
Advancing Clostridia to Clinical Trial: Past Lessons and Recent Progress

 , ,
, ,  , ,
, , 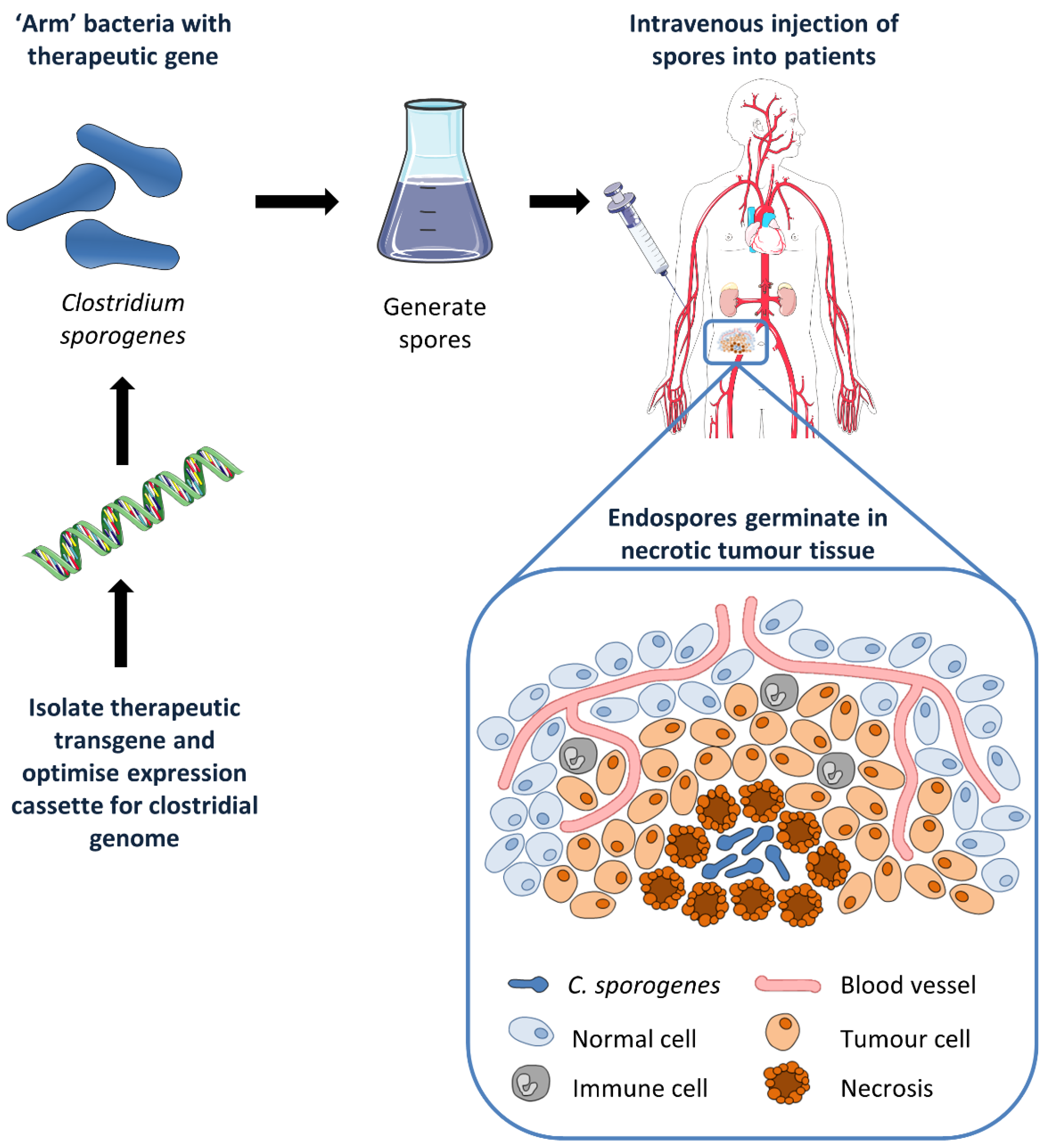{kind=link}
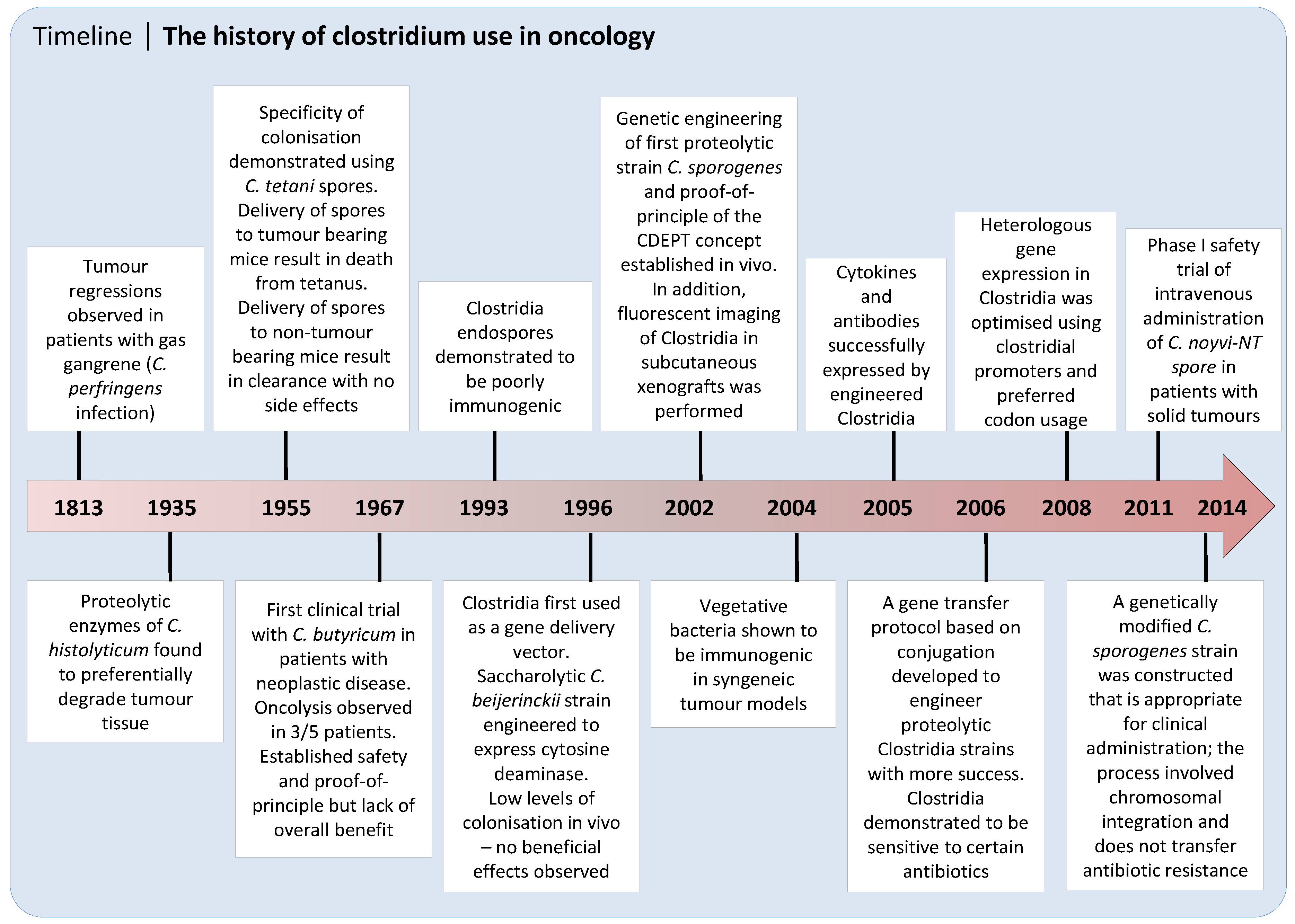{kind=link}
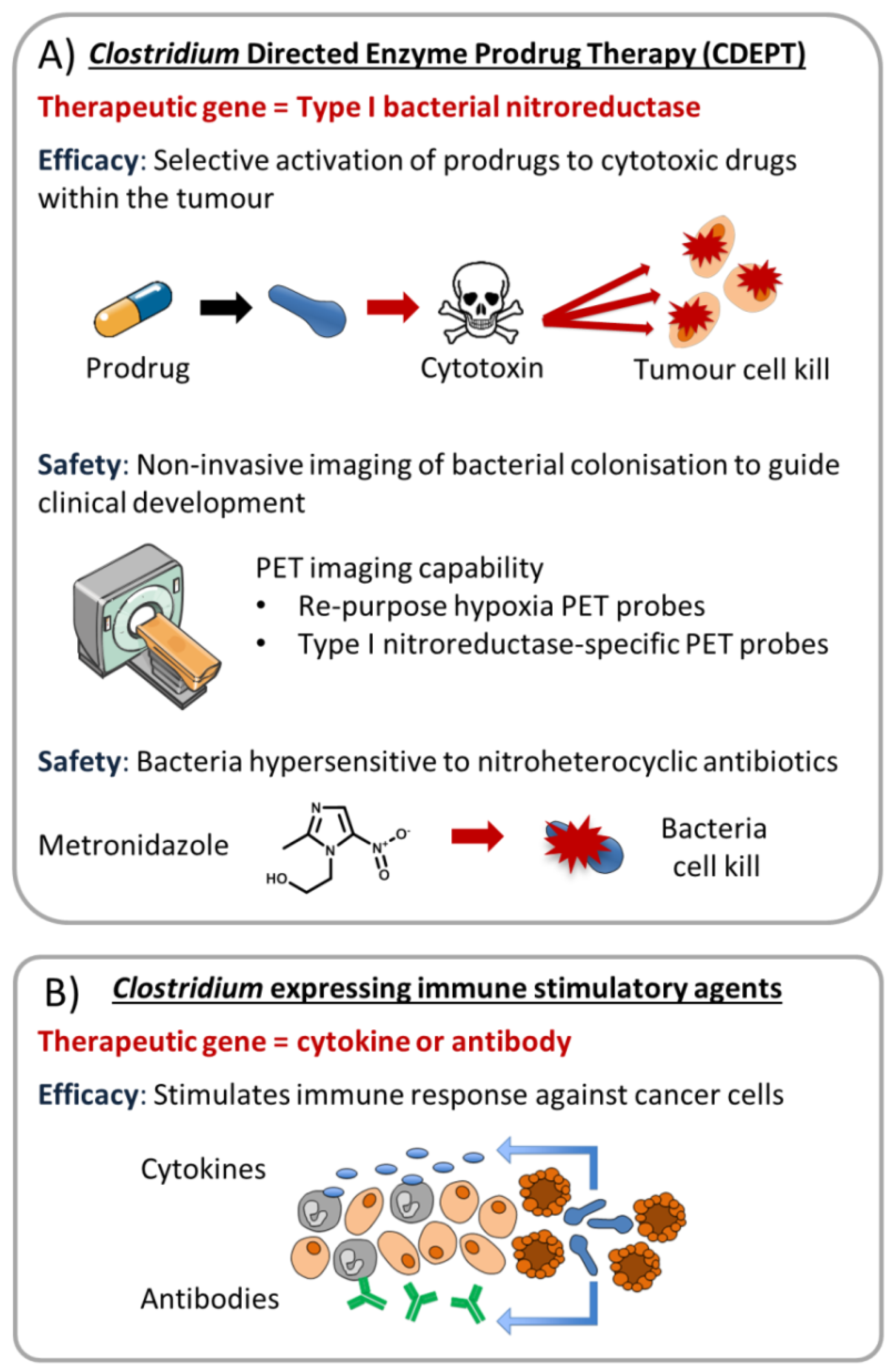{kind=link}
Abstract
:1. Tumor Necrosis as a Target for Cancer Therapy
2. Discovery and Early Development of Clostridium as an Anticancer Agent
3. Recent Progress on the Use of Unarmed Clostridia to Treat Cancer
4. Clostridium Directed Enzyme Prodrug Therapy (CDEPT)
4.1. CDEPT Principle and Proof-of-Concept
4.2. Advantages of Clostridial Vectors over Viral Delivery Systems
5. The Requirement for Non-invasive Imaging to Expedite Clinical Development
5.1. Imaging of Necrosis and Colonisation with C. sporogenes in Preclinical Animal Models
5.2. Non-Invasive Imaging of CDEPT Using Positron Emission Tomography
6. Clostridia and Immunotherapy
7. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Umer, B.; Good, D.; Anne, J.; Duan, W.; Wei, M.Q. Clostridial spores for cancer therapy: Targeting solid tumour microenvironment. J. Toxicol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Giaccia, A.J. The unique physiology of solid tumors: Opportunities (and problems) for cancer therapy. Cancer Res. 1998, 58, 1408–1416. [Google Scholar] [PubMed]
- Richards, C.H.; Mohammed, Z.; Qayyum, T.; Horgan, P.G.; McMillan, D.C. The prognostic value of histological tumor necrosis in solid organ malignant disease: A systematic review. Future Oncol. 2011, 7, 1223–1235. [Google Scholar] [CrossRef] [PubMed]
- Martens, K.; Meyners, T.; Rades, D.; Tronnier, V.; Bonsanto, M.M.; Petersen, D.; Dunst, J.; Dellas, K. The prognostic value of tumor necrosis in patients undergoing stereotactic radiosurgery of brain metastases. Radiat. Oncol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Fisher, E.R.; Paleka, A.; Rockette, H.; Redmond, C.; Fisher, B. Pathologic findings from the national surgical ddjuvant breast project (protocol No. 4) V. significance of axillary nodal micro- and macrometastases. Cancer 1978, 42, 2032–2038. [Google Scholar] [CrossRef]
- Pollheimer, M.J.; Kornprat, P.; Lindtner, R.A.; Harbaum, L.; Schlemmer, A.; Rehak, P.; Langner, C. Tumor necrosis is a new promising prognostic factor in colorectal cancer. Hum. Pathol. 2010, 41, 1749–1757. [Google Scholar] [CrossRef] [PubMed]
- Ladstein, R.G.; Bachmann, I.M.; Straume, O.; Akslen, L.A. Tumor necrosis is a prognostic factor in thick cutaneous melanoma. Am. J. Surg. Pathol. 2012, 36, 1477–1482. [Google Scholar] [CrossRef] [PubMed]
- Swinson, D.E.; Jones, J.L.; Richardson, D.; Cox, G.; Edwards, J.G.; O′Byrne, K.J. Tumour necrosis is an independent prognostic marker in non-small cell lung cancer: Correlation with biological variables. Lung Cancer 2002, 37, 235–240. [Google Scholar] [CrossRef]
- Hiraoka, N.; Ino, Y.; Sekine, S.; Tsuda, H.; Shimada, K.; Kosuge, T.; Zavada, J.; Yoshida, M.; Yamada, K.; Koyama, T.; et al. Tumour necrosis is a postoperative prognostic marker for pancreatic cancer patients with a high interobserver reproducibility in histological evaluation. Br. J. Cancer 2010, 103, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Lohse, C.; Leibovich, B.; Frank, I.; Thompson, R.; Webster, W.; Zincke, H.; Blute, M.L.; Cheville, J.C.; Kwon, E.D. Histologic coagulative tumor necrosis as a prognostic indicator of renal cell carcinoma aggressiveness. Cancer 2005, 104, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Barbe, S.; van Mellaert, L.; Anne, J. The use of clostridial spores for cancer treatment. J. Appl. Microbiol. 2006, 101, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Minton, N.P. Clostridia in cancer therapy. Nat. Rev. Microbiol. 2003, 1, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Ryan, R.M.; Green, J.; Lewis, C.E. Use of bacteria in anti-cancer therapies. Bioessays 2006, 28, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Torrey, J.C.; Kahn, M.C. The treatment of Flexner-Jobling rat carcinomas with bacterial proteolytic ferments. J. Can. Res. 1927, 11, 334–376. [Google Scholar]
- Connell, H.C. The study and treatment of cancer by proteolytic enzymes: A preliminary report. Can. Med. Assoc. J. 1935, 33, 364–370. [Google Scholar] [PubMed]
- Parker, R.C.; Plummer, H.C.; Siebenmann, C.O.; Chapman, M.G. Effect of Histolyticus Infection and Toxin on Transplantable Mouse Tumors. Proc. Soc. Exp. Biol. Med. 1947, 66, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Malmgren, R.A.; Flanigan, C.C. Localization of the vegetative form of Clostridium tetani in mouse tumors following intravenous spore administration. Cancer Res. 1955, 15, 473–478. [Google Scholar] [PubMed]
- Mose, J.R.; Mose, G. Oncolysis by clostridia. I. Activity of Clostridium butyricum (M-55) and other non pathogenic clostridia against the Erlich carcinoma. Cancer Res. 1964, 24, 212–216. [Google Scholar]
- Thiele, E.H.; Arison, R.N.; Boxer, G.E. Oncolysis by clostridia. III. Effects of clostridia and chemotherapeutic agents on rodent tumours. Cancer Res. 1964, 24, 222–233. [Google Scholar]
- Engelbart, K.; Gericke, D. Oncolysis by clostridia. V. Transplanted tumors of the hamster. Cancer Res. 1964, 24, 239–243. [Google Scholar] [PubMed]
- Mohr, U.; Hondius, B.W.; Emminger, A.; Behagel, H.A. Oncolysis by a New Strain of Clostridium. Cancer Res. 1972, 32, 1122–1128. [Google Scholar] [PubMed]
- Carey, R.W.; Holland, J.F.; Whang, H.Y.; Neter, E.; Bryant, B. Clostridial oncolysis in man. Eur. J. Cancer 1967, 3, 37–46. [Google Scholar] [CrossRef]
- Heppner, F.; Mose, J.R. The liquefaction (oncolysis) of malignant gliomas by a non pathogenic Clostridium. Acta Neurochir. 1978, 42, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.H.; Bettegowda, C.; Huso, D.L.; Kinzler, K.W.; Vogelstein, B. Combination bacteriolytic therapy for the treatment of experimental tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 15155–15160. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Bettegowda, C.; Cheong, I.; Geshwind, J.; Drake, C.G.; Hipkiss, E.L.; Tatsumi, M.; Dang, L.H.; Diaz, L.A.; Pomper, M.; et al. Bacteriolytic therapy can generate a potent immune response against experimental tumors. Proc. Natl. Acad. Sci. USA 2004, 101, 15172–15177. [Google Scholar] [CrossRef] [PubMed]
- Krick, E.L.; Sorenmo, K.U.; Rankin, S.U.; Cheong, I.; Kobrin, B.; Thornton, K.; Kinzler, K.W.; Vogelstein, B.; Zhou, S.; Diaz, L.A., Jr.; et al. Evaluation of Clostridium novyi-NT spores in dogs with naturally occurring tumors. Am. J. Vet. Res. 2012, 73, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Roberts, N.J.; Zhang, L.; Janku, F.; Collins, A.; Bai, R.Y.; Staedtke, V.; Rusk, A.W.; Tung, D.; Miller, M.; Roix, J.; et al. Intratumoral injection of Clostridium novyi-NT spores induces antitumor responses. Sci. Transl. Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- McNeish, I.A.; Searle, P.F.; Young, L.S.; Kerr, D.J. Gene directed enzyme prodrug therapy for cancer. Adv. Drug. Del. Rev. 1997, 26, 173–184. [Google Scholar] [CrossRef]
- Freytag, S.O.; Stricker, H.; Pegg, J.; Paielli, D.; Pradhan, D.G.; Peabody, J.; de Peralta-Venturina, M.; Xia, X.; Brown, S.; Lu, M.; et al. Phase I study of replication-competent adenovirus-mediated double-suicide gene therapy in combination with conventional-dose three-dimensional conformal radiation therapy for the treatment of newly diagnosed, intermediate- to high-risk prostate cancer. Cancer Res. 2003, 63, 7497–7506. [Google Scholar] [PubMed]
- Patel, P.; Young, J.G.; Mautner, V.; Ashdown, D.; Bonney, S.; Pineda, R.G.; Collins, S.I.; Searle, P.F.; Hull, D.; Peers, E.; et al. A phase I/II clinical trial in localized prostate cancer of an adenovirus expressing nitroreductase with CB1954. Mol. Ther. 2009, 17, 1292–1299. [Google Scholar] [CrossRef] [PubMed]
- Fox, M.E.; Lemmon, M.J.; Mauchline, M.L.; Davis, T.O.; Giaccia, A.J.; Minton, N.P.; Brown, J.M. Anaerobic bacteria as a delivery system for cancer gene therapy: In vitro activation of 5-fluorocytosine by genetically engineered clostridia. Gene Ther. 1996, 3, 173–178. [Google Scholar] [PubMed]
- Theys, J.; Landuyt, W.; Nuyts, S.; van Mellaert, L.; van Oosterom, A.; Lambin, P.; Annea, J. Specific targeting of cytosine deaminase to solid tumors by engineered Clostridium acetobutylicum. Cancer Gene Ther. 2001, 8, 294–297. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.J.; van Zijl, P.; Fox, M.E.; Mauchline, M.L.; Giaccia, A.J.; Minton, N.P.; Brown, J.M. Anaerobic bacteria as a gene delivery system that is controlled by the tumor microenvironment. Gene Ther. 1997, 4, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.C.; Minton, N.P.; Giaccia, A.J.; Brown, J.M. Anticancer efficacy of systemically delivered anaerobic bacteria as gene therapy vectors targeting tumor hypoxia/necrosis. Gene Ther. 2002, 9, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Theys, J.; Pennington, O.; Dubois, L.; Anlezark, G.M.; Vaughan, T.; Mengesha, A.; Landuyt, W.; Anne, J.; Burke, P.J.; Durre, P.; et al. Repeated cycles of Clostridium-directed enzyme prodrug therapy result in sustained antitumour effects in vivo. Br. J. Cancer 2006, 95, 1212–1219. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.C.; Ahn, G.O.; Kioi, M.; Dorie, M.J.; Patterson, A.V.; Brown, J.M. Optimised Clostridium-directed enzyme prodrug therapy improves the antitumor activity of the novel DNA crosslinking agent PR-104. Cancer Res. 2008, 68, 7995–8003. [Google Scholar] [CrossRef] [PubMed]
- Brekken, R.A.; Li, C.; Kumar, S. Strategies for vascular targeting in tumors. Int. J. Cancer 2002, 100, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Heap, J.T.; Ehsaan, M.; Cooksley, C.M.; Ng, Y.K.; Cartman, S.T.; Winzer, K.; Minton, N.P. Integration of DNA into bacterial chromosomes from plasmids without a counter-selection marker. Nucleic. Acids Res. 2012. [Google Scholar] [CrossRef] [PubMed]
- Heap, J.T.; Theys, J.; Ehsaan, M.; Kubiak, A.M.; Dubois, L.; Paesmans, K.; van Mellaert, L.; Knox, R.; Kuehne, S.A.; Lambin, P.; et al. Spores of Clostridium engineered for clinical efficacy and safety cause regression and cure of tumours in vivo. Oncotarget 2014, 5, 1761–1769. [Google Scholar] [CrossRef] [PubMed]
- Vile, R.; Ando, D.; Kirn, D. The oncolytic virotherapy treatment platform for cancer: unique biological and biosafety points to consider. Cancer Gene Ther. 2002, 9, 1062–1067. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, R.; Miest, T.; Shashkova, E.V.; Barry, M.A. Reprogrammed viruses as cancer therapeutics: targeted, armed and shielded. Nat. Rev. Microbiol. 2008, 6, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Parato, K.A.; Senger, D.; Forsyth, P.A.; Bell, J.C. Recent progress in the battle between oncolytic viruses and tumours. Nat. Rev. Cancer 2005, 5, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.R.; Kratzke, R.A. Oncolytic virus therapy for cancer: The first wave of translational clinical trials. Transl. Res. 2013, 161, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, L.K.; Lemoine, N.R.; Kirn, D. Oncolytic biotherapy: a novel therapeutic platform. Lancet Oncol. 2002, 3, 17–26. [Google Scholar] [CrossRef]
- Wei, M.Q.; Mengesha, A.; Good, D.; Anne, J. Bacterial targeted tumour therapy—Dawn of a new era? Cancer Lett. 2008, 259, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Egeland, T.A.M.; Gaustad, J.V.; Galante, Y.M.; Rofstad, E.K. Magnetic resonance imaging of tumour necrosis. Acta Oncol. 2011, 50, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Kirn, D.; Martuza, R.L.; Zwiebel, J. Replication-selective virotherapy for cancer: Biological principles, risk management and future directions. Nat. Med. 2001, 7, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Gambhir, S.S. Molecular imaging of cancer with positron emission tomography. Nat. Rev. Cancer 2002, 2, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Min, J.J.; Gambhir, S.S. Gene therapy progress and prospects: noninvasive imaging of gene therapy in living subjects. Gene Ther. 2004, 11, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B. Sodium Iodide Symporter for Nuclear Molecular Imaging and Gene Therapy: From Bedside to Bench and Back. Theranostics 2012, 2, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Penheiter, A.R.; Russell, S.J.; Carlson, S.K. The Sodium Iodide Symporter (NIS) as an Imaging Reporter for Gene, Viral, and Cell-based Therapies. Curr. Gene Ther. 2012, 12, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Barton, K.N.; Stricker, H.; Brown, S.L.; Elshaikh, M.; Aref, I.; Lu, M.; Pegg, J.; Zhang, Y.; Karvelis, K.C.; Siddiqui, F.; et al. Phase I study of noninvasive imaging of adenovirus-mediated gene expression in the human prostate. Mol. Ther. 2008, 16, 1761–1769. [Google Scholar] [CrossRef] [PubMed]
- Msaouel, P.; Dispenzieri, A.; Galanis, E. Clinical testing of engineered oncolytic measles virus strains in the treatment of cancer: An overview. Curr. Opin. Mol. Ther. 2009, 11, 43–53. [Google Scholar] [PubMed]
- Bhaumik, S. Advances in Imaging Gene-Directed Enzyme Prodrug Therapy. Curr. Pharm. Biotechnol. 2011, 12, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Kuruppu, D.; Brownell, A.L.; Zhu, A.; Yu, M.; Wang, X.; Kulu, Y.; Fuchs, B.C.; Kawasaki, H.; Tanabe, K.K. Positron Emission Tomography of Herpes Simplex Virus 1 Oncolysis. Cancer Res. 2007, 67, 3295–3300. [Google Scholar] [CrossRef] [PubMed]
- Yaghoubi, S.S.; Couto, M.A.; Chen, C.C.; Polavaram, L.; Cui, G.; Sen, L.; Gambhir, S.S. Preclinical safety evaluation of 18F-FHBG: A pet reporter probe for imaging herpes simplex virus type 1 thymidine kinase (HSV1-tk) or mutant HSV1-sr39tk’s expression. J. Nucl. Med. 2006, 47, 706–715. [Google Scholar] [PubMed]
- Penuelas, I.; Mazzolini, G.; Boan, J.F.; Sangro, B.; Marti-Climent, J.; Ruiz, M.; Ruiz, J.; Satyamurthy, N.; Qian, C.; Barrio, J.R.; et al. Positron emission tomography imaging of adenoviral-mediated transgene expression in liver cancer patients. Gastroenterology 2005, 128, 1787–1795. [Google Scholar] [CrossRef] [PubMed]
- Peeters, S.G.; Zegers, C.M.; Lieuwes, N.G.; van Elmpt, W.; Eriksson, J.; van Dongen, G.A.; Dubois, L.; Lambin, P. A comparative study of the hypoxia PET tracers [(1)(8)F]HX4, [(1)(8)F]FAZA, and [(1)(8)F]FMISO in a preclinical tumor model. Int. J. Radiat. Oncol. Biol. Phys. 2015, 91, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Min, J.J.; Gambhir, S.S. Molecular imaging of PET reporter gene expression. Handb. Exp. Pharmacol. 2008, 185, 277–303. [Google Scholar] [PubMed]
- Williams, E.M.; Little, R.F.; Mowday, A.M.; Rich, M.H.; Chan-Hyams, J.V.; Copp, J.N.; Smaill, J.B.; Patterson, A.V.; Ackerley, D.F. Nitroreductase gene-directed enzyme prodrug therapy: Insights and advances toward clinical utility. Biochem. J. 2015, 471, 131–153. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O′Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Eng. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N. Eng. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Fabricius, E.M.; Schneeweiss, U.; Schau, H.P.; Schmidt, W.; Benedix, A. Quantitative investigations into the elimination of in vitro-obtained spores of the non-pathogenic Clostridium butyricum strain CNRZ 528, and their persistence in organs of different species following intravenous spore administration. Res. Microbiol. 1993, 144, 741–753. [Google Scholar] [CrossRef]
- Nuyts, S.; van Mellaert, L.; Theys, J.; Landuyt, W.; Bosmans, E.; Anne, J.; Lambin, P. Radio-responsive recA promoter significantly increases TNFalpha production in recombinant clostridia after 2 Gy irradiation. Gene Ther. 2001, 15, 1197–1201. [Google Scholar] [CrossRef] [PubMed]
- Antony, G.K.; Dudek, A.Z. Interleukin 2 in cancer therapy. Curr. Med. Chem. 2010, 17, 3297–3302. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high dose bolus IL-2. JAMA 1994, 271, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Lotze, M.T.; Matory, Y.L.; Rayner, A.A.; Ettinghausen, S.E.; Vetto, J.T.; Seipp, C.A.; Rosenberg, S.A. Clinical effects and toxicities of IL-2 in patients with cancer. Cancer 1986, 58, 2764–2772. [Google Scholar] [CrossRef]
- Zegers, C.M.; Rekers, N.H.; Quaden, D.H.; Lieuwes, N.G.; Yaromina, A.; Germeraad, W.T.; Wieten, L.; Biessen, E.A.; Boon, L.; Neri, D.; et al. Radiotherapy combined with the immunocytokine L19-IL2 provides long-lasting antitumor effects. Clin. Cancer Res. 2015, 21, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Theys, J.; Landuyt, W.; Nuyts, S.; van Mellaert, L.; Bosmans, E.; Rijnders, A.; van Den Bogaert, W.; van Oosterom, A.; Anne, J.; Lambin, P. Improvement of clostridium tumour targeting vectors evaluated in rat rhabdomyosarcomas. FEMS Immunol. Med. Microbiol. 2001, 30, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Barbe, S.; van Mellaert, L.; Theys, J.; Geukens, N.; Lammertyn, E.; Lambin, P.; Anne, J. Secretory production of biologically active rat interleukin-2 by Clostridium acetobutylicum DSM792 as a tool for anti-tumor treatment. FEMS Microbiol. Lett. 2005, 246, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Groot, A.J.; Mengesha, A.; van der Wall, E.; van Diest, P.J.; Theys, J.; Vooijs, M. Functional antibodies produced by oncolytic clostridia. Biochem. Biophys. Res. Commun. 2007, 364, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Hasan, A.; Mazzone, M. Sixty shades of oxygen—An attractive opportunity for cancer immunotherapy. Ann. Transl. Med. 2015. [Google Scholar] [CrossRef]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mowday, A.M.; Guise, C.P.; Ackerley, D.F.; Minton, N.P.; Lambin, P.; Dubois, L.J.; Theys, J.; Smaill, J.B.; Patterson, A.V. Advancing Clostridia to Clinical Trial: Past Lessons and Recent Progress. Cancers 2016, 8, 63. https://doi.org/10.3390/cancers8070063
Mowday AM, Guise CP, Ackerley DF, Minton NP, Lambin P, Dubois LJ, Theys J, Smaill JB, Patterson AV. Advancing Clostridia to Clinical Trial: Past Lessons and Recent Progress. Cancers. 2016; 8(7):63. https://doi.org/10.3390/cancers8070063
Chicago/Turabian StyleMowday, Alexandra M., Christopher P. Guise, David F. Ackerley, Nigel P. Minton, Philippe Lambin, Ludwig J. Dubois, Jan Theys, Jeff B. Smaill, and Adam V. Patterson. 2016. "Advancing Clostridia to Clinical Trial: Past Lessons and Recent Progress" Cancers 8, no. 7: 63. https://doi.org/10.3390/cancers8070063