Hydrogenation of Anthracene in Supercritical Carbon Dioxide Solvent Using Ni Supported on Hβ-Zeolite Catalyst


Abstract
:1. Introduction
2. Results and Discussion
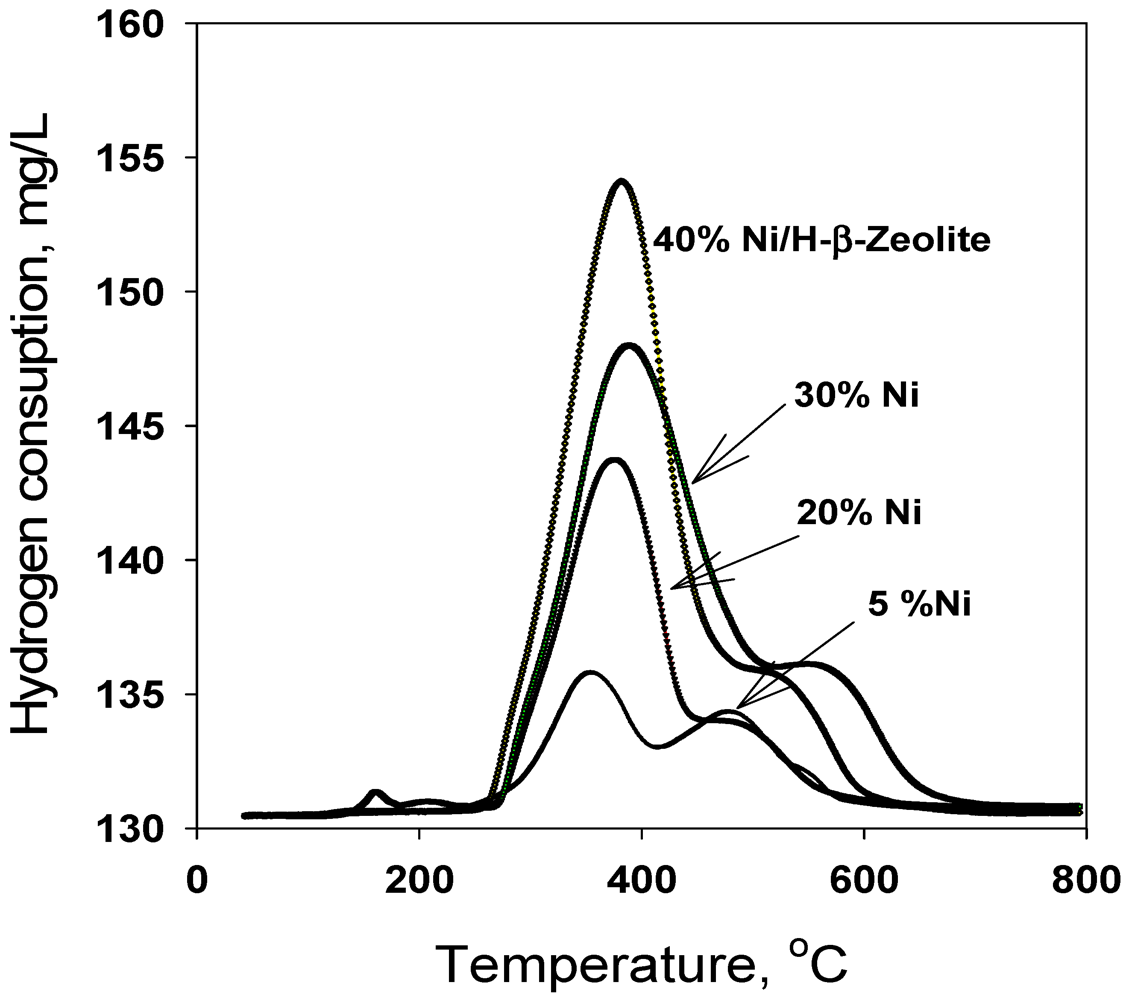
2.1. Catalyst Characterization
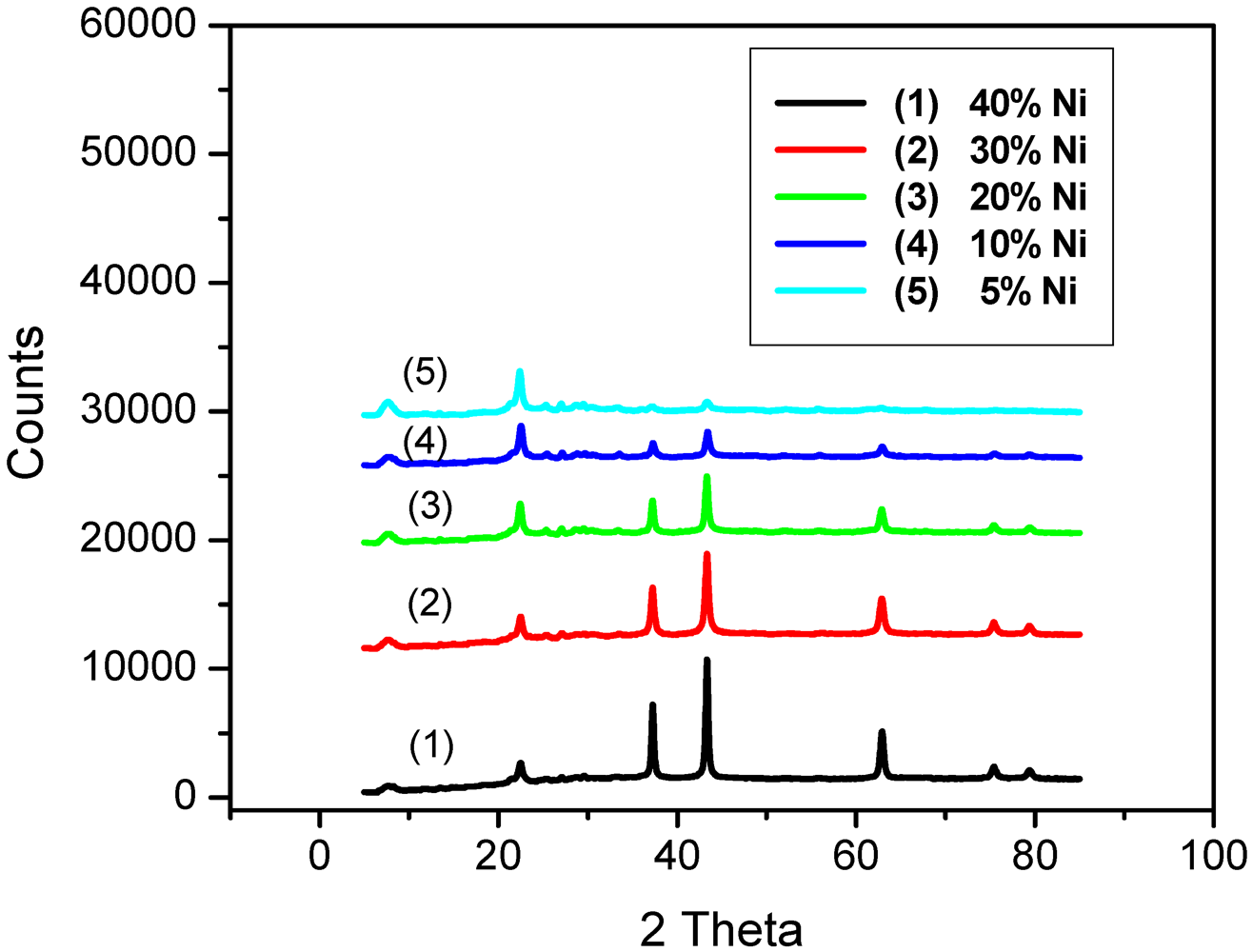

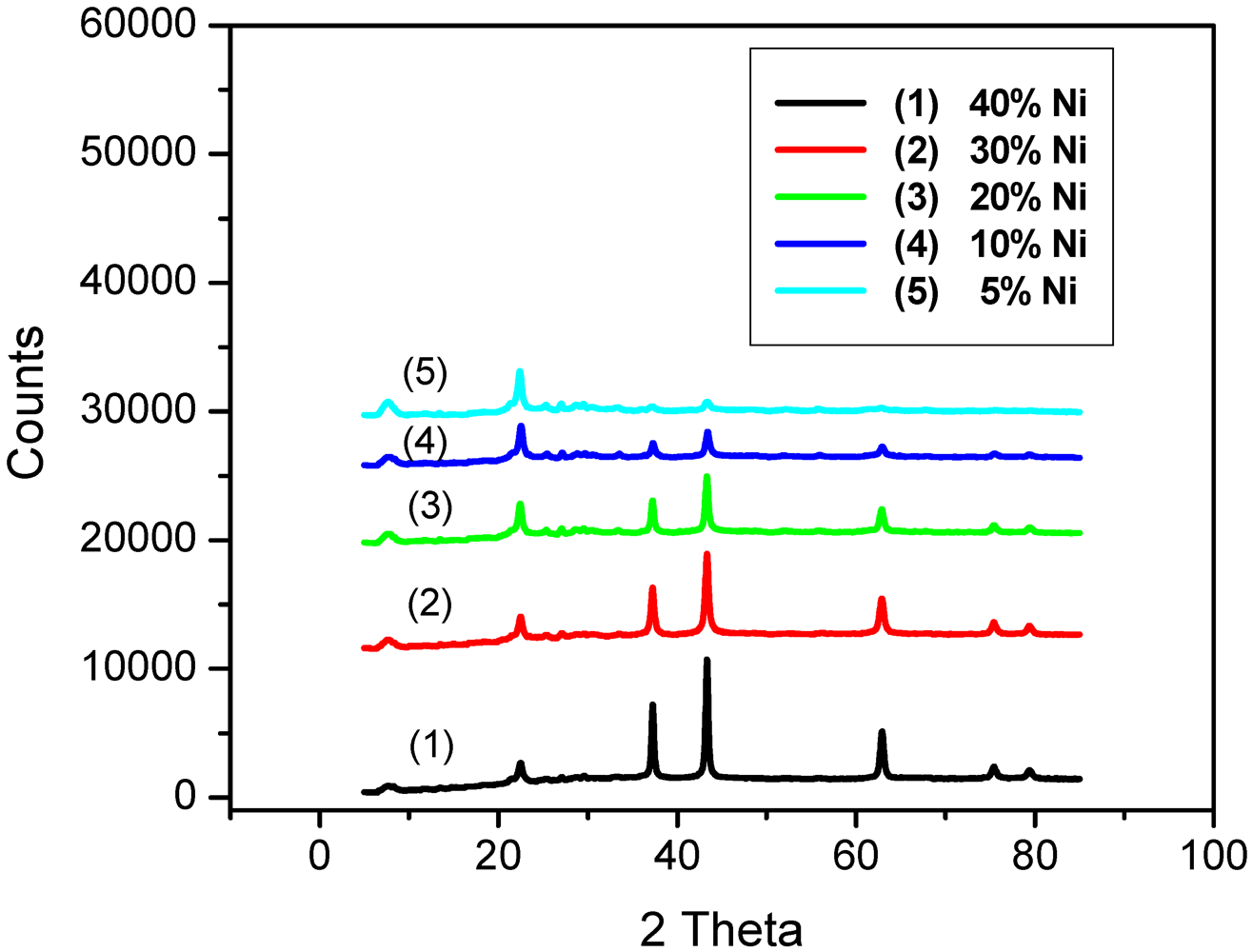{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Number | Catalyst | BET-SA (m2·g−1) | Tmax | H2 uptake | Tmax | H2 uptake | Total H2 uptake |
|---|---|---|---|---|---|---|---|
| °C | mMol | °C | mMol | mMol | |||
| 1 | 5% Ni-Hβ | 258 | 354 | 118 | 478 | 95 | 214 |
| 2 | 10% Ni-Hβ | 163.65 | 217372 | 41178 | 525 | 102 | 321 |
| 3 | 20% Ni-Hβ | 141.6 | 376 | 416 | 498 | 86 | 502 |
| 4 | 30% Ni-Hβ | 95.6 | 389 | 716 | 577 | 33 | 749 |
| 5 | 40% Ni-Hβ | 50.6 | 382 | 801 | 530 | 36 | 838 |

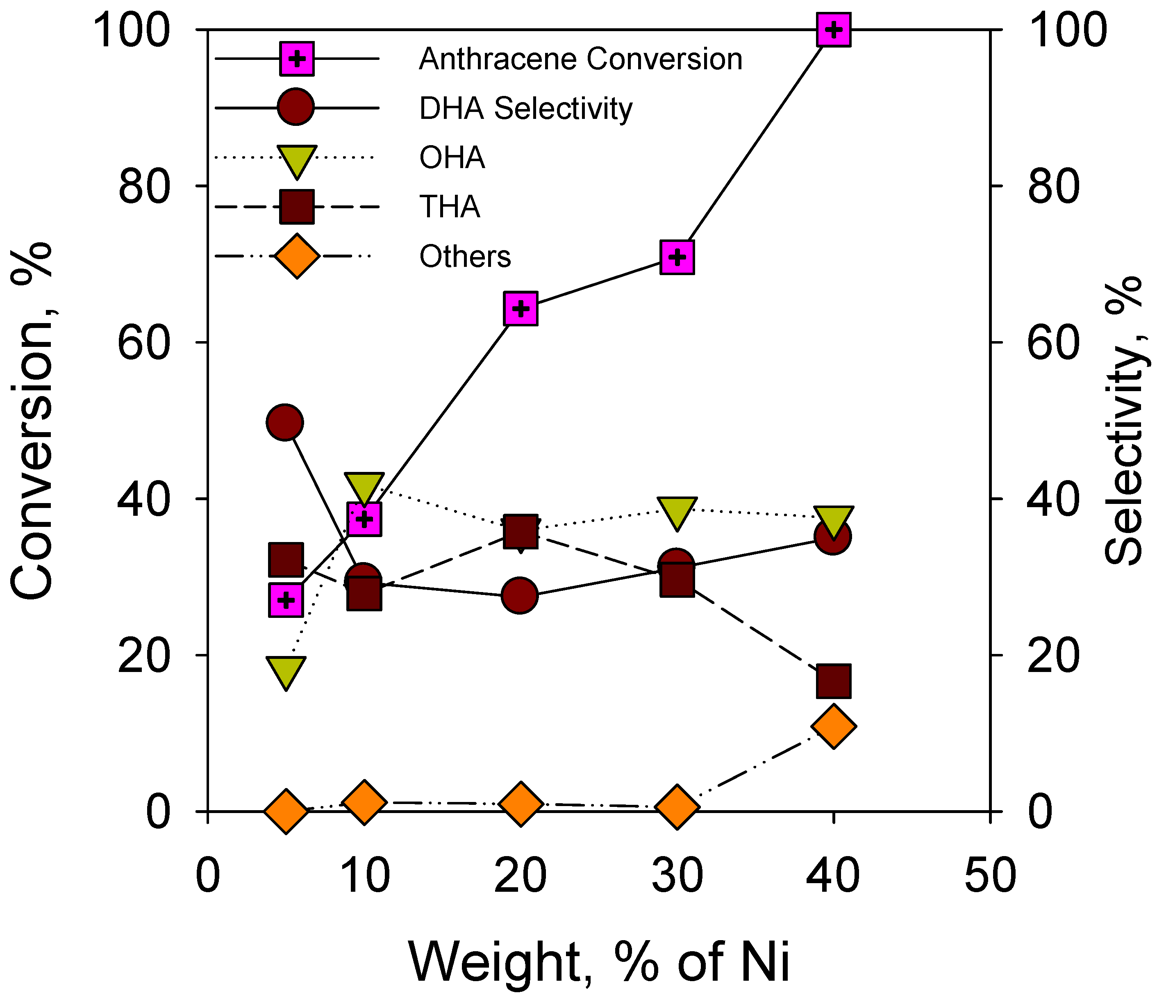
2.2. Catalytic Activity
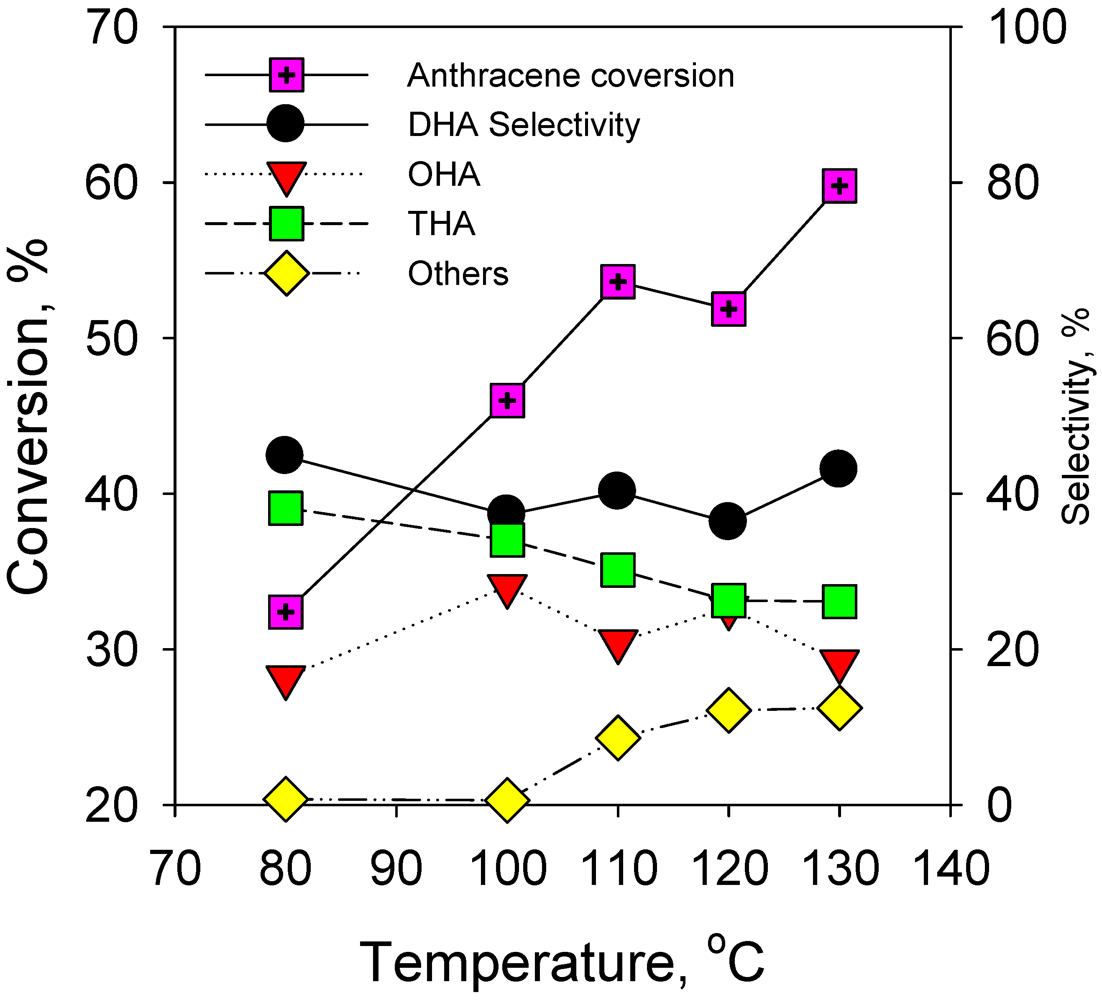
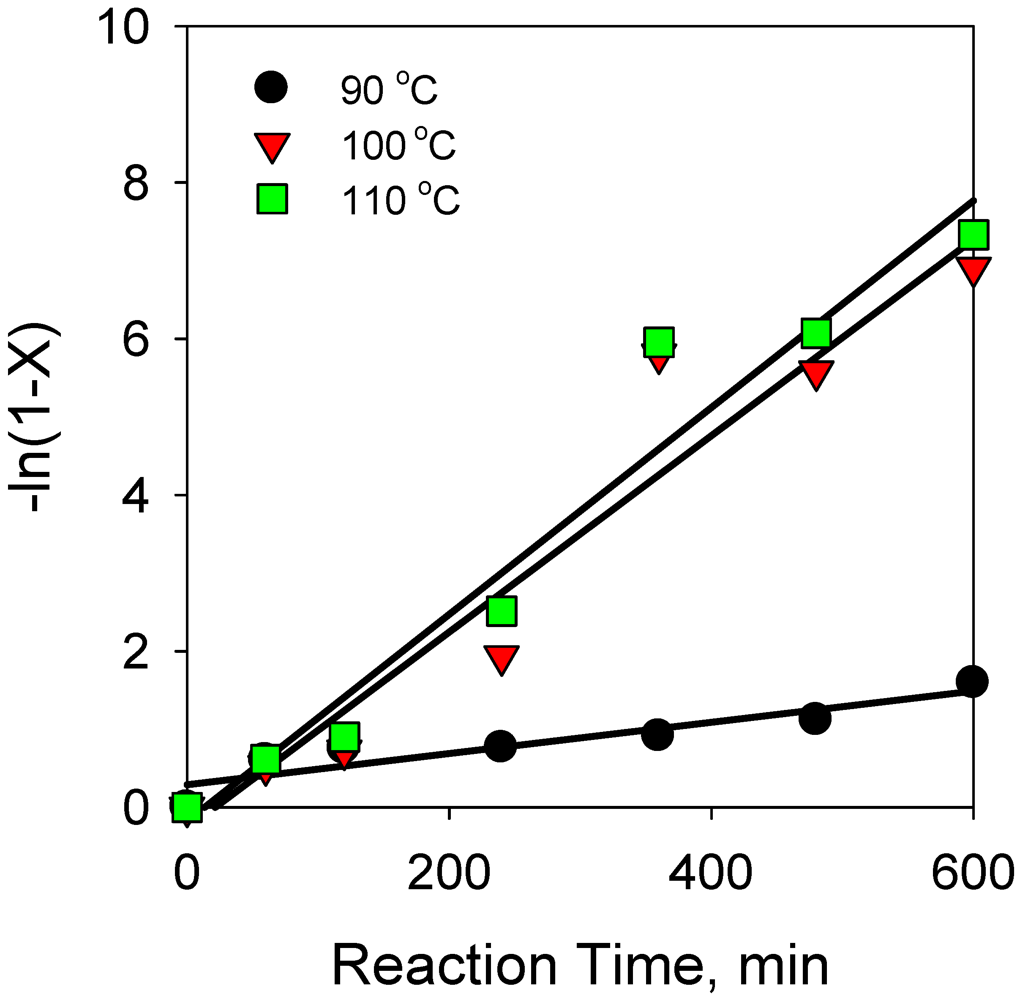
2.2.1. Effect of Reaction Temperature on Hydrogenation of Anthracene
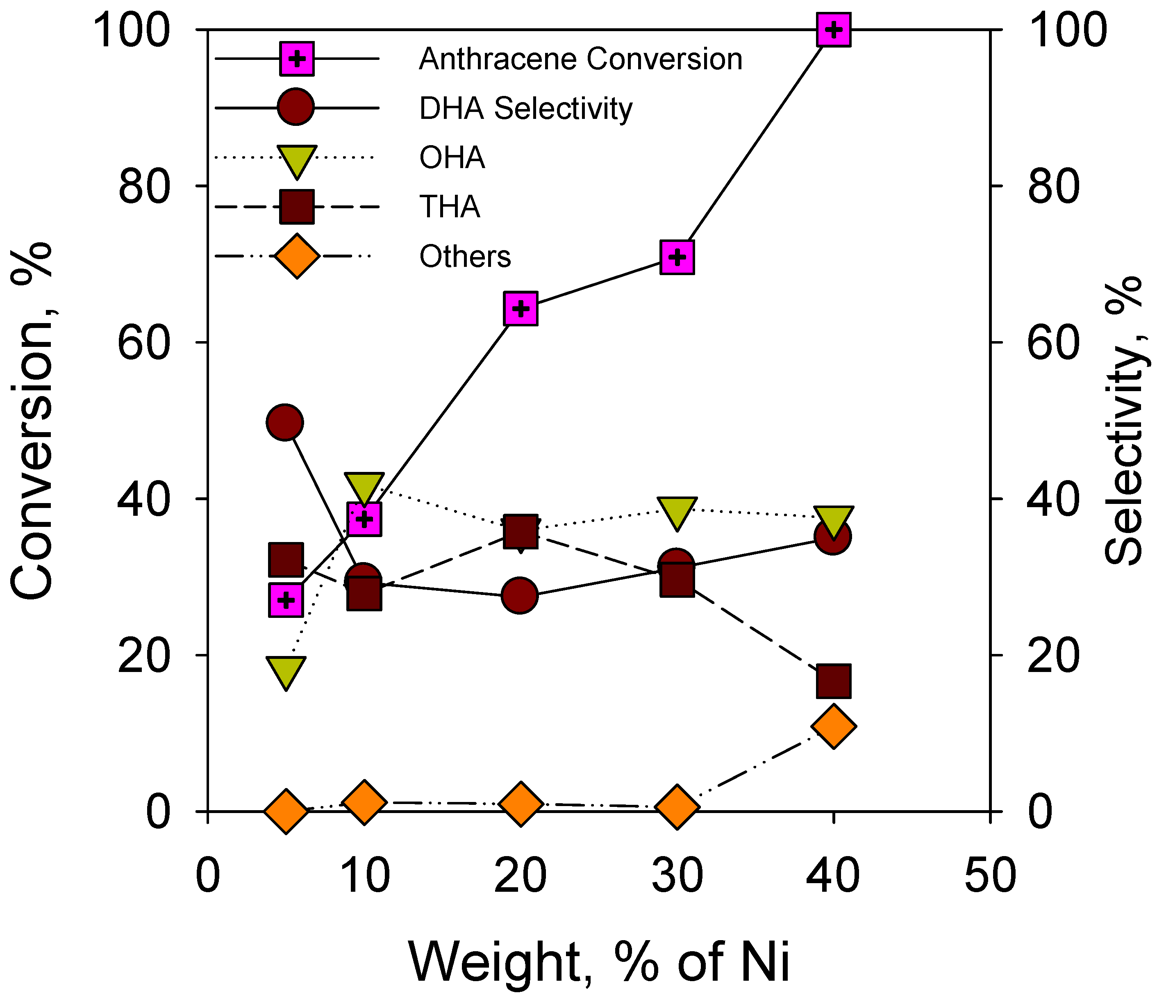
2.2.2. Effect of Nickel Loading on Hydrogenation of Anthracene
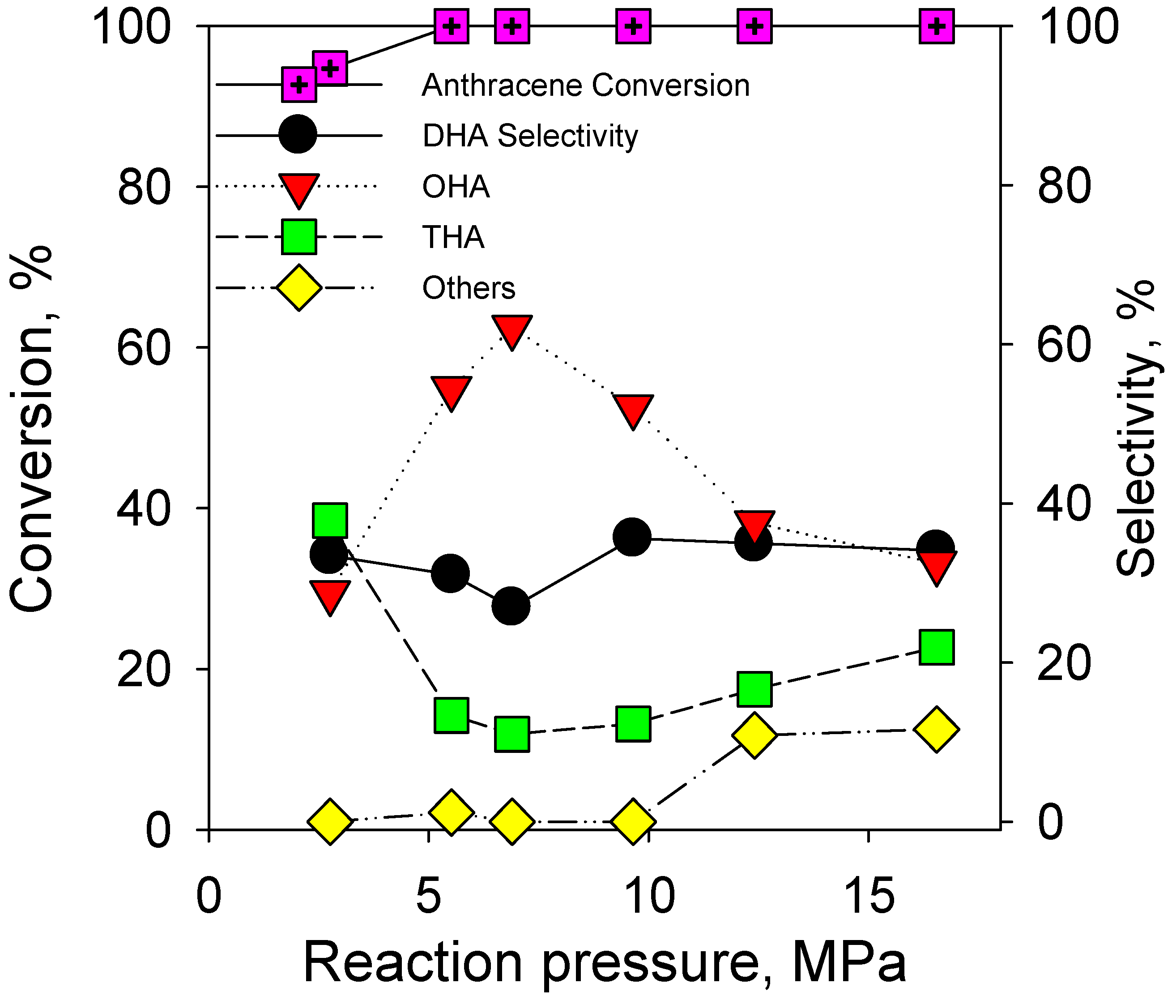
2.2.3. Effect of Reaction Pressure on Hydrogenation of Anthracene

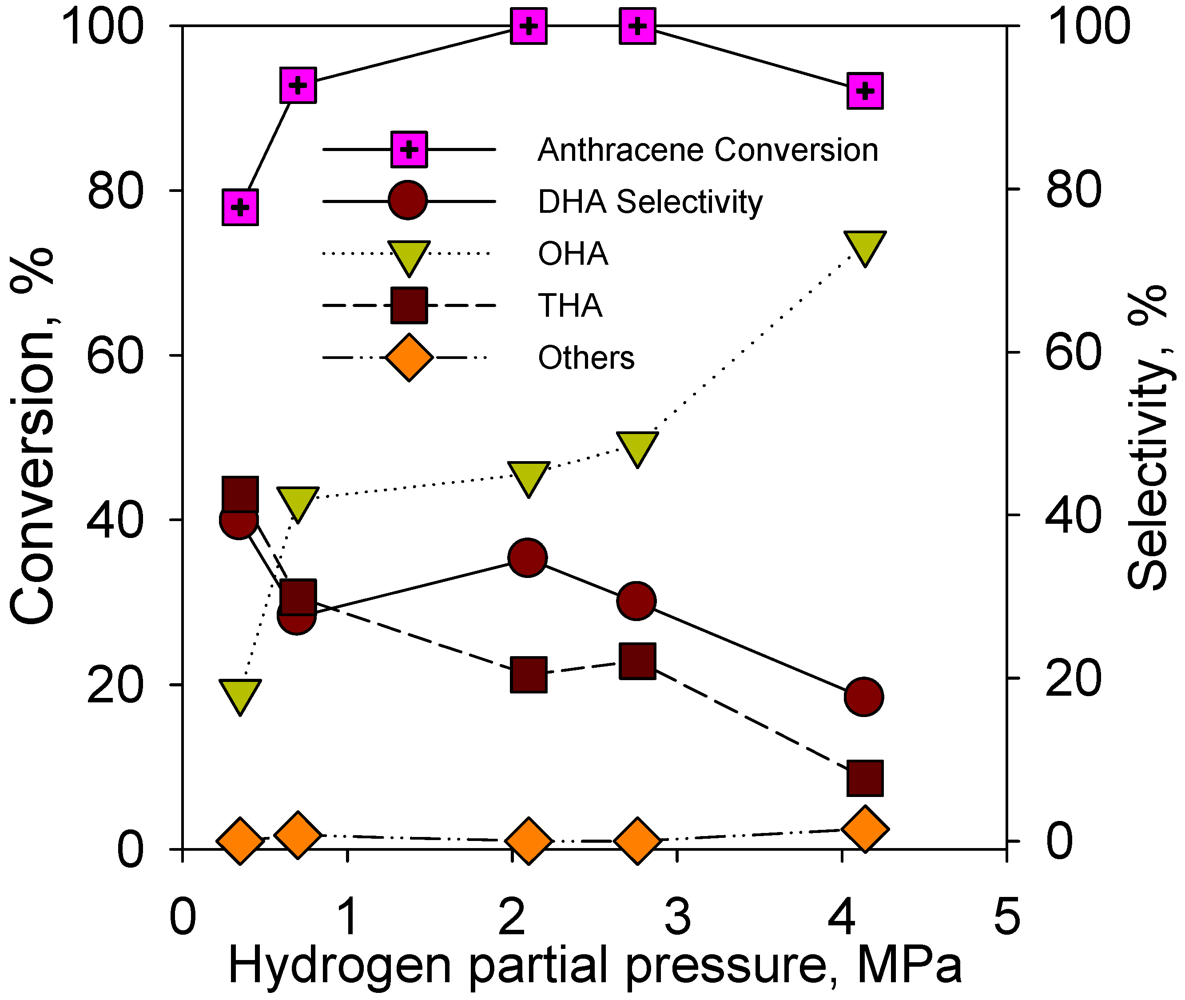
2.2.4. Effect of Hydrogen Partial Pressure on Hydrogenation of Anthracene
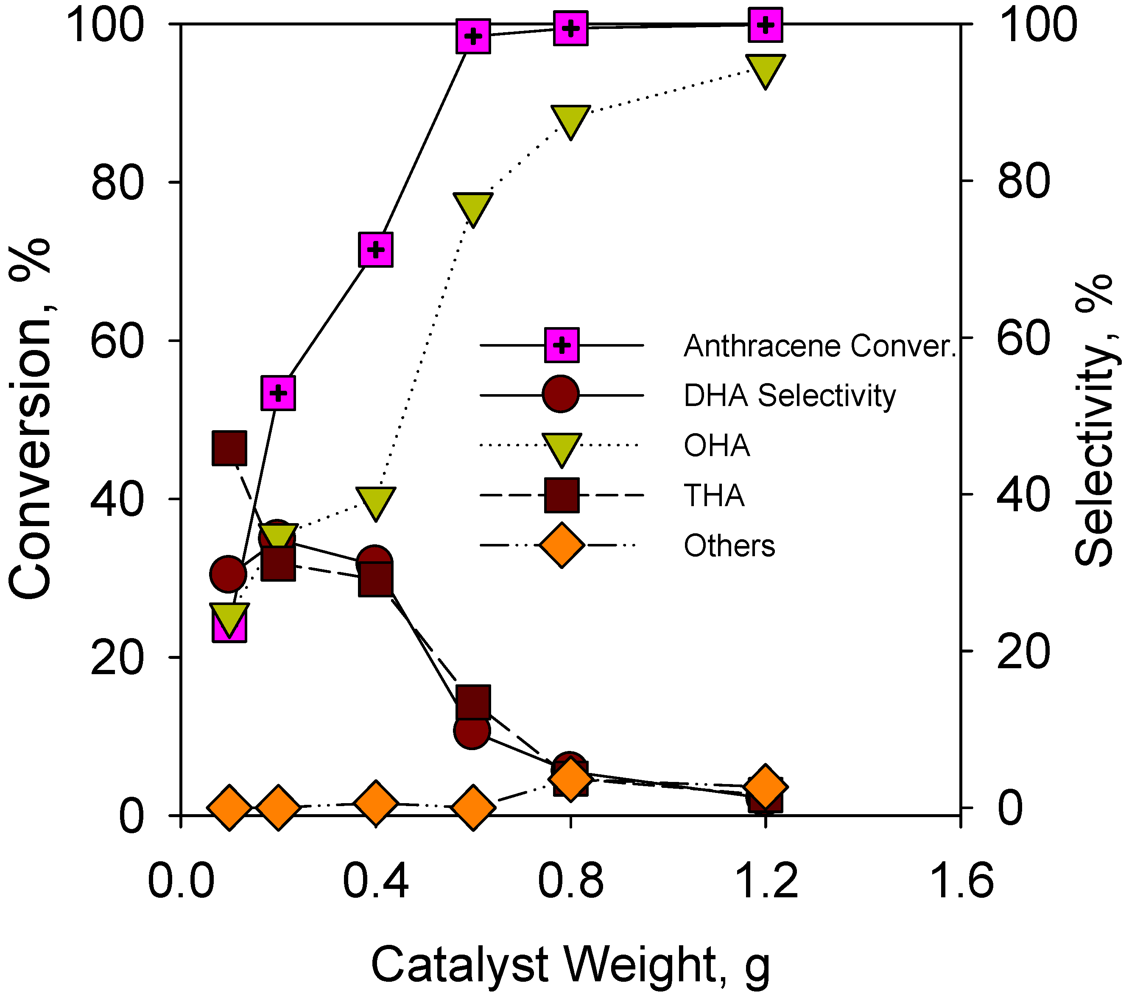
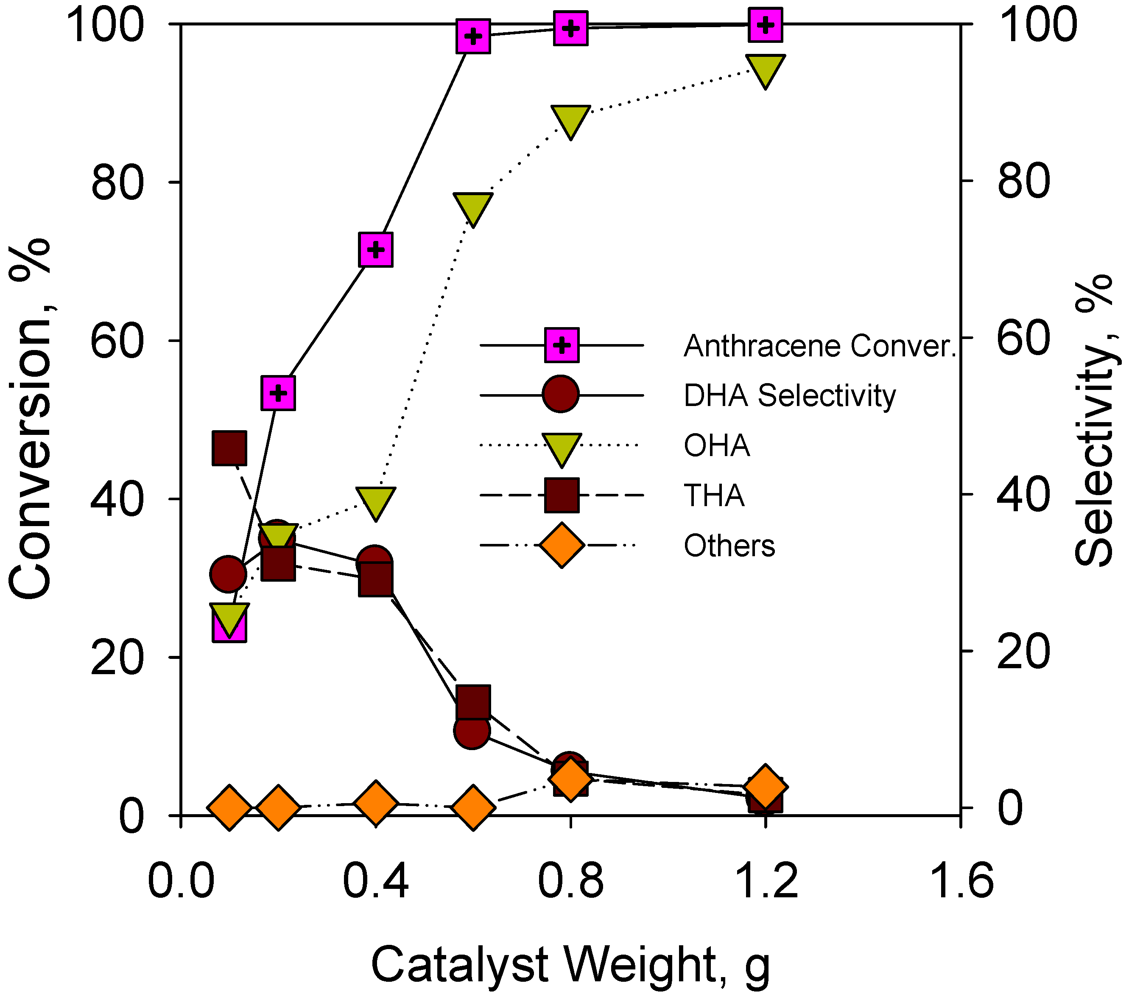
2.2.5. Effect of Catalyst Weight on Hydrogenation of Anthracene

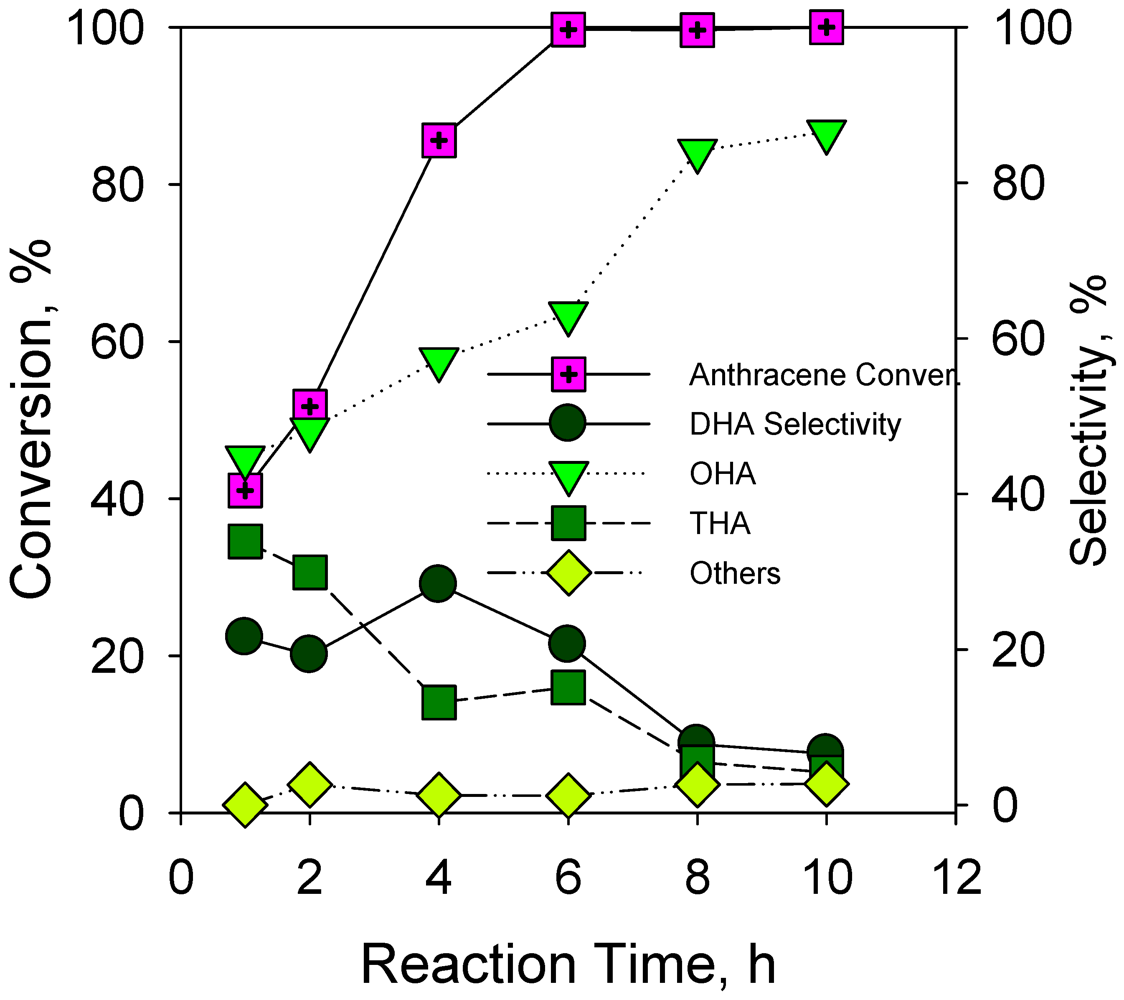
2.2.6. Effect of Reaction Time on Hydrogenation of Anthracene

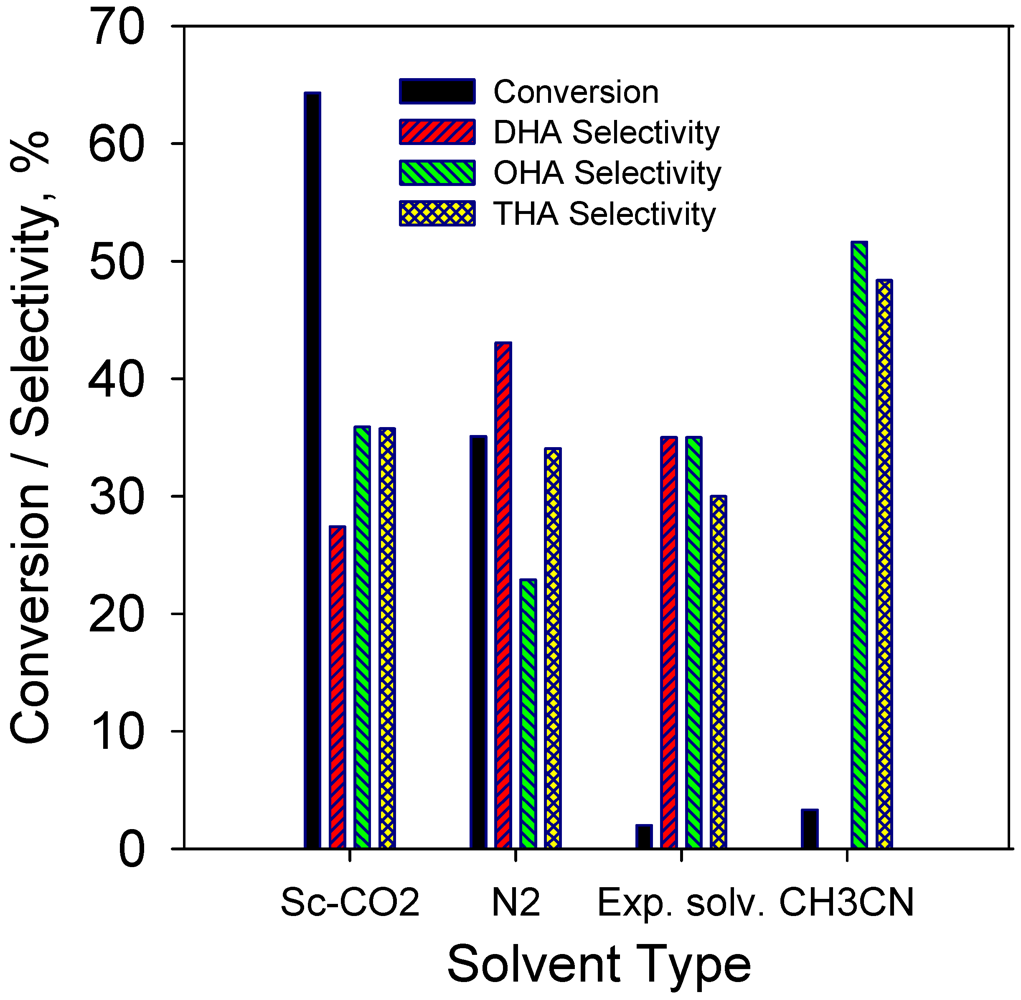
2.2.7. Effect of Solvent on Hydrogenation of Anthracene

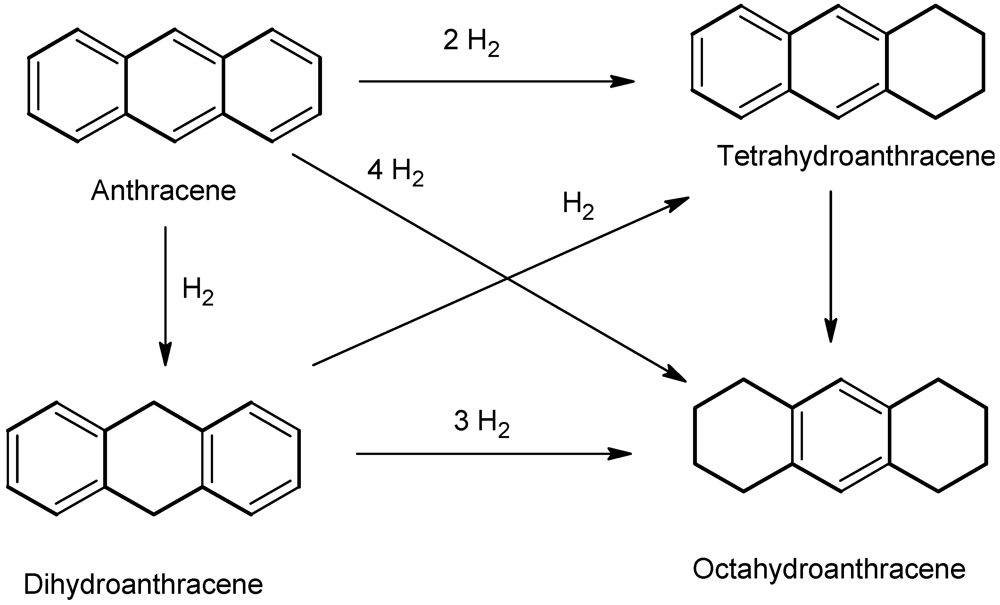
2.2.8. Reaction Mechanism of Anthracene Hydrogenation

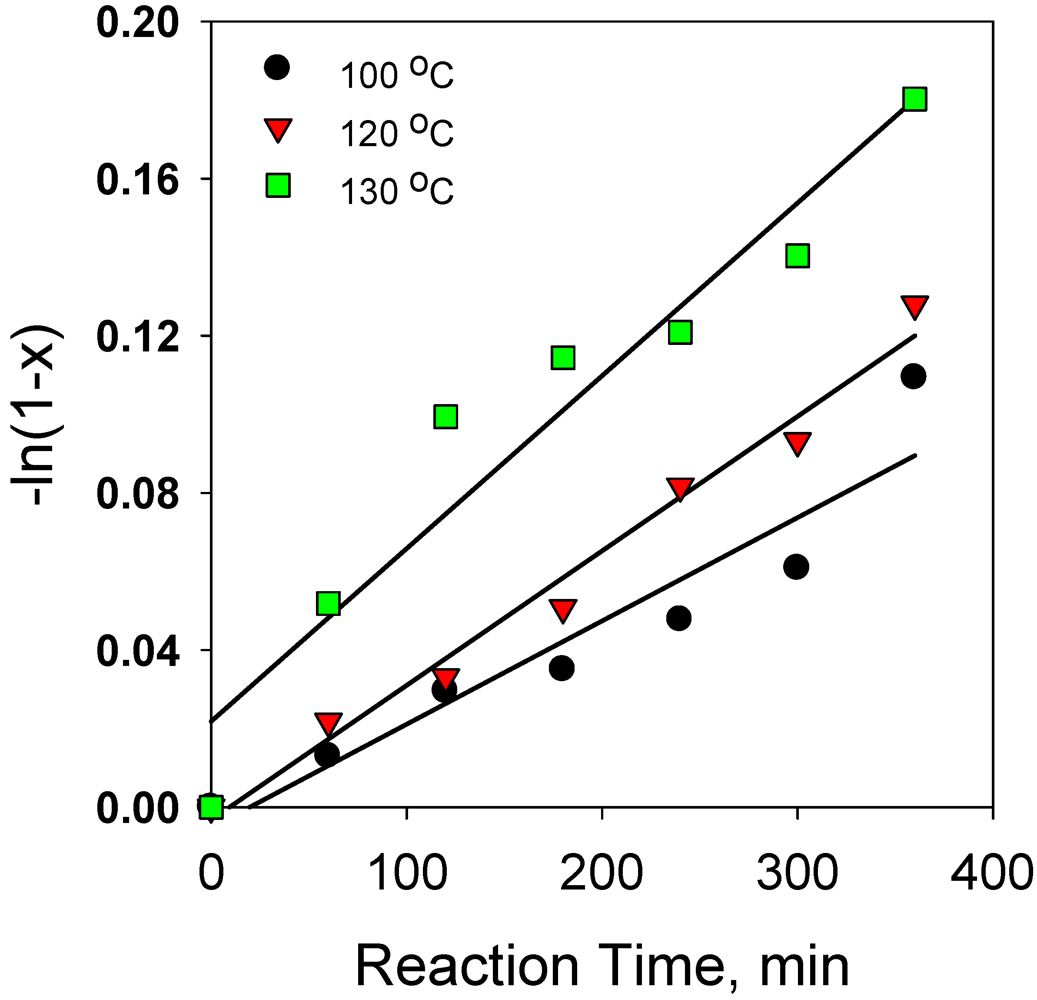
2.2.9. Comparison of Reaction Kinetics of Hydrogenation Anthracene with Phenanthrene and Naphthalene
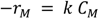
3. Experimental Section
3.1. Catalyst Preparation
3.2. Catalyst Characterization
3.3. Catalytic Reactions
4. Conclusions
References
- Alhumaidan, F.; Cresswell, D.; Garforth, A. Hydrogen storage in liquid organic hydride: Producing hydrogen catalytically from methylcyclohexane. Energy Fuels 2011, 25, 4217–4234. [Google Scholar] [CrossRef]
- Hodoshima, S.; Arai, H.; Takaiwa, S.; Saito, Y. Catalytic decalin dehydrogenation/naphthalene hydrogenation pair as a hydrogen source for fuel-cell vehicle. Int. J. Hydrog. Energy 2003, 28, 1255–1262. [Google Scholar] [CrossRef]
- Kariya, N.; Fukuoka, A.; Ichikawa, M. Efficient evolution of hydrogen from liquid cycloalkanes over pt-containing catalysts supported on active carbons under “wet-dry multiphase conditions”. Appl. Catal. A 2002, 233, 91–102. [Google Scholar] [CrossRef]
- Wang, Y.; Shah, N.; Huffman, G.P. Pure hydrogen production by partial dehydrogenation of cyclohexane and methylcyclohexane over nanotube-supported pt and pd catalysts. Energy Fuels 2004, 18, 1429–1433. [Google Scholar] [CrossRef]
- Zieliński, M.; Pietrowski, M.; Wojciechowska, M. New promising iridium catalyst for toluene hydrogenation. ChemCatChem 2011, 3, 1653–1658. [Google Scholar] [CrossRef]
- Cooper, B.H.; Donnis, B.B.L. Aromatic saturation of distillates: An overview. Appl. Catal. A 1996, 137, 203–223. [Google Scholar] [CrossRef]
- Huang, T.-C.; Kang, B.-C. Naphthalene hydrogenation over pt/al2o3 catalyst in a trickle bed reactor. Ind. Eng. Chem. Res. 1995, 34, 2349–2357. [Google Scholar] [CrossRef]
- Qian, W.; Shirai, H.; Ifuku, M.; Ishihara, A.; Kabe, T. Reactions of tetralin with tritiated molecular hydrogen on pt/al2o3, pd/al2o3, and pt-pd/al2o3 catalysts. Energy Fuels 2000, 14, 1205–1211. [Google Scholar] [CrossRef]
- Keith, L.H.; Telliard, W.A. Priority pollutants. I. A perspective view. Environ. Sci. Technol. 1979, 13, 416–423. [Google Scholar] [CrossRef]
- Agency for Toxic Substances and Disease Registry (ATSDR), Toxicology Profile for Polyaromatic Hydrocarbons (CD-ROM); CRC Press: Boca Raton, FL, USA, 2005.
- Sun, Z.; Zhang, H.; An, G.; Yang, G.; Liu, Z. Supercritical CO2-facilitating large-scale synthesis of ceo2 nanowires and their application for solvent-free selective hydrogenation of nitroarenes. J. Mater. Chem. 2010, 20, 1947–1952. [Google Scholar]
- List, G.R.; King, J.W. Hydrogenation of Fats and Oils: Theory and Practice; AOCS Press: Urbana, IL, USA, 2011. [Google Scholar]
- Kröcher, O.; Köppel, R.A.; Baiker, A. Sol-gel derived hybrid materials as heterogeneous catalysts for the synthesis of n,n-dimethylformamide from supercritical carbon dioxide. Chem. Commun. 1996, 1497–1498. [Google Scholar]
- Sahle-Demessie, E.; Gonzalez, M.A.; Enriquez, J.; Zhao, Q. Selective oxidation in supercritical carbon dioxide using clean oxidants. Ind. Eng. Chem. Res. 2000, 39, 4858–4864. [Google Scholar] [CrossRef]
- Stevens, J.G.; Bourne, R.A.; Twigg, M.V.; Poliakoff, M. Real-time product switching using a twin catalyst system for the hydrogenation of furfural in supercritical CO2. Angew. Chem. Int. Ed. 2010, 49, 8856–8859. [Google Scholar] [CrossRef]
- Hitzler, M.G.; Smail, F.R.; Ross, S.K.; Poliakoff, M. Selective catalytic hydrogenation of organic compounds in supercritical fluids as a continuous process. Org. Process Res. Dev. 1998, 2, 137–146. [Google Scholar] [CrossRef]
- Arunajatesan, V.; Subramaniam, B.; Hutchenson, K.W.; Herkes, F.E. Fixed-bed hydrogenation of organic compounds in supercritical carbon dioxide. Chem. Eng. Sci. 2001, 56, 1363–1369. [Google Scholar] [CrossRef]
- Shirai, M.; Rode, C.V.; Mine, E.; Sasaki, A.; Sato, O.; Hiyoshi, N. Ring hydrogenation of naphthalene and 1-naphthol over supported metal catalysts in supercritical carbon dioxide solvent. Catal. Today 2006, 115, 248–253. [Google Scholar]
- Hiyoshi, N.; Inoue, T.; Rode, C.V.; Sato, O.; Shirai, M. Tuning cis-decalin selectivity in naphthalene hydrogenation over carbon-supported rhodium catalyst under supercritical carbon dioxide. Catal. Lett. 2006, 106, 133–138. [Google Scholar] [CrossRef]
- Yuan, T.; Marshall, W.D. Catalytic hydrogenation of polycyclic aromatic hydrocarbons over palladium/γ-Al2O3 under mild conditions. J. Hazard. Mater. 2005, 126, 149–157. [Google Scholar] [CrossRef]
- Nelkenbaum, E.; Dror, I.; Berkowitz, B. Reductive hydrogenation of polycyclic aromatic hydrocarbons catalyzed by metalloporphyrins. Chemosphere 2007, 68, 210–217. [Google Scholar] [CrossRef]
- Houndonougbo, Y.; Jin, H.; Rajagopalan, B.; Wong, K.; Kuczera, K.; Subramaniam, B.; Laird, B. Phase equilibria in carbon dioxide expanded solvents: Experiments and molecular simulations. J. Phys. Chem. B 2006, 110, 13195–13202. [Google Scholar]
- Cheng, Z.X.; Zhao, X.G.; Li, J.L.; Zhu, Q.M. Role of support in CO2 reforming of CH4 over a Ni/γ-Al2O3 catalyst. Appl. Catal. A 2001, 205, 31–36. [Google Scholar] [CrossRef]
- Li, C.; Chen, Y.-W. Temperature-programmed reduction studies of nickel oxide/alumina catalysts: Effects of the preparation method. Thermochim. Acta 1995, 256. [Google Scholar] [CrossRef]
- Rynkowski, J.M.; Paryjczak, T.; Lenik, M. On the nature of oxidic phases in NiO/γ-Al2O3 catalysts. Appl. Catal. A 1993, 106, 73–82. [Google Scholar] [CrossRef]
- Wang, S.; Lu, G.Q. Reforming of methane with carbon dioxide over Ni/Al2O3 catalysts: Effect of nickel precursor. Appl. Catal. A 1998, 169, 271–280. [Google Scholar] [CrossRef]
- Mattos, M.; Souza, V.M.; Schmal, M. Supported Nickel catalysts for steam reforming of Methane. In Proceedings of the 2nd Mercosur Congress on Chemical Engineering4th Mercosur Congress on Process Systems Engineering, Rio de Janeiro, Brazil, 14–18 August 2005. ISBN 85-7650-043-4.
- Pendrosa, A.M.G.; Souxa, M.J.B.; Melo, D.M.A.; Araujo, A.S. Cobalt and nickel supported on HY zeolite: Synthesis, characterization and catalytic properties. Res. Bull. 2006, 41, 1105–1111. [Google Scholar] [CrossRef]
- Hampson, J.W. A recirculating equilibrium procedure for determining organic compound solubility in supercritical fluids. Anthracene in carbon dioxide. J. Chem. Eng. Data 1996, 41, 97–100. [Google Scholar] [CrossRef]
- Ashraf-Khorassani, M.; Combs, M.T.; Taylor, L.T.; Schweighardt, F.K.; Mathias, P.S. Solubility study of sulfamethazine and sulfadimethoxine in supercritical carbon dioxide, fluoroform, and subcritical freon 134A. J. Chem. Eng. Data 1997, 42, 636–640. [Google Scholar] [CrossRef]
- Hiyoshi, N.; Rode, C.V.; Sato, O.; Shirai, M. Biphenyl hydrogenation over supported transition metal catalysts under supercritical carbon dioxide solvent. Appl. Catal. A 2005, 288, 43–47. [Google Scholar] [CrossRef]
- Aizawa, T.; Janttarakeeree, S.; Ikushima, Y.; Saitoh, N.; Arai, K.; Smith, R., Jr. Cosolvent effect on enhancement of reaction rate constant in near-critical region. J. Supercrit. Fluids 2003, 27, 247–253. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sahle-Demessie, E.; Devulapelli, V.G.; Hassan, A.A. Hydrogenation of Anthracene in Supercritical Carbon Dioxide Solvent Using Ni Supported on Hβ-Zeolite Catalyst. Catalysts 2012, 2, 85-100. https://doi.org/10.3390/catal2010085
Sahle-Demessie E, Devulapelli VG, Hassan AA. Hydrogenation of Anthracene in Supercritical Carbon Dioxide Solvent Using Ni Supported on Hβ-Zeolite Catalyst. Catalysts. 2012; 2(1):85-100. https://doi.org/10.3390/catal2010085
Chicago/Turabian StyleSahle-Demessie, Endalkachew, Venu Gopal Devulapelli, and Ashraf Aly Hassan. 2012. "Hydrogenation of Anthracene in Supercritical Carbon Dioxide Solvent Using Ni Supported on Hβ-Zeolite Catalyst" Catalysts 2, no. 1: 85-100. https://doi.org/10.3390/catal2010085