STAT3 Activities and Energy Metabolism: Dangerous Liaisons

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
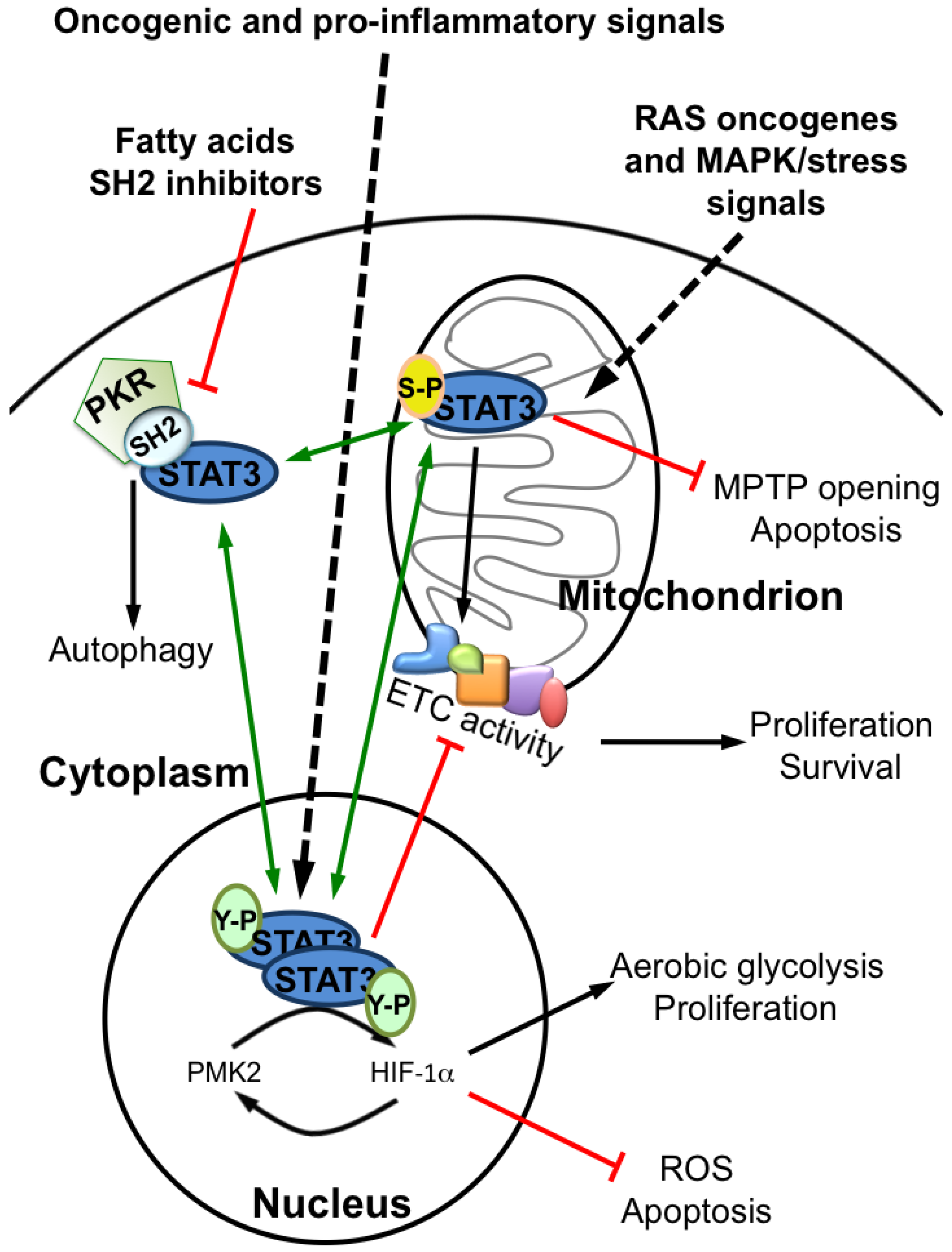
1.1. STAT3 Differentially Modified Forms and Cell Metabolism
2. Results and Discussion
2.1. STAT3 Constitutive Activation Elicits Pre-Oncogenic Features in Stat3C/C MEFs.
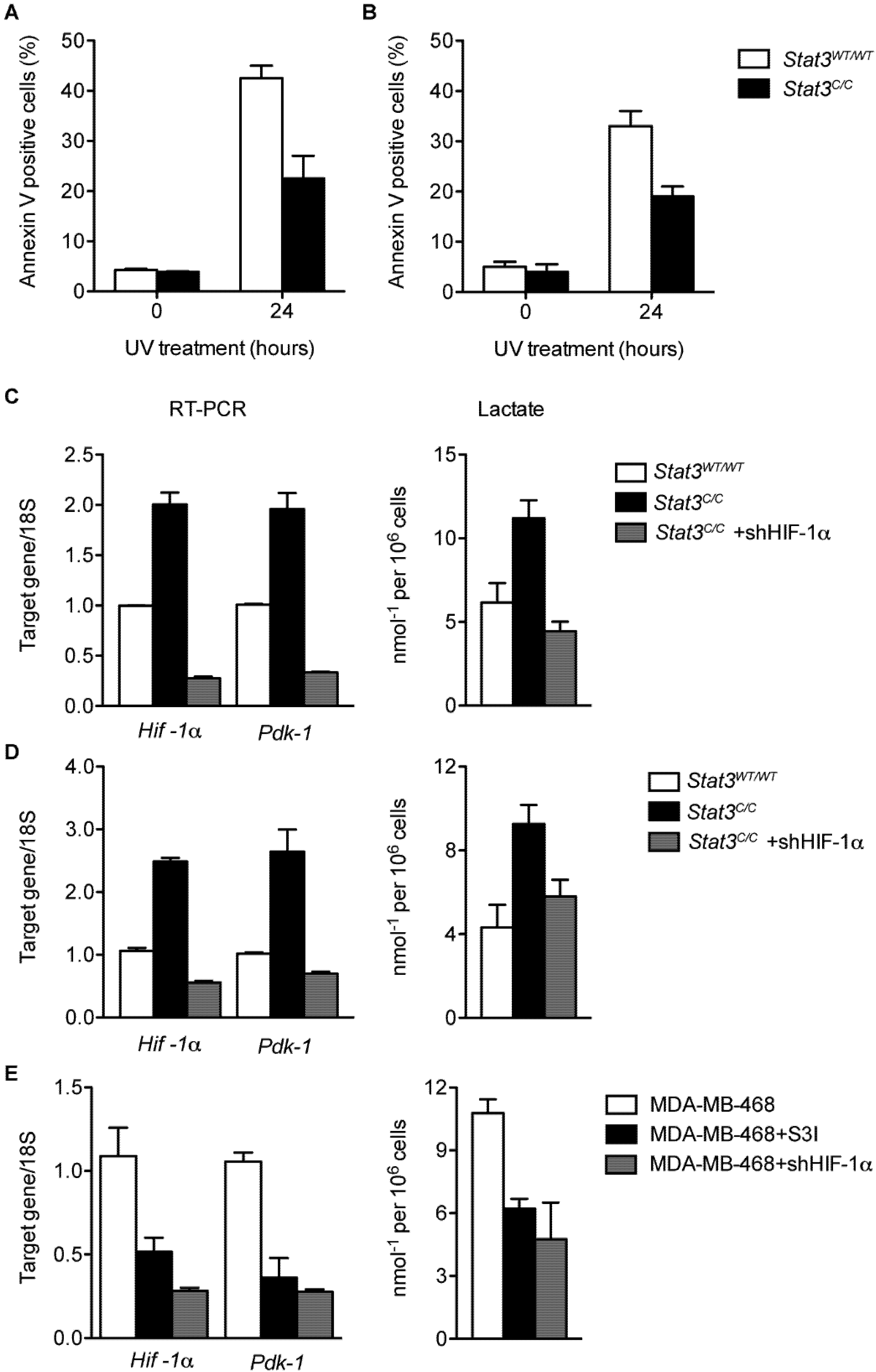
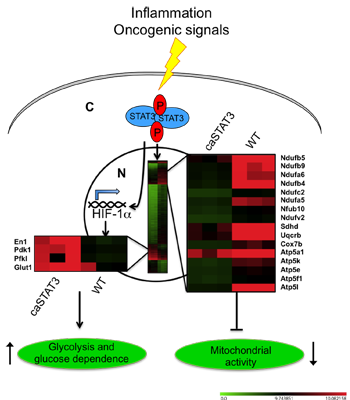
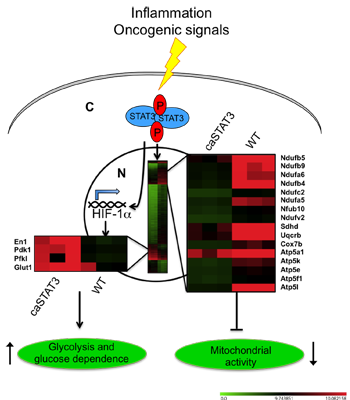
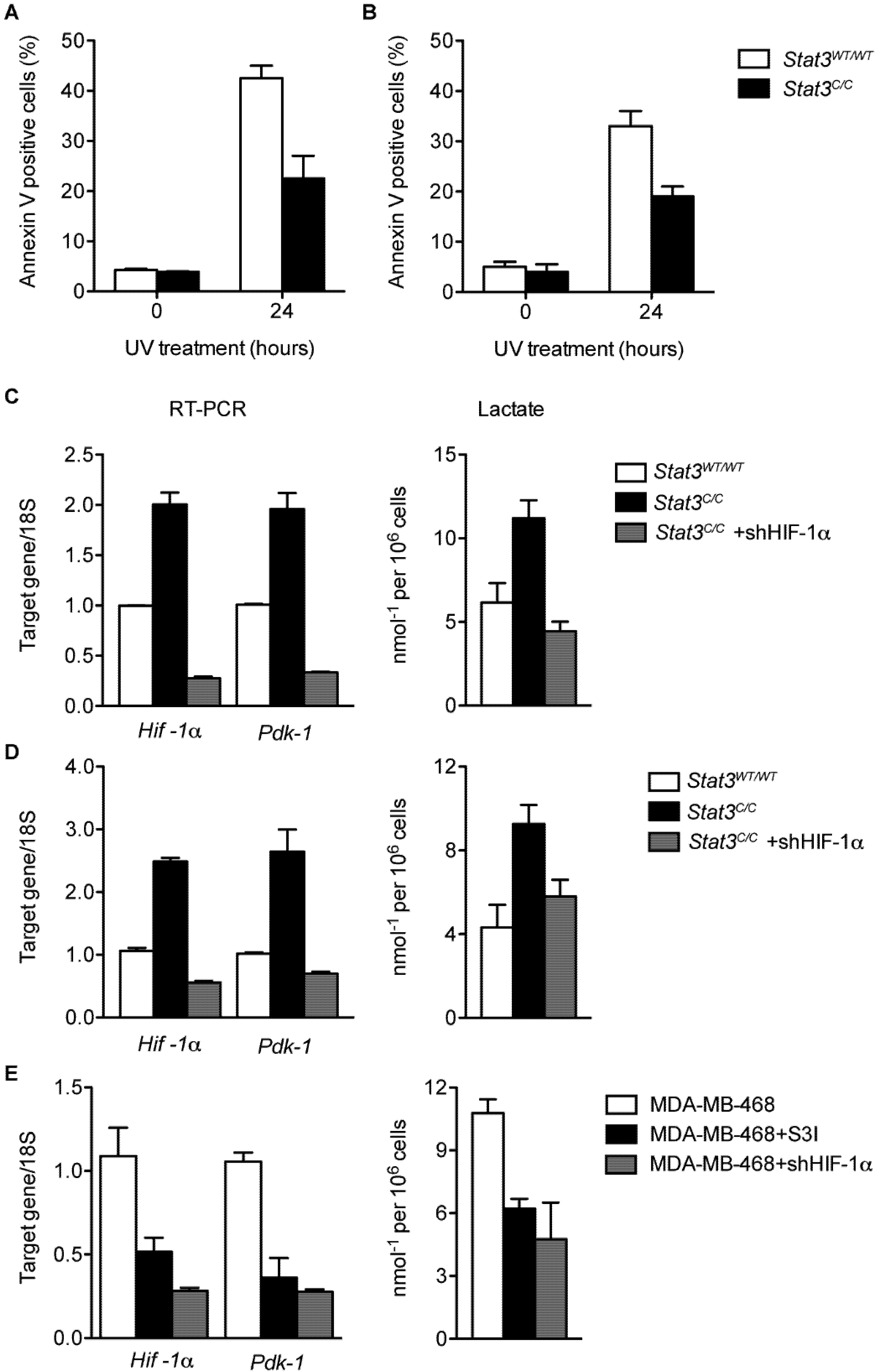
2.2. Constitutive STAT3 Activity Induces a Metabolic Switch to Aerobic Glycolysis in MEFs and Tumor Cell Lines

2.3. Reduced Mitochondrial Activity in Cells with a Constitutive Activation of STAT3
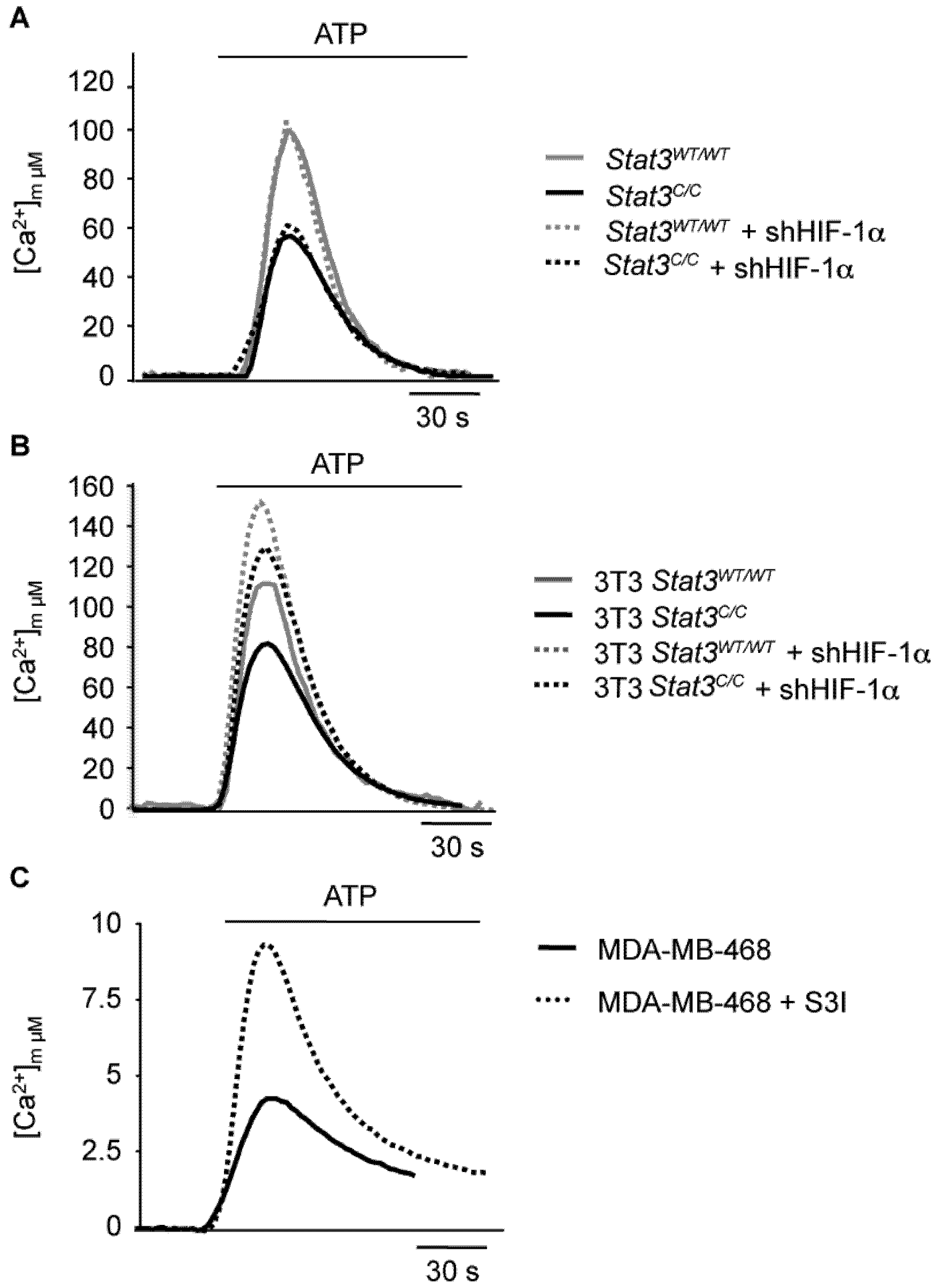
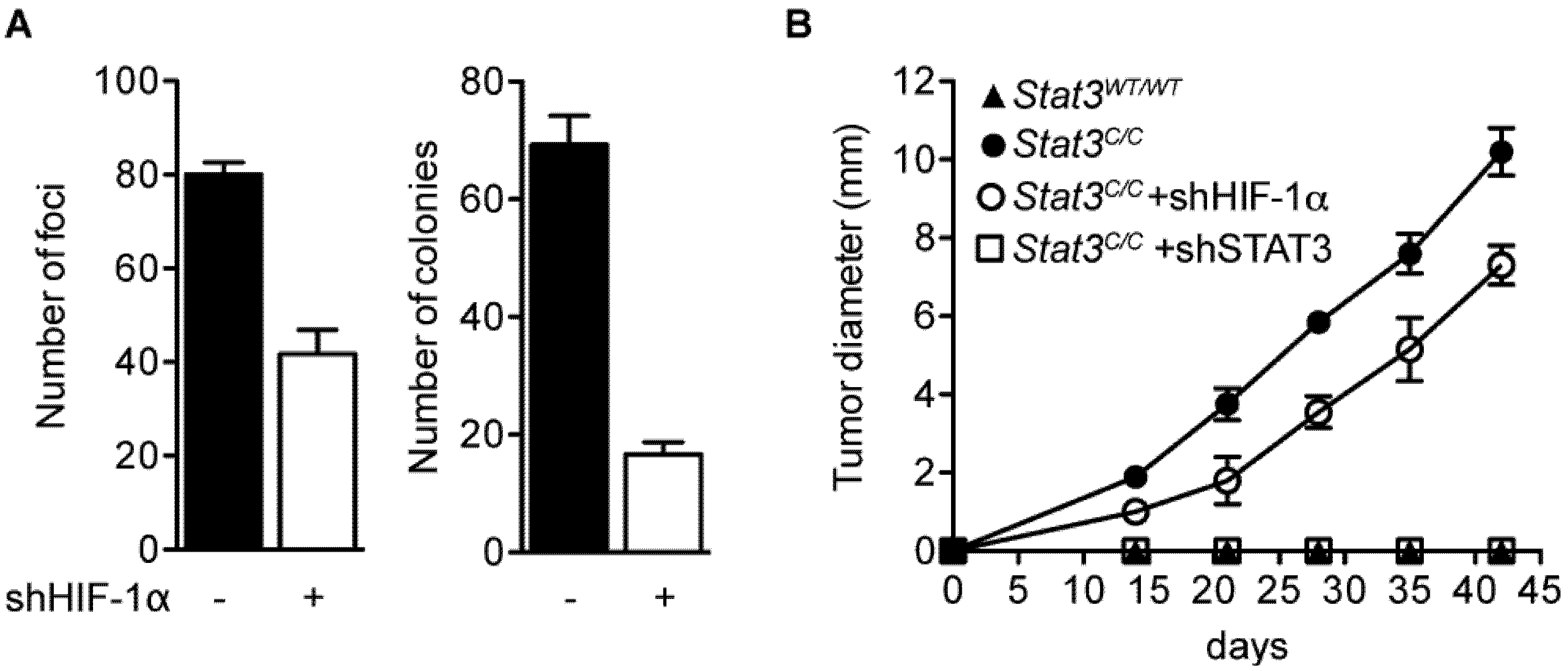
2.4. STAT3C Triggers Tumorigenic Transformation in Immortalized MEFs
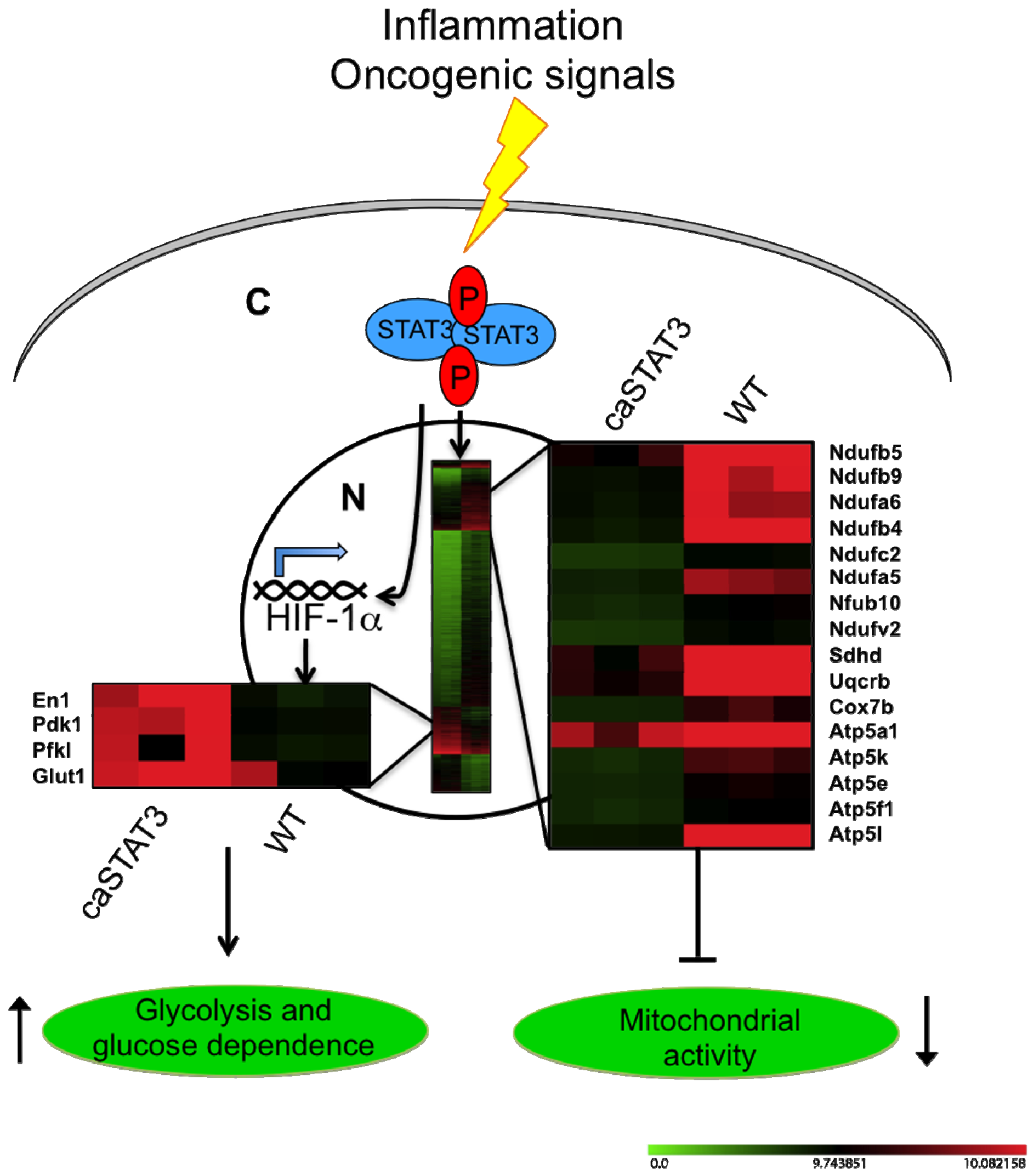
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chung, J.; Uchida, E.; Grammer, T.C.; Blenis, J. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol. Cell. Biol. 1997, 17, 6508–6516. [Google Scholar]
- Yokogami, K.; Wakisaka, S.; Avruch, J.; Reeves, S.A. Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr. Biol. 2000, 10, 47–50. [Google Scholar] [CrossRef]
- Aznar, S.; Valeron, P.F.; del Rincon, S.V.; Perez, L.F.; Perona, R.; Lacal, J.C. Simultaneous tyrosine and serine phosphorylation of STAT3 transcription factor is involved in Rho A GTPase oncogenic transformation. Mol. Biol. Cell 2001, 12, 3282–3294. [Google Scholar] [CrossRef]
- Kubo, M.; Hanada, T.; Yoshimura, A. Suppressors of cytokine signaling and immunity. Nat. Immunol. 2003, 4, 1169–1176. [Google Scholar] [CrossRef]
- Shuai, K.; Liu, B. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat. Rev. Immunol. 2005, 5, 593–605. [Google Scholar] [CrossRef]
- Haura, E.B.; Turkson, J.; Jove, R. Mechanisms of disease: Insights into the emerging role of signal transducers and activators of transcription in cancer. Nat. Clin. Pract. Oncol. 2005, 2, 315–324. [Google Scholar] [CrossRef]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. STAT3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Alvarez, J.V.; Febbo, P.G.; Ramaswamy, S.; Loda, M.; Richardson, A.; Frank, D.A. Identification of a genetic signature of activated signal transducer and activator of transcription 3 in human tumors. Cancer Res. 2005, 65, 5054–5062. [Google Scholar] [CrossRef]
- Chiarle, R.; Simmons, W.J.; Cai, H.; Dhall, G.; Zamo, A.; Raz, R.; Karras, J.G.; Levy, D.E.; Inghirami, G. STAT3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat. Med. 2005, 11, 623–629. [Google Scholar] [CrossRef]
- Weinstein, I.B. Cancer. Addiction to oncogenes—The Achilles heal of cancer. Science 2002, 297, 63–64. [Google Scholar] [CrossRef]
- Yu, H.; Jove, R. The STATs of cancer—New molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef]
- Bromberg, J.; Wang, T.C. Inflammation and cancer: IL-6 and STAT3 complete the link. Cancer Cell 2009, 15, 79–80. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Demaria, M.; Camporeale, A.; Poli, V. STAT3 and metabolism: How many ways to use a single molecule? Int. J. Cancer 2014. [Google Scholar] [CrossRef]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial STAT3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef]
- Boengler, K.; Hilfiker-Kleiner, D.; Heusch, G.; Schulz, R. Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res. Cardiol. 2010, 105, 771–785. [Google Scholar]
- Tammineni, P.; Anugula, C.; Mohammed, F.; Anjaneyulu, M.; Larner, A.C.; Sepuri, N.B. The import of the transcription factor STAT3 into mitochondria depends on GRIM-19, a component of the electron transport chain. J. Biol. Chem. 2013, 288, 4723–4732. [Google Scholar] [CrossRef]
- Zheng, H.; Li, S.; Hsu, P.; Qu, C.K. Induction of a tumor-associated activating mutation in protein tyrosine phosphatase Ptpn11 (Shp2) enhances mitochondrial metabolism, leading to oxidative stress and senescence. J. Biol. Chem. 2013, 288, 25727–25738. [Google Scholar] [CrossRef]
- Demaria, M.; Giorgi, C.; Lebiedzinska, M.; Esposito, G.; D’Angeli, L.; Bartoli, A.; Gough, D.J.; Turkson, J.; Levy, D.E.; Watson, C.J.; et al. A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging (Albany NY) 2010, 2, 823–842. [Google Scholar]
- Pawlus, M.R.; Wang, L.; Murakami, A.; Dai, G.; Hu, C.J. STAT3 or USF2 contributes to Hif target gene specificity. PLoS One 2013, 8, e72358. [Google Scholar]
- Demaria, M.; Misale, S.; Giorgi, C.; Miano, V.; Camporeale, A.; Campisi, J.; Pinton, P.; Poli, V. STAT3 can serve as a hit in the process of malignant transformation of primary cells. Cell Death Differ. 2012, 19, 1390–1397. [Google Scholar] [CrossRef]
- Gao, X.; Wang, H.; Yang, J.J.; Liu, X.; Liu, Z.R. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol. Cell 2012, 45, 598–609. [Google Scholar] [CrossRef]
- Demaria, M.; Poli, V. PKM2, STAT3 and Hif-1alpha: The Warburg’s vicious circle. JAKSTAT 2012, 1, 194–196. [Google Scholar]
- Zhao, S.; Xu, W.; Jiang, W.; Yu, W.; Lin, Y.; Zhang, T.; Yao, J.; Zhou, L.; Zeng, Y.; Li, H.; et al. Regulation of cellular metabolism by protein lysine acetylation. Science 2010, 327, 1000–1004. [Google Scholar] [CrossRef]
- Yuan, Z.L.; Guan, Y.J.; Chatterjee, D.; Chin, Y.E. STAT3 dimerization regulated by reversible acetylation of a single lysine residue. Science 2005, 307, 269–273. [Google Scholar] [CrossRef]
- Lee, J.L.; Wang, M.J.; Chen, J.Y. Acetylation and activation of STAT3 mediated by nuclear translocation of CD44. J. Cell Biol. 2009, 185, 949–957. [Google Scholar] [CrossRef]
- Lee, H.; Zhang, P.; Herrmann, A.; Yang, C.; Xin, H.; Wang, Z.; Hoon, D.S.; Forman, S.J.; Jove, R.; Riggs, A.D.; et al. Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc. Natl. Acad. Sci. USA 2012, 109, 7765–7769. [Google Scholar] [CrossRef]
- Nie, Y.; Erion, D.M.; Yuan, Z.; Dietrich, M.; Shulman, G.I.; Horvath, T.L.; Gao, Q. STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat. Cell Biol. 2009, 11, 492–500. [Google Scholar] [CrossRef]
- Bernier, M.; Paul, R.K.; Martin-Montalvo, A.; Scheibye-Knudsen, M.; Song, S.; He, H.J.; Armour, S.M.; Hubbard, B.P.; Bohr, V.A.; Wang, L.; et al. Negative regulation of STAT3 protein-mediated cellular respiration by SIRT1 protein. J. Biol. Chem. 2011, 286, 19270–19279. [Google Scholar] [CrossRef]
- Finley, L.W.; Carracedo, A.; Lee, J.; Souza, A.; Egia, A.; Zhang, J.; Teruya-Feldstein, J.; Moreira, P.I.; Cardoso, S.M.; Clish, C.B.; et al. SIRT3 opposes reprogramming of cancer cell metabolism through Hif 1alpha destabilization. Cancer Cell 2011, 19, 416–428. [Google Scholar] [CrossRef]
- Horak, P.; Crawford, A.R.; Vadysirisack, D.D.; Nash, Z.M.; deYoung, M.P.; Sgroi, D.; Ellisen, L.W. Negative feedback control of Hif-1 through REDD1-regulated ROS suppresses tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 4675–4680. [Google Scholar]
- Yang, F.; Zhang, W.; Li, D.; Zhan, Q. Gadd45a suppresses tumor angiogenesis via inhibition of the mTOR/STAT3 protein pathway. J. Biol. Chem. 2013, 288, 6552–6560. [Google Scholar] [CrossRef]
- Goncharova, E.A.; Goncharov, D.A.; Damera, G.; Tliba, O.; Amrani, Y.; Panettieri, R.A., Jr.; Krymskaya, V.P. Signal transducer and activator of transcription 3 is required for abnormal proliferation and survival of TSC2-deficient cells: Relevance to pulmonary lymphangioleiomyomatosis. Mol. Pharm. 2009, 76, 766–777. [Google Scholar] [CrossRef]
- Zhou, J.; Wulfkuhle, J.; Zhang, H.; Gu, P.; Yang, Y.; Deng, J.; Margolick, J.B.; Liotta, L.A.; Petricoin, E., III; Zhang, Y. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc. Natl. Acad. Sci. USA 2007, 104, 16158–16163. [Google Scholar] [CrossRef]
- Roy, S.; Debnath, J. Autophagy and tumorigenesis. Semin. Immunopathol. 2010, 32, 383–396. [Google Scholar] [CrossRef]
- Shen, S.; Niso-Santano, M.; Adjemian, S.; Takehara, T.; Malik, S.A.; Minoux, H.; Souquere, S.; Marino, G.; Lachkar, S.; Senovilla, L.; et al. Cytoplasmic STAT3 represses autophagy by inhibiting PKR activity. Mol. Cell 2012, 48, 667–680. [Google Scholar] [CrossRef]
- Barbieri, I.; Pensa, S.; Pannellini, T.; Quaglino, E.; Maritano, D.; Demaria, M.; Voster, A.; Turkson, J.; Cavallo, F.; Watson, C.J.; et al. Constitutively active STAT3 enhances neu-mediated migration and metastasis in mammary tumors via upregulation of Cten. Cancer Res. 2010, 70, 2558–2567. [Google Scholar] [CrossRef]
- Niu, G.; Briggs, J.; Deng, J.; Ma, Y.; Lee, H.; Kortylewski, M.; Kujawski, M.; Kay, H.; Cress, W.D.; Jove, R.; et al. Signal transducer and activator of transcription 3 is required for hypoxia-inducible factor-1alpha RNA expression in both tumor cells and tumor-associated myeloid cells. Mol. Cancer Res. 2008, 6, 1099–1105. [Google Scholar] [CrossRef]
- Salceda, S.; Caro, J. Hypoxia-inducible factor 1alpha (Hif-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J. Biol. Chem. 1997, 272, 22642–22647. [Google Scholar] [CrossRef]
- Dolled-Filhart, M.; Camp, R.L.; Kowalski, D.P.; Smith, B.L.; Rimm, D.L. Tissue microarray analysis of signal transducers and activators of transcription 3 (STAT3) and phospho-STAT3 (Tyr705) in node-negative breast cancer shows nuclear localization is associated with a better prognosis. Clin. Cancer Res. 2003, 9, 594–600. [Google Scholar]
- Pectasides, E.; Egloff, A.M.; Sasaki, C.; Kountourakis, P.; Burtness, B.; Fountzilas, G.; Dafni, U.; Zaramboukas, T.; Rampias, T.; Rimm, D.; et al. Nuclear localization of signal transducer and activator of transcription 3 in head and neck squamous cell carcinoma is associated with a better prognosis. Clin. Cancer Res. 2010, 16, 2427–2434. [Google Scholar] [CrossRef]
- Musteanu, M.; Blaas, L.; Mair, M.; Schlederer, M.; Bilban, M.; Tauber, S.; Esterbauer, H.; Mueller, M.; Casanova, E.; Kenner, L.; et al. STAT3 is a negative regulator of intestinal tumor progression in Apc(Min) mice. Gastroenterology 2010, 138, 1003–1011. [Google Scholar] [CrossRef]
- Couto, J.P.; Daly, L.; Almeida, A.; Knauf, J.A.; Fagin, J.A.; Sobrinho-Simoes, M.; Lima, J.; Maximo, V.; Soares, P.; Lyden, D.; et al. STAT3 negatively regulates thyroid tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, E2361–E2370. [Google Scholar] [CrossRef]
- Bertout, J.A.; Patel, S.A.; Simon, M.C. The impact of O2 availability on human cancer. Nat. Rev. Cancer 2008, 8, 967–975. [Google Scholar] [CrossRef]
- Marchi, S.; Pinton, P. The mitochondrial calcium uniporter complex: Molecular components, structure and physiopathological implications. J. Physiol. 2014, 592, Pt 5. 829–839. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef]
- Giorgi, C.; de Stefani, D.; Bononi, A.; Rizzuto, R.; Pinton, P. Structural and functional link between the mitochondrial network and the endoplasmic reticulum. Int. J. Biochem. Cell Biol. 2009, 41, 1817–1827. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pozzan, T. Microdomains of intracellular Ca2+: Molecular determinants and functional consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef]
- Marchi, S.; Patergnani, S.; Pinton, P. The endoplasmic reticulum-mitochondria connection: One touch, multiple functions. Biochim. Biophys. Acta 2014, 1837, 461–469. [Google Scholar] [CrossRef]
- Giorgi, C.; Baldassari, F.; Bononi, A.; Bonora, M.; de Marchi, E.; Marchi, S.; Missiroli, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; et al. Mitochondrial Ca2+ and apoptosis. Cell Calcium 2012, 52, 36–43. [Google Scholar] [CrossRef]
- Bonora, M.; Bononi, A.; de Marchi, E.; Giorgi, C.; Lebiedzinska, M.; Marchi, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; Wojtala, A.; et al. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle 2013, 12, 674–683. [Google Scholar] [CrossRef]
- Bonora, M.; Giorgi, C.; Bononi, A.; Marchi, S.; Patergnani, S.; Rimessi, A.; Rizzuto, R.; Pinton, P. Subcellular calcium measurements in mammalian cells using jellyfish photoprotein aequorin-based probes. Nat. Protoc. 2013, 8, 2105–2118. [Google Scholar] [CrossRef]
- Monteleone, M.; Camporeale, A.; Poli, V.; Molecular Biotechnology Center and Department of Molecular Biotechnology and Life Sciences, University of Turin, Turin, Italy. Personal observation. 2014.
- Wang, B.; Hsu, S.H.; Frankel, W.; Ghoshal, K.; Jacob, S.T. STAT3-mediated activation of microRNA-23a suppresses gluconeogenesis in hepatocellular carcinoma by down-regulating glucose-6-phosphatase and peroxisome proliferator-activated receptor gamma, coactivator 1 alpha. Hepatology 2012, 56, 186–197. [Google Scholar]
- Austin, S.; Klimcakova, E.; St-Pierre, J. Impact of PGC-1alpha on the topology and rate of superoxide production by the mitochondrial electron transport chain. Free Radic. Biol. Med. 2011, 51, 2243–2248. [Google Scholar] [CrossRef]
- Wenz, T.; Rossi, S.G.; Rotundo, R.L.; Spiegelman, B.M.; Moraes, C.T. Increased muscle PGC-1alpha expression protects from sarcopenia and metabolic disease during aging. Proc. Natl. Acad. Sci. USA 2009, 106, 20405–20410. [Google Scholar] [CrossRef]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jager, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef]
- Poli, V.; Wieckowski, M.R.; Molecular Biotechnology Center and Department of Molecular Biotechnology and Life Sciences, University of Turin, Turin, Italy (V.P.); Nencki Institute of Experimental Biology, Department of Biochemistry, Warsaw, Poland (M.R.W.). Personal observation. 2014.
- Li, F.; Wang, Y.; Zeller, K.I.; Potter, J.J.; Wonsey, D.R.; O’Donnell, K.A.; Kim, J.W.; Yustein, J.T.; Lee, L.A.; Dang, C.V. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol. Cell Biol. 2005, 25, 6225–6234. [Google Scholar] [CrossRef]
- Todaro, G.J.; Green, H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol. 1963, 17, 299–313. [Google Scholar] [CrossRef]
- Krontiris, T.G.; Cooper, G.M. Transforming activity of human tumor DNAs. Proc. Natl. Acad. Sci. USA 1981, 78, 1181–1184. [Google Scholar] [CrossRef]
- Hodge, D.R.; Hurt, E.M.; Farrar, W.L. The role of IL-6 and STAT3 in inflammation and cancer. Eur. J. Cancer 2005, 41, 2502–2512. [Google Scholar] [CrossRef]
- Kiuchi, N.; Nakajima, K.; Ichiba, M.; Fukada, T.; Narimatsu, M.; Mizuno, K.; Hibi, M.; Hirano, T. STAT3 is required for the gp130-mediated full activation of the c-myc gene. J. Exp. Med. 1999, 189, 63–73. [Google Scholar] [CrossRef]
- Avalle, L.; Pensa, S.; Regis, G.; Novelli, F.; Poli, V. STAT1 and STAT3 in tumorigenesis: A matter of balance. JAKSTAT 2012, 1, 65–72. [Google Scholar]
- Bromberg, J.F.; Horvath, C.M.; Besser, D.; Lathem, W.W.; Darnell, J.E., Jr. STAT3 activation is required for cellular transformation by v-src. Mol. Cell Biol. 1998, 18, 2553–2558. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Camporeale, A.; Demaria, M.; Monteleone, E.; Giorgi, C.; Wieckowski, M.R.; Pinton, P.; Poli, V. STAT3 Activities and Energy Metabolism: Dangerous Liaisons. Cancers 2014, 6, 1579-1596. https://doi.org/10.3390/cancers6031579
Camporeale A, Demaria M, Monteleone E, Giorgi C, Wieckowski MR, Pinton P, Poli V. STAT3 Activities and Energy Metabolism: Dangerous Liaisons. Cancers. 2014; 6(3):1579-1596. https://doi.org/10.3390/cancers6031579
Chicago/Turabian StyleCamporeale, Annalisa, Marco Demaria, Emanuele Monteleone, Carlotta Giorgi, Mariusz R. Wieckowski, Paolo Pinton, and Valeria Poli. 2014. "STAT3 Activities and Energy Metabolism: Dangerous Liaisons" Cancers 6, no. 3: 1579-1596. https://doi.org/10.3390/cancers6031579