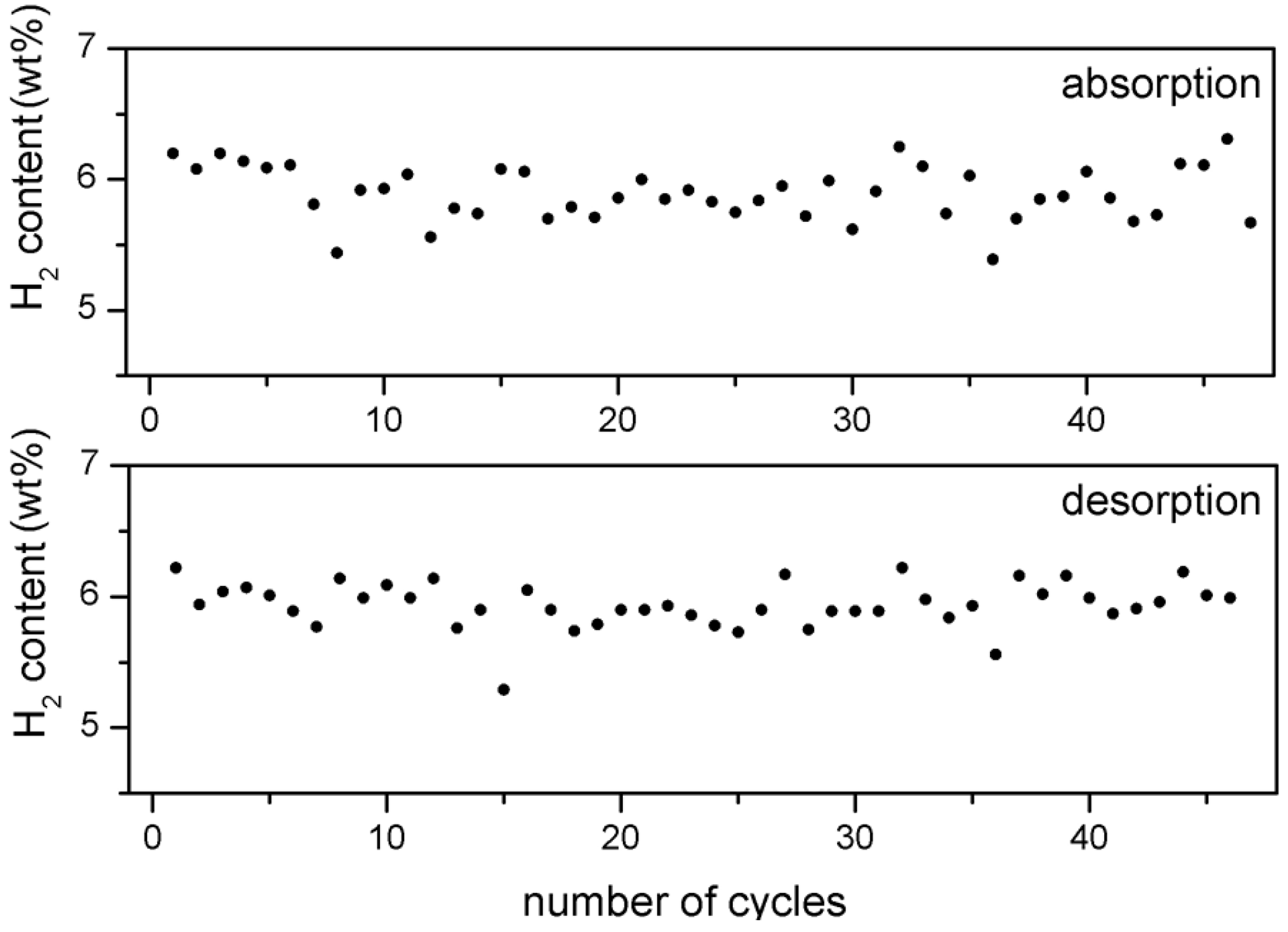
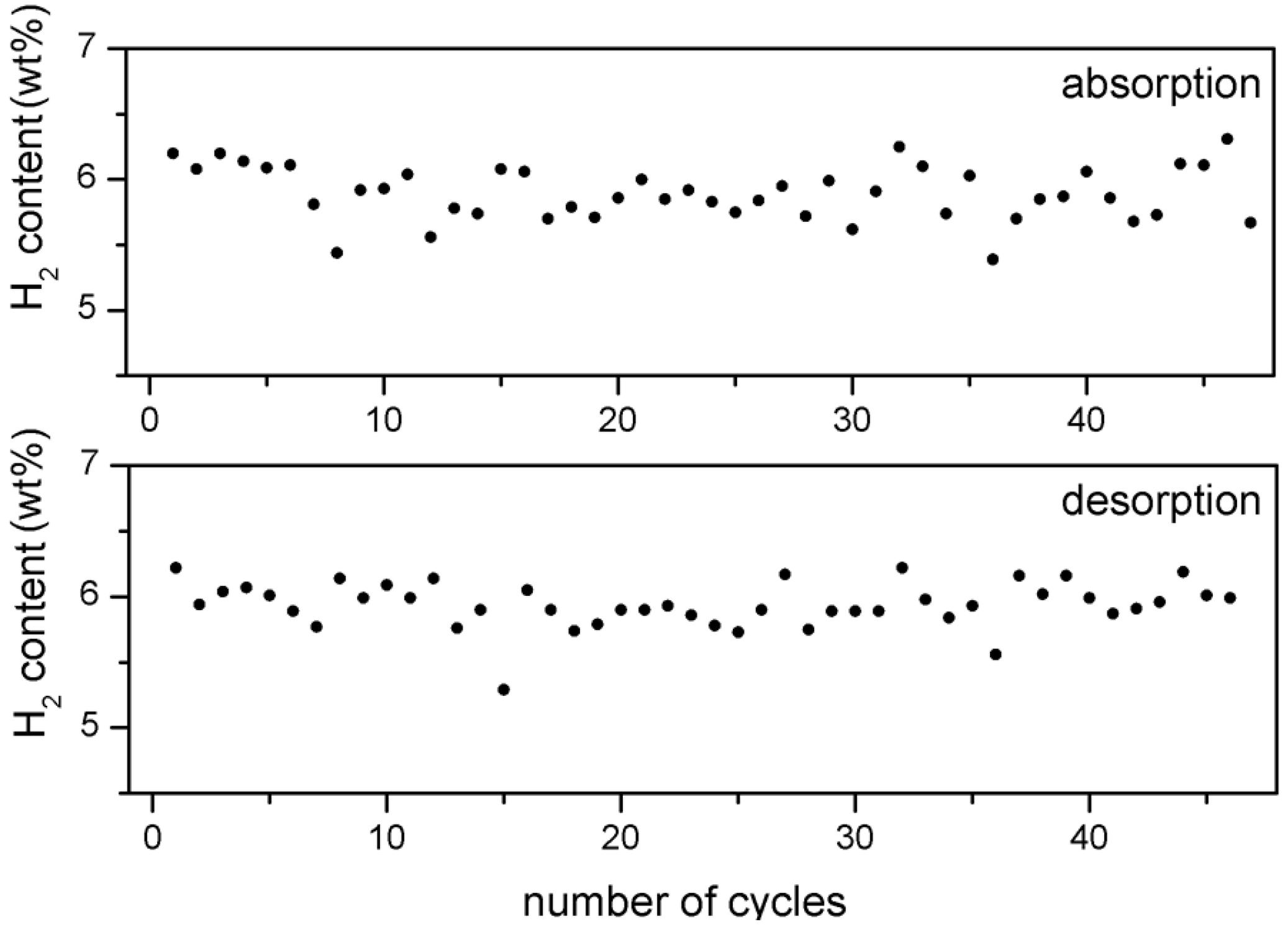
Despite this effect, the hydrogen storage capacity of about 6 wt% remains largely unaffected (
Figure 3).
Figure 3.
H2 storage capacity of 5 wt% Fe-doped MgH2 after each absorption and desorption cycle versus number of cycles.
Figure 3.
H2 storage capacity of 5 wt% Fe-doped MgH2 after each absorption and desorption cycle versus number of cycles.
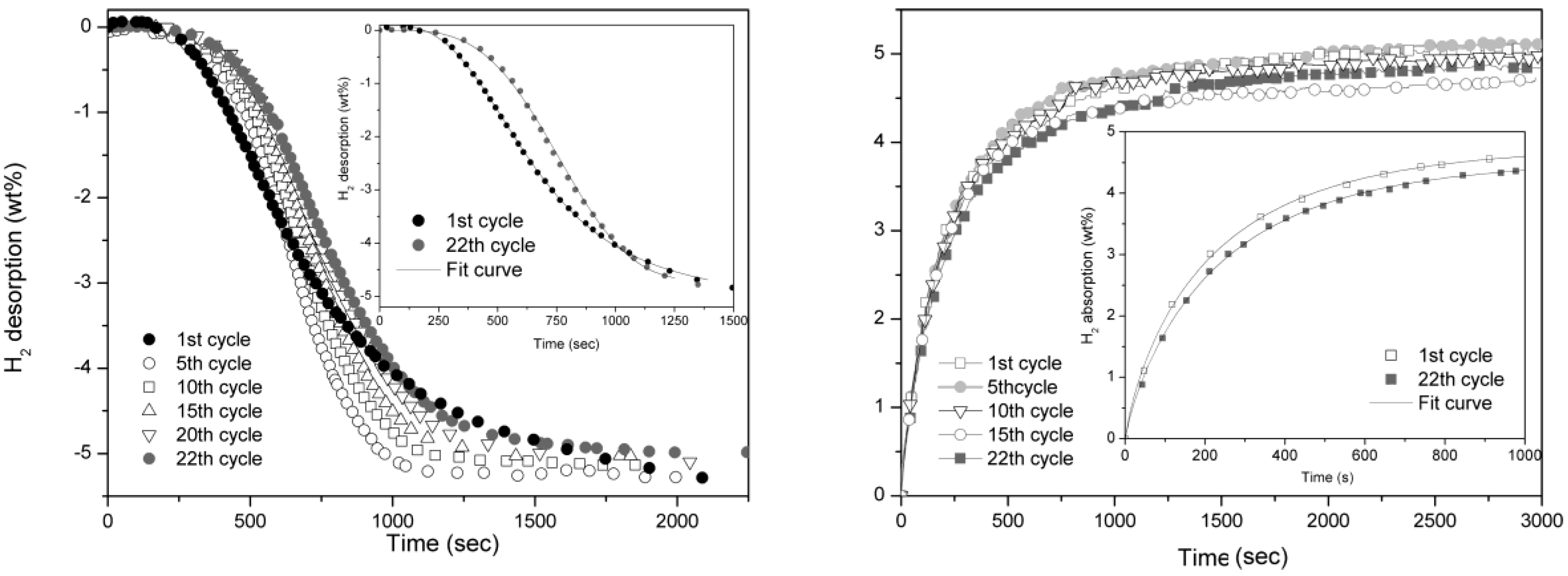
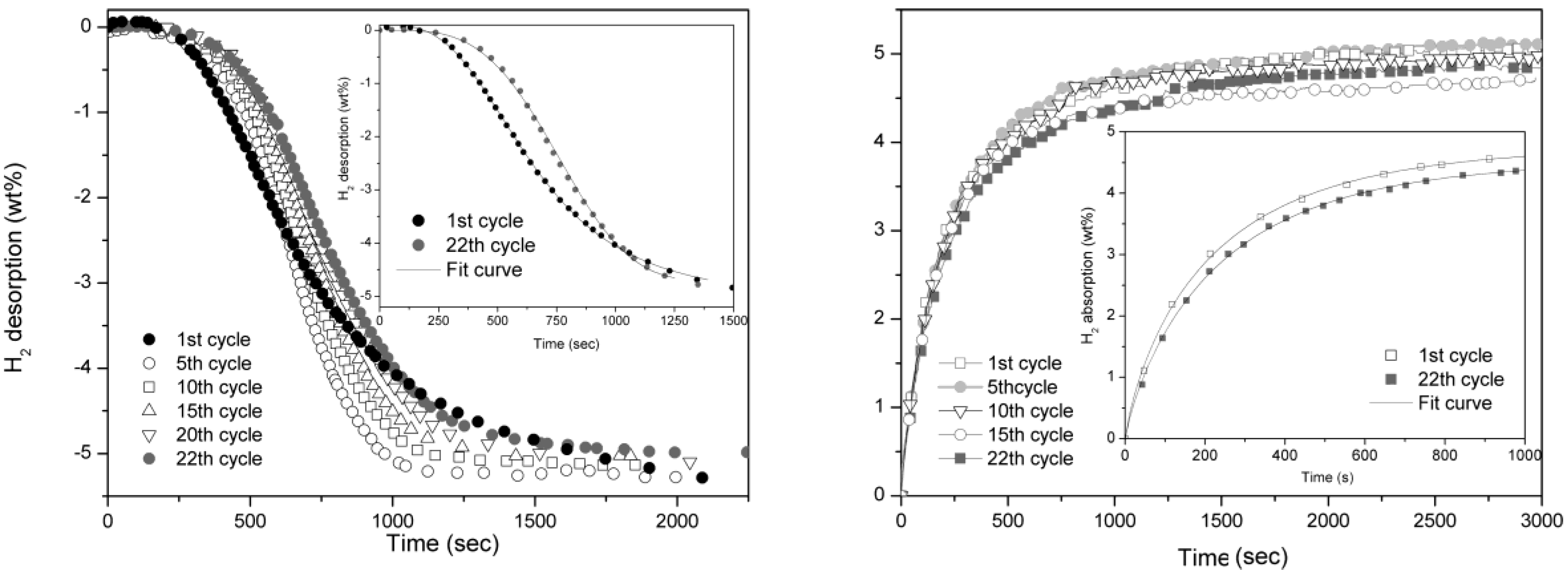
As reference, pure MgH
2 kinetics is reported in
Figure 4. In this case the reaction is much slower, in fact, to have a comparable reaction time, the isothermal cycling was performed at 350 °C. In both desorption and absorption reactions, the absence of catalyst implies a gradual degradation of reaction rate even if slightly evident in the former one. The Avrami exponent,
n, for desorption reaction evolves from a value of 4 observed at first cycle to about 3.5 after 22 cycles. In the case of H
2 absorption,
n is 0.8 and does not show any evolution upon cycling. The hydrogen storage capacity remains unaffected also in MgH
2 sample after 22 cycles.
From the comparison of the curve fitting between Fe-MgH
2 and pure MgH
2, the rate-limiting step of the absorption reaction is controlled by the same mechanism. The value of
n in the range from 0.5 to 1 suggests a transformation at constant density of MgH
2 nuclei and controlled by limited hydrogen flux at the powder particle surface [
22]. In the case of desorption reaction, the process is influenced by the presence of Fe. In fact, in the case of pure MgH
2 the reaction is interface-controlled with a three-dimensional growth at constant nucleation rate while, in the case of Fe-MgH
2, the desorption reaction is diffusion-controlled.
Figure 4.
Desorption (left) and absorption (right) curves of pure MgH2 taken at 350 °C and 0.2 bar for desorption and 10 bar for absorption.
Figure 4.
Desorption (left) and absorption (right) curves of pure MgH2 taken at 350 °C and 0.2 bar for desorption and 10 bar for absorption.
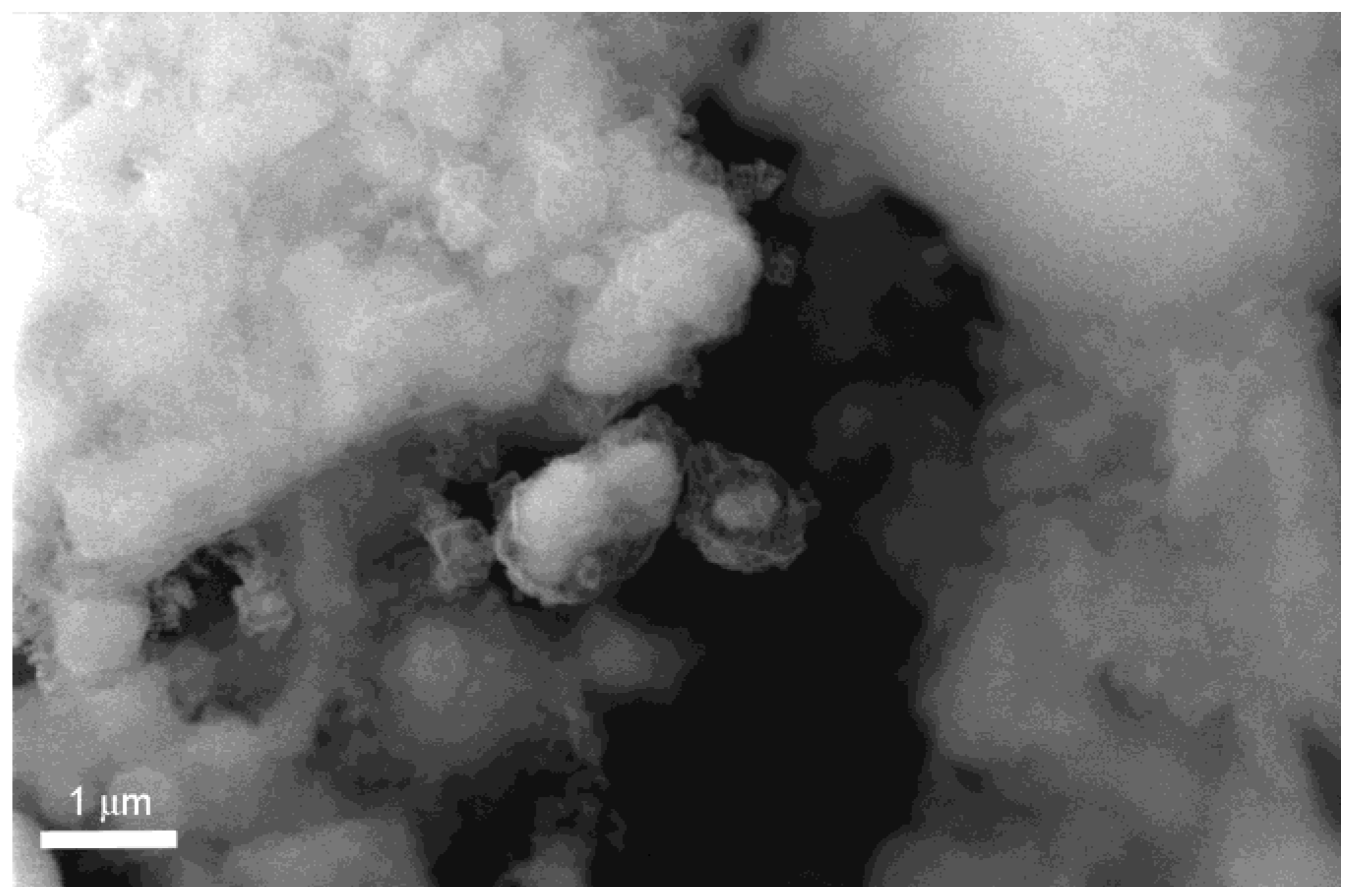
SEM observations confirm that major modifications of sample microstructure occur during the cycling experiment. Backscattered electron (BSE) images of MgH
2 with 5 wt% of Fe at different number of cycling (
Figure 5a–c) clearly show the localization of the catalyst particles visible as white dots. From the morphologic point of view, upon cycling the material evolves from a typical structure of as milled powder, characterized by irregular shape and broad particle size distribution function and still maintained after the first cycle [
23] (
Figure 5a), to a powder constituted by larger agglomerates, where surface protrusions can be observed (
Figure 5b,c). After several cycling, the internal material appears to come out from the particle surface, giving rise to worm-like structures where no catalyst particles are present. Similar microstructural evolution is observed for as milled [
22] and cycled pure MgH
2 powder (
Figure 5d). In particular the extent of protrusion is comparable to the catalyzed sample with the same number of cycles as well as the size of the agglomerates.
Figure 5.
SEM-BSE images of 5 wt% Fe-doped MgH2, ball milled for 10 h, after (a) 1 cycle; (b) 22 cycles and (c) 47 cycles at 300 °C; (d) SEM-BSE images of pure MgH2, ball milled for 10 h after 22 cycles, at 350 °C.
Figure 5.
SEM-BSE images of 5 wt% Fe-doped MgH2, ball milled for 10 h, after (a) 1 cycle; (b) 22 cycles and (c) 47 cycles at 300 °C; (d) SEM-BSE images of pure MgH2, ball milled for 10 h after 22 cycles, at 350 °C.
Besides particle coalescence, which can be expected considering the relatively high processing temperature, some other important effect seems to be directly related to the H
2 cycling. This hypothesis is confirmed by the observations of the internal structure reported in
Figure 6.
Figure 6.
SEM-BSE image of 5 wt% Fe-doped MgH2, ball milled for 10 h, after 47 cycles (cross section). The white spot.
Figure 6.
SEM-BSE image of 5 wt% Fe-doped MgH2, ball milled for 10 h, after 47 cycles (cross section). The white spot.
As it is possible to noticing in
Figure 6, the particle structure is assuming a core shell configuration with the catalyst particles concentrated in the core structure and an outer shell where only Mg without Fe particles is observed.
A similar evolution can be found in the images reported by Malka
et al. [
24] in an analogous experiment.
Another important effect related to the H
2 cycling, besides the particle coarsening, is observed in the morphology of pure MgH
2 after 22 cycles in
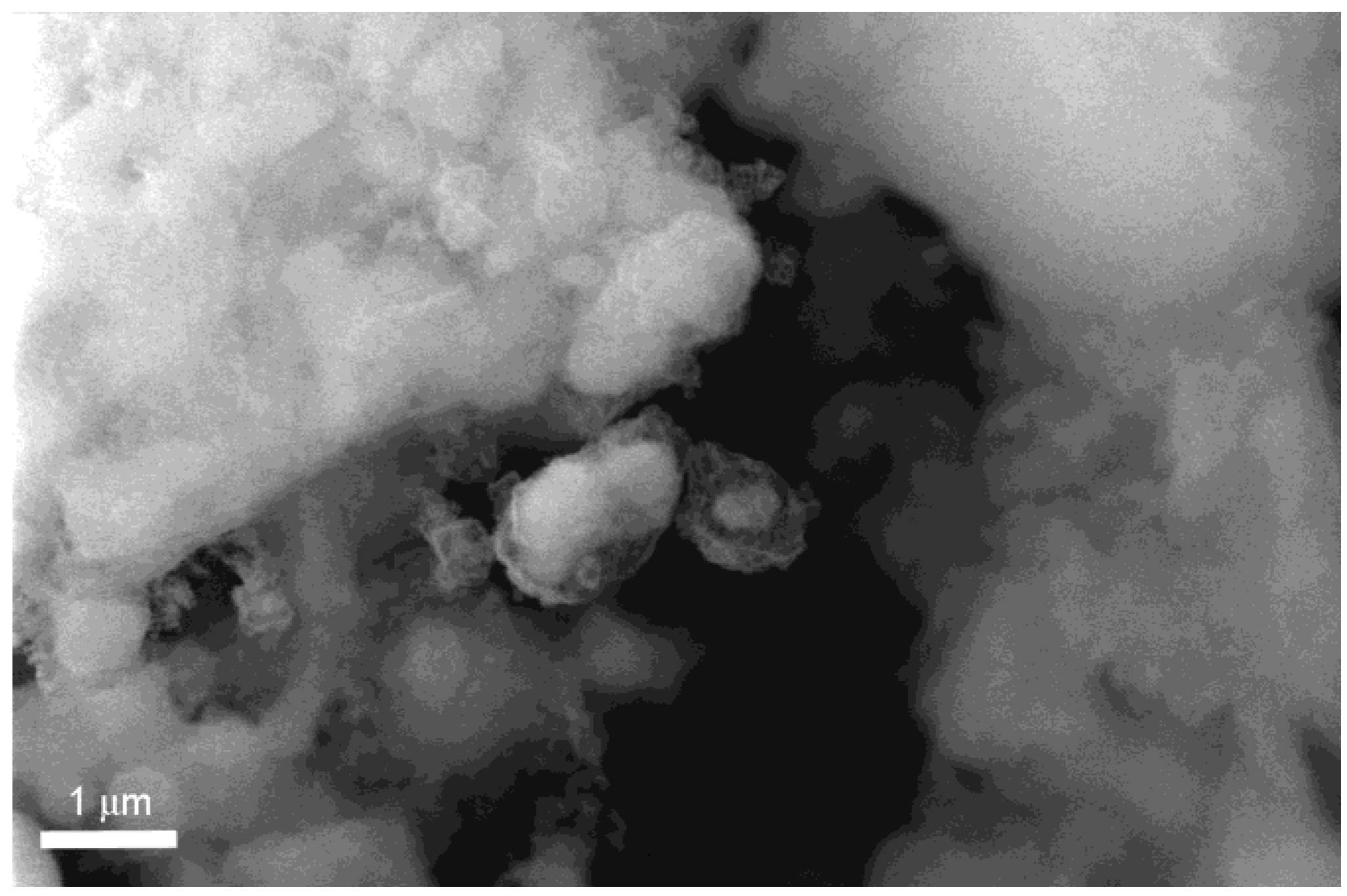
Figure 7. Also in this case Mg appears to exit from the MgO crust surrounding the particles. This effect is particularly evident in the smaller particles where partially empty structures can be seen in the higher magnification Secondary Electron (SE) image.
Figure 7.
SEM-SE image of pure MgH2 after 22 cycles.
Figure 7.
SEM-SE image of pure MgH2 after 22 cycles.
2.2. Discussion
The repeated phase transformation between the MgH2 and Mg induces major modifications to the powder material. Concerning the catalyzed sample, both the structure and the mechanisms underlying the hydrogen absorption and release appear strongly modified by this processing.
The main morphological and structural effect is constituted, besides a noticeable coarsening of the powder structure, by the presence of the material, which protrudes out of the powder particles and which is constituted by only pure Mg without the catalyst particles.
Morphological details of ball milled powder particles involved in the reaction with hydrogen gas can provide a tentative explanation for this effect. It is known from literature [
25] that the powder particles have a core-shell structure with a metallic core surrounded by a surface layer, in the nm range thickness, constituted by Mg oxide or hydroxide, resulting from the exposure of the just milled material to the atmosphere. When the Mg based particles react with hydrogen to form the MgH
2 phase, this layer separates the two reacting species so that, in order for the reaction to occur, one or both elements have to diffuse through this MgO membrane. It is quite evident that the amount of the product phase formed on either side of the membrane is related to the diffusion fluxes of the reacting elements. This means that the product phase will be located on a single face of the MgO membrane only if the diffusive flux of one of the reactants through this separation layer is negligible with respect to the other. In a simplified example, if hydrogen is the only mobile specie in the MgO layer, the product phase is expected to be synthesized inside the particle while in the opposite case, if only Mg is mobile, MgH
2 is expected to form outside the particle. In a general case, if both the diffusive fluxes are of the same order of magnitude, the product phase will be formed on either side of the membrane in a fraction inversely proportional to the diffusivity of the elements present on that side at the beginning of the reaction. The driving force for diffusion process is provided by the difference in the Gibbs free energy between the reacting elements and the product phase, which generates a chemical potential gradient across the surface membrane for both the reacting species [
26].
During the inverse reaction, hydrogen is released by the decomposition of the MgH2. Considering that MgH2 can be located either inside the MgO shell surrounding the particles or outside, the hydrogen released in the former case has to cross the MgO layer while this step is not necessary in the latter case. In the former case the driving force inducing the diffusive flux through the membrane is provided by the difference between the internal pressure, which can be considered of the order of the equilibrium pressure at the operating temperature and the external pressure which is kept at a lower value in order to induce the hydride decomposition. It is important to notice that this scenario provides a driving force only for hydrogen diffusion and no driving force is present to assist Mg mobility. In particular there is not any driving force forcing the Mg atoms backward inside the powder particle, so that the net result of a full charge-discharge cycle is that a fraction of the Mg atoms are extracted from the MgO shell. Considering that this effect is operative during each cycle, the fraction of Mg present outside the original powder grains is expected to increase with the number of cycles.
Due to the catalytic activity, Fe does not react with hydrogen but assists the phase transformation and, as a consequence, is expected to remain trapped inside the MgO crust as noticed in
Figure 5 and
Figure 6.
Most of the experimental observations reported in this paper can be accounted for by the delineated hypothesis. In fact the Mg is observed to protrude out of the powder particles only at localized sites where, probably the structure of the MgO layer is supporting a particularly fast diffusion of Mg atoms. The Fe catalyst particle are not observed to participate in the process and are observed to concentrate, in many cases, in a defined volume of the material leaving space for areas where only pure Mg is observed. These findings are supported by the experimental results concerning the cycling of pure Mg. In this case the cycling temperature has to be increased to 350 °C in order to compensate for a lower reactivity of the sample. The experimental result reported in
Figure 7, where many empty MgO shells can be observed, confirms that also in this case Mg is extracted from the MgO shell by the hydrogen cycling process. Moreover, in this case, the process appears to be more efficient since particles completely free of the Mg core are observed after only 22 cycles; this enhancement of the extraction efficiency can be related to the higher operating temperature which can favor Mg diffusivity with respect to the H ion one.
The experimental results reported in this paper can have important practical applications. In fact the process can be interpreted as an in situ cleaning of Mg which, after cycling, can be present in a hydrogen tank without the MgO crust since the process is carried out completely under an hydrogen atmosphere and the fresh Mg surface can be stable in this environment. We want to notice that, within the sensitivity of our methods no further MgO is formed during cycling, since the total hydrogen storage capacity of the material remains constant. Considering that the surface reactivity of Mg has been often considered as an important drawback for technological application in the hydrogen storage, our observation can open new routes for producing tank devices filled with clean Mg base material.
However, these observations highlight quite complex phenomena, which require a deeper investigation for a full explanation. In fact, even if the progressive extraction of Mg from the MgO shell can account for the variation in the reaction kinetics observed upon cycling, the complex structure of the powder particles is still playing a role in defining the reaction kinetics. The behavior of these samples after a higher number of cycles is under investigation.
The behavior of doped Mg is different from what displayed by pure Mg even after cycling, so that we can infer that the catalyst particles, even if trapped inside the MgO shell, are still playing a role speeding the reaction kinetics with respect to pure Mg. On the other hand, the evolution of the microstructure has to be taken into account when defining the way of distributing the catalyst particles in the Mg material, and probably most of the studies on the efficiency of different kind of catalyst have to be reconsidered in the light of possible structural evolution during the life of technological devices.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}