
Hepatocyte Growth Factor, a Key Tumor-Promoting Factor in the Tumor Microenvironment

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
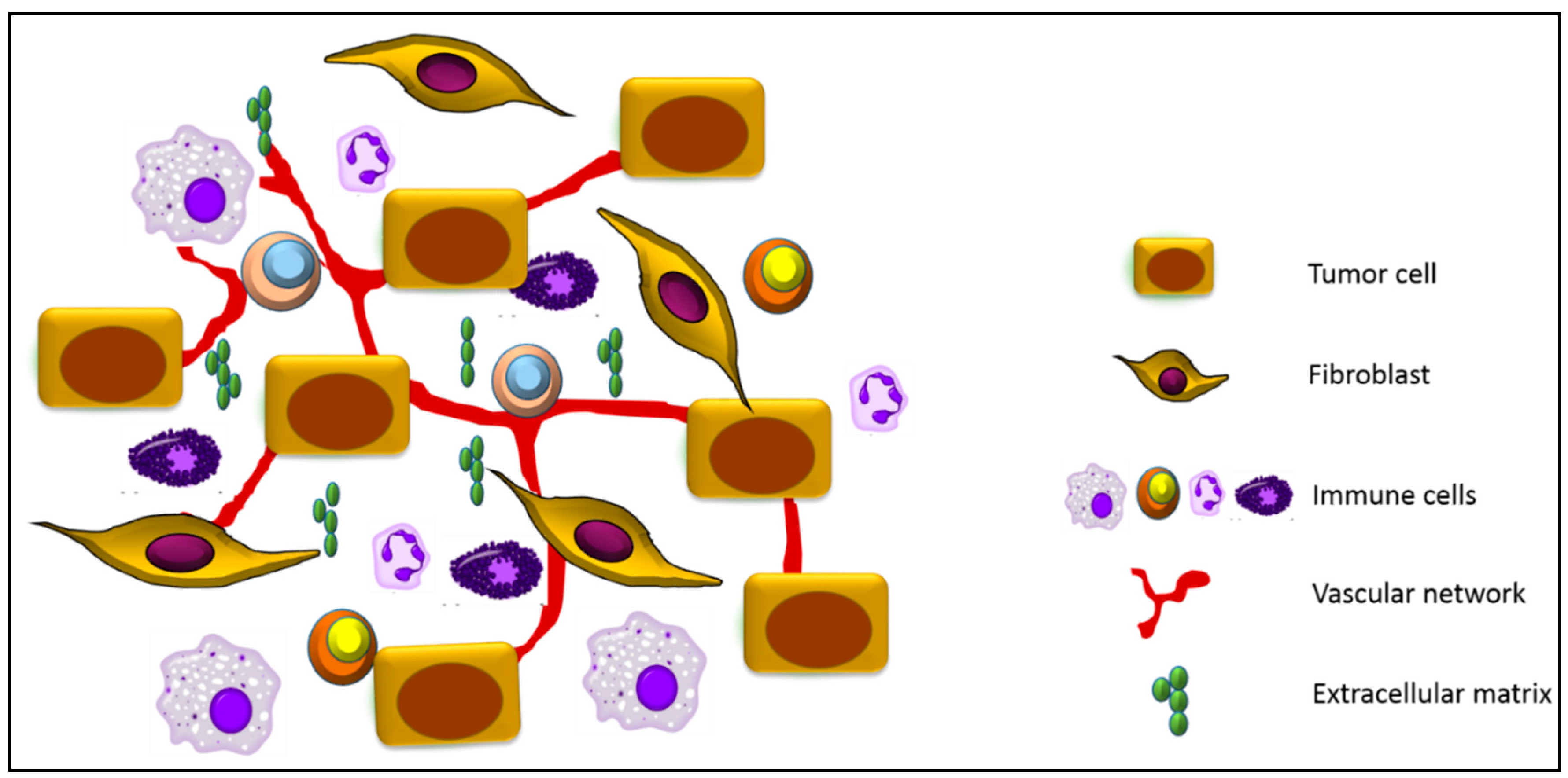
:1. Tumor Microenvironment
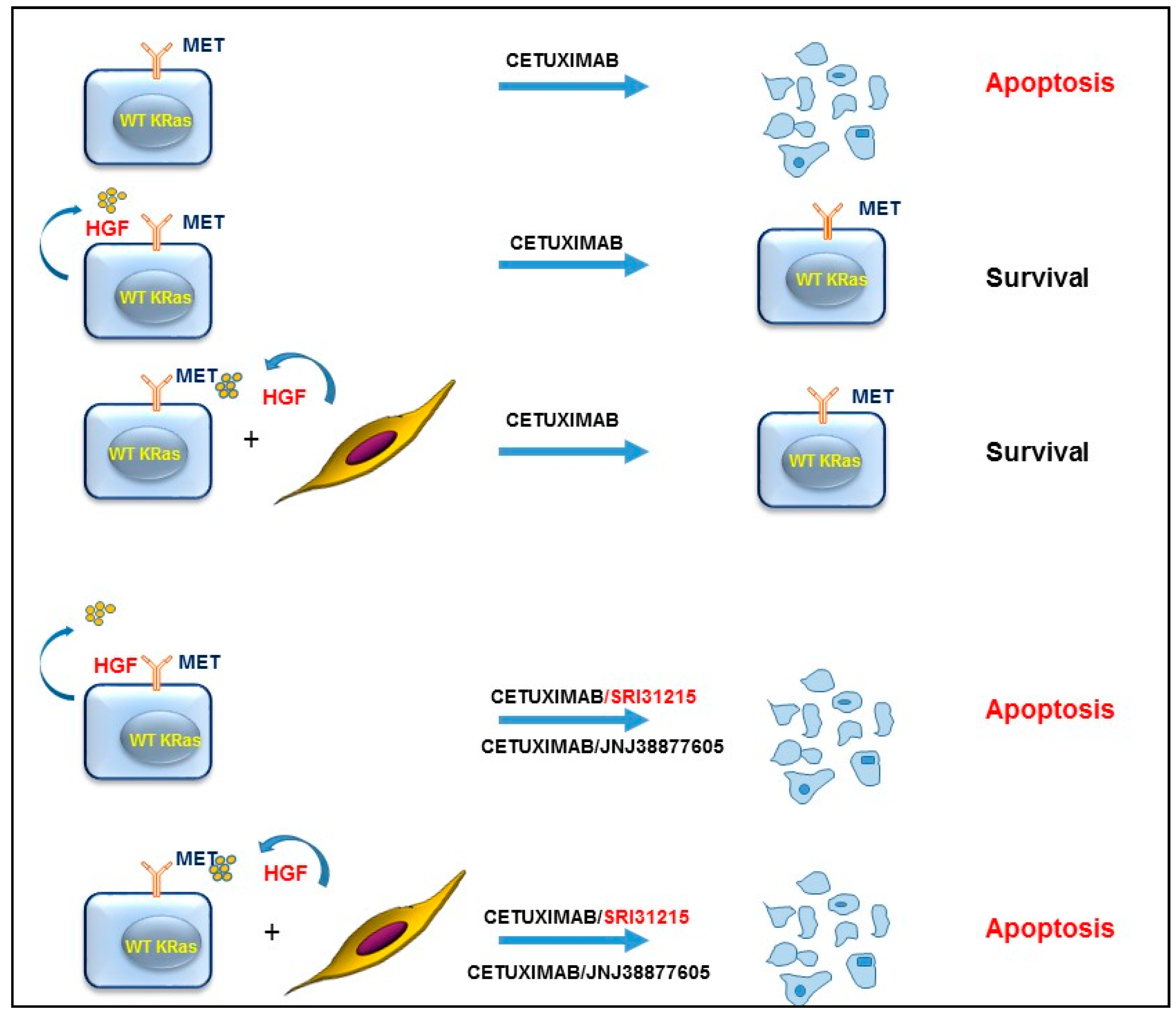
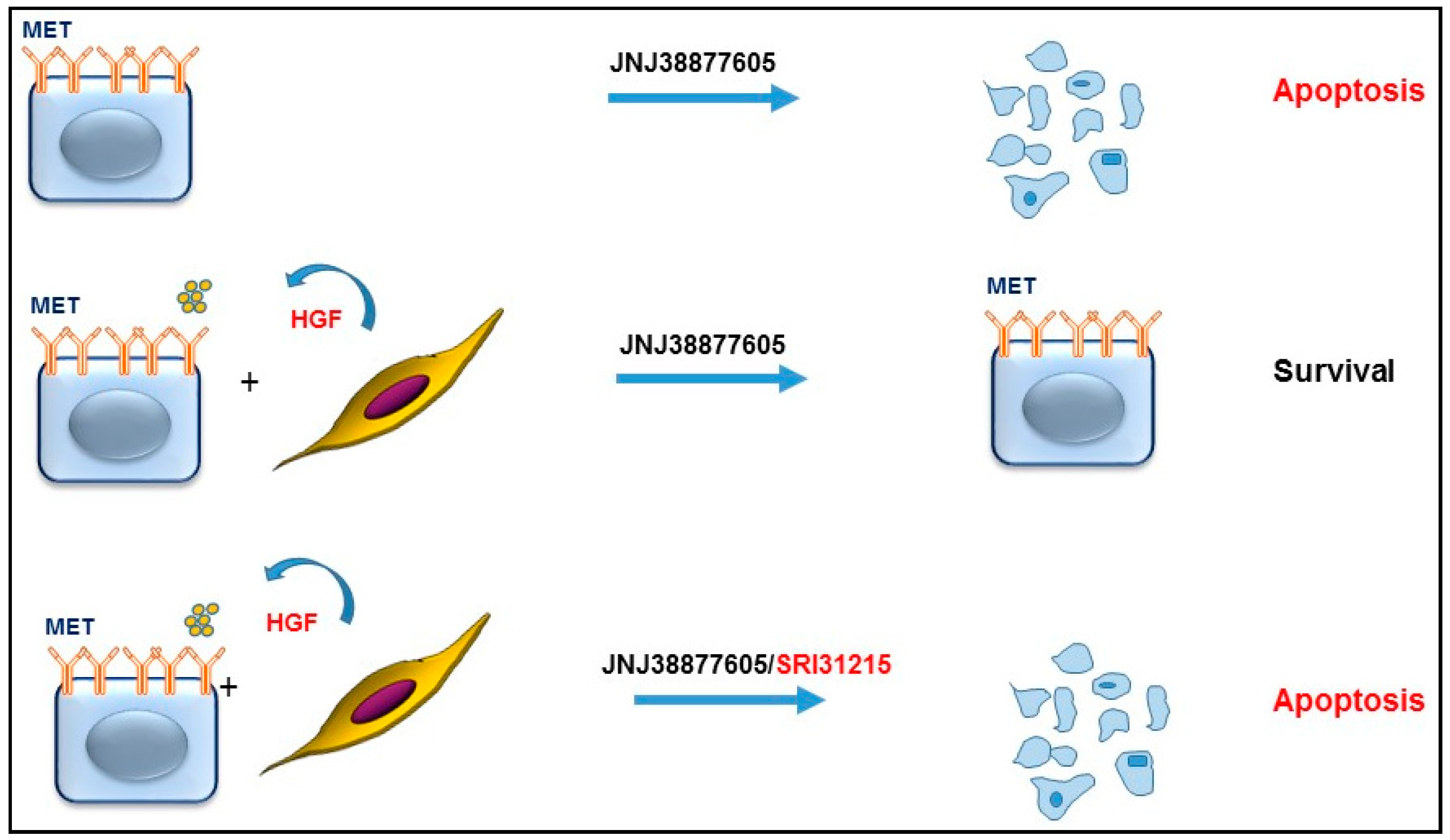
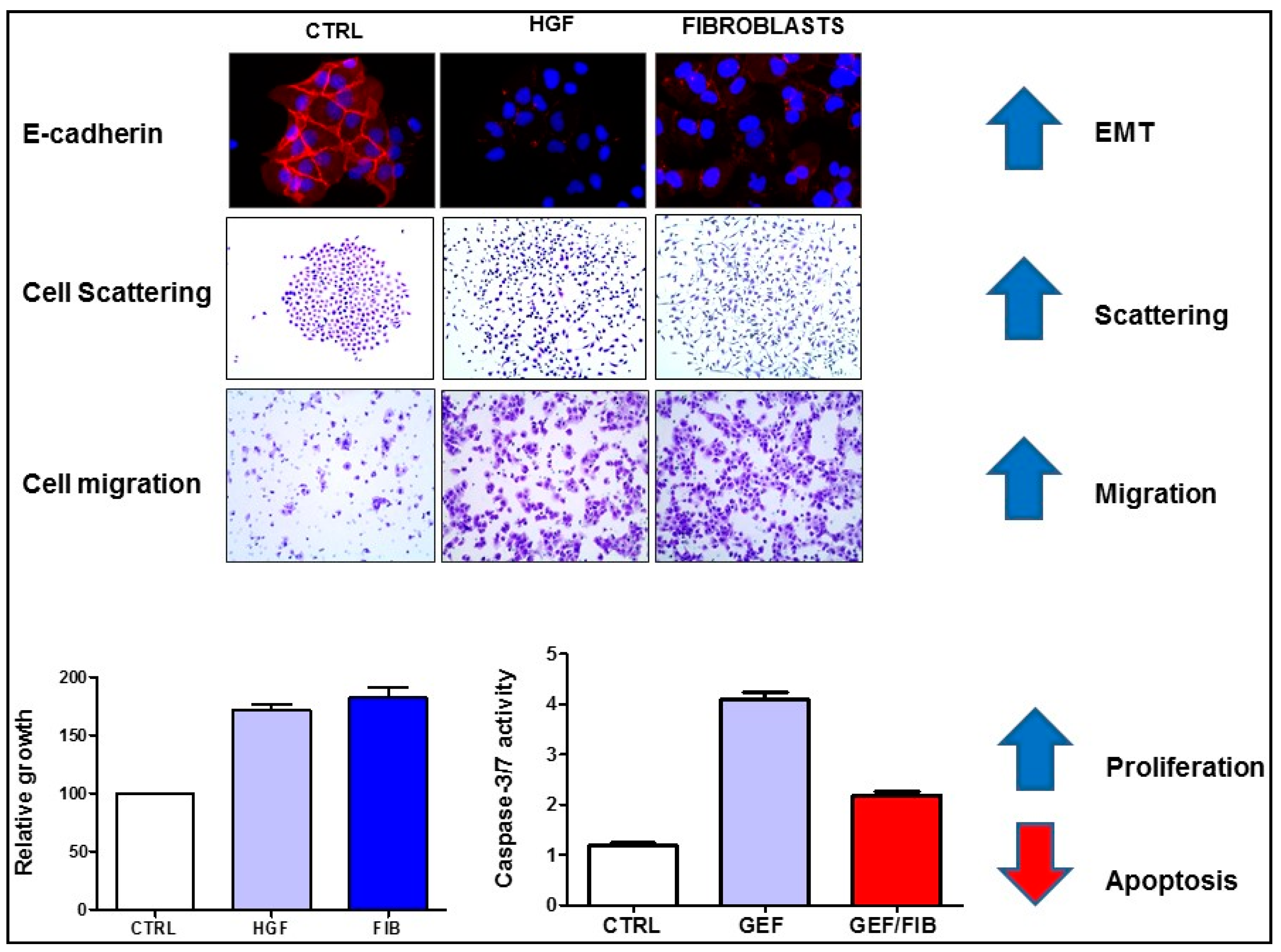
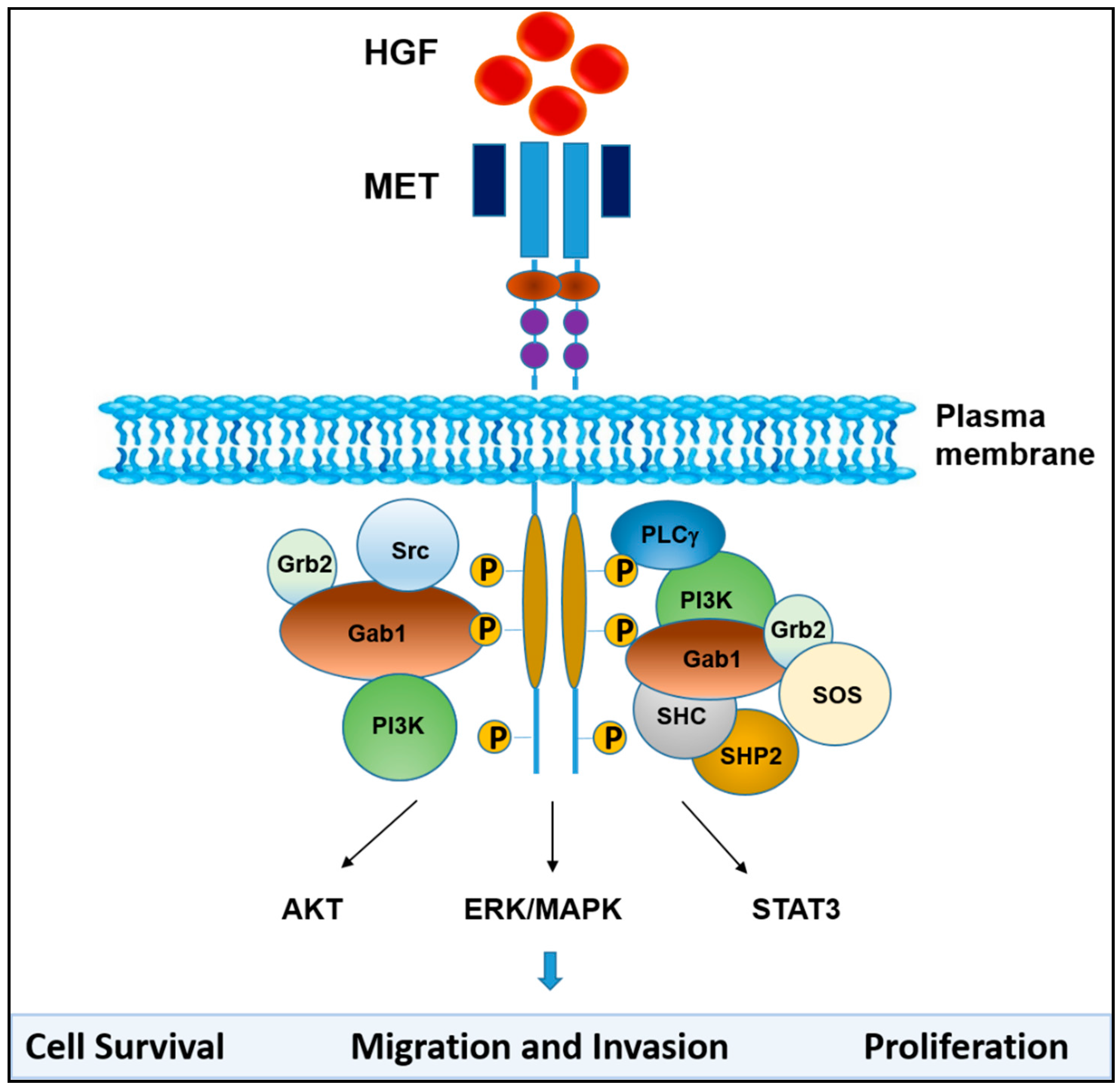
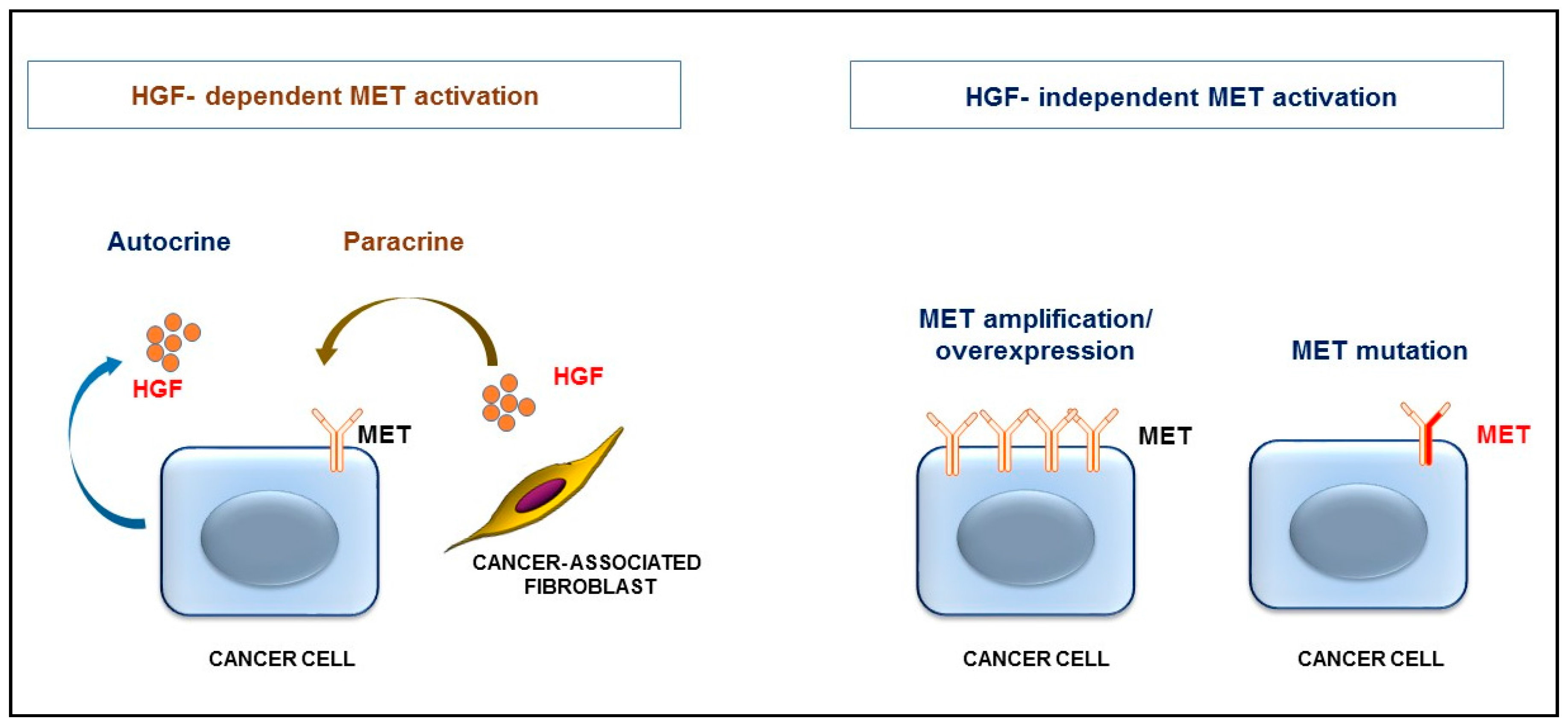
2. HGF/MET Signaling in the Tumor Microenvironment
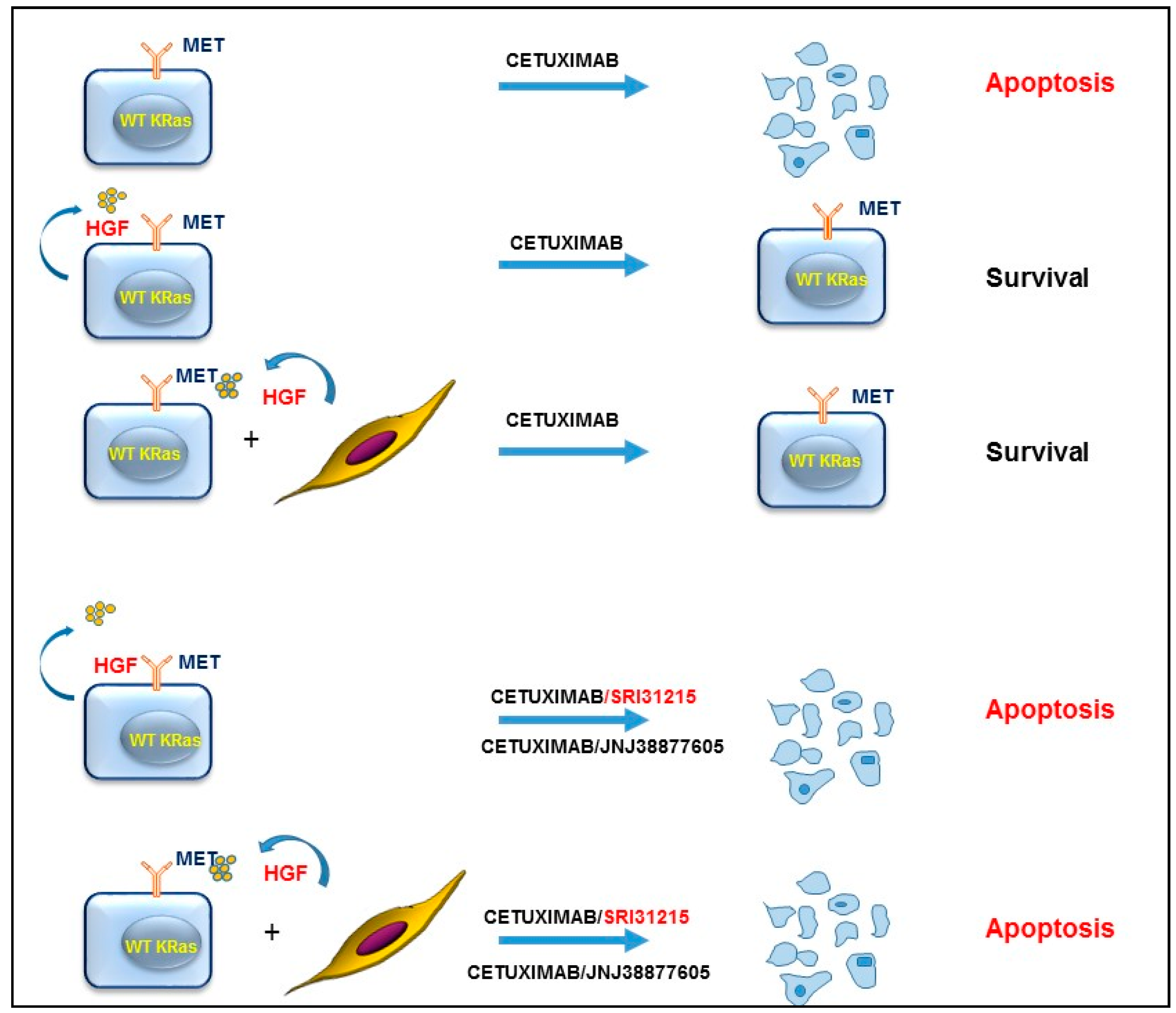
3. HGF/MET Signaling Is a Hallmark of Therapeutic Resistance
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Egeblad, M.; Nakasone, E.S.; Werb, Z. Tumors as organs: Complex tissues that interface with the entire organism. Dev. Cell 2010, 18, 884–901. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Fan, X.; Houghton, J. Tumor microenvironment: The role of the tumor stroma in cancer. J. Cell. Biochem. 2007, 101, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y. Tumor microenvironment and cancer therapy resistance. Cancer Lett. 2016, 380, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef] [PubMed]
- Lisanti, M.P.; Martinez-Outschoorn, U.E.; Sotgia, F. Oncogenes induce the cancer-associated fibroblast phenotype: Metabolic symbiosis and “fibroblast addiction” are new therapeutic targets for drug discovery. Cell Cycle 2013, 12, 2723–2732. [Google Scholar] [CrossRef] [PubMed]
- Dong-Le Bourhis, X.; Berthois, Y.; Millot, G.; Degeorges, A.; Sylvi, M.; Martin, P.M.; Calvo, F. Effect of stromal and epithelial cells derived from normal and tumorous breast tissue on the proliferation of human breast cancer cell lines in co-culture. Int. J. Cancer J. Int. Cancer 1997, 71, 42–48. [Google Scholar]
- Zitvogel, L.; Tesniere, A.; Kroemer, G. Cancer despite immunosurveillance: Immunoselection and immunosubversion. Nat. Rev. Immunol. 2006, 6, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Bissell, M.J.; Hines, W.C. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 2011, 17, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; Affara, N.I.; Coussens, L.M. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012, 33, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; DeNardo, D.G.; Affara, N.I.; Coussens, L.M. Lymphocytes in cancer development: Polarization towards pro-tumor immunity. Cytokine Growth Factor Rev. 2010, 21, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Gudjonsson, T.; Ronnov-Jessen, L.; Villadsen, R.; Rank, F.; Bissell, M.J.; Petersen, O.W. Normal and tumor-derived myoepithelial cells differ in their ability to interact with luminal breast epithelial cells for polarity and basement membrane deposition. J. Cell Sci. 2002, 115, 39–50. [Google Scholar] [PubMed]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Eng. J. Med. 1986, 315, 1650–1659. [Google Scholar]
- Cruz-Correa, M.; Hylind, L.M.; Romans, K.E.; Booker, S.V.; Giardiello, F.M. Long-term treatment with sulindac in familial adenomatous polyposis: A prospective cohort study. Gastroenterology 2002, 122, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Lochhead, P.; Giovannucci, E.; Meyerhardt, J.A.; Fuchs, C.S.; Chan, A.T. Discovery of colorectal cancer PIK3CA mutation as potential predictive biomarker: Power and promise of molecular pathological epidemiology. Oncogene 2014, 33, 2949–2955. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; De Sousa, E.M.F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Owusu, B.Y.; Bansal, N.; Venukadasula, P.K.; Ross, L.J.; Messick, T.E.; Goel, S.; Galemmo, R.A.; Klampfer, L. Inhibition of pro-HGF activation by SRI31215, a novel approach to block oncogenic HGF/MET signaling. Oncotarget 2016, 7, 29492–29506. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, E.M.F.; Wang, X.; Jansen, M.; Fessler, E.; Trinh, A.; de Rooij, L.P.; de Jong, J.H.; de Boer, O.J.; van Leersum, R.; Bijlsma, M.F.; et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat. Med. 2013, 19, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Marisa, L.; de Reynies, A.; Duval, A.; Selves, J.; Gaub, M.P.; Vescovo, L.; Etienne-Grimaldi, M.C.; Schiappa, R.; Guenot, D.; Ayadi, M.; et al. Gene expression classification of colon cancer into molecular subtypes: Characterization, validation, and prognostic value. PLoS Med. 2013, 10, e1001453. [Google Scholar] [CrossRef] [PubMed]
- Sadanandam, A.; Lyssiotis, C.A.; Homicsko, K.; Collisson, E.A.; Gibb, W.J.; Wullschleger, S.; Ostos, L.C.; Lannon, W.A.; Grotzinger, C.; Del Rio, M.; et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat. Med. 2013, 19, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Calon, A.; Lonardo, E.; Berenguer-Llergo, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.; Byrom, D.; et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet. 2015, 47, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Isella, C.; Terrasi, A.; Bellomo, S.E.; Petti, C.; Galatola, G.; Muratore, A.; Mellano, A.; Senetta, R.; Cassenti, A.; Sonetto, C.; et al. Stromal contribution to the colorectal cancer transcriptome. Nat. Genet. 2015, 47, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Weidner, K.M.; Arakaki, N.; Hartmann, G.; Vandekerckhove, J.; Weingart, S.; Rieder, H.; Fonatsch, C.; Tsubouchi, H.; Hishida, T.; Daikuhara, Y.; et al. Evidence for the identity of human scatter factor and human hepatocyte growth factor. Proc. Natl. Acad. Sci. USA 1991, 88, 7001–7005. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Nishizawa, T.; Hagiya, M.; Seki, T.; Shimonishi, M.; Sugimura, A.; Tashiro, K.; Shimizu, S. Molecular cloning and expression of human hepatocyte growth factor. Nature 1989, 342, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L.; Weidner, K.M.; Vigna, E.; Gaudino, G.; Bardelli, A.; Ponzetto, C.; Narsimhan, R.P.; Hartmann, G.; Zarnegar, R.; Michalopoulos, G.K.; et al. Scatter factor and hepatocyte growth factor are indistinguishable ligands for the met receptor. EMBO J. 1991, 10, 2867–2878. [Google Scholar] [PubMed]
- Stoker, M.; Gherardi, E.; Perryman, M.; Gray, J. Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature 1987, 327, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Dulak, A.M.; Gubish, C.T.; Stabile, L.P.; Henry, C.; Siegfried, J.M. HGF-independent potentiation of EGFR action by c-MET. Oncogene 2011, 30, 3625–3635. [Google Scholar] [CrossRef] [PubMed]
- Pennacchietti, S.; Cazzanti, M.; Bertotti, A.; Rideout, W.M., 3rd; Han, M.; Gyuris, J.; Perera, T.; Comoglio, P.M.; Trusolino, L.; Michieli, P. Microenvironment-derived HGF overcomes genetically determined sensitivity to anti-MET drugs. Cancer Res. 2014, 74, 6598–6609. [Google Scholar] [CrossRef] [PubMed]
- Lesko, E.; Majka, M. The biological role of HGF-MET axis in tumor growth and development of metastasis. Front. Biosci. J. Virtual Libr. 2008, 13, 1271–1280. [Google Scholar] [CrossRef]
- Liska, D.; Chen, C.T.; Bachleitner-Hofmann, T.; Christensen, J.G.; Weiser, M.R. HGF rescues colorectal cancer cells from EGFR inhibition via MET activation. Clin. Cancer Res. 2011, 17, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Comoglio, P.M.; Giordano, S.; Trusolino, L. Drug development of MET inhibitors: Targeting oncogene addiction and expedience. Nat. Rev. Drug Discov. 2008, 7, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Sierra, J.R.; Tsao, M.S. C-MET as a potential therapeutic target and biomarker in cancer. Ther. Adv. Med. Oncol. 2011, 3, S21–S35. [Google Scholar] [CrossRef] [PubMed]
- Fukuura, T.; Miki, C.; Inoue, T.; Matsumoto, K.; Suzuki, H. Serum hepatocyte growth factor as an index of disease status of patients with colorectal carcinoma. Br. J. Cancer 1998, 78, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Toiyama, Y.; Miki, C.; Inoue, Y.; Okugawa, Y.; Tanaka, K.; Kusunoki, M. Serum hepatocyte growth factor as a prognostic marker for stage II or III colorectal cancer patients. Int. J. Cancer. J. Int. Cancer 2009, 125, 1657–1662. [Google Scholar] [CrossRef] [PubMed]
- Toi, M.; Taniguchi, T.; Ueno, T.; Asano, M.; Funata, N.; Sekiguchi, K.; Iwanari, H.; Tominaga, T. Significance of circulating hepatocyte growth factor level as a prognostic indicator in primary breast cancer. Clin. Cancer Res. 1998, 4, 659–664. [Google Scholar] [PubMed]
- Taniguchi, T.; Toi, M.; Inada, K.; Imazawa, T.; Yamamoto, Y.; Tominaga, T. Serum concentrations of hepatocyte growth factor in breast cancer patients. Clin. Cancer Res. 1995, 1, 1031–1034. [Google Scholar] [PubMed]
- Seidel, C.; Borset, M.; Turesson, I.; Abildgaard, N.; Sundan, A.; Waage, A. Elevated serum concentrations of hepatocyte growth factor in patients with multiple myeloma. The nordic myeloma study group. Blood 1998, 91, 806–812. [Google Scholar] [PubMed]
- Verstovsek, S.; Kantarjian, H.; Estey, E.; Aguayo, A.; Giles, F.J.; Manshouri, T.; Koller, C.; Estrov, Z.; Freireich, E.; Keating, M.; et al. Plasma hepatocyte growth factor is a prognostic factor in patients with acute myeloid leukemia but not in patients with myelodysplastic syndrome. Leukemia 2001, 15, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Seneviratne, D.; Ma, J.; Tan, X.; Kwon, Y.K.; Muhammad, E.; Melhem, M.; DeFrances, M.C.; Zarnegar, R. Genomic instability causes HGF gene activation in colon cancer cells, promoting their resistance to necroptosis. Gastroenterology 2015, 148, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; DeFrances, M.C.; Zou, C.; Johnson, C.; Ferrell, R.; Zarnegar, R. Somatic mutation and functional polymorphism of a novel regulatory element in the HGF gene promoter causes its aberrant expression in human breast cancer. J. Clin. Investig. 2009, 119, 478–491. [Google Scholar] [CrossRef] [PubMed]
- Hino, M.; Inaba, M.; Goto, H.; Nishizawa, Y.; Tatsumi, N.; Nishino, T.; Morii, H. Hepatocyte growth factor levels in bone marrow plasma of patients with leukaemia and its gene expression in leukaemic blast cells. Br. J. Cancer 1996, 73, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Kentsis, A.; Reed, C.; Rice, K.L.; Sanda, T.; Rodig, S.J.; Tholouli, E.; Christie, A.; Valk, P.J.; Delwel, R.; Ngo, V.; et al. Autocrine activation of the MET receptor tyrosine kinase in acute myeloid leukemia. Nat. Med. 2012, 18, 1118–1122. [Google Scholar] [CrossRef] [PubMed]
- Reikvam, H.; Nepstad, I.; Bruserud, O.; Hatfield, K.J. Pharmacological targeting of the PI3K/mtor pathway alters the release of angioregulatory mediators both from primary human acute myeloid leukemia cells and their neighboring stromal cells. Oncotarget 2013, 4, 830–843. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Pallangyo, C.K.; Ziegler, P.K.; Greten, F.R. IKKβ acts as a tumor suppressor in cancer-associated fibroblasts during intestinal tumorigenesis. J. Exp. Med. 2015, 212, 2253–2266. [Google Scholar] [CrossRef] [PubMed]
- Luraghi, P.; Reato, G.; Cipriano, E.; Sassi, F.; Orzan, F.; Bigatto, V.; De Bacco, F.; Menietti, E.; Han, M.; Rideout, W.M., 3rd; et al. MET signaling in colon cancer stem-like cells blunts the therapeutic response to egfr inhibitors. Cancer Res. 2014, 74, 1857–1869. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.C.; Wang, T.T.; Liu, W.; Fu, B.S.; Hua, X.; Wang, G.Y.; Li, T.J.; Li, X.; Wu, X.Y.; Tai, Y.; et al. Cancer-associated fibroblasts from hepatocellular carcinoma promote malignant cell proliferation by hgf secretion. PLoS ONE 2013, 8, e63243. [Google Scholar] [CrossRef] [PubMed]
- Swietlicki, E.A.; Bala, S.; Lu, J.; Shaker, A.; Kularatna, G.; Levin, M.S.; Rubin, D.C. Epimorphin deletion inhibits polyposis in the APCmin/+ mouse model of colon carcinogenesis via decreased myofibroblast HGF secretion. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G564–G572. [Google Scholar] [CrossRef] [PubMed]
- Koliaraki, V.; Roulis, M.; Kollias, G. TPL2 regulates intestinal myofibroblast hgf release to suppress colitis-associated tumorigenesis. J. Clin. Investig. 2012, 122, 4231–4242. [Google Scholar] [CrossRef] [PubMed]
- Serebrennikova, O.B.; Tsatsanis, C.; Mao, C.; Gounaris, E.; Ren, W.; Siracusa, L.D.; Eliopoulos, A.G.; Khazaie, K.; Tsichlis, P.N. TPL2 ablation promotes intestinal inflammation and tumorigenesis in apcmin mice by inhibiting IL-10 secretion and regulatory T-cell generation. Proc. Natl. Acad. Sci. USA 2012, 109, E1082–E1091. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, M.; Basilico, C.; Cavassa, S.; Pennacchietti, S.; Risio, M.; Naldini, L.; Comoglio, P.M.; Michieli, P. An uncleavable form of pro-scatter factor suppresses tumor growth and dissemination in mice. J. Clin. Investig. 2004, 114, 1418–1432. [Google Scholar] [CrossRef] [PubMed]
- Forbs, D.; Thiel, S.; Stella, M.C.; Sturzebecher, A.; Schweinitz, A.; Steinmetzer, T.; Sturzebecher, J.; Uhland, K. In vitro inhibition of matriptase prevents invasive growth of cell lines of prostate and colon carcinoma. Int. J. Oncol. 2005, 27, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Herter, S.; Piper, D.E.; Aaron, W.; Gabriele, T.; Cutler, G.; Cao, P.; Bhatt, A.S.; Choe, Y.; Craik, C.S.; Walker, N.; et al. Hepatocyte growth factor is a preferred in vitro substrate for human hepsin, a membrane-anchored serine protease implicated in prostate and ovarian cancers. Biochem. J. 2005, 390, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, H.; Hamasuna, R.; Itoh, H.; Kitamura, N.; Koono, M. Activation of hepatocyte growth factor/scatter factor in colorectal carcinoma. Cancer Res. 2000, 60, 6148–6159. [Google Scholar] [PubMed]
- Lee, S.L.; Dickson, R.B.; Lin, C.Y. Activation of hepatocyte growth factor and urokinase/plasminogen activator by matriptase, an epithelial membrane serine protease. J. Biol. Chem. 2000, 275, 36720–36725. [Google Scholar] [CrossRef] [PubMed]
- Owen, K.A.; Qiu, D.; Alves, J.; Schumacher, A.M.; Kilpatrick, L.M.; Li, J.; Harris, J.L.; Ellis, V. Pericellular activation of hepatocyte growth factor by the transmembrane serine proteases matriptase and hepsin, but not by the membrane-associated protease UPA. Biochem. J. 2010, 426, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Parr, C.; Watkins, G.; Mansel, R.E.; Jiang, W.G. The hepatocyte growth factor regulatory factors in human breast cancer. Clin. Cancer Res. 2004, 10, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Szabo, R.; Rasmussen, A.L.; Moyer, A.B.; Kosa, P.; Schafer, J.M.; Molinolo, A.A.; Gutkind, J.S.; Bugge, T.H. c-MET-induced epithelial carcinogenesis is initiated by the serine protease matriptase. Oncogene 2011, 30, 2003–2016. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, M.; Kataoka, H. Mechanisms of hepatocyte growth factor activation in cancer tissues. Cancers 2014, 6, 1890–1904. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L.; Vigna, E.; Bardelli, A.; Follenzi, A.; Galimi, F.; Comoglio, P.M. Biological activation of pro-HGF (hepatocyte growth factor) by urokinase is controlled by a stoichiometric reaction. J. Biol. Chem. 1995, 270, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Kawaguchi, M.; Haruyama, Y.; Kanemaru, A.; Fukushima, T.; Yamamoto, K.; Lin, C.Y.; Kataoka, H. Loss of hepatocyte growth factor activator inhibitor type 1 participates in metastatic spreading of human pancreatic cancer cells in a mouse orthotopic transplantation model. Cancer Sci. 2014, 105, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, M.; Takeda, N.; Hoshiko, S.; Yorita, K.; Baba, T.; Sawaguchi, A.; Nezu, Y.; Yoshikawa, T.; Fukushima, T.; Kataoka, H. Membrane-bound serine protease inhibitor HAI-1 is required for maintenance of intestinal epithelial integrity. Am. J. Pathol. 2011, 179, 1815–1826. [Google Scholar] [CrossRef] [PubMed]
- Hoshiko, S.; Kawaguchi, M.; Fukushima, T.; Haruyama, Y.; Yorita, K.; Tanaka, H.; Seiki, M.; Inatsu, H.; Kitamura, K.; Kataoka, H. Hepatocyte growth factor activator inhibitor type 1 is a suppressor of intestinal tumorigenesis. Cancer Res. 2013, 73, 2659–2670. [Google Scholar] [CrossRef] [PubMed]
- Saleem, M.; Adhami, V.M.; Zhong, W.; Longley, B.J.; Lin, C.Y.; Dickson, R.B.; Reagan-Shaw, S.; Jarrard, D.F.; Mukhtar, H. A novel biomarker for staging human prostate adenocarcinoma: Overexpression of matriptase with concomitant loss of its inhibitor, hepatocyte growth factor activator inhibitor-1. Cancer Epidemiol. Biomark. Prev. 2006, 15, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Oberst, M.D.; Johnson, M.D.; Dickson, R.B.; Lin, C.Y.; Singh, B.; Stewart, M.; Williams, A.; Al-Nafussi, A.; Smyth, J.F.; Gabra, H.; et al. Expression of the serine protease matriptase and its inhibitor HAI-1 in epithelial ovarian cancer: Correlation with clinical outcome and tumor clinicopathological parameters. Clin. Cancer Res. 2002, 8, 1101–1107. [Google Scholar] [PubMed]
- Zeng, L.; Cao, J.; Zhang, X. Expression of serine protease SNC19/matriptase and its inhibitor hepatocyte growth factor activator inhibitor type 1 in normal and malignant tissues of gastrointestinal tract. World J. Gastroenterol. WJG 2005, 11, 6202–6207. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Abarzua, F.; Kodama, J.; Hongo, A.; Nasu, Y.; Kumon, H.; Hiramatsu, Y. Expression of hepatocyte growth factor activator inhibitors (HAI-1 and HAI-2) in ovarian cancer. Int. J. Oncol. 2009, 34, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Hamasuna, R.; Kataoka, H.; Meng, J.Y.; Itoh, H.; Moriyama, T.; Wakisaka, S.; Koono, M. Reduced expression of hepatocyte growth factor activator inhibitor type-2/placental bikunin (HAI-2/pb) in human glioblastomas: Implication for anti-invasive role of HAI-2/pb in glioblastoma cells. Int. J. Cancer 2001, 93, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.R.; Gentle, D.; Abdulrahman, M.; Maina, E.N.; Gupta, K.; Banks, R.E.; Wiesener, M.S.; Kishida, T.; Yao, M.; Teh, B.; et al. Tumor suppressor activity and epigenetic inactivation of hepatocyte growth factor activator inhibitor type 2/SPINT2 in papillary and clear cell renal cell carcinoma. Cancer Res. 2005, 65, 4598–4606. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; Vande Woude, G. Targeting met in cancer: Rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Harris, P.K.; Karmakar, P.; Kim, T.; Owusu, B.Y.; Wildman, S.A.; Klampfer, L.; Janetka, J.W. Alpha-ketobenzothiazole serine protease inhibitors of aberrant HGF/c-MET and MSP/RON kinase pathway signaling in cancer. ChemMedChem 2016. [Google Scholar] [CrossRef]
- Venukadasula, P.K.; Owusu, B.Y.; Bansal, N.; Ross, L.J.; Hobrath, J.V.; Bao, D.; Truss, J.W.; Stackhouse, M.; Messick, T.E.; Klampfer, L.; et al. Design and synthesis of nonpeptide inhibitors of hepatocyte growth factor activation. ACS Med. Chem. Lett. 2016, 7, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Harris, P.K.; Jones, D.E.; Chugani, R.; Kim, T.; Agarwal, M.; Shen, W.; Wildman, S.A.; Janetka, J.W. Inhibitors of HGFA, matriptase, and hepsin serine proteases: A nonkinase strategy to block cell signaling in cancer. ACS Med. Chem. Lett. 2014, 5, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.; Duh, F.M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; Scherer, S.W.; Zhuang, Z.; Lubensky, I.; Dean, M.; et al. Germline and somatic mutations in the tyrosine kinase domain of the met proto-oncogene in papillary renal carcinomas. Nat. Genet. 1997, 16, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Neklason, D.W.; Done, M.W.; Sargent, N.R.; Schwartz, A.G.; Anton-Culver, H.; Griffin, C.A.; Ahnen, D.J.; Schildkraut, J.M.; Tomlinson, G.E.; Strong, L.C.; et al. Activating mutation in MET oncogene in familial colorectal cancer. BMC Cancer 2011, 11, 424. [Google Scholar] [CrossRef] [PubMed]
- Di Renzo, M.F.; Olivero, M.; Giacomini, A.; Porte, H.; Chastre, E.; Mirossay, L.; Nordlinger, B.; Bretti, S.; Bottardi, S.; Giordano, S.; et al. Overexpression and amplification of the MET/HGF receptor gene during the progression of colorectal cancer. Clin. Cancer Res. 1995, 1, 147–154. [Google Scholar] [PubMed]
- Xing, F.; Liu, Y.; Sharma, S.; Wu, K.; Chan, M.D.; Lo, H.W.; Carpenter, R.L.; Metheny-Barlow, L.J.; Zhou, X.; Qasem, S.A.; et al. Activation of the c-MET pathway mobilizes an inflammatory network in the brain microenvironment to promote brain metastasis of breast cancer. Cancer Res. 2016, 76, 4970–4980. [Google Scholar] [CrossRef] [PubMed]
- Bardelli, A.; Corso, S.; Bertotti, A.; Hobor, S.; Valtorta, E.; Siravegna, G.; Sartore-Bianchi, A.; Scala, E.; Cassingena, A.; Zecchin, D.; et al. Amplification of the met receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013, 3, 658–673. [Google Scholar] [CrossRef] [PubMed]
- Turke, A.B.; Zejnullahu, K.; Wu, Y.L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and clonal selection of MET amplification in egfr mutant NSCLC. Cancer Cell 2010, 17, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, A.A.; Aranda, V.; Bardelli, A.; Blanpain, C.; Bock, C.; Borowski, C.; Caldas, C.; Califano, A.; Doherty, M.; Elsner, M.; et al. Toward understanding and exploiting tumor heterogeneity. Nat. Med. 2015, 21, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Campbell, L.L.; Polyak, K. Breast tumor heterogeneity: Cancer stem cells or clonal evolution? Cell Cycle 2007, 6, 2332–2338. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.R.; Fridlyand, J.; Yan, Y.; Penuel, E.; Burton, L.; Chan, E.; Peng, J.; Lin, E.; Wang, Y.; Sosman, J.; et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012, 487, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.N.; Bhowmick, N.A. Role of EMT in metastasis and therapy resistance. J. Clin. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, H.; Ren, G. Epithelial-mesenchymal transition and drug resistance in breast cancer (review). Int. J. Oncol. 2015, 47, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Witta, S.E.; Gemmill, R.M.; Hirsch, F.R.; Coldren, C.D.; Hedman, K.; Ravdel, L.; Helfrich, B.; Dziadziuszko, R.; Chan, D.C.; Sugita, M.; et al. Restoring e-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res. 2006, 66, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Grotegut, S.; von Schweinitz, D.; Christofori, G.; Lehembre, F. Hepatocyte growth factor induces cell scattering through MAPK/EGR-1-mediated upregulation of snail. EMBO J. 2006, 25, 3534–3545. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Luo, Y.; Wang, Y.; Chen, Y.; Li, M.; Jiang, Y. Hepatocyte growth factor increases the invasive potential of pc-3 human prostate cancer cells via an ERK/MAPK and ZEB-1 signaling pathway. Oncol. Lett. 2016, 11, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Vega, S.; Morales, A.V.; Ocana, O.H.; Valdes, F.; Fabregat, I.; Nieto, M.A. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004, 18, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Kaler, P.; Augenlicht, L.; Klampfer, L. Activating mutations in beta-catenin in colon cancer cells alter their interaction with macrophages; the role of snail. PLoS ONE 2012, 7, e45462. [Google Scholar] [CrossRef] [PubMed]
- Kaler, P.; Galea, V.; Augenlicht, L.; Klampfer, L. Tumor associated macrophages protect colon cancer cells from trail-induced apoptosis through IL-1beta-dependent stabilization of snail in tumor cells. PLoS ONE 2010, 5, e11700. [Google Scholar] [CrossRef] [PubMed]
- Kudo-Saito, C.; Shirako, H.; Takeuchi, T.; Kawakami, Y. Cancer metastasis is accelerated through immunosuppression during snail-induced emt of cancer cells. Cancer Cell 2009, 15, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhao, X.; Xiao, Q.; Liu, Q.; Ding, K.; Yu, F.; Zhang, R.; Zhu, T.; Ge, G. Snail and slug mediate tamoxifen resistance in breast cancer cells through activation of EGFR-ERK independent of epithelial-mesenchymal transition. J. Mol. Cell Biol. 2014, 6, 352–354. [Google Scholar] [CrossRef] [PubMed]
- Ware, K.E.; Somarelli, J.A.; Schaeffer, D.; Li, J.; Zhang, T.; Park, S.; Patierno, S.R.; Freedman, J.; Foo, W.C.; Garcia-Blanco, M.A.; et al. Snail promotes resistance to enzalutamide through regulation of androgen receptor activity in prostate cancer. Oncotarget 2016, 7, 50507–50521. [Google Scholar] [CrossRef] [PubMed]
- De Bacco, F.; D’Ambrosio, A.; Casanova, E.; Orzan, F.; Neggia, R.; Albano, R.; Verginelli, F.; Cominelli, M.; Poliani, P.L.; Luraghi, P.; et al. Met inhibition overcomes radiation resistance of glioblastoma stem-like cells. EMBO Mol. Mead. 2016, 8, 550–568. [Google Scholar] [CrossRef] [PubMed]
- Boccaccio, C.; Comoglio, P.M. The met oncogene in glioblastoma stem cells: Implications as a diagnostic marker and a therapeutic target. Cancer Res. 2013, 73, 3193–3199. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Liu, S.; Duan, S.Z.; Zhang, L.; Zhou, H.; Hu, Y.; Zhou, X.; Shi, C.; Zhou, R.; Zhang, Z. Targeting the c-MET/FZD8 signaling axis eliminates patient-derived cancer stem-like cells in head and neck squamous carcinomas. Cancer Res. 2014, 74, 7546–7559. [Google Scholar] [CrossRef] [PubMed]
- Van Leenders, G.J.; Sookhlall, R.; Teubel, W.J.; de Ridder, C.M.; Reneman, S.; Sacchetti, A.; Vissers, K.J.; van Weerden, W.; Jenster, G. Activation of c-MET induces a stem-like phenotype in human prostate cancer. PLoS ONE 2011, 6, e26753. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wu, J.J.; Hynes, M.; Dosch, J.; Sarkar, B.; Welling, T.H.; Pasca di Magliano, M.; Simeone, D.M. C-MET is a marker of pancreatic cancer stem cells and therapeutic target. Gastroenterology 2011, 141, 2218–2227. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Yamada, Y.; Furuta, K.; Honma, Y.; Iwasa, S.; Takashima, A.; Kato, K.; Hamaguchi, T.; Shimada, Y. Serum levels of hepatocyte growth factor and epiregulin are associated with the prognosis on anti-EGFR antibody treatment in KRAS wild-type metastatic colorectal cancer. Br. J. Cancer 2014, 110, 2716–2727. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, K.; Arao, T.; Sakai, K.; Matsumoto, K.; Sakai, A.; Kimura, H.; Sone, T.; Horiike, A.; Nishio, M.; Ohira, T.; et al. Impact of serum hepatocyte growth factor on treatment response to epidermal growth factor receptor tyrosine kinase inhibitors in patients with non-small cell lung adenocarcinoma. Clin. Cancer Res. 2010, 16, 4616–4624. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Janne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [PubMed]
- Bean, J.; Brennan, C.; Shih, J.Y.; Riely, G.; Viale, A.; Wang, L.; Chitale, D.; Motoi, N.; Szoke, J.; Broderick, S.; et al. Met amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. USA 2007, 104, 20932–20937. [Google Scholar] [CrossRef] [PubMed]
- Smolen, G.A.; Sordella, R.; Muir, B.; Mohapatra, G.; Barmettler, A.; Archibald, H.; Kim, W.J.; Okimoto, R.A.; Bell, D.W.; Sgroi, D.C.; et al. Amplification of met may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA-665752. Proc. Natl. Acad. Sci. USA 2006, 103, 2316–2321. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Li, J.; Hao, K.; Yan, J.; Zhou, H. The efficacy and risk profile of c-MET inhibitors in Non-small Cell Lung Cancer: A meta-analysis. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Pupo, E.; Ducano, N.; Lupo, B.; Vigna, E.; Avanzato, D.; Perera, T.; Trusolino, L.; Lanzetti, L.; Comoglio, P.M. Rebound effects caused by withdrawal of met kinase inhibitor are quenched by a met therapeutic antibody. Cancer Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- McDermott, U.; Pusapati, R.V.; Christensen, J.G.; Gray, N.S.; Settleman, J. Acquired resistance of non-small cell lung cancer cells to MET kinase inhibition is mediated by a switch to epidermal growth factor receptor dependency. Cancer Res. 2010, 70, 1625–1634. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.W.; Staal, B.; Essenburg, C.; Lewis, S.; Kaufman, D.; Vande Woude, G.F. Strengthening context-dependent anticancer effects on non-small cell lung carcinoma by inhibition of both MET and EGFR. Mol. Cancer Ther. 2013, 12, 1429–1441. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, T.; Ohmori, T.; Ohba, M.; Arata, S.; Kishino, Y.; Murata, Y.; Kusumoto, S.; Ishida, H.; Shirai, T.; Hirose, T.; et al. Acquired resistance mechanisms to combination MET-TKI/EGFR-TKI exposure in MET-amplified EGFR-TKI resistant lung adenocarcinoma harboring an activating EGFR mutation. Mol. Cancer Ther. 2016, 15, 3040–3054. [Google Scholar] [CrossRef] [PubMed]
- Owusu, B.Y.; Thomas, S.; Venukadasula, P.; Han, Z.; Janetka, J.; Galemmo, R.; Klampfer, L. Targeting the tumor-promoting microenvironment in met-amplified NSCLC cells with a novel inhibitor of pro-HGF activation. Oncotarget 2017. Under review. [Google Scholar]
- Michieli, P.; Basilico, C.; Pennacchietti, S.; Maffe, A.; Tamagnone, L.; Giordano, S.; Bardelli, A.; Comoglio, P.M. Mutant MET-mediated transformation is ligand-dependent and can be inhibited by HGF antagonists. Oncogene 1999, 18, 5221–5231. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Owusu, B.Y.; Galemmo, R.; Janetka, J.; Klampfer, L. Hepatocyte Growth Factor, a Key Tumor-Promoting Factor in the Tumor Microenvironment. Cancers 2017, 9, 35. https://doi.org/10.3390/cancers9040035
Owusu BY, Galemmo R, Janetka J, Klampfer L. Hepatocyte Growth Factor, a Key Tumor-Promoting Factor in the Tumor Microenvironment. Cancers. 2017; 9(4):35. https://doi.org/10.3390/cancers9040035
Chicago/Turabian StyleOwusu, Benjamin Yaw, Robert Galemmo, James Janetka, and Lidija Klampfer. 2017. "Hepatocyte Growth Factor, a Key Tumor-Promoting Factor in the Tumor Microenvironment" Cancers 9, no. 4: 35. https://doi.org/10.3390/cancers9040035