STAT3 in Cancer—Friend or Foe?


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. The Normal Functions of STAT3
2. The Oncogenic Potential of STAT3
2.1. An Overview
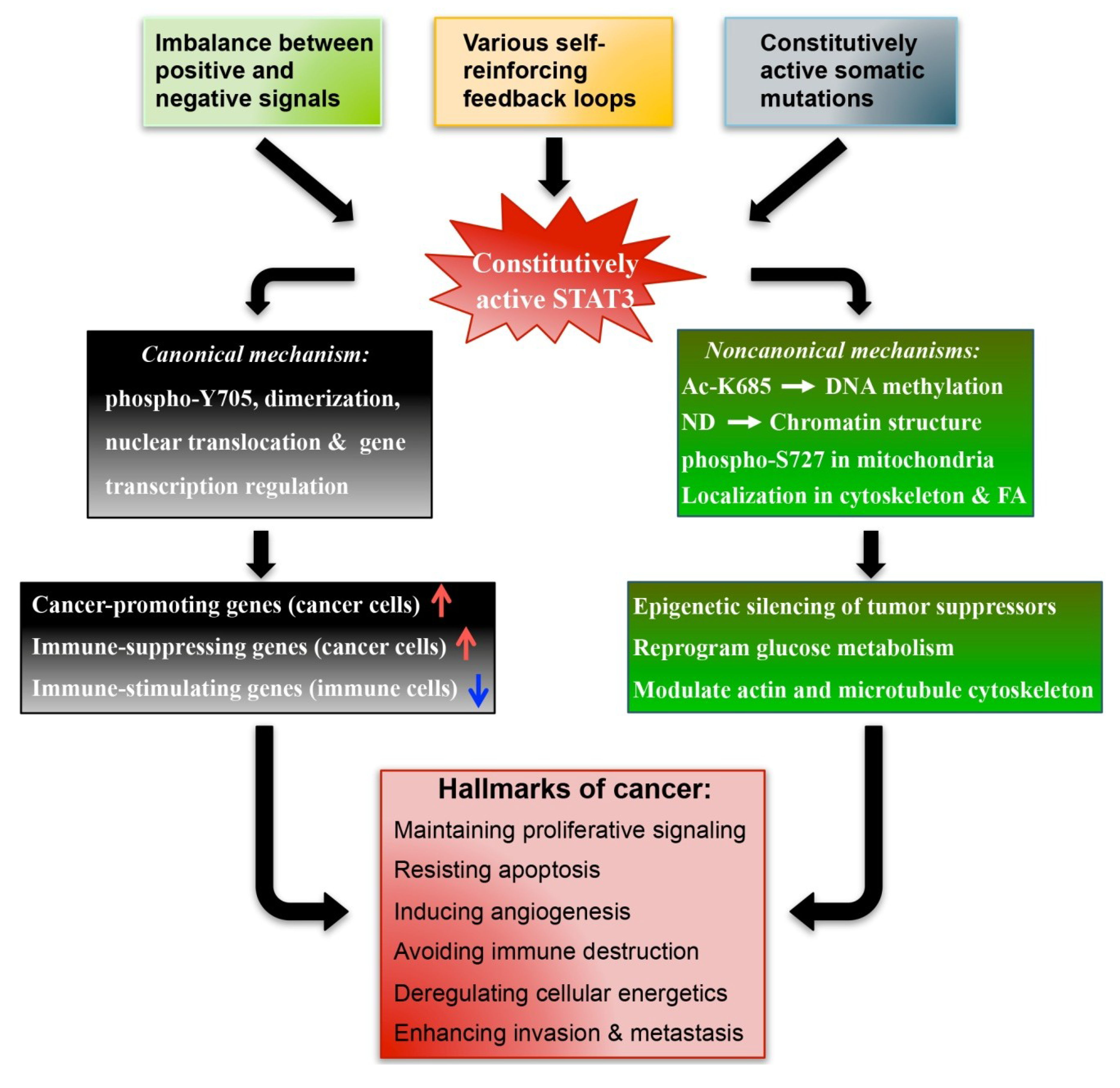
2.2. Mechanisms Underlying the Constitutive Activation of STAT3 in Cancer
2.2.1. Loss of the Negative Regulation of STAT3
2.2.2. Excessive Stimulation of STAT3
2.2.3. Positive Feedback Loops that Sustain Persistent STAT3 Activation
2.2.4. Constitutively Active Somatic STAT3 Mutations
2.3. The Canonical and Non-Canonical STAT3 Pathways in Cancer
2.3.1. Canonical Mechanisms

2.3.2. Non-Canonical Mechanisms
3. The Tumor Suppressor Functions of STAT3
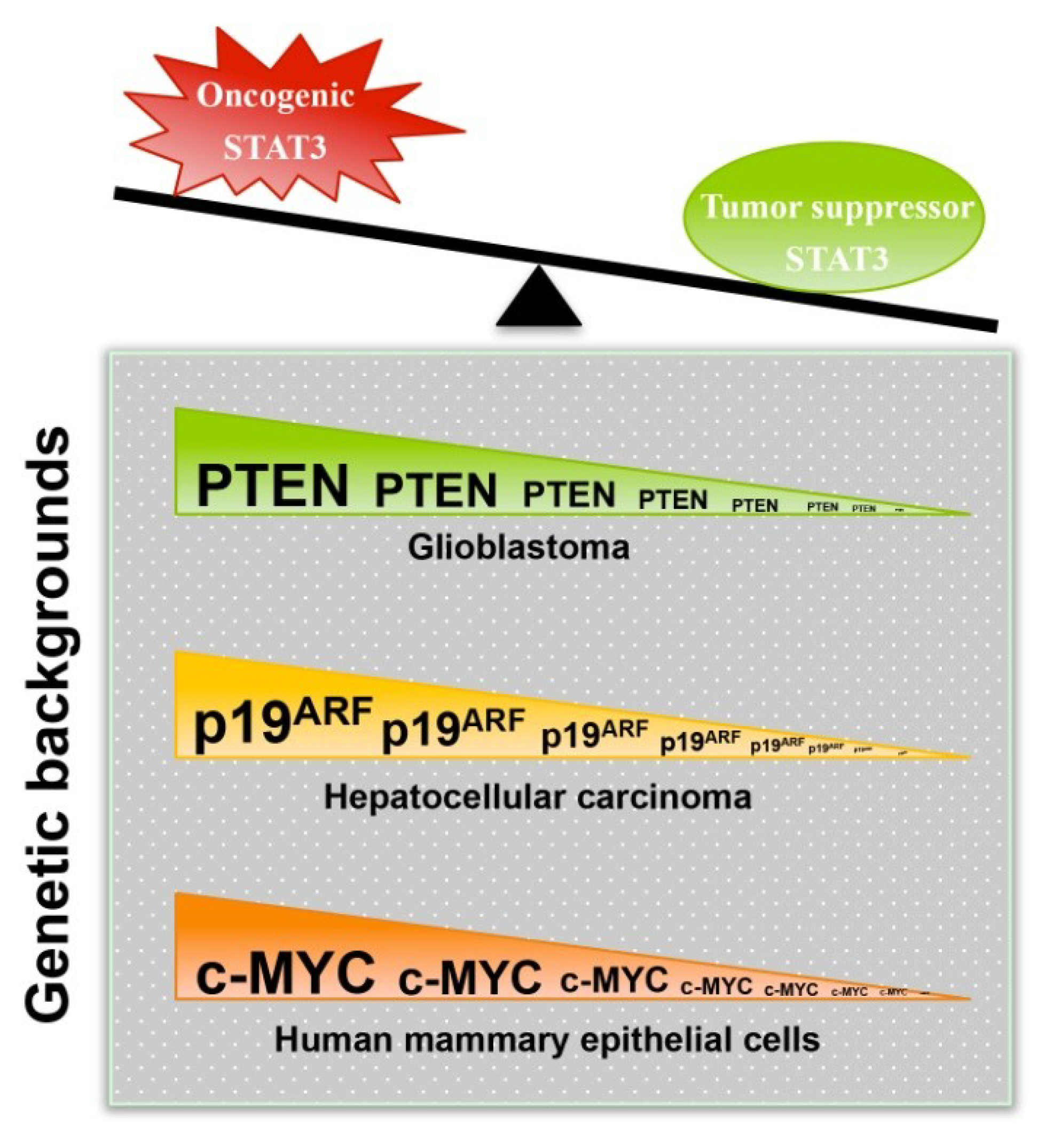
3.1. STAT3 Can Exert Tumor Suppressor Effects
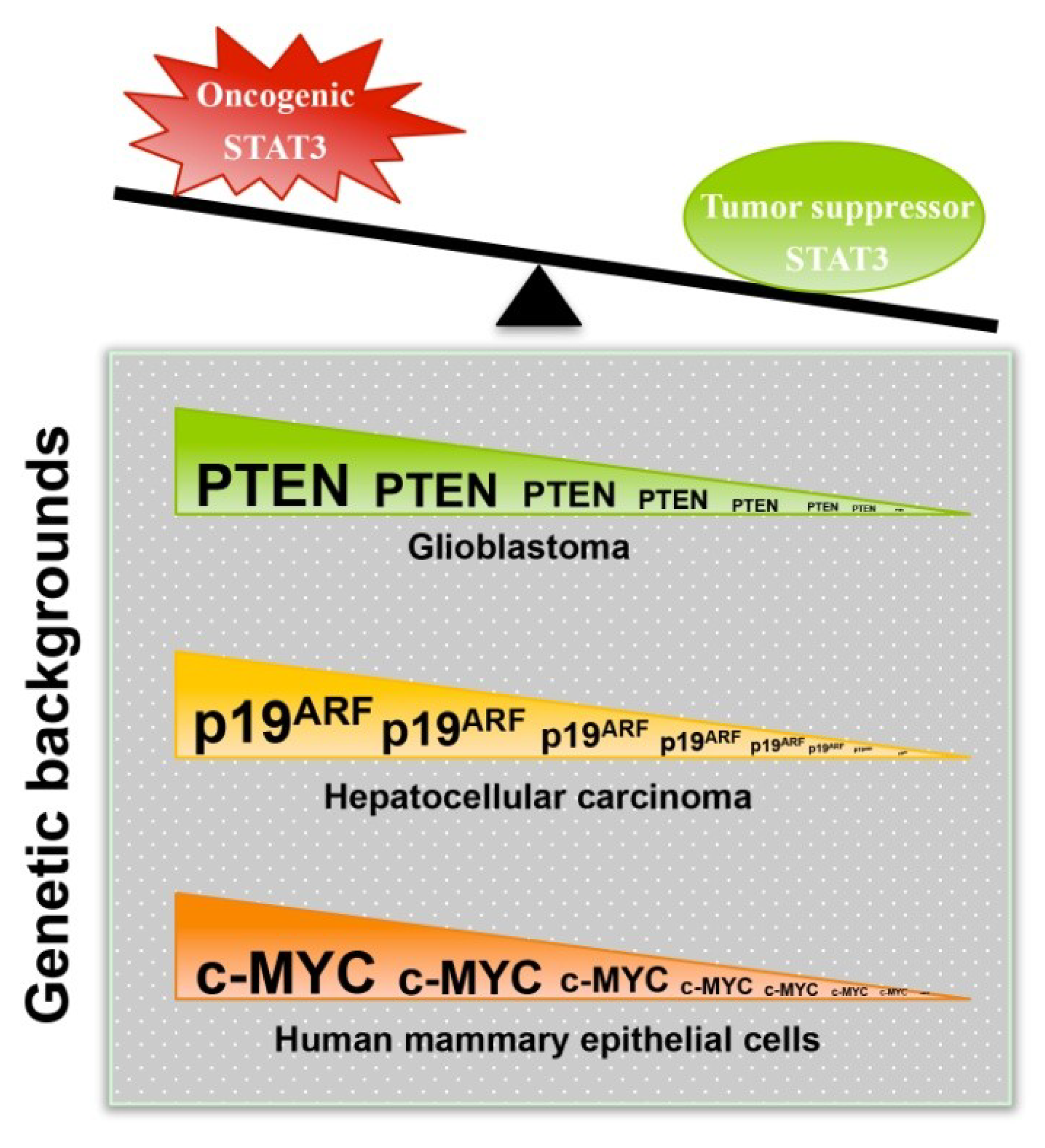
3.2. Mechanisms That Mediate or Regulate the Tumor Suppressor Function of STAT3
3.3. Clinical Observations Supporting the Tumor Suppressor Role of STAT3
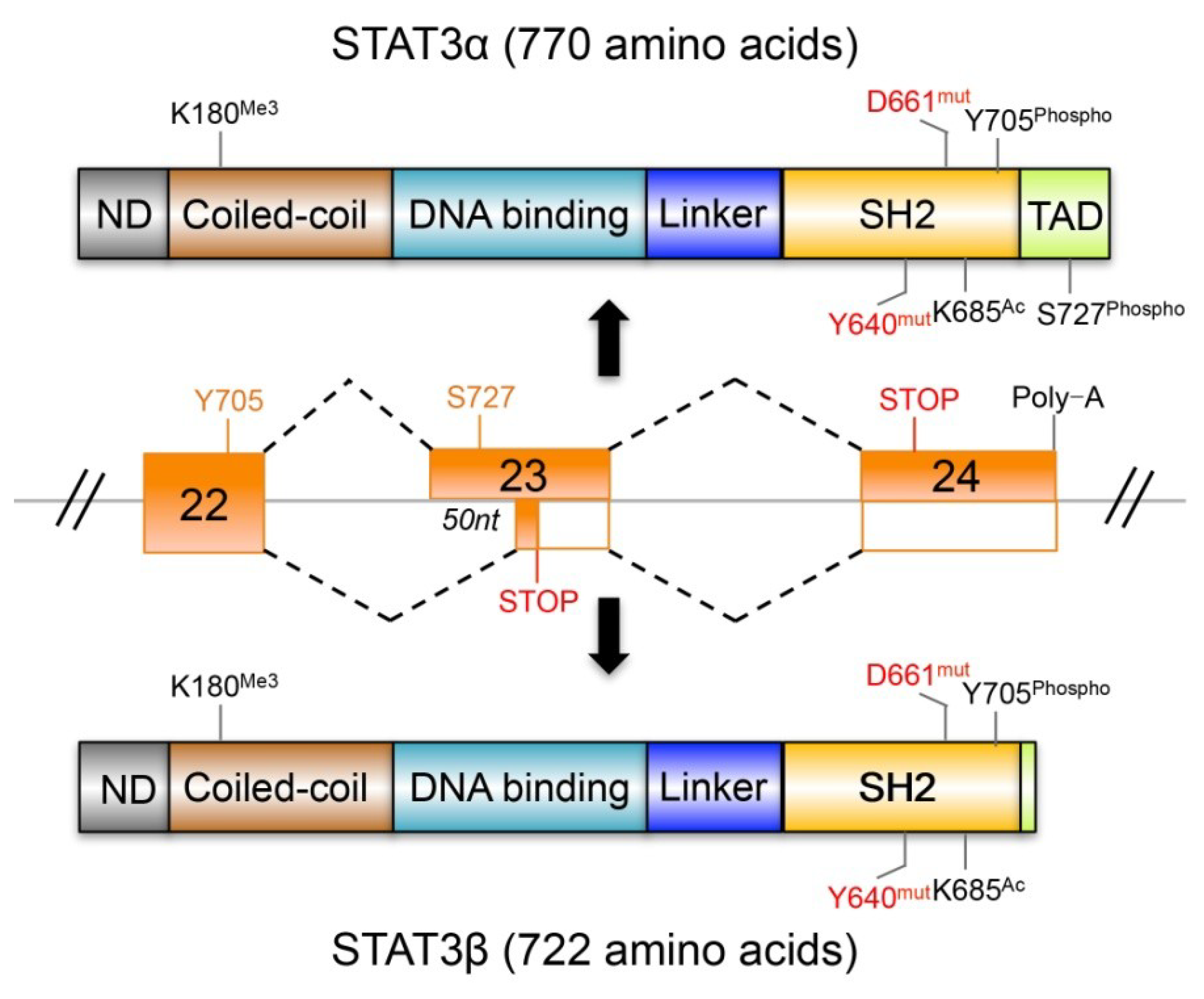
4. STAT3β and the Tumor Suppressor Effects of STAT3
4.1. STAT3β Has Biochemical and Biological Features Different from STAT3α
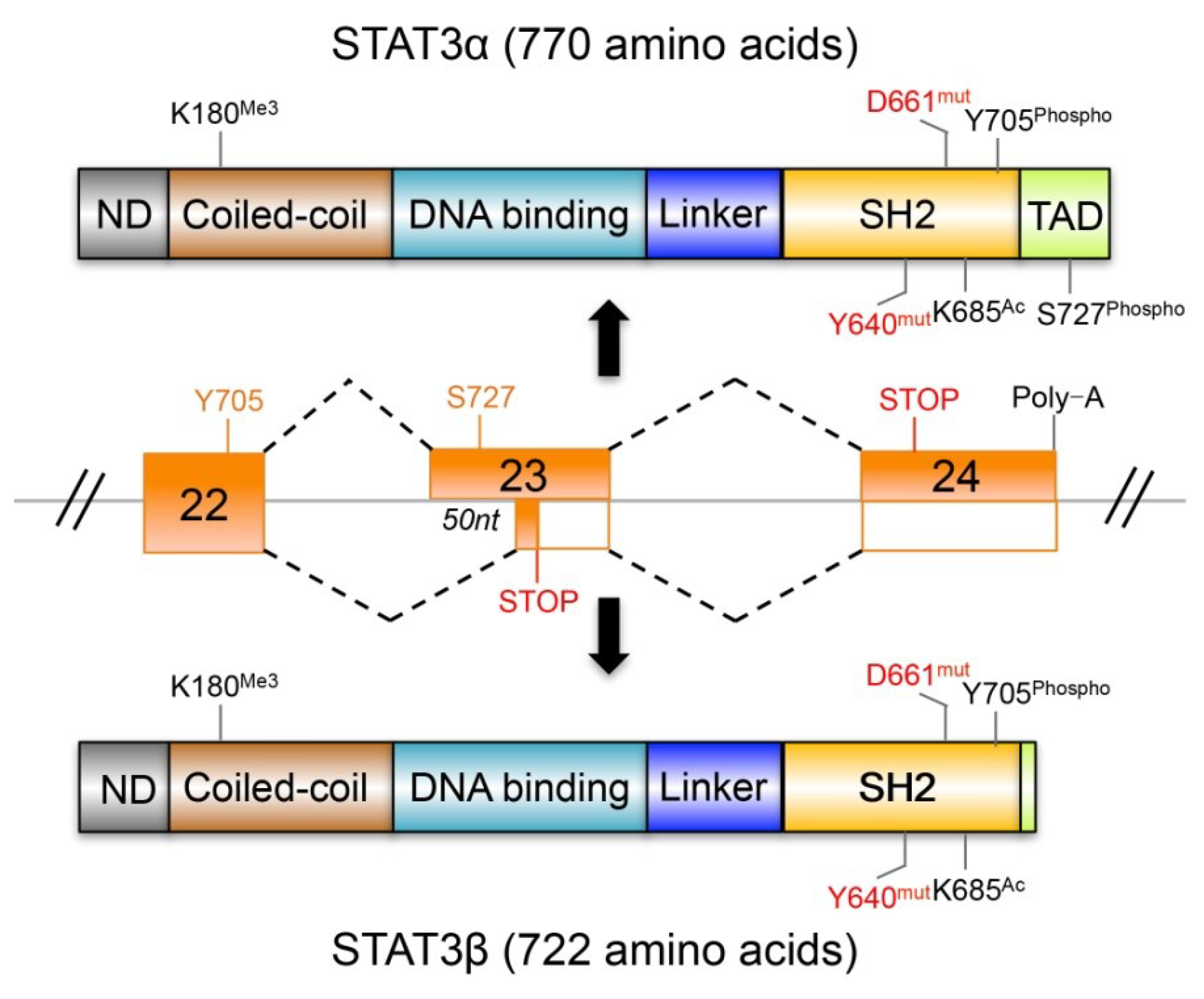
4.2. The Dominant Negative Role of STAT3β
4.3. STAT3β Regulates a Gene Set That Is Distinct from That of STAT3α
4.4. STAT3β Regulates the Phosphorylation Dynamics of STAT3α
4.5. Is STAT3β Responsible for the Tumor Suppressor Function of STAT3?
5. Evaluation of STAT3 Expression in Patient Samples
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef]
- Yu, H.; Jove, R. The stats of cancer—New molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef]
- Yue, P.; Turkson, J. Targeting STAT3 in cancer: How successful are we? Expert Opin. Investig. Drugs 2009, 18, 45–56. [Google Scholar] [CrossRef]
- Pensa, S.; Regis, G.; Boselli, D.; Novelli, F.; Poli, V. Stat1 and STAT3 in Tumorigenesis: Two Sides of the Same Coin? Landes Bioscience: Austin, TX, USA, 2009. [Google Scholar]
- Sellier, H.; Rebillard, A.; Guette, C.; Barre, B.; Coqueret, O. How should we define STAT3 as an oncogene and as a potential target for therapy? JAK-STAT 2013, 2, e24716. [Google Scholar] [CrossRef] [Green Version]
- Levy, D.E.; Lee, C.K. What does STAT3 do? J. Clin. Investig. 2002, 109, 1143–1148. [Google Scholar] [CrossRef]
- Akira, S. Roles of STAT3 defined by tissue-specific gene targeting. Oncogene 2000, 19, 2607–2611. [Google Scholar] [CrossRef]
- Akira, S. Functional roles of STAT family proteins: Lessons from knockout mice. Stem Cells 1999, 17, 138–146. [Google Scholar] [CrossRef]
- Takeda, K.; Noguchi, K.; Shi, W.; Tanaka, T.; Matsumoto, M.; Yoshida, N.; Kishimoto, T.; Akira, S. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc. Natl. Acad. Sci. USA 1997, 94, 3801–3804. [Google Scholar]
- Sano, S.; Itami, S.; Takeda, K.; Tarutani, M.; Yamaguchi, Y.; Miura, H.; Yoshikawa, K.; Akira, S.; Takeda, J. Keratinocyte-specific ablation of Stat3 exhibits impaired skin remodeling, but does not affect skin morphogenesis. EMBO J. 1999, 18, 4657–4668. [Google Scholar] [CrossRef]
- Chapman, R.S.; Lourenco, P.C.; Tonner, E.; Flint, D.J.; Selbert, S.; Takeda, K.; Akira, S.; Clarke, A.R.; Watson, C.J. Suppression of epithelial apoptosis and delayed mammary gland involution in mice with a conditional knockout of Stat3. Genes Dev. 1999, 13, 2604–2616. [Google Scholar] [CrossRef]
- Sano, S.; Takahama, Y.; Sugawara, T.; Kosaka, H.; Itami, S.; Yoshikawa, K.; Miyazaki, J.; van Ewijk, W.; Takeda, J. Stat3 in thymic epithelial cells is essential for postnatal maintenance of thymic architecture and thymocyte survival. Immunity 2001, 15, 261–273. [Google Scholar] [CrossRef]
- Takeda, K.; Kaisho, T.; Yoshida, N.; Takeda, J.; Kishimoto, T.; Akira, S. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: Generation and characterization of T cell-specific Stat3-deficient mice. J. Immunol. 1998, 161, 4652–4660. [Google Scholar]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. STAT3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Dechow, T.N.; Pedranzini, L.; Leitch, A.; Leslie, K.; Gerald, W.L.; Linkov, I.; Bromberg, J.F. Requirement of matrix metalloproteinase-9 for the transformation of human mammary epithelial cells by Stat3-C. Proc. Natl. Acad. Sci. USA 2004, 101, 10602–10607. [Google Scholar] [CrossRef]
- Azare, J.; Leslie, K.; Al-Ahmadie, H.; Gerald, W.; Weinreb, P.H.; Violette, S.M.; Bromberg, J. Constitutively activated Stat3 induces tumorigenesis and enhances cell motility of prostate epithelial cells through integrin beta 6. Mol. Cell. Biol. 2007, 27, 4444–4453. [Google Scholar] [CrossRef]
- Kortylewski, M.; Yu, H. Stat3 as a potential target for cancer immunotherapy. J. Immunother. 2007, 30, 131–139. [Google Scholar] [CrossRef]
- Sherry, M.M.; Reeves, A.; Wu, J.K.; Cochran, B.H. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells 2009, 27, 2383–2392. [Google Scholar] [CrossRef]
- Guryanova, O.A.; Wu, Q.; Cheng, L.; Lathia, J.D.; Huang, Z.; Yang, J.; MacSwords, J.; Eyler, C.E.; McLendon, R.E.; Heddleston, J.M.; et al. Nonreceptor tyrosine kinase bmx maintains self-renewal and tumorigenic potential of glioblastoma stem cells by activating STAT3. Cancer Cell 2011, 19, 498–511. [Google Scholar] [CrossRef]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(−) stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef]
- Kim, E.; Kim, M.; Woo, D.H.; Shin, Y.; Shin, J.; Chang, N.; Oh, Y.T.; Kim, H.; Rheey, J.; Nakano, I.; et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013, 23, 839–852. [Google Scholar] [CrossRef]
- Liu, K.; Jiang, M.; Lu, Y.; Chen, H.; Sun, J.; Wu, S.; Ku, W.Y.; Nakagawa, H.; Kita, Y.; Natsugoe, S.; et al. Sox2 cooperates with inflammation-mediated Stat3 activation in the malignant transformation of foregut basal progenitor cells. Cell Stem Cell 2013, 12, 304–315. [Google Scholar] [CrossRef]
- Kaptein, A.; Paillard, V.; Saunders, M. Dominant negative stat3 mutant inhibits interleukin-6-induced Jak-STAT signal transduction. J. Biol. Chem. 1996, 271, 5961–5964. [Google Scholar] [CrossRef]
- Chen, C.L.; Cen, L.; Kohout, J.; Hutzen, B.; Chan, C.; Hsieh, F.C.; Loy, A.; Huang, V.; Cheng, G.; Lin, J. Signal transducer and activator of transcription 3 activation is associated with bladder cancer cell growth and survival. Mol. Cancer 2008, 7, 78. [Google Scholar] [CrossRef]
- Rivat, C.; de Wever, O.; Bruyneel, E.; Mareel, M.; Gespach, C.; Attoub, S. Disruption of STAT3 signaling leads to tumor cell invasion through alterations of homotypic cell-cell adhesion complexes. Oncogene 2004, 23, 3317–3327. [Google Scholar] [CrossRef]
- Wang, X.; Crowe, P.J.; Goldstein, D.; Yang, J.L. STAT3 inhibition, a novel approach to enhancing targeted therapy in human cancers (review). Int. J. Oncol. 2012, 41, 1181–1191. [Google Scholar]
- Bromberg, J. Stat proteins and oncogenesis. J. Clin. Investig. 2002, 109, 1139–1142. [Google Scholar] [CrossRef]
- Xiong, H.; Du, W.; Wang, J.L.; Wang, Y.C.; Tang, J.T.; Hong, J.; Fang, J.Y. Constitutive activation of STAT3 is predictive of poor prognosis in human gastric cancer. J. Mol. Med. (Berl.) 2012, 90, 1037–1046. [Google Scholar] [CrossRef]
- Kusaba, T.; Nakayama, T.; Yamazumi, K.; Yakata, Y.; Yoshizaki, A.; Inoue, K.; Nagayasu, T.; Sekine, I. Activation of STAT3 is a marker of poor prognosis in human colorectal cancer. Oncol. Rep. 2006, 15, 1445–1451. [Google Scholar]
- Huang, X.; Meng, B.; Iqbal, J.; Ding, B.B.; Perry, A.M.; Cao, W.; Smith, L.M.; Bi, C.; Jiang, C.; Greiner, T.C.; et al. Activation of the STAT3 signaling pathway is associated with poor survival in diffuse large B-cell lymphoma treated with R-CHOP. J. Clin. Oncol. 2013, 31, 4520–4528. [Google Scholar] [CrossRef]
- Takemoto, S.; Ushijima, K.; Kawano, K.; Yamaguchi, T.; Terada, A.; Fujiyoshi, N.; Nishio, S.; Tsuda, N.; Ijichi, M.; Kakuma, T.; et al. Expression of activated signal transducer and activator of transcription-3 predicts poor prognosis in cervical squamous-cell carcinoma. Br. J. Cancer 2009, 101, 967–972. [Google Scholar] [CrossRef]
- Schoppmann, S.F.; Jesch, B.; Friedrich, J.; Jomrich, G.; Maroske, F.; Birner, P. Phosphorylation of signal transducer and activator of transcription 3 (STAT3) correlates with Her-2 status, carbonic anhydrase 9 expression and prognosis in esophageal cancer. Clin. Exp. Metastasis 2012, 29, 615–624. [Google Scholar] [CrossRef]
- You, Z.; Xu, D.; Ji, J.; Guo, W.; Zhu, W.; He, J. JAK/STAT signal pathway activation promotes progression and survival of human oesophageal squamous cell carcinoma. Clin. Transl. Oncol. 2012, 14, 143–149. [Google Scholar] [CrossRef]
- Macha, M.A.; Matta, A.; Kaur, J.; Chauhan, S.S.; Thakar, A.; Shukla, N.K.; Gupta, S.D.; Ralhan, R. Prognostic significance of nuclear pSTAT3 in oral cancer. Head Neck 2011, 33, 482–489. [Google Scholar] [CrossRef]
- Shah, N.G.; Trivedi, T.I.; Tankshali, R.A.; Goswami, J.V.; Jetly, D.H.; Shukla, S.N.; Shah, P.M.; Verma, R.J. Prognostic significance of molecular markers in oral squamous cell carcinoma: A multivariate analysis. Head Neck 2009, 31, 1544–1556. [Google Scholar] [CrossRef]
- Li, C.; Wang, Z.; Liu, Y.; Wang, P.; Zhang, R. STAT3 expression correlates with prognosis of thymic epithelial tumors. J. Cardiothorac. Surg. 2013, 8, 92. [Google Scholar] [CrossRef]
- Auernhammer, C.J.; Bousquet, C.; Melmed, S. Autoregulation of pituitary corticotroph SOCS-3 expression: Characterization of the murine SOCS-3 promoter. Proc. Natl. Acad. Sci. USA 1999, 96, 6964–6969. [Google Scholar] [CrossRef]
- Yoshimura, A.; Naka, T.; Kubo, M. SOCS proteins, cytokine signalling and immune regulation. Nat. Rev. Immunol. 2007, 7, 454–465. [Google Scholar] [CrossRef]
- Ostman, A.; Hellberg, C.; Bohmer, F.D. Protein-tyrosine phosphatases and cancer. Nat. Rev. Cancer 2006, 6, 307–320. [Google Scholar] [CrossRef]
- Julien, S.G.; Dube, N.; Hardy, S.; Tremblay, M.L. Inside the human cancer tyrosine phosphatome. Nat. Rev. Cancer 2011, 11, 35–49. [Google Scholar] [CrossRef]
- Barry, A.C.; O’Sullivan, L.A.; Ward, A.C. Suppressors of Cytokine Signaling: Functions in Normal Biology and Roles in Disease; Landes Bioscience: Austin, TX, USA, 2009. [Google Scholar]
- Inagaki-Ohara, K.; Kondo, T.; Ito, M.; Yoshimura, A. SOCS, inflammation, and cancer. JAK-STAT 2013, 2, e24053. [Google Scholar] [CrossRef]
- Weber, A.; Hengge, U.R.; Bardenheuer, W.; Tischoff, I.; Sommerer, F.; Markwarth, A.; Dietz, A.; Wittekind, C.; Tannapfel, A. SOCS-3 is frequently methylated in head and neck squamous cell carcinoma and its precursor lesions and causes growth inhibition. Oncogene 2005, 24, 6699–6708. [Google Scholar] [CrossRef]
- Tischoff, I.; Hengge, U.R.; Vieth, M.; Ell, C.; Stolte, M.; Weber, A.; Schmidt, W.E.; Tannapfel, A. Methylation of SOCS-3 and SOCS-1 in the carcinogenesis of barrett’s adenocarcinoma. Gut 2007, 56, 1047–1053. [Google Scholar] [CrossRef]
- Niwa, Y.; Kanda, H.; Shikauchi, Y.; Saiura, A.; Matsubara, K.; Kitagawa, T.; Yamamoto, J.; Kubo, T.; Yoshikawa, H. Methylation silencing of SOCS-3 promotes cell growth and migration by enhancing jak/stat and fak signalings in human hepatocellular carcinoma. Oncogene 2005, 24, 6406–6417. [Google Scholar]
- He, B.; You, L.; Uematsu, K.; Zang, K.; Xu, Z.; Lee, A.Y.; Costello, J.F.; McCormick, F.; Jablons, D.M. SOCS-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 14133–14138. [Google Scholar] [CrossRef]
- Yoshikawa, H.; Matsubara, K.; Qian, G.S.; Jackson, P.; Groopman, J.D.; Manning, J.E.; Harris, C.C.; Herman, J.G. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nat. Genet. 2001, 28, 29–35. [Google Scholar]
- Galm, O.; Yoshikawa, H.; Esteller, M.; Osieka, R.; Herman, J.G. SOCS-1, a negative regulator of cytokine signaling, is frequently silenced by methylation in multiple myeloma. Blood 2003, 101, 2784–2788. [Google Scholar] [CrossRef]
- Chim, C.S.; Fung, T.K.; Cheung, W.C.; Liang, R.; Kwong, Y.L. SOCS1 and SHP1 hypermethylation in multiple myeloma: Implications for epigenetic activation of the JAK/STAT pathway. Blood 2004, 103, 4630–4635. [Google Scholar] [CrossRef]
- To, K.F.; Chan, M.W.; Leung, W.K.; Ng, E.K.; Yu, J.; Bai, A.H.; Lo, A.W.; Chu, S.H.; Tong, J.H.; Lo, K.W.; et al. Constitutional activation of IL-6-mediated JAK/STAT pathway through hypermethylation of SOCS-1 in human gastric cancer cell line. Br. J. Cancer 2004, 91, 1335–1341. [Google Scholar] [CrossRef]
- Alonso, A.; Sasin, J.; Bottini, N.; Friedberg, I.; Osterman, A.; Godzik, A.; Hunter, T.; Dixon, J.; Mustelin, T. Protein tyrosine phosphatases in the human genome. Cell 2004, 117, 699–711. [Google Scholar] [CrossRef]
- Oka, T.; Ouchida, M.; Koyama, M.; Ogama, Y.; Takada, S.; Nakatani, Y.; Tanaka, T.; Yoshino, T.; Hayashi, K.; Ohara, N.; et al. Gene silencing of the tyrosine phosphatase SHP1 gene by aberrant methylation in leukemias/lymphomas. Cancer Res. 2002, 62, 6390–6394. [Google Scholar]
- Zhang, Q.; Wang, H.Y.; Marzec, M.; Raghunath, P.N.; Nagasawa, T.; Wasik, M.A. STAT3- and DNA methyltransferase 1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. Proc. Natl. Acad. Sci. USA 2005, 102, 6948–6953. [Google Scholar]
- Zhang, Q.; Raghunath, P.N.; Vonderheid, E.; Odum, N.; Wasik, M.A. Lack of phosphotyrosine phosphatase SHP-1 expression in malignant T-cell lymphoma cells results from methylation of the SHP-1 promoter. Am. J. Pathol. 2000, 157, 1137–1146. [Google Scholar] [CrossRef]
- Brantley, E.C.; Nabors, L.B.; Gillespie, G.Y.; Choi, Y.H.; Palmer, C.A.; Harrison, K.; Roarty, K.; Benveniste, E.N. Loss of protein inhibitors of activated STAT-3 expression in glioblastoma multiforme tumors: Implications for STAT-3 activation and gene expression. Clin. Cancer Res. 2008, 14, 4694–4704. [Google Scholar] [CrossRef]
- Ogata, Y.; Osaki, T.; Naka, T.; Iwahori, K.; Furukawa, M.; Nagatomo, I.; Kijima, T.; Kumagai, T.; Yoshida, M.; Tachibana, I.; et al. Overexpression of PIAS3 suppresses cell growth and restores the drug sensitivity of human lung cancer cells in association with PI3-K/Akt inactivation. Neoplasia 2006, 8, 817–825. [Google Scholar] [CrossRef]
- Kluge, A.; Dabir, S.; Vlassenbroeck, I.; Eisenberg, R.; Dowlati, A. Protein inhibitor of activated STAT3 expression in lung cancer. Mol. Oncol. 2011, 5, 256–264. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Catlett-Falcone, R.; Landowski, T.H.; Oshiro, M.M.; Turkson, J.; Levitzki, A.; Savino, R.; Ciliberto, G.; Moscinski, L.; Fernandez-Luna, J.L.; Nunez, G.; et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 1999, 10, 105–115. [Google Scholar] [CrossRef]
- Shain, K.H.; Yarde, D.N.; Meads, M.B.; Huang, M.; Jove, R.; Hazlehurst, L.A.; Dalton, W.S. Beta1 integrin adhesion enhances IL-6-mediated STAT3 signaling in myeloma cells: Implications for microenvironment influence on tumor survival and proliferation. Cancer Res. 2009, 69, 1009–1015. [Google Scholar] [CrossRef]
- Frame, M.C. Src in cancer: Deregulation and consequences for cell behaviour. Biochim. Biophys. Acta 2002, 1602, 114–130. [Google Scholar]
- Alvarez, R.H.; Kantarjian, H.M.; Cortes, J.E. The role of Src in solid and hematologic malignancies: Development of new-generation src inhibitors. Cancer 2006, 107, 1918–1929. [Google Scholar] [CrossRef]
- Turkson, J.; Bowman, T.; Garcia, R.; Caldenhoven, E.; de Groot, R.P.; Jove, R. Stat3 activation by Src induces specific gene regulation and is required for cell transformation. Mol. Cell. Biol. 1998, 18, 2545–2552. [Google Scholar]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar]
- Shiota, M.; Fujimoto, J.; Semba, T.; Satoh, H.; Yamamoto, T.; Mori, S. Hyperphosphorylation of a novel 80 kDa protein-tyrosine kinase similar to Ltk in a human Ki-1 lymphoma cell line, AMS3. Oncogene 1994, 9, 1567–1574. [Google Scholar]
- Chiarle, R.; Simmons, W.J.; Cai, H.; Dhall, G.; Zamo, A.; Raz, R.; Karras, J.G.; Levy, D.E.; Inghirami, G. Stat3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat. Med. 2005, 11, 623–629. [Google Scholar] [CrossRef]
- Zamo, A.; Chiarle, R.; Piva, R.; Howes, J.; Fan, Y.; Chilosi, M.; Levy, D.E.; Inghirami, G. Anaplastic lymphoma kinase (ALK) activates Stat3 and protects hematopoietic cells from cell death. Oncogene 2002, 21, 1038–1047. [Google Scholar] [CrossRef]
- Zhang, Q.; Raghunath, P.N.; Xue, L.; Majewski, M.; Carpentieri, D.F.; Odum, N.; Morris, S.; Skorski, T.; Wasik, M.A. Multilevel dysregulation of STAT3 activation in anaplastic lymphoma kinase-positive T/null-cell lymphoma. J. Immunol. 2002, 168, 466–474. [Google Scholar] [CrossRef]
- Lai, R.; Ingham, R.J. The pathobiology of the oncogenic tyrosine kinase NPM-ALK: A brief update. Ther. Adv. Hematol. 2013, 4, 119–131. [Google Scholar] [CrossRef]
- Gabler, K.; Behrmann, I.; Haan, C. JAK2 mutants (e.g., JAK2V617F) and their importance as drug targets in myeloproliferative neoplasms. JAK-STAT 2013, 2, e25025. [Google Scholar] [CrossRef]
- Oku, S.; Takenaka, K.; Kuriyama, T.; Shide, K.; Kumano, T.; Kikushige, Y.; Urata, S.; Yamauchi, T.; Iwamoto, C.; Shimoda, H.K.; et al. JAK2 V617F uses distinct signalling pathways to induce cell proliferation and neutrophil activation. Br. J. Haematol. 2010, 150, 334–344. [Google Scholar] [CrossRef]
- Quintas-Cardama, A.; Vaddi, K.; Liu, P.; Manshouri, T.; Li, J.; Scherle, P.A.; Caulder, E.; Wen, X.; Li, Y.; Waeltz, P.; et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: Therapeutic implications for the treatment of myeloproliferative neoplasms. Blood 2010, 115, 3109–3117. [Google Scholar] [CrossRef]
- Mughal, T.I.; Girnius, S.; Rosen, S.T.; Kumar, S.; Wiestner, A.; Abdel-Wahab, O.; Kiladjian, J.J.; Wilson, W.H.; van Etten, R.A. Emerging therapeutic paradigms to target the dysregulated Janus kinase/signal transducer and activator of transcription pathway in hematological malignancies. Leuk. Lymphoma 2014. [Google Scholar] [CrossRef]
- Gao, S.P.; Mark, K.G.; Leslie, K.; Pao, W.; Motoi, N.; Gerald, W.L.; Travis, W.D.; Bornmann, W.; Veach, D.; Clarkson, B.; et al. Mutations in the egfr kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J. Clin. Investig. 2007, 117, 3846–3856. [Google Scholar] [CrossRef]
- Inda, M.M.; Bonavia, R.; Mukasa, A.; Narita, Y.; Sah, D.W.; Vandenberg, S.; Brennan, C.; Johns, T.G.; Bachoo, R.; Hadwiger, P.; et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev. 2010, 24, 1731–1745. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Jaeger, S.A.; Hirsch, H.A.; Bulyk, M.L.; Struhl, K. STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol. Cell 2010, 39, 493–506. [Google Scholar] [CrossRef]
- Lee, H.; Deng, J.; Kujawski, M.; Yang, C.; Liu, Y.; Herrmann, A.; Kortylewski, M.; Horne, D.; Somlo, G.; Forman, S.; et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat. Med. 2010, 16, 1421–1428. [Google Scholar] [CrossRef]
- Deng, J.; Liu, Y.; Lee, H.; Herrmann, A.; Zhang, W.; Zhang, C.; Shen, S.; Priceman, S.J.; Kujawski, M.; Pal, S.K.; et al. S1PR1-STAT3 signaling is crucial for myeloid cell colonization at future metastatic sites. Cancer Cell 2012, 21, 642–654. [Google Scholar] [CrossRef]
- Chang, Q.; Bournazou, E.; Sansone, P.; Berishaj, M.; Gao, S.P.; Daly, L.; Wels, J.; Theilen, T.; Granitto, S.; Zhang, X.; et al. The IL-6/JAK/STAT3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia 2013, 15, 848–862. [Google Scholar]
- Demaria, M.; Poli, V. PKM2, STAT3 and HIF-1alpha: The warburg’s vicious circle. JAK-STAT 2012, 1, 194–196. [Google Scholar] [CrossRef]
- Semenova, G.; Chernoff, J. PKM2 enters the morpheein academy. Mol. Cell 2012, 45, 583–584. [Google Scholar] [CrossRef]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Demaria, M.; Giorgi, C.; Lebiedzinska, M.; Esposito, G.; D’Angeli, L.; Bartoli, A.; Gough, D.J.; Turkson, J.; Levy, D.E.; Watson, C.J.; et al. A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging 2010, 2, 823–842. [Google Scholar]
- Demaria, M.; Poli, V. From the nucleus to the mitochondria and back: The odyssey of a multitask STAT3. Cell Cycle 2011, 10, 3221–3222. [Google Scholar] [CrossRef]
- Marzec, M.; Liu, X.; Wong, W.; Yang, Y.; Pasha, T.; Kantekure, K.; Zhang, P.; Woetmann, A.; Cheng, M.; Odum, N.; et al. Oncogenic kinase NPM/ALK induces expression of HIF1alpha mRNA. Oncogene 2011, 30, 1372–1378. [Google Scholar] [CrossRef]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef]
- Zhang, Y.; Du, X.L.; Wang, C.J.; Lin, D.C.; Ruan, X.; Feng, Y.B.; Huo, Y.Q.; Peng, H.; Cui, J.L.; Zhang, T.T.; et al. Reciprocal activation between PLK1 and Stat3 contributes to survival and proliferation of esophageal cancer cells. Gastroenterology 2012, 142, 521–530.e3. [Google Scholar] [CrossRef]
- Xiong, H.; Du, W.; Sun, T.T.; Lin, Y.W.; Wang, J.L.; Hong, J.; Fang, J.Y. A positive feedback loop between STAT3 and cyclooxygenase-2 gene may contribute to helicobacter pylori-associated human gastric tumorigenesis. Int. J. Cancer 2013, 134, 2030–2040. [Google Scholar]
- Pilati, C.; Amessou, M.; Bihl, M.P.; Balabaud, C.; Nhieu, J.T.; Paradis, V.; Nault, J.C.; Izard, T.; Bioulac-Sage, P.; Couchy, G.; et al. Somatic mutations activating STAT3 in human inflammatory hepatocellular adenomas. J. Exp. Med. 2011, 208, 1359–1366. [Google Scholar] [CrossRef] [Green Version]
- Koskela, H.L.; Eldfors, S.; Ellonen, P.; van Adrichem, A.J.; Kuusanmaki, H.; Andersson, E.I.; Lagstrom, S.; Clemente, M.J.; Olson, T.; Jalkanen, S.E.; et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N. Engl. J. Med. 2012, 366, 1905–1913. [Google Scholar] [CrossRef]
- Jerez, A.; Clemente, M.J.; Makishima, H.; Koskela, H.; Leblanc, F.; Peng Ng, K.; Olson, T.; Przychodzen, B.; Afable, M.; Gomez-Segui, I.; et al. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte leukemia. Blood 2012, 120, 3048–3057. [Google Scholar] [CrossRef]
- Ohgami, R.S.; Ma, L.; Merker, J.D.; Martinez, B.; Zehnder, J.L.; Arber, D.A. STAT3 mutations are frequent in CD30+ T-cell lymphomas and T-cell large granular lymphocytic leukemia. Leukemia 2013, 27, 2244–2247. [Google Scholar] [CrossRef]
- Fasan, A.; Kern, W.; Grossmann, V.; Haferlach, C.; Haferlach, T.; Schnittger, S. STAT3 mutations are highly specific for large granular lymphocytic leukemia. Leukemia 2013, 27, 1598–1600. [Google Scholar] [CrossRef]
- Hu, G.; Witzig, T.E.; Gupta, M. A novel missense (M206K) STAT3 mutation in diffuse large B cell lymphoma deregulates STAT3 signaling. PLoS One 2013, 8, e67851. [Google Scholar]
- Couronne, L.; Scourzic, L.; Pilati, C.; Della Valle, V.; Duffourd, Y.; Solary, E.; Vainchenker, W.; Merlio, J.P.; Beylot-Barry, M.; Damm, F.; et al. STAT3 mutations identified in human hematologic neoplasms induce myeloid malignancies in a mouse bone marrow transplantation model. Haematologica 2013, 98, 1748–1752. [Google Scholar] [CrossRef]
- Levy, D.E.; Darnell, J.E., Jr. Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef]
- Jing, N.; Tweardy, D.J. Targeting Stat3 in cancer therapy. Anti-Cancer Drugs 2005, 16, 601–607. [Google Scholar] [CrossRef]
- Masuda, M.; Suzui, M.; Yasumasstu, R.; Nakashima, T.; Kuratomi, Y.; Azuma, K.; Tomita, K.; Komiyama, S.; Weinstein, I.B. Constitutive activation of signal transducers and activators of transcription 3 correlates with cyclin D1 overexpression and may provide a novel prognostic marker in head and neck squamous cell carcinoma. Cancer Res. 2002, 62, 3351–3355. [Google Scholar]
- Ivanov, V.N.; Bhoumik, A.; Krasilnikov, M.; Raz, R.; Owen-Schaub, L.B.; Levy, D.; Horvath, C.M.; Ronai, Z. Cooperation between STAT3 and c-jun suppresses Fas transcription. Mol. Cell 2001, 7, 517–528. [Google Scholar] [CrossRef]
- Ivanov, V.N.; Krasilnikov, M.; Ronai, Z. Regulation of fas expression by STAT3 and c-jun is mediated by phosphatidylinositol 3-kinase-AKT signaling. J. Biol. Chem. 2002, 277, 4932–4944. [Google Scholar]
- Kunigal, S.; Lakka, S.S.; Sodadasu, P.K.; Estes, N.; Rao, J.S. STAT3-siRNA induces fas-mediated apoptosis in vitro and in vivo in breast cancer. Int. J. Oncol. 2009, 34, 1209–1220. [Google Scholar]
- Niu, G.; Wright, K.L.; Ma, Y.; Wright, G.M.; Huang, M.; Irby, R.; Briggs, J.; Karras, J.; Cress, W.D.; Pardoll, D.; et al. Role of Stat3 in regulating p53 expression and function. Mol. Cell. Biol. 2005, 25, 7432–7440. [Google Scholar] [CrossRef]
- Wei, L.H.; Kuo, M.L.; Chen, C.A.; Chou, C.H.; Lai, K.B.; Lee, C.N.; Hsieh, C.Y. Interleukin-6 promotes cervical tumor growth by VEGF-dependent angiogenesis via a stat3 pathway. Oncogene 2003, 22, 1517–1527. [Google Scholar]
- Wei, D.; Le, X.; Zheng, L.; Wang, L.; Frey, J.A.; Gao, A.C.; Peng, Z.; Huang, S.; Xiong, H.Q.; Abbruzzese, J.L.; et al. STAT3 activation regulates the expression of vascular endothelial growth factor and human pancreatic cancer angiogenesis and metastasis. Oncogene 2003, 22, 319–329. [Google Scholar] [CrossRef]
- Niu, G.; Wright, K.L.; Huang, M.; Song, L.; Haura, E.; Turkson, J.; Zhang, S.; Wang, T.; Sinibaldi, D.; Coppola, D.; et al. Constitutive STAT3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 2002, 21, 2000–2008. [Google Scholar] [CrossRef]
- Xu, Q.; Briggs, J.; Park, S.; Niu, G.; Kortylewski, M.; Zhang, S.; Gritsko, T.; Turkson, J.; Kay, H.; Semenza, G.L.; et al. Targeting STAT3 blocks both HIF-1 and vegf expression induced by multiple oncogenic growth signaling pathways. Oncogene 2005, 24, 5552–5560. [Google Scholar] [CrossRef]
- Kujawski, M.; Kortylewski, M.; Lee, H.; Herrmann, A.; Kay, H.; Yu, H. STAT3 mediates myeloid cell-dependent tumor angiogenesis in mice. J. Clin. Investig. 2008, 118, 3367–3377. [Google Scholar] [CrossRef]
- Jung, J.E.; Kim, H.S.; Lee, C.S.; Park, D.H.; Kim, Y.N.; Lee, M.J.; Lee, J.W.; Park, J.W.; Kim, M.S.; Ye, S.K.; et al. Caffeic acid and its synthetic derivative cadpe suppress tumor angiogenesis by blocking STAT3-mediated vegf expression in human renal carcinoma cells. Carcinogenesis 2007, 28, 1780–1787. [Google Scholar] [CrossRef]
- Yahata, Y.; Shirakata, Y.; Tokumaru, S.; Yamasaki, K.; Sayama, K.; Hanakawa, Y.; Detmar, M.; Hashimoto, K. Nuclear translocation of phosphorylated STAT3 is essential for vascular endothelial growth factor-induced human dermal microvascular endothelial cell migration and tube formation. J. Biol. Chem. 2003, 278, 40026–40031. [Google Scholar] [CrossRef]
- Bartoli, M.; Platt, D.; Lemtalsi, T.; Gu, X.; Brooks, S.E.; Marrero, M.B.; Caldwell, R.B. VEGF differentially activates STAT3 in microvascular endothelial cells. FASEB J. 2003, 17, 1562–1564. [Google Scholar]
- Yadav, A.; Kumar, B.; Datta, J.; Teknos, T.N.; Kumar, P. IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-snail signaling pathway. Mol. Cancer Res. 2011, 9, 1658–1667. [Google Scholar] [CrossRef]
- Lo, H.W.; Hsu, S.C.; Xia, W.; Cao, X.; Shih, J.Y.; Wei, Y.; Abbruzzese, J.L.; Hortobagyi, G.N.; Hung, M.C. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of twist gene expression. Cancer Res. 2007, 67, 9066–9076. [Google Scholar] [CrossRef]
- Guo, L.; Chen, C.; Shi, M.; Wang, F.; Chen, X.; Diao, D.; Hu, M.; Yu, M.; Qian, L.; Guo, N. STAT3-coordinated Lin-28-let-7-HMGA2 and miR-200-ZEB1 circuits initiate and maintain oncostatin M-driven epithelial-mesenchymal transition. Oncogene 2013, 32, 5272–5282. [Google Scholar] [CrossRef]
- Xiong, H.; Hong, J.; Du, W.; Lin, Y.W.; Ren, L.L.; Wang, Y.C.; Su, W.Y.; Wang, J.L.; Cui, Y.; Wang, Z.H.; et al. Roles of STAT3 and ZEB1 proteins in e-cadherin down-regulation and human colorectal cancer epithelial-mesenchymal transition. J. Biol. Chem. 2012, 287, 5819–5832. [Google Scholar] [CrossRef]
- Itoh, M.; Murata, T.; Suzuki, T.; Shindoh, M.; Nakajima, K.; Imai, K.; Yoshida, K. Requirement of stat3 activation for maximal collagenase-1 (MMP-1) induction by epidermal growth factor and malignant characteristics in t24 bladder cancer cells. Oncogene 2006, 25, 1195–1204. [Google Scholar] [CrossRef]
- Song, Y.; Qian, L.; Song, S.; Chen, L.; Zhang, Y.; Yuan, G.; Zhang, H.; Xia, Q.; Hu, M.; Yu, M.; et al. Fra-1 and STAT3 synergistically regulate activation of human mmp-9 gene. Mol. Immunol. 2008, 45, 137–143. [Google Scholar] [CrossRef]
- Tsareva, S.A.; Moriggl, R.; Corvinus, F.M.; Wiederanders, B.; Schutz, A.; Kovacic, B.; Friedrich, K. Signal transducer and activator of transcription 3 activation promotes invasive growth of colon carcinomas through matrix metalloproteinase induction. Neoplasia 2007, 9, 279–291. [Google Scholar] [CrossRef]
- Xie, T.X.; Wei, D.; Liu, M.; Gao, A.C.; Ali-Osman, F.; Sawaya, R.; Huang, S. STAT3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis. Oncogene 2004, 23, 3550–3560. [Google Scholar]
- Fukuda, A.; Wang, S.C.; Morris, J.P.T.; Folias, A.E.; Liou, A.; Kim, G.E.; Akira, S.; Boucher, K.M.; Firpo, M.A.; Mulvihill, S.J.; et al. STAT3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell 2011, 19, 441–455. [Google Scholar] [CrossRef]
- Li, H.; Huang, C.; Huang, K.; Wu, W.; Jiang, T.; Cao, J.; Feng, Z.; Qiu, Z. STAT3 knockdown reduces pancreatic cancer cell invasiveness and matrix metalloproteinase-7 expression in nude mice. PLoS One 2011, 6, e25941. [Google Scholar]
- Barbieri, I.; Pensa, S.; Pannellini, T.; Quaglino, E.; Maritano, D.; Demaria, M.; Voster, A.; Turkson, J.; Cavallo, F.; Watson, C.J.; et al. Constitutively active STAT3 enhances neu-mediated migration and metastasis in mammary tumors via upregulation of cten. Cancer Res. 2010, 70, 2558–2567. [Google Scholar] [CrossRef]
- McLean, G.W.; Carragher, N.O.; Avizienyte, E.; Evans, J.; Brunton, V.G.; Frame, M.C. The role of focal-adhesion kinase in cancer—A new therapeutic opportunity. Nat. Rev. Cancer 2005, 5, 505–515. [Google Scholar] [CrossRef]
- Nagano, M.; Hoshino, D.; Koshikawa, N.; Akizawa, T.; Seiki, M. Turnover of focal adhesions and cancer cell migration. Int. J. Cell Biol. 2012. [Google Scholar] [CrossRef]
- Niu, G.; Heller, R.; Catlett-Falcone, R.; Coppola, D.; Jaroszeski, M.; Dalton, W.; Jove, R.; Yu, H. Gene therapy with dominant-negative STAT3 suppresses growth of the murine melanoma B16 tumor in vivo. Cancer Res. 1999, 59, 5059–5063. [Google Scholar]
- Sun, Z.; Yao, Z.; Liu, S.; Tang, H.; Yan, X. An oligonucleotide decoy for STAT3 activates the immune response of macrophages to breast cancer. Immunobiology 2006, 211, 199–209. [Google Scholar] [CrossRef]
- Takeda, K.; Clausen, B.E.; Kaisho, T.; Tsujimura, T.; Terada, N.; Forster, I.; Akira, S. Enhanced th1 activity and development of chronic enterocolitis in mice devoid of STAT3 in macrophages and neutrophils. Immunity 1999, 10, 39–49. [Google Scholar] [CrossRef]
- Lee, H.; Zhang, P.; Herrmann, A.; Yang, C.; Xin, H.; Wang, Z.; Hoon, D.S.; Forman, S.J.; Jove, R.; Riggs, A.D.; et al. Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc. Natl. Acad. Sci. USA 2012, 109, 7765–7769. [Google Scholar] [CrossRef]
- Yuan, Z.L.; Guan, Y.J.; Chatterjee, D.; Chin, Y.E. STAT3 dimerization regulated by reversible acetylation of a single lysine residue. Science 2005, 307, 269–273. [Google Scholar] [CrossRef]
- Lee, J.L.; Wang, M.J.; Chen, J.Y. Acetylation and activation of STAT3 mediated by nuclear translocation of cd44. J. Cell Biol. 2009, 185, 949–957. [Google Scholar] [CrossRef]
- Liu, L.; McBride, K.M.; Reich, N.C. STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-alpha3. Proc. Natl. Acad. Sci. USA 2005, 102, 8150–8155. [Google Scholar] [CrossRef]
- Yang, J.; Chatterjee-Kishore, M.; Staugaitis, S.M.; Nguyen, H.; Schlessinger, K.; Levy, D.E.; Stark, G.R. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 2005, 65, 939–947. [Google Scholar]
- Yang, J.; Liao, X.; Agarwal, M.K.; Barnes, L.; Auron, P.E.; Stark, G.R. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappab. Genes Dev. 2007, 21, 1396–1408. [Google Scholar] [CrossRef]
- Hazan-Halevy, I.; Harris, D.; Liu, Z.; Liu, J.; Li, P.; Chen, X.; Shanker, S.; Ferrajoli, A.; Keating, M.J.; Estrov, Z. STAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in cll cells. Blood 2010, 115, 2852–2863. [Google Scholar] [CrossRef]
- Qin, H.R.; Kim, H.J.; Kim, J.Y.; Hurt, E.M.; Klarmann, G.J.; Kawasaki, B.T.; Duhagon Serrat, M.A.; Farrar, W.L. Activation of signal transducer and activator of transcription 3 through a phosphomimetic serine 727 promotes prostate tumorigenesis independent of tyrosine 705 phosphorylation. Cancer Res. 2008, 68, 7736–7741. [Google Scholar] [CrossRef]
- Timofeeva, O.A.; Tarasova, N.I.; Zhang, X.; Chasovskikh, S.; Cheema, A.K.; Wang, H.; Brown, M.L.; Dritschilo, A. STAT3 suppresses transcription of proapoptotic genes in cancer cells with the involvement of its N-terminal domain. Proc. Natl. Acad. Sci. USA 2013, 110, 1267–1272. [Google Scholar] [CrossRef]
- Timofeeva, O.A.; Chasovskikh, S.; Lonskaya, I.; Tarasova, N.I.; Khavrutskii, L.; Tarasov, S.G.; Zhang, X.; Korostyshevskiy, V.R.; Cheema, A.; Zhang, L.; et al. Mechanisms of unphosphorylated STAT3 transcription factor binding to DNA. J. Biol. Chem. 2012, 287, 14192–14200. [Google Scholar] [CrossRef]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial STAT3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef]
- Zhang, Q.; Raje, V.; Yakovlev, V.A.; Yacoub, A.; Szczepanek, K.; Meier, J.; Derecka, M.; Chen, Q.; Hu, Y.; Sisler, J.; et al. Mitochondrial localized STAT3 promotes breast cancer growth via phosphorylation of serine 727. J. Biol. Chem. 2013, 288, 31280–31288. [Google Scholar] [CrossRef]
- Badgwell, D.B.; Lu, Z.; Le, K.; Gao, F.; Yang, M.; Suh, G.K.; Bao, J.J.; Das, P.; Andreeff, M.; Chen, W.; et al. The tumor-suppressor gene arhi (DIRAS3) suppresses ovarian cancer cell migration through inhibition of the Stat3 and FAK/Rho signaling pathways. Oncogene 2012, 31, 68–79. [Google Scholar] [CrossRef]
- Silver, D.L.; Naora, H.; Liu, J.; Cheng, W.; Montell, D.J. Activated signal transducer and activator of transcription (STAT) 3: Localization in focal adhesions and function in ovarian cancer cell motility. Cancer Res. 2004, 64, 3550–3558. [Google Scholar] [CrossRef]
- Abdulghani, J.; Gu, L.; Dagvadorj, A.; Lutz, J.; Leiby, B.; Bonuccelli, G.; Lisanti, M.P.; Zellweger, T.; Alanen, K.; Mirtti, T.; et al. Stat3 promotes metastatic progression of prostate cancer. Am. J. Pathol. 2008, 172, 1717–1728. [Google Scholar] [CrossRef]
- Gu, L.; Dagvadorj, A.; Lutz, J.; Leiby, B.; Bonuccelli, G.; Lisanti, M.P.; Addya, S.; Fortina, P.; Dasgupta, A.; Hyslop, T.; et al. Transcription factor Stat3 stimulates metastatic behavior of human prostate cancer cells in vivo, whereas Stat5b has a preferential role in the promotion of prostate cancer cell viability and tumor growth. Am. J. Pathol. 2010, 176, 1959–1972. [Google Scholar] [CrossRef]
- Wei, Z.; Jiang, X.; Qiao, H.; Zhai, B.; Zhang, L.; Zhang, Q.; Wu, Y.; Jiang, H.; Sun, X. STAT3 interacts with skp2/p27/p21 pathway to regulate the motility and invasion of gastric cancer cells. Cell. Signal. 2013, 25, 931–938. [Google Scholar] [CrossRef]
- Ng, D.C.; Lin, B.H.; Lim, C.P.; Huang, G.; Zhang, T.; Poli, V.; Cao, X. Stat3 regulates microtubules by antagonizing the depolymerization activity of stathmin. J. Cell Biol. 2006, 172, 245–257. [Google Scholar] [CrossRef]
- Verma, N.K.; Dourlat, J.; Davies, A.M.; Long, A.; Liu, W.Q.; Garbay, C.; Kelleher, D.; Volkov, Y. Stat3-stathmin interactions control microtubule dynamics in migrating T-cells. J. Biol. Chem. 2009, 284, 12349–12362. [Google Scholar]
- De la Iglesia, N.; Konopka, G.; Puram, S.V.; Chan, J.A.; Bachoo, R.M.; You, M.J.; Levy, D.E.; Depinho, R.A.; Bonni, A. Identification of a pten-regulated STAT3 brain tumor suppressor pathway. Genes Dev. 2008, 22, 449–462. [Google Scholar] [CrossRef]
- Schneller, D.; Machat, G.; Sousek, A.; Proell, V.; van Zijl, F.; Zulehner, G.; Huber, H.; Mair, M.; Muellner, M.K.; Nijman, S.M.; et al. P19(ARF) /p14(ARF) controls oncogenic functions of signal transducer and activator of transcription 3 in hepatocellular carcinoma. Hepatology 2011, 54, 164–172. [Google Scholar]
- Musteanu, M.; Blaas, L.; Mair, M.; Schlederer, M.; Bilban, M.; Tauber, S.; Esterbauer, H.; Mueller, M.; Casanova, E.; Kenner, L.; et al. Stat3 is a negative regulator of intestinal tumor progression in Apcmin mice. Gastroenterology 2010, 138, 1003–1011. [Google Scholar] [CrossRef]
- Wang, H.; Lafdil, F.; Wang, L.; Park, O.; Yin, S.; Niu, J.; Miller, A.M.; Sun, Z.; Gao, B. Hepatoprotective versus oncogenic functions of STAT3 in liver tumorigenesis. Am. J. Pathol. 2011, 179, 714–724. [Google Scholar] [CrossRef]
- Verna, L.; Whysner, J.; Williams, G.M. N-Nitrosodiethylamine mechanistic data and risk assessment: Bioactivation, DNA-adduct formation, mutagenicity, and tumor initiation. Pharmacol. Ther. 1996, 71, 57–81. [Google Scholar] [CrossRef]
- Couto, J.P.; Daly, L.; Almeida, A.; Knauf, J.A.; Fagin, J.A.; Sobrinho-Simoes, M.; Lima, J.; Maximo, V.; Soares, P.; Lyden, D.; et al. STAT3 negatively regulates thyroid tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, E2361–E2370. [Google Scholar] [CrossRef]
- Ecker, A.; Simma, O.; Hoelbl, A.; Kenner, L.; Beug, H.; Moriggl, R.; Sexl, V. The dark and the bright side of Stat3: Proto-oncogene and tumor-suppressor. Front. Biosci. (Landmark Ed.) 2009, 14, 2944–2958. [Google Scholar]
- Kan, C.E.; Cipriano, R.; Jackson, M.W. C-myc functions as a molecular switch to alter the response of human mammary epithelial cells to oncostatin m. Cancer Res. 2011, 71, 6930–6939. [Google Scholar] [CrossRef]
- Lee, J.; Kim, J.C.; Lee, S.E.; Quinley, C.; Kim, H.; Herdman, S.; Corr, M.; Raz, E. Signal transducer and activator of transcription 3 (STAT3) protein suppresses adenoma-to-carcinoma transition in APCmin/+ mice via regulation of Snail-1 (SNAI) protein stability. J. Biol. Chem. 2012, 287, 18182–18189. [Google Scholar]
- Pectasides, E.; Egloff, A.M.; Sasaki, C.; Kountourakis, P.; Burtness, B.; Fountzilas, G.; Dafni, U.; Zaramboukas, T.; Rampias, T.; Rimm, D.; et al. Nuclear localization of signal transducer and activator of transcription 3 in head and neck squamous cell carcinoma is associated with a better prognosis. Clin. Cancer Res. 2010, 16, 2427–2434. [Google Scholar] [CrossRef]
- Ettl, T.; Stiegler, C.; Zeitler, K.; Agaimy, A.; Zenk, J.; Reichert, T.E.; Gosau, M.; Kuhnel, T.; Brockhoff, G.; Schwarz, S. EGFR, HER2, survivin, and loss of pSTAT3 characterize high-grade malignancy in salivary gland cancer with impact on prognosis. Hum. Pathol. 2012, 43, 921–931. [Google Scholar] [CrossRef]
- Dolled-Filhart, M.; Camp, R.L.; Kowalski, D.P.; Smith, B.L.; Rimm, D.L. Tissue microarray analysis of signal transducers and activators of transcription 3 (Stat3) and phospho-Stat3 (Tyr705) in node-negative breast cancer shows nuclear localization is associated with a better prognosis. Clin. Cancer Res. 2003, 9, 594–600. [Google Scholar]
- Sato, T.; Neilson, L.M.; Peck, A.R.; Liu, C.; Tran, T.H.; Witkiewicz, A.; Hyslop, T.; Nevalainen, M.T.; Sauter, G.; Rui, H. Signal transducer and activator of transcription-3 and breast cancer prognosis. Am. J. Cancer Res. 2011, 1, 347–355. [Google Scholar]
- Lai, S.Y.; Johnson, F.M. Defining the role of the JAK-STAT pathway in head and neck and thoracic malignancies: Implications for future therapeutic approaches. Drug Resist. Updat. 2010, 13, 67–78. [Google Scholar] [CrossRef]
- Leeman, R.J.; Lui, V.W.; Grandis, J.R. STAT3 as a therapeutic target in head and neck cancer. Expert Opin. Biol. Ther. 2006, 6, 231–241. [Google Scholar] [CrossRef]
- Song, J.I.; Grandis, J.R. STAT signaling in head and neck cancer. Oncogene 2000, 19, 2489–2495. [Google Scholar] [CrossRef]
- Gritsko, T.; Williams, A.; Turkson, J.; Kaneko, S.; Bowman, T.; Huang, M.; Nam, S.; Eweis, I.; Diaz, N.; Sullivan, D.; et al. Persistent activation of stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clin. Cancer Res. 2006, 12, 11–19. [Google Scholar] [CrossRef]
- Caldenhoven, E.; van Dijk, T.B.; Solari, R.; Armstrong, J.; Raaijmakers, J.A.; Lammers, J.W.; Koenderman, L.; de Groot, R.P. STAT3beta, a splice variant of transcription factor STAT3, is a dominant negative regulator of transcription. J. Biol. Chem. 1996, 271, 13221–13227. [Google Scholar]
- Schaefer, T.S.; Sanders, L.K.; Nathans, D. Cooperative transcriptional activity of jun and Stat3 beta, a short form of Stat3. Proc. Natl. Acad. Sci. USA 1995, 92, 9097–9101. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Huso, D.L.; Nathans, D.; Desiderio, S. Specific ablation of Stat3beta distorts the pattern of Stat3-responsive gene expression and impairs recovery from endotoxic shock. Cell 2002, 108, 331–344. [Google Scholar] [CrossRef]
- Biethahn, S.; Alves, F.; Wilde, S.; Hiddemann, W.; Spiekermann, K. Expression of granulocyte colony-stimulating factor- and granulocyte-macrophage colony-stimulating factor-associated signal transduction proteins of the JAK/STAT pathway in normal granulopoiesis and in blast cells of acute myelogenous leukemia. Exp. Hematol. 1999, 27, 885–894. [Google Scholar] [CrossRef]
- Chakraborty, A.; White, S.M.; Schaefer, T.S.; Ball, E.D.; Dyer, K.F.; Tweardy, D.J. Granulocyte colony-stimulating factor activation of Stat3 alpha and Stat3 beta in immature normal and leukemic human myeloid cells. Blood 1996, 88, 2442–2449. [Google Scholar]
- Hevehan, D.L.; Miller, W.M.; Papoutsakis, E.T. Differential expression and phosphorylation of distinct STAT3 proteins during granulocytic differentiation. Blood 2002, 99, 1627–1637. [Google Scholar] [CrossRef]
- Huang, Y.; Qiu, J.; Dong, S.; Redell, M.S.; Poli, V.; Mancini, M.A.; Tweardy, D.J. Stat3 isoforms, alpha and beta, demonstrate distinct intracellular dynamics with prolonged nuclear retention of Stat3beta mapping to its unique C-terminal end. J. Biol. Chem. 2007, 282, 34958–34967. [Google Scholar]
- Ng, I.H.; Ng, D.C.; Jans, D.A.; Bogoyevitch, M.A. Selective STAT3-alpha or -beta expression reveals spliceform-specific phosphorylation kinetics, nuclear retention and distinct gene expression outcomes. Biochem. J. 2012, 447, 125–136. [Google Scholar] [CrossRef]
- Schaefer, T.S.; Sanders, L.K.; Park, O.K.; Nathans, D. Functional differences between Stat3alpha and Stat3beta. Mol. Cell. Biol. 1997, 17, 5307–5316. [Google Scholar]
- Park, O.K.; Schaefer, L.K.; Wang, W.; Schaefer, T.S. Dimer stability as a determinant of differential DNA binding activity of Stat3 isoforms. J. Biol. Chem. 2000, 275, 32244–32249. [Google Scholar] [CrossRef]
- Epling-Burnette, P.K.; Liu, J.H.; Catlett-Falcone, R.; Turkson, J.; Oshiro, M.; Kothapalli, R.; Li, Y.; Wang, J.M.; Yang-Yen, H.F.; Karras, J.; et al. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased mcl-1 expression. J. Clin. Investig. 2001, 107, 351–362. [Google Scholar] [CrossRef]
- Karni, R.; Jove, R.; Levitzki, A. Inhibition of pp60c-Src reduces Bcl-XL expression and reverses the transformed phenotype of cells overexpressing EGF and HER-2 receptors. Oncogene 1999, 18, 4654–4662. [Google Scholar] [CrossRef]
- Sinibaldi, D.; Wharton, W.; Turkson, J.; Bowman, T.; Pledger, W.J.; Jove, R. Induction of p21WAF1/CIP1 and cyclin d1 expression by the src oncoprotein in mouse fibroblasts: Role of activated STAT3 signaling. Oncogene 2000, 19, 5419–5427. [Google Scholar] [CrossRef]
- Niu, G.; Shain, K.H.; Huang, M.; Ravi, R.; Bedi, A.; Dalton, W.S.; Jove, R.; Yu, H. Overexpression of a dominant-negative signal transducer and activator of transcription 3 variant in tumor cells leads to production of soluble factors that induce apoptosis and cell cycle arrest. Cancer Res. 2001, 61, 3276–3280. [Google Scholar]
- Xu, G.; Zhang, C.; Zhang, J. Dominant negative STAT3 suppresses the growth and invasion capability of human lung cancer cells. Mol. Med. Rep. 2009, 2, 819–824. [Google Scholar]
- Alonzi, T.; Maritano, D.; Gorgoni, B.; Rizzuto, G.; Libert, C.; Poli, V. Essential role of STAT3 in the control of the acute-phase response as revealed by inducible gene inactivation [correction of activation] in the liver. Mol. Cell. Biol. 2001, 21, 1621–1632. [Google Scholar] [CrossRef]
- Maritano, D.; Sugrue, M.L.; Tininini, S.; Dewilde, S.; Strobl, B.; Fu, X.; Murray-Tait, V.; Chiarle, R.; Poli, V. The STAT3 isoforms alpha and beta have unique and specific functions. Nat. Immunol. 2004, 5, 401–409. [Google Scholar] [CrossRef]
- Zammarchi, F.; de Stanchina, E.; Bournazou, E.; Supakorndej, T.; Martires, K.; Riedel, E.; Corben, A.D.; Bromberg, J.F.; Cartegni, L. Antitumorigenic potential of STAT3 alternative splicing modulation. Proc. Natl. Acad. Sci. USA 2011, 108, 17779–17784. [Google Scholar] [CrossRef]
- Ivanov, V.N.; Zhou, H.; Partridge, M.A.; Hei, T.K. Inhibition of ataxia telangiectasia mutated kinase activity enhances trail-mediated apoptosis in human melanoma cells. Cancer Res. 2009, 69, 3510–3519. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, H.-F.; Lai, R. STAT3 in Cancer—Friend or Foe? Cancers 2014, 6, 1408-1440. https://doi.org/10.3390/cancers6031408
Zhang H-F, Lai R. STAT3 in Cancer—Friend or Foe? Cancers. 2014; 6(3):1408-1440. https://doi.org/10.3390/cancers6031408
Chicago/Turabian StyleZhang, Hai-Feng, and Raymond Lai. 2014. "STAT3 in Cancer—Friend or Foe?" Cancers 6, no. 3: 1408-1440. https://doi.org/10.3390/cancers6031408