
Exploring the Mechanisms of Gastrointestinal Cancer Development Using Deep Sequencing Analysis

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Deep Sequencing of Premalignant Inflamed Tissues Using NGS Technologies
2.1. Importance of Studying Genetic Alterations in Inflamed Noncancerous Tissues
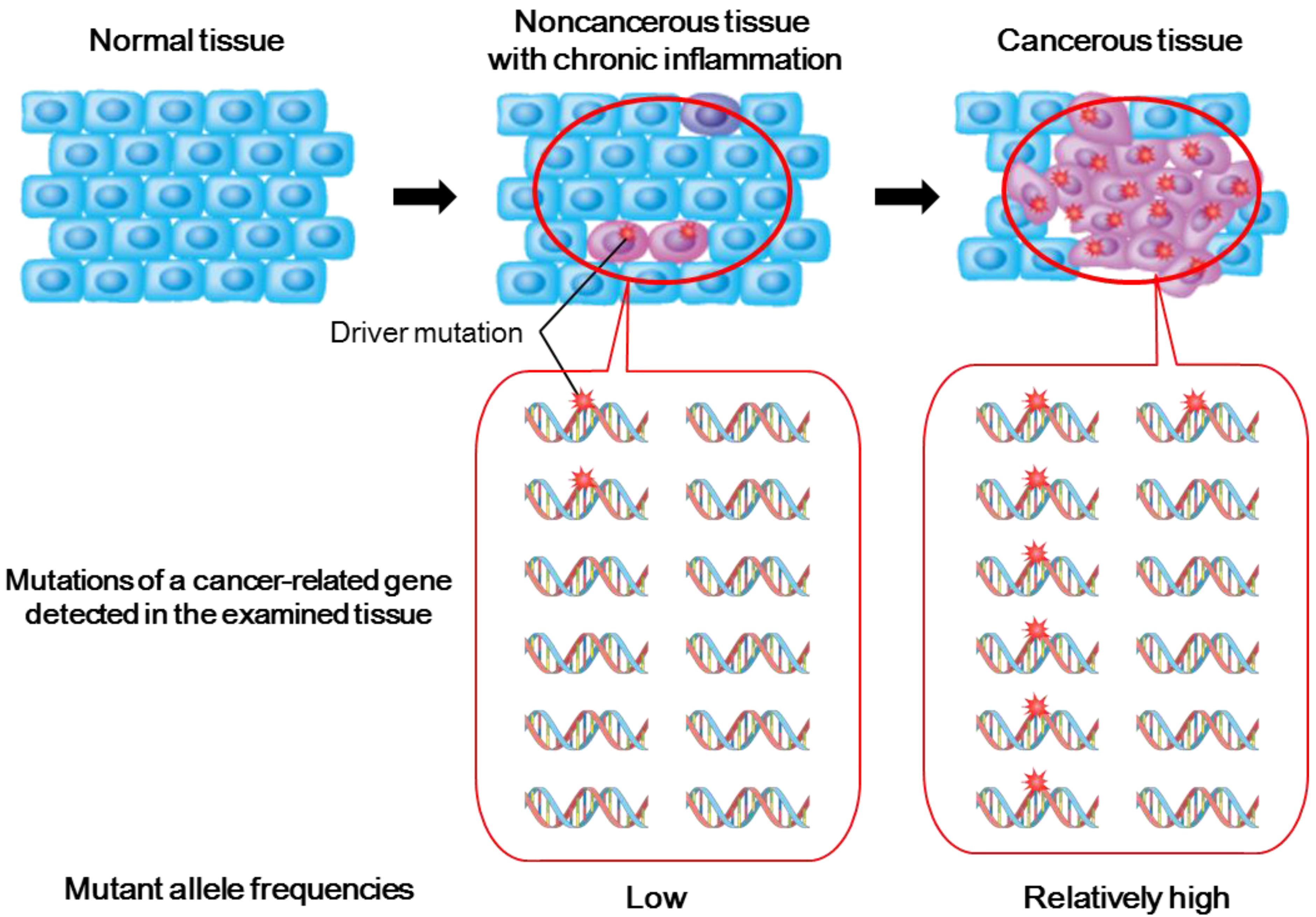
2.2. Genetic Alterations in Inflamed Noncancerous Tissues Determined by Conventional Sanger Sequencing
2.3. Deep Sequencing Analysis of Inflamed Noncancerous Tissues
3. Mutation Signatures Provide Clues to Predict Tumorigenic Mechanisms
3.1. Mutational Signatures Specific to Various Types of Cancers
3.2. Extrinsic and Intrinsic Mutagens and Mutational Signatures
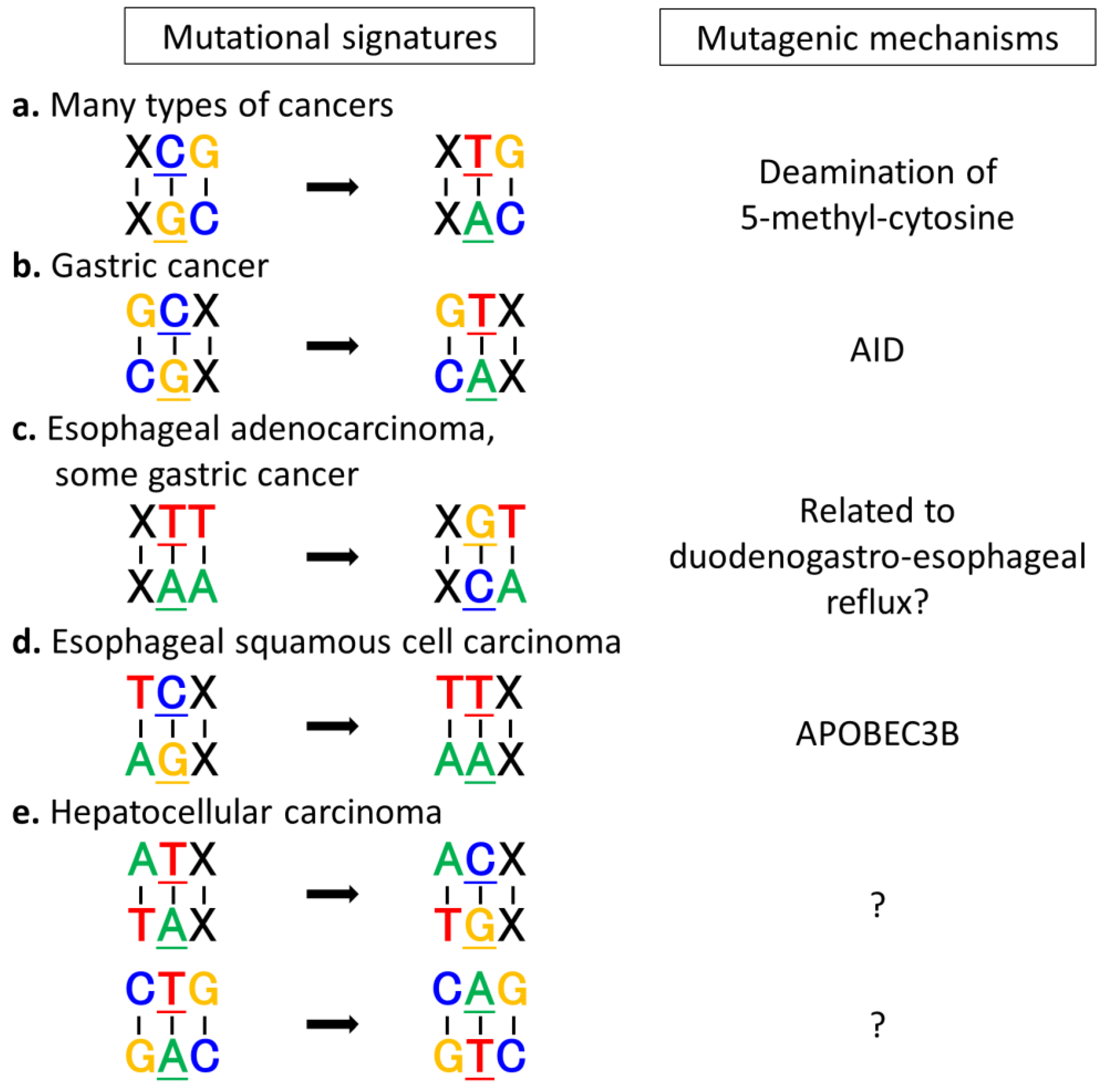
3.3. Exploring Carcinogenic Mechanisms by Analyzing Mutational Signatures
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef]
- Watson, I.R.; Takahashi, K.; Futreal, P.A.; Chin, L. Emerging patterns of somatic mutations in cancer. Nat. Rev. Genet. 2013, 14, 703–718. [Google Scholar] [CrossRef]
- Wang, K.; Kan, J.; Yuen, S.T.; Shi, S.T.; Chu, K.M.; Law, S.; Chan, T.L.; Kan, Z.; Chan, A.S.Y.; Tsui, W.Y.; et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat. Genet. 2011, 43, 1219–1223. [Google Scholar] [CrossRef]
- Zang, Z.J.; Cutcutache, I.; Poon, S.L.; Zhang, S.L.; McPherson, J.R.; Tao, J.; Rajasegaran, V.; Heng, H.L.; Deng, N.; Gan, A.; et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat. Genet. 2012, 44, 570–574. [Google Scholar] [CrossRef]
- Wang, K.; Yuen, S.T.; Xu, J.; Lee, S.P.; Yan, H.H.N.; Shi, S.T.; Siu, H.C.; Deng, S.; Chu, K.M.; Law, S.; et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat. Genet. 2014, 46, 573–582. [Google Scholar] [CrossRef]
- Nagarajan, N.; Bertrand, D.; Hillmer, A.M.; Zang, Z.J.; Yao, F.; Jacques, P.-É.; Teo, A.S.M.; Cutcutache, I.; Zhang, Z.; Lee, W.H.; et al. Whole-genome reconstruction and mutational signatures in gastric cancer. Genome Biol. 2012, 13, R115. [Google Scholar] [CrossRef]
- Shimizu, T.; Marusawa, H.; Matsumoto, Y.; Inuzuka, T.; Ikeda, A.; Fujii, Y.; Minamiguchi, S.; Miyamoto, S.; Kou, T.; Sakai, Y.; et al. Accumulation of somatic mutations in TP53 in gastric epithelium with Helicobacter pylori infection. Gastroenterology 2014, 147, 407–417. [Google Scholar] [CrossRef] [Green Version]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, A.; Totoki, Y.; Abe, T.; Boroevich, K.A.; Hosoda, F.; Nguyen, H.H.; Aoki, M.; Hosono, N.; Kubo, M.; Miya, F.; et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 2012, 44, 760–764. [Google Scholar] [CrossRef]
- Nakagawa, H.; Shibata, T. Comprehensive genome sequencing of the liver cancer genome. Cancer Lett. 2013, 340, 234–240. [Google Scholar] [CrossRef]
- Barrett, M.T.; Sanchez, C.A.; Prevo, L.J.; Wong, D.J.; Galipeau, P.C.; Paulson, T.G.; Rabinovitch, P.S.; Reid, B.J. Evolution of neoplastic cell lineages in Barrett oesophagus. Nat. Genet. 1999, 22, 106–109. [Google Scholar]
- Leedham, S.J.; Graham, T.A.; Oukrif, D.; McDonald, S.A.C.; Rodriguez-Justo, M.; Harrison, R.F.; Shepherd, N.A.; Novelli, M.R.; Jankowski, J.A.Z.; Wright, N.A. Clonality, founder mutations, and field cancerization in human ulcerative colitis-associated neoplasia. Gastroenterology 2009, 136, 542–550. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef]
- Hecht, S.S. Tobacco carcinogens, their biomarkers and tobacco-induced cancer. Nat. Rev. Cancer 2003, 3, 733–744. [Google Scholar] [CrossRef]
- Brash, D.E.; Rudolph, J.A.; Simon, J.A.; Lin, A.; McKenna, G.J.; Baden, H.P.; Halperin, A.J.; Pontén, J. A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 1991, 88, 10124–10128. [Google Scholar] [CrossRef]
- Pfeifer, G.P.; You, Y.-H.; Besaratinia, A. Mutations induced by ultraviolet light. Mutat. Res. 2005, 571, 19–31. [Google Scholar] [CrossRef]
- Foulkes, W.D. Inherited susceptibility to common cancers. N. Engl. J. Med. 2008, 359, 2143–2153. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Chiba, T.; Marusawa, H.; Ushijima, T. Inflammation-associated cancer development in digestive organs: Mechanisms and roles for genetic and epigenetic modulation. Gastroenterology 2012, 143, 550–563. [Google Scholar] [CrossRef] [Green Version]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Brentnall, T.A.; Haggitt, R.C.; Rabinovitch, P.S.; Kimmey, M.B.; Bronner, M.P.; Levine, D.S.; Kowdley, K.V.; Stevens, A.C.; Crispin, D.A.; Emond, M.; et al. Risk and natural history of colonic neoplasia in patients with primary sclerosing cholangitis and ulcerative colitis. Gastroenterology 1996, 110, 331–338. [Google Scholar] [CrossRef]
- Hussain, S.P.; Amstad, P.; Raja, K.; Ambs, S.; Nagashima, M.; Bennett, W.P.; Shields, P.G.; Ham, A.J.; Swenberg, J.A.; Marrogi, A.J.; et al. Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: A cancer-prone chronic inflammatory disease. Cancer Res. 2000, 60, 3333–3337. [Google Scholar] [PubMed]
- Kou, T.; Marusawa, H.; Kinoshita, K.; Endo, Y.; Okazaki, I.-M.; Ueda, Y.; Kodama, Y.; Haga, H.; Ikai, I.; Chiba, T. Expression of activation-induced cytidine deaminase in human hepatocytes during hepatocarcinogenesis. Int. J. Cancer 2007, 120, 469–476. [Google Scholar] [CrossRef]
- Hamada, S.; Masamune, A.; Shimosegawa, T. Inflammation and pancreatic cancer: Disease promoter and new therapeutic target. J. Gastroenterol. 2014, 49, 605–617. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef]
- Yoshida, K.; Sanada, M.; Ogawa, S. Deep sequencing in cancer research. Jpn. J. Clin. Oncol. 2013, 43, 110–115. [Google Scholar] [CrossRef]
- Bass, A.J.; Thorsson, V.; Shmulevich, I.; Reynolds, S.M.; Miller, M.; Bernard, B.; Hinoue, T.; Laird, P.W.; Curtis, C.; Shen, H.; et al. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef]
- Agrawal, N.; Jiao, Y.; Bettegowda, C.; Hutfless, S.M.; Wang, Y.; David, S.; Cheng, Y.; Twaddell, W.S.; Latt, N.L.; Shin, E.J.; et al. Comparative genomic analysis of esophageal adenocarcinoma and squamous cell carcinoma. Cancer Discov. 2012, 2, 899–905. [Google Scholar] [CrossRef]
- Streppel, M.M.; Lata, S.; Delabastide, M.; Montgomery, E.A.; Wang, J.S.; Canto, M.I.; Macgregor-Das, A.M.; Pai, S.; Morsink, F.H.M.; Offerhaus, G.J.; et al. Next-generation sequencing of endoscopic biopsies identifies ARID1A as a tumor-suppressor gene in Barrett’s esophagus. Oncogene 2014, 33, 347–357. [Google Scholar] [CrossRef]
- Weaver, J.M.J.; Ross-Innes, C.S.; Shannon, N.; Lynch, A.G.; Forshew, T.; Barbera, M.; Murtaza, M.; Ong, C.-A.J.; Lao-Sirieix, P.; Dunning, M.J.; et al. Ordering of mutations in preinvasive disease stages of esophageal carcinogenesis. Nat. Genet. 2014, 46, 837–843. [Google Scholar] [CrossRef]
- Ikeda, A.; Shimizu, T.; Matsumoto, Y.; Fujii, Y.; Eso, Y.; Inuzuka, T.; Mizuguchi, A.; Shimizu, K.; Hatano, E.; Uemoto, S.; et al. Leptin receptor somatic mutations are frequent in HCV-infected cirrhotic liver and associated with hepatocellular carcinoma. Gastroenterology 2014, 146, 222–232. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.H.; Liu, Y.F.; Ke, A.W.; Gu, F.M.; Yu, Y.; Dai, Z.; Gao, Q.; Shi, G.M.; Liao, B.Y.; Xie, Y.H.; et al. Clinical significance of the ubiquitin ligase UBE3C in hepatocellular carcinoma revealed by exome sequencing. Hepatology 2014, 59, 2216–2227. [Google Scholar] [CrossRef]
- Burns, M.B.; Temiz, N.A.; Harris, R.S. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 2013, 45, 977–983. [Google Scholar] [CrossRef]
- Poon, S.; McPherson, J.R.; Tan, P.; Teh, B.; Rozen, S.G. Mutation signatures of carcinogen exposure: Genome-wide detection and new opportunities for cancer prevention. Genome Med. 2014, 6, 24. [Google Scholar] [CrossRef]
- Wang, K.; Yuen, S.T.; Xu, J.; Lee, S.P.; Yan, H.H.N.; Shi, S.T.; Siu, H.C.; Deng, S.; Chu, K.M.; Law, S.; et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat. Genet. 2014, 46, 573–582. [Google Scholar] [CrossRef]
- Dulak, A.M.; Stojanov, P.; Peng, S.; Lawrence, M.S.; Fox, C.; Stewart, C.; Bandla, S.; Imamura, Y.; Schumacher, S.E.; Shefler, E.; et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat. Genet. 2013, 45, 478–486. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, Z.; Li, J.; Hu, X.; Shi, X.; Sun, Z.; Zhang, F.; Zhao, Z.; Li, Z.; Liu, Z.; et al. Genetic landscape of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 1097–1102. [Google Scholar] [CrossRef]
- Lin, D.-C.; Hao, J.-J.; Nagata, Y.; Xu, L.; Shang, L.; Meng, X.; Sato, Y.; Okuno, Y.; Varela, A.M.; Ding, L.-W.; et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 467–473. [Google Scholar] [CrossRef]
- Song, Y.; Li, L.; Ou, Y.; Gao, Z.; Li, E.; Li, X.; Zhang, W.; Wang, J.; Xu, L.; Zhou, Y.; et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature 2014, 509, 91–95. [Google Scholar] [CrossRef]
- Totoki, Y.; Tatsuno, K.; Yamamoto, S.; Arai, Y.; Hosoda, F.; Ishikawa, S.; Tsutsumi, S.; Sonoda, K.; Totsuka, H.; Shirakihara, T.; et al. High-resolution characterization of a hepatocellular carcinoma genome. Nat. Genet. 2011, 43, 464–469. [Google Scholar] [CrossRef]
- Totoki, Y.; Tatsuno, K.; Covington, K.R.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; Donehower, L.A.; Slagle, B.L.; Nakamura, H.; et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 2014, 46, 1267–1273. [Google Scholar] [CrossRef]
- Chan-On, W.; Nairismägi, M.-L.; Ong, C.K.; Lim, W.K.; Dima, S.; Pairojkul, C.; Lim, K.H.; McPherson, J.R.; Cutcutache, I.; Heng, H.L.; et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat. Genet. 2013, 45, 1474–1478. [Google Scholar] [CrossRef]
- Jiao, Y.; Pawlik, T.M.; Anders, R.A.; Selaru, F.M.; Streppel, M.M.; Lucas, D.J.; Niknafs, N.; Guthrie, V.B.; Maitra, A.; Argani, P.; et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat. Genet. 2013, 45, 1470–1473. [Google Scholar] [CrossRef]
- Olivier, M.; Weninger, A.; Ardin, M.; Huskova, H.; Castells, X.; Vallée, M.P.; McKay, J.; Nedelko, T.; Muehlbauer, K.-R.; Marusawa, H.; et al. Modelling mutational landscapes of human cancers in vitro. Sci. Rep. 2014, 4, 4482. [Google Scholar] [CrossRef]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.V.; Carter, S.L.; Saksena, G.; et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar] [CrossRef]
- Peltomäki, P. Role of DNA mismatch repair defects in the pathogenesis of human cancer. J. Clin. Oncol. 2003, 21, 1174–1179. [Google Scholar] [CrossRef]
- Imai, K.; Yamamoto, H. Carcinogenesis and microsatellite instability: The interrelationship between genetics and epigenetics. Carcinogenesis 2008, 29, 673–680. [Google Scholar] [CrossRef]
- Shah, S.N.; Hile, S.E.; Eckert, K.A. Defective mismatch repair, microsatellite mutation bias, and variability in clinical cancer phenotypes. Cancer Res. 2010, 70, 431–435. [Google Scholar] [CrossRef]
- Heitzer, E.; Tomlinson, I. Replicative DNA polymerase mutations in cancer. Curr. Opin. Genet. Dev. 2014, 24, 107–113. [Google Scholar] [CrossRef]
- Palles, C.; Cazier, J.-B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Guarino Almeida, E.; et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013, 45, 136–144. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef] [Green Version]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef]
- Jayaraman, S.S.; Rayhan, D.J.; Hazany, S.; Kolodney, M.S. Mutational landscape of basal cell carcinomas by whole-exome sequencing. J. Invest. Dermatol. 2014, 134, 213–220. [Google Scholar] [CrossRef]
- Pfeifer, G.P.; Denissenko, M.F.; Olivier, M.; Tretyakova, N.; Hecht, S.S.; Hainaut, P. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene 2002, 21, 7435–7451. [Google Scholar] [CrossRef]
- Shimizu, T.; Marusawa, H.; Endo, Y.; Chiba, T. Inflammation-mediated genomic instability: Roles of activation-induced cytidine deaminase in carcinogenesis. Cancer Sci. 2012, 103, 1201–1206. [Google Scholar] [CrossRef]
- Takai, A.; Marusawa, H.; Chiba, T. Acquisition of Genetic Aberrations by Activation-Induced Cytidine Deaminase (AID) during Inflammation-Associated Carcinogenesis. Cancers 2011, 3, 2750–2766. [Google Scholar] [CrossRef]
- Muramatsu, M.; Kinoshita, K.; Fagarasan, S.; Yamada, S.; Shinkai, Y.; Honjo, T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 2000, 102, 553–563. [Google Scholar] [CrossRef]
- Honjo, T.; Kinoshita, K.; Muramatsu, M. Molecular mechanism of class switch recombination: Linkage with somatic hypermutation. Annu. Rev. Immunol. 2002, 20, 165–196. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Marusawa, H.; Kinoshita, K.; Endo, Y.; Kou, T.; Morisawa, T.; Azuma, T.; Okazaki, I.-M.; Honjo, T.; Chiba, T. Helicobacter pylori infection triggers aberrant expression of activation-induced cytidine deaminase in gastric epithelium. Nat. Med. 2007, 13, 470–476. [Google Scholar] [CrossRef]
- Endo, Y.; Marusawa, H.; Kinoshita, K.; Morisawa, T.; Sakurai, T.; Okazaki, I.-M.; Watashi, K.; Shimotohno, K.; Honjo, T.; Chiba, T. Expression of activation-induced cytidine deaminase in human hepatocytes via NF-kappaB signaling. Oncogene 2007, 26, 5587–5595. [Google Scholar] [CrossRef]
- Liu, M.; Schatz, D.G. Balancing AID and DNA repair during somatic hypermutation. Trends Immunol. 2009, 30, 173–181. [Google Scholar] [CrossRef]
- Kim, S.K.; Nasu, A.; Komori, J.; Shimizu, T.; Matsumoto, Y.; Minaki, Y.; Kohno, K.; Shimizu, K.; Uemoto, S.; Chiba, T.; et al. A model of liver carcinogenesis originating from hepatic progenitor cells with accumulation of genetic alterations. Int. J. Cancer 2014, 134, 1067–1076. [Google Scholar] [CrossRef] [Green Version]
- Shinohara, M.; Io, K.; Shindo, K.; Matsui, M.; Sakamoto, T.; Tada, K.; Kobayashi, M.; Kadowaki, N.; Takaori-Kondo, A. APOBEC3B can impair genomic stability by inducing base substitutions in genomic DNA in human cells. Sci. Rep. 2012, 2, 806. [Google Scholar] [CrossRef] [Green Version]
- Morisawa, T.; Marusawa, H.; Ueda, Y.; Iwai, A.; Okazaki, I.; Honjo, T.; Chiba, T. Organ-specific profiles of genetic changes in cancers caused by activation-induced cytidine deaminase expression. Int. J. Cancer 2008, 123, 2735–2740. [Google Scholar] [CrossRef]
- Shen, J.C.; Rideout, W.M.; Jones, P.A. The rate of hydrolytic deamination of 5-methylcytosine in double-stranded DNA. Nucleic Acids Res. 1994, 22, 972–976. [Google Scholar] [CrossRef]
- Wood, A.R.; Tuke, M.A.; Nalls, M.; Hernandez, D.; Gibbs, J.R.; Lin, H.; Xu, C.S.; Li, Q.; Shen, J.; Jun, G.; et al. Whole-genome sequencing to understand the genetic architecture of common gene expression and biomarker phenotypes. Hum. Mol. Genet. 2014, 24, 1504–1512. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsumoto, T.; Shimizu, T.; Takai, A.; Marusawa, H. Exploring the Mechanisms of Gastrointestinal Cancer Development Using Deep Sequencing Analysis. Cancers 2015, 7, 1037-1051. https://doi.org/10.3390/cancers7020823
Matsumoto T, Shimizu T, Takai A, Marusawa H. Exploring the Mechanisms of Gastrointestinal Cancer Development Using Deep Sequencing Analysis. Cancers. 2015; 7(2):1037-1051. https://doi.org/10.3390/cancers7020823
Chicago/Turabian StyleMatsumoto, Tomonori, Takahiro Shimizu, Atsushi Takai, and Hiroyuki Marusawa. 2015. "Exploring the Mechanisms of Gastrointestinal Cancer Development Using Deep Sequencing Analysis" Cancers 7, no. 2: 1037-1051. https://doi.org/10.3390/cancers7020823