
The Therapeutic Targeting of HGF/c-Met Signaling in Hepatocellular Carcinoma: Alternative Approaches

{kind=link}
Abstract
:1. Introduction
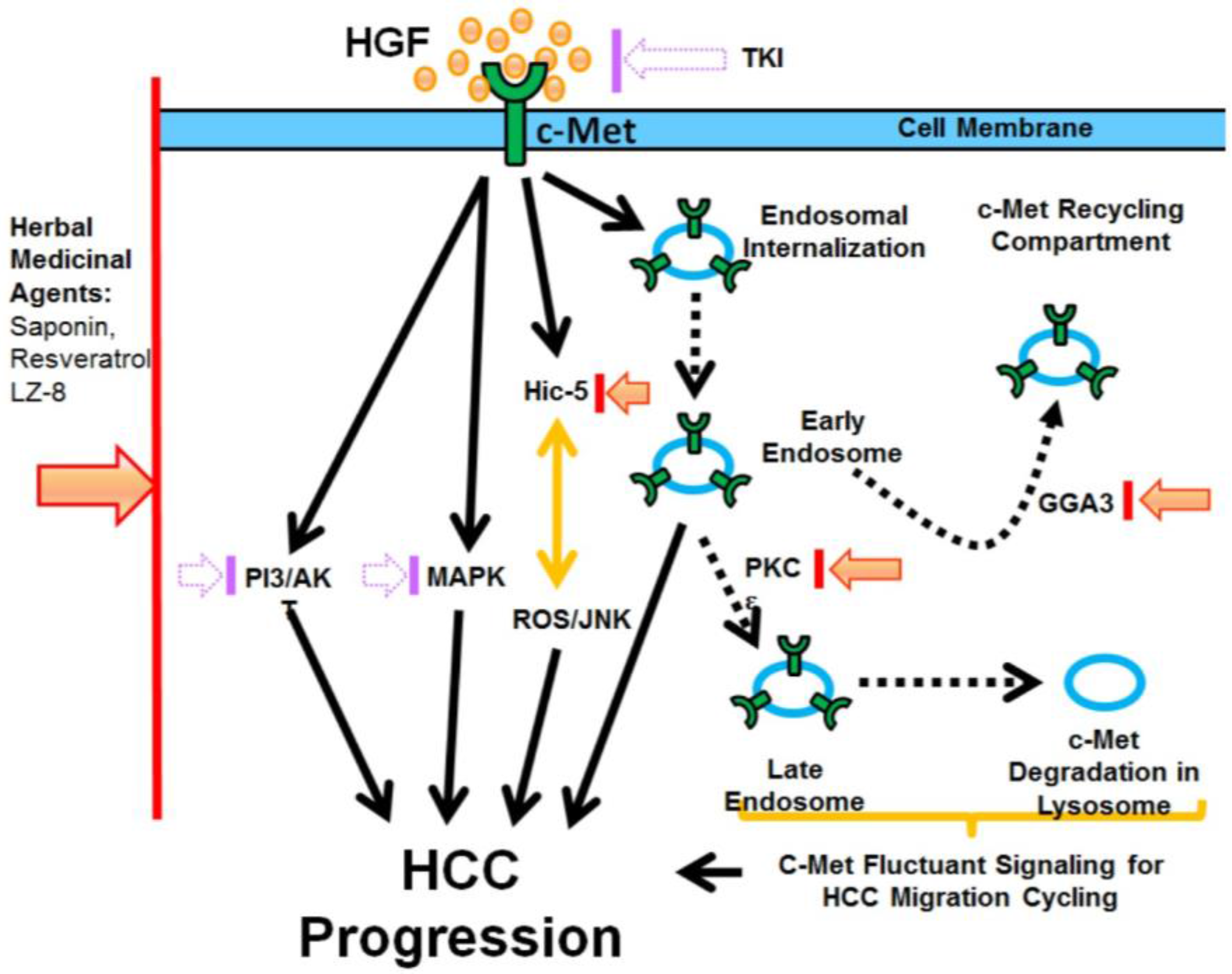
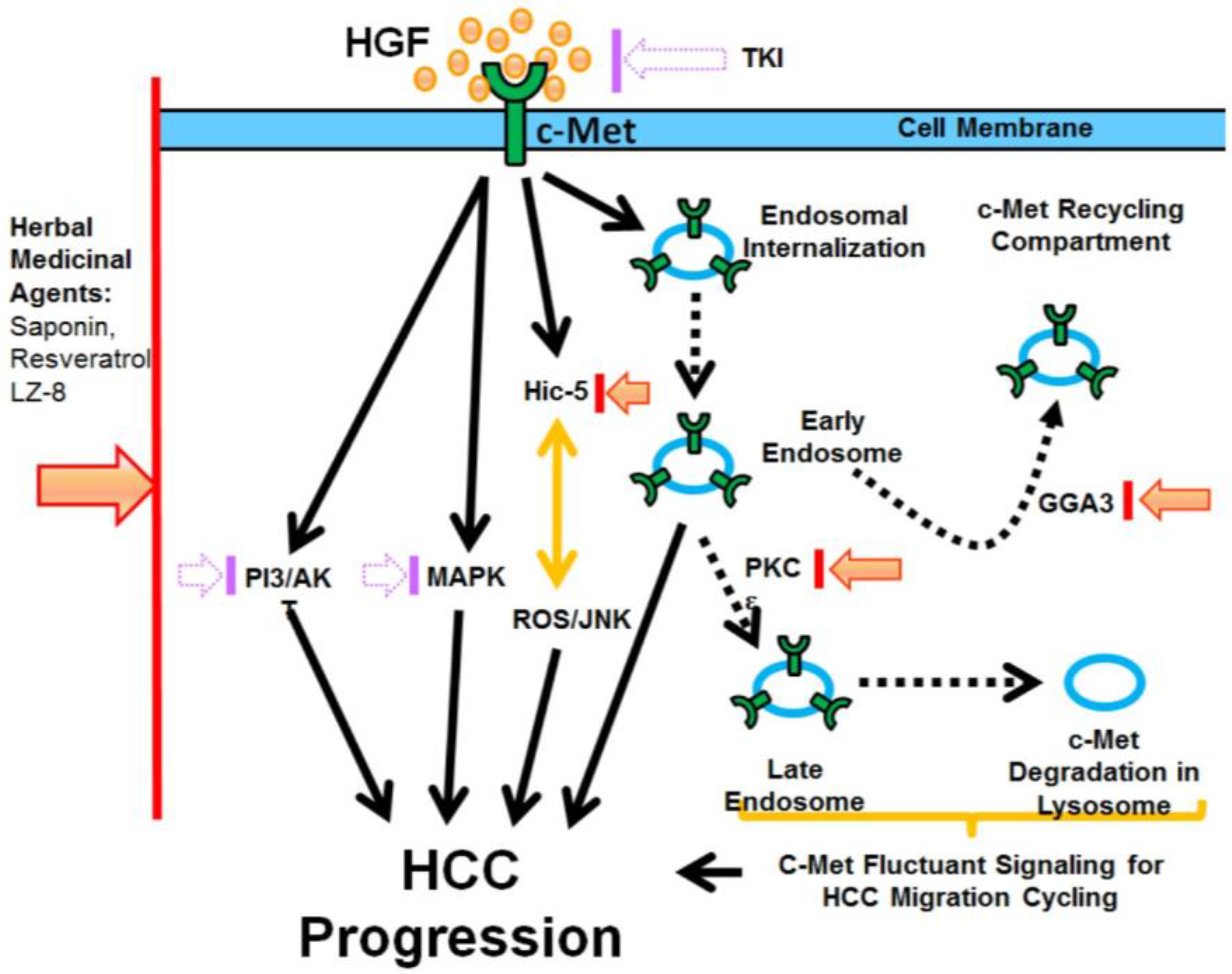
2. HGF-c-Met Signaling Mediates Tumor Progression
3. Target Therapy Aiming at c-Met against HCC Progression
4. Resistance in c-Met Targeting
5. Side Effects in c-Met Targeting
6. Targeting Downstream Effector of c-Met Improves c-Met Target Therapy
6.1. Hic-5 as a Specific Downstream Target of c-Met Pathway
6.2. Targeting c-Met Endosomal Signaling of c-Met
7. Using Herbal Medicinal Antagonists of c-Met to Achieve More Effective and Safer c-Met Targeting
8. Conclusions
Conflicts of Interest
References
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. Met signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.Y.; Pon, Y.L.; Wong, A.S. Hgf/met signaling in ovarian cancer. Curr. Mol. Med. 2008, 8, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Nakamura, T.; Sakai, K.; Nakamura, T. Hepatocyte growth factor and met in tumor biology and therapeutic approach with nk4. Proteomics 2008, 8, 3360–3370. [Google Scholar] [CrossRef] [PubMed]
- Benvenuti, S.; Comoglio, P.M. The met receptor tyrosine kinase in invasion and metastasis. J. Cell. Physiol. 2007, 213, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Lesko, E.; Majka, M. The biological role of hgf-met axis in tumor growth and development of metastasis. Front. Biosci. 2008, 13, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Quesnelle, K.M.; Boehm, A.L.; Grandis, J.R. Stat-mediated egfr signaling in cancer. J. Cell. Biochem. 2007, 102, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (egfr) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Carotenuto, A.; Rachiglio, A.; Gallo, M.; Maiello, M.R.; Aldinucci, D.; Pinto, A.; Normanno, N. The role of the egfr signaling in tumor microenvironment. J. Cell. Physiol. 2008, 214, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Osada, S.; Kanematsu, M.; Imai, H.; Goshima, S. Clinical significance of serum hgf and c-met expression in tumor tissue for evaluation of properties and treatment of hepatocellular carcinoma. Hepato-Gastroenterology 2008, 55, 544–549. [Google Scholar] [PubMed]
- Ogunwobi, O.O.; Liu, C. Hepatocyte growth factor upregulation promotes carcinogenesis and epithelial-mesenchymal transition in hepatocellular carcinoma via akt and cox-2 pathways. Clin. Exp. Metastasis 2008, 28, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Xu, W.; Lu, J.; He, F.; Yang, X. Invasiveness of hepatocellular carcinoma cell lines: Contribution of hepatocyte growth factor, c-met, and transcription factor ets-1. Biochem. Biophys. Res. Commun. 2001, 286, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Ogunwobi, O.O.; Wang, T.; Zhang, L.; Liu, C. Cyclooxygenase-2 and akt mediate multiple growth-factor-induced epithelial-mesenchymal transition in human hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2008, 27, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Lau, E.Y.; Lo, J.; Cheng, B.Y.; Ma, M.K.; Lee, J.M.; Ng, J.K.; Chai, S.; Lin, C.H.; Tsang, S.Y.; Ma, S.; et al. Cancer-Associated Fibroblasts Regulate Tumor-Initiating Cell Plasticity in Hepatocellular Carcinoma through c-Met/FRA1/HEY1 Signaling. Cell Rep. 2016, 15, 1175–1189. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.C.; Wang, T.T.; Liu, W.; Fu, B.S.; Hua, X.; Wang, G.Y.; Li, T.J.; Li, X.; Wu, X.Y.; Tai, Y.; et al. Cancer-associated fibroblasts from hepatocellular carcinoma promote malignant cell proliferation by HGF secretion. PLoS ONE 2013, 8, e63243. [Google Scholar] [CrossRef] [PubMed]
- Poste, G.; Fidler, I.J. The pathogenesis of cancer metastasis. Nature 1980, 283, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Roussos, E.T.; Condeelis, J.S.; Patsialou, A. Chemotaxis in cancer. Nat. Rev. Cancer 2011, 11, 573–587. [Google Scholar] [CrossRef] [PubMed]
- Condeelis, J.; Segall, J.E. Intravital imaging of cell movement in tumours. Nat. Rev. Cancer 2003, 3, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, S.; Marais, R.; Zhu, A.X. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene 2010, 29, 4989–5005. [Google Scholar] [CrossRef] [PubMed]
- Maroun, C.R.; Rowlands, T. The met receptor tyrosine kinase: A key player in oncogenesis and drug resistance. Pharmacol. Ther. 2014, 142, 316–338. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Coranguez, M.; Segovia, J.; Lopez-Ornelas, A.; Puerta-Guardo, H.; Ludert, J.; Chavez, B.; Meraz-Cruz, N.; Gonzalez-Mariscal, L. Transmigration of neural stem cells across the blood brain barrier induced by glioma cells. PLoS ONE 2013, 8, e60655. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Ding, W.; Dang, H.; Jiang, Y.; Rountree, C.B. c-Met represents a potential therapeutic target for personalized treatment in hepatocellular carcinoma. Hepatology 2011, 54, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Inagaki, Y.; Song, P.; Qu, X.; Kokudo, N.; Tang, W. Targeting c-met as a promising strategy for the treatment of hepatocellular carcinoma. Pharmacol. Res. 2012, 65, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Blagotinsek, K.; Rozman, D. Targeting signalling pathways in hepatocellular carcinoma. Curr. Pharm. Des. 2016, 23, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.; Columbano, A. Met as a therapeutic target in hcc: Facts and hopes. J. Hepatol. 2014, 60, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Goyal, L.; Muzumdar, M.D.; Zhu, A.X. Targeting the hgf/c-met pathway in hepatocellular carcinoma. Clin. Cancer Res. 2013, 19, 2310–2318. [Google Scholar] [CrossRef] [PubMed]
- Graveel, C.R.; Tolbert, D.; Vande Woude, G.F. Met: A critical player in tumorigenesis and therapeutic target. Cold Spring Harb. Perspect. Biol. 2013, 5, a009209. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Deng, G.; Liu, W.; Zhou, K.; Li, M. Resveratrol suppresses human hepatocellular carcinoma via targeting HGF-c-Met signaling pathway. Oncol Rep. 2017, 37, 1203–1211. [Google Scholar] [CrossRef] [PubMed]
- Ponzetto, C.; Bardelli, A.; Zhen, Z.; Maina, F.; dalla Zonca, P.; Giordano, S.; Graziani, A.; Panayotou, G.; Comoglio, P.M. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell 1994, 77, 261–271. [Google Scholar] [CrossRef]
- Pelicci, G.; Giordano, S.; Zhen, Z.; Salcini, A.E.; Lanfrancone, L.; Bardelli, A.; Panayotou, G.; Waterfield, M.D.; Ponzetto, C.; Pelicci, P.G.; et al. The motogenic and mitogenic responses to hgf are amplified by the shc adaptor protein. Oncogene 1995, 10, 1631–1638. [Google Scholar] [PubMed]
- Zeng, Q.; Chen, S.; You, Z.; Yang, F.; Carey, T.E.; Saims, D.; Wang, C.Y. Hepatocyte growth factor inhibits anoikis in head and neck squamous cell carcinoma cells by activation of erk and akt signaling independent of nfkappa b. J. Biol. Chem. 2002, 277, 25203–25208. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Hyun, M.S.; Kim, J.R. Growth factor-dependent activation of the mapk pathway in human pancreatic cancer: Mek/erk and p38 map kinase interaction in upa synthesis. Clin. Exp. Metastasis 2003, 20, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Recio, J.A.; Merlino, G. Hepatocyte growth factor/scatter factor activates proliferation in melanoma cells through p38 mapk, atf-2 and cyclin d1. Oncogene 2002, 21, 1000–1008. [Google Scholar] [CrossRef] [PubMed]
- Kermorgant, S.; Parker, P.J. C-met signalling: Spatio-temporal decisions. Cell Cycle 2005, 4, 352–355. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Dou, C.; Lu, Z.; Zheng, X.; Liu, Q. Macc1 suppresses cell apoptosis in hepatocellular carcinoma by targeting the hgf/c-met/akt pathway. Cell Physiol. Biochem. 2015, 35, 983–996. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, X.F.; Lu, X.; Jia, H.L.; Liang, L.; Dong, Q.Z.; Ye, Q.H.; Qin, L.X. Microrna-26a suppresses angiogenesis in human hepatocellular carcinoma by targeting hepatocyte growth factor-cmet pathway. Hepatology 2014, 59, 1874–1885. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Li, R.; Ding, K.; Lobie, P.E.; Zhu, T. Mir-198 inhibits migration and invasion of hepatocellular carcinoma cells by targeting the hgf/c-met pathway. FEBS Lett. 2011, 585, 2229–2234. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.; Yeganeh, M.; Donates, Y.C.; Tobelaim, W.S.; Chababi, W.; Mayhue, M.; Yoshimura, A.; Ramanathan, S.; Saucier, C.; Ilangumaran, S. Regulation of met receptor tyrosine kinase signaling by suppressor of cytokine signaling 1 in hepatocellular carcinoma. Oncogene 2015, 34, 5718–5728. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Li, H.; Jiang, X.; Zhai, B.; Tan, G.; Zhao, D.; Qiao, H.; Liu, B.; Jiang, H.; Sun, X. Dual inhibition of akt and c-met as a second-line therapy following acquired resistance to sorafenib in hepatocellular carcinoma cells. Mol. Oncol. 2017, 11, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Firtina Karagonlar, Z.; Koc, D.; Iscan, E.; Erdal, E.; Atabey, N. Elevated hepatocyte growth factor expression as an autocrine c-met activation mechanism in acquired resistance to sorafenib in hepatocellular carcinoma cells. Cancer Sci. 2016, 107, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.J.; Inagaki, Y.; Xue, X.; Qu, X.J.; Tang, W. c-Met: A potential therapeutic target for hepatocellular carcinoma. Drug Discov. Ther. 2011, 5, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Heideman, D.A.; Overmeer, R.M.; van Beusechem, V.W.; Lamers, W.H.; Hakvoort, T.B.; Snijders, P.J.; Craanen, M.E.; Offerhaus, G.J.; Meijer, C.J.; Gerritsen, W.R. Inhibition of angiogenesis and hgf-cmet-elicited malignant processes in human hepatocellular carcinoma cells using adenoviral vector-mediated nk4 gene therapy. Cancer Gene Ther. 2005, 12, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Xing, R.; Chen, P.; Gou, Y.; Li, S.; Xiao, J.; Dong, J. Down-regulation of c-met expression inhibits human hcc cells growth and invasion by rna interference. J. Surg. Res. 2010, 162, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Okuma, H.S.; Kondo, S. Trends in the development of met inhibitors for hepatocellular carcinoma. Future Oncol. 2016, 12, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.S.; Guo, X.Z.; Han, G.H.; Li, H.Y.; Chen, J. MET inhibitors for treatment of advanced hepatocellular carcinoma: A review. World J. Gastroenterol. 2015, 21, 5445–5453. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.Y.; Yoo, S.Y.; Heo, J. New chemical treatment options in second-line hepatocellular carcinoma: What to do when sorafenib fails? Expert Opin. Pharmacother. 2016, 18, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Nishio, M.; Horiike, A.; Nokihara, H.; Horinouchi, H.; Nakamichi, S.; Wakui, H.; Ohyanagi, F.; Kudo, K.; Yanagitani, N.; Takahashi, S.; et al. Phase I study of the anti-MET antibody onartuzumab in patients with solid tumors and MET-positive lung cancer. Investig. New Drugs 2015, 33, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Elez, M.E.; Herranz, M.; Rico, I.; Prudkin, L.; Andreu, J.; Mateos, J.; Carreras, M.J.; Han, M.; Gifford, J.; et al. A pharmacodynamic/pharmacokinetic study of ficlatuzumab in patients with advanced solid tumors and liver metastases. Clin. Cancer Res. 2014, 20, 2793–2804. [Google Scholar] [CrossRef] [PubMed]
- Karagonlar, Z.F.; Korhan, P.; Atabey, N. Targeting c-Met in Cancer by MicroRNAs: Potential Therapeutic Applications in Hepatocellular Carcinoma. Drug Dev. Res. 2015, 76, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Cepero, V.; Sierra, J.R.; Corso, S.; Ghiso, E.; Casorzo, L.; Perera, T.; Comoglio, P.M.; Giordano, S. Met and kras gene amplification mediates acquired resistance to met tyrosine kinase inhibitors. Cancer Res. 2010, 70, 7580–7590. [Google Scholar] [CrossRef] [PubMed]
- Blivet-Van Eggelpoel, M.J.; Chettouh, H.; Fartoux, L.; Aoudjehane, L.; Barbu, V.; Rey, C.; Priam, S.; Housset, C.; Rosmorduc, O.; Desbois-Mouthon, C. Epidermal growth factor receptor and her-3 restrict cell response to sorafenib in hepatocellular carcinoma cells. J. Hepatol. 2012, 57, 108–115. [Google Scholar] [CrossRef] [PubMed]
- McDermott, U.; Pusapati, R.V.; Christensen, J.G.; Gray, N.S.; Settleman, J. Acquired resistance of non-small cell lung cancer cells to met kinase inhibition is mediated by a switch to epidermal growth factor receptor dependency. Cancer Res. 2010, 70, 1625–1634. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; McTigue, M.A.; Rogers, A.; Lifshits, E.; Christensen, J.G.; Janne, P.A.; Engelman, J.A. Multiple mutations and bypass mechanisms can contribute to development of acquired resistance to met inhibitors. Cancer Res. 2011, 71, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, J.W.; Lu, L.G.; Shao, P.J.; Hu, B.S.; Huang, G.M.; Wei, Z.G.; Zhang, L. Clinical analysis of the treatment: Transcatheter arterial chemoembolization combined with sorafenib in advanced hepatocellular carcinoma. Zhonghua Yi Xue Za Zhi 2010, 90, 2187–2192. [Google Scholar] [PubMed]
- Suzuki, M.; Shiraha, H.; Fujikawa, T.; Takaoka, N.; Ueda, N.; Nakanishi, Y.; Koike, K.; Takaki, A.; Shiratori, Y. Des-gamma-carboxy prothrombin is a potential autologous growth factor for hepatocellular carcinoma. J. Biol. Chem. 2005, 280, 6409–6415. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Ferrell, L.D.; Faouzi, S.; Maher, J.J.; Bishop, J.M. Activation of the met receptor by cell attachment induces and sustains hepatocellular carcinomas in transgenic mice. J. Cell Boil. 2001, 153, 1023–1034. [Google Scholar] [CrossRef]
- Steinway, S.N.; Dang, H.; You, H.; Rountree, C.B.; Ding, W. The egfr/erbb3 pathway acts as a compensatory survival mechanism upon c-met inhibition in human c-met+ hepatocellular carcinoma. PLoS ONE 2015, 10, e0128159. [Google Scholar] [CrossRef] [PubMed]
- Diaz, D.; Ford, K.A.; Hartley, D.P.; Harstad, E.B.; Cain, G.R.; Achilles-Poon, K.; Nguyen, T.; Peng, J.; Zheng, Z.; Merchant, M.; et al. Pharmacokinetic drivers of toxicity for basic molecules: Strategy to lower pka results in decreased tissue exposure and toxicity for a small molecule met inhibitor. Toxicol. Appl. Pharmacol. 2013, 266, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.J.; Shen, H.; Tran-Dube, M.; Nambu, M.; McTigue, M.; Grodsky, N.; Ryan, K.; Yamazaki, S.; Aguirre, S.; Parker, M.; et al. Lessons from (s)-6-(1-(6-(1-methyl-1h-pyrazol-4-yl)-[1,2,4]triazolo[4,3-b]pyridazin-3-yl)ethyl)quinoline (pf-04254644), an inhibitor of receptor tyrosine kinase c-met with high protein kinase selectivity but broad phosphodiesterase family inhibition leading to myocardial degeneration in rats. J. Med. Chem. 2013, 56, 6651–6665. [Google Scholar] [PubMed]
- Santoro, A.; Simonelli, M.; Rodriguez-Lope, C.; Zucali, P.; Camacho, L.H.; Granito, A.; Senzer, N.; Rimassa, L.; Abbadessa, G.; Schwartz, B.; et al. A phase-1b study of tivantinib (arq 197) in adult patients with hepatocellular carcinoma and cirrhosis. Br. J. Cancer 2013, 108, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Vaishampayan, U.; Rosenberg, J.E.; Logan, T.F.; Harzstark, A.L.; Bukowski, R.M.; Rini, B.I.; Srinivas, S.; Stein, M.N.; Adams, L.M.; et al. Phase ii and biomarker study of the dual met/vegfr2 inhibitor foretinib in patients with papillary renal cell carcinoma. J. Clin. Oncol. 2013, 31, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; Vande Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003, 4, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Atabey, N.; Gao, Y.; Yao, Z.J.; Breckenridge, D.; Soon, L.; Soriano, J.V.; Burke, T.R., Jr.; Bottaro, D.P. Potent blockade of hepatocyte growth factor-stimulated cell motility, matrix invasion and branching morphogenesis by antagonists of grb2 src homology 2 domain interactions. J. Biol. Chem. 2001, 276, 14308–14314. [Google Scholar] [CrossRef] [PubMed]
- Shibanuma, M.; Mashimo, J.; Kuroki, T.; Nose, K. Characterization of the tgf beta 1-inducible hic-5 gene that encodes a putative novel zinc finger protein and its possible involvement in cellular senescence. J. Biol. Chem. 1994, 269, 26767–26774. [Google Scholar] [PubMed]
- Brown, M.C.; Turner, C.E. Paxillin: Adapting to change. Physiol. Rev. 2004, 84, 1315–1339. [Google Scholar] [CrossRef] [PubMed]
- Deakin, N.O.; Turner, C.E. Distinct roles for paxillin and hic-5 in regulating breast cancer cell morphology, invasion, and metastasis. Mol. Biol. Cell 2011, 22, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.R.; Hu, C.T.; You, R.I.; Pan, S.M.; Cheng, C.C.; Lee, M.C.; Wu, C.C.; Chang, Y.J.; Lin, S.C.; Chen, C.S.; et al. Hydrogen peroxide inducible clone-5 mediates reactive oxygen species signaling for hepatocellular carcinoma progression. Oncotarget 2015, 6, 32526–32544. [Google Scholar] [PubMed]
- Hu, C.T.; Wu, J.R.; Wu, W.S. The role of endosomal signaling triggered by metastatic growth factors in tumor progression. Cell. Signal. 2013, 25, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Sorkin, A.; von Zastrow, M. Endocytosis and signalling: Intertwining molecular networks. Nat. Rev. Mol. Cell Biol. 2009, 10, 609–622. [Google Scholar] [CrossRef] [PubMed]
- McMahon, H.T.; Boucrot, E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2011, 12, 517–533. [Google Scholar] [CrossRef] [PubMed]
- Polo, S.; Di Fiore, P.P. Endocytosis conducts the cell signaling orchestra. Cell 2006, 124, 897–900. [Google Scholar] [CrossRef] [PubMed]
- Parachoniak, C.A.; Luo, Y.; Abella, J.V.; Keen, J.H.; Park, M. Gga3 functions as a switch to promote met receptor recycling, essential for sustained erk and cell migration. Dev. Cell 2011, 20, 751–763. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J. Met receptor: A moving target. Sci. Signal. 2011, 4, pe40. [Google Scholar] [CrossRef] [PubMed]
- Alvi, F.; Idkowiak-Baldys, J.; Baldys, A.; Raymond, J.R.; Hannun, Y.A. Regulation of membrane trafficking and endocytosis by protein kinase c: Emerging role of the pericentrion, a novel protein kinase c-dependent subset of recycling endosomes. Cell. Mol. Life Sci. 2007, 64, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Kermorgant, S.; Zicha, D.; Parker, P.J. Protein kinase c controls microtubule-based traffic but not proteasomal degradation of c-met. J. Biol. Chem. 2003, 278, 28921–28929. [Google Scholar] [CrossRef] [PubMed]
- Kermorgant, S.; Zicha, D.; Parker, P.J. Pkc controls hgf-dependent c-met traffic, signalling and cell migration. EMBO J. 2004, 23, 3721–3734. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.T.; Cheng, C.C.; Wu, J.R.; Pan, S.M.; Wu, W.S. Pkcepsilon-mediated c-met endosomal processing directs fluctuant c-met-jnk-paxillin signaling for tumor progression of hepg2. Cell. Signal. 2015, 27, 1544–1555. [Google Scholar] [CrossRef] [PubMed]
- Joffre, C.; Barrow, R.; Menard, L.; Calleja, V.; Hart, I.R.; Kermorgant, S. A direct role for met endocytosis in tumorigenesis. Nat. Cell Biol. 2011, 13, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Barrow-McGee, R.; Kermorgant, S. Met endosomal signalling: In the right place, at the right time. Int. J. Biochem. Cell Biol. 2014, 49, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.H.; Yang, C.W.; Wang, J.Y.; Tsai, Y.F.; Tseng, L.M.; King, K.L.; Chen, W.S.; Chiu, J.H.; Shyr, Y.M. Timosaponin aiii suppresses hepatocyte growth factor-induced invasive activity through sustained erk activation in breast cancer mda-mb-231 cells. Evid. Based Complement. Alternat. Med. 2013, 2013, 421051. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Deng, G.; Liu, W.; Zhou, K.; Li, M. Resveratrol suppresses human hepatocellular carcinoma via targeting hgf-c-met signaling pathway. Oncol. Rep. 2017, 37, 1203–1211. [Google Scholar] [CrossRef] [PubMed]
- Amitani, M.; Amitani, H.; Sloan, R.A.; Suzuki, H.; Sameshima, N.; Asakawa, A.; Nerome, Y.; Owaki, T.; Inui, A.; Hoshino, E. The translational aspect of complementary and alternative medicine for cancer with particular emphasis on kampo. Front. Pharmacol. 2015, 6, 150. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.R.; Hu, C.T.; You, R.I.; Ma, P.L.; Pan, S.M.; Lee, M.C.; Wu, W.S. Preclinical trials for prevention of tumor progression of hepatocellular carcinoma by lz-8 targeting c-met dependent and independent pathways. PLoS ONE 2015, 10, e0114495. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, C.-T.; Wu, J.-R.; Cheng, C.-C.; Wu, W.-S. The Therapeutic Targeting of HGF/c-Met Signaling in Hepatocellular Carcinoma: Alternative Approaches. Cancers 2017, 9, 58. https://doi.org/10.3390/cancers9060058
Hu C-T, Wu J-R, Cheng C-C, Wu W-S. The Therapeutic Targeting of HGF/c-Met Signaling in Hepatocellular Carcinoma: Alternative Approaches. Cancers. 2017; 9(6):58. https://doi.org/10.3390/cancers9060058
Chicago/Turabian StyleHu, Chi-Tan, Jia-Ru Wu, Chuan-Chu Cheng, and Wen-Sheng Wu. 2017. "The Therapeutic Targeting of HGF/c-Met Signaling in Hepatocellular Carcinoma: Alternative Approaches" Cancers 9, no. 6: 58. https://doi.org/10.3390/cancers9060058