Merkel Cell Polyomavirus: Molecular Insights into the Most Recently Discovered Human Tumour Virus


 and
and
Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VIRUS | YEAR | FAMILY | GENOME | ONCOGENES | CANCER ASSOCIATION |
|---|---|---|---|---|---|
| Epstein-Barr Virus (EBV) | 1964 | Herpesviridae | dsDNA | LMP1 | Including Burkitt’s lymphoma, nasopharyngeal carcinoma, and some other lymphoproliferative disorders |
| Hepatitis B virus (HBV) | 1965 | Hepadnaviridae | ssDNA and dsDNA | HBx | Some hepatocellular carcinomas |
| Human T-lymphotropic virus-I (HTLV-I) | 1980 | Retroviridae | ssRNA | Tax | Adult T cell leukaemia |
| Human papillomaviruses (HPV) 16 and 18 | 1983-1984 | Papillomaviridae | dsDNA | E5, E6, E7 | Most cervical cancer and penile cancers. |
| Hepatitis C virus (HCV) | 1989 | Hepaciviridae | (+)ssRNA | NS5A | Some hepatocellular carcinomas and lymphomas |
| Kaposi’s sarcoma herpesvirus (KSHV) | 1994 | Herpesviridae | dsDNA | LANA, vflip, and vBcl-2, among others | Kaposi’s sarcoma and primary effusion lymphoma |
| Merkel Cell Polyomavirus (MCPyV) | 2008 | Polyomaviridae | dsDNA | T antigens | Most Merkel cell carcinomas, |
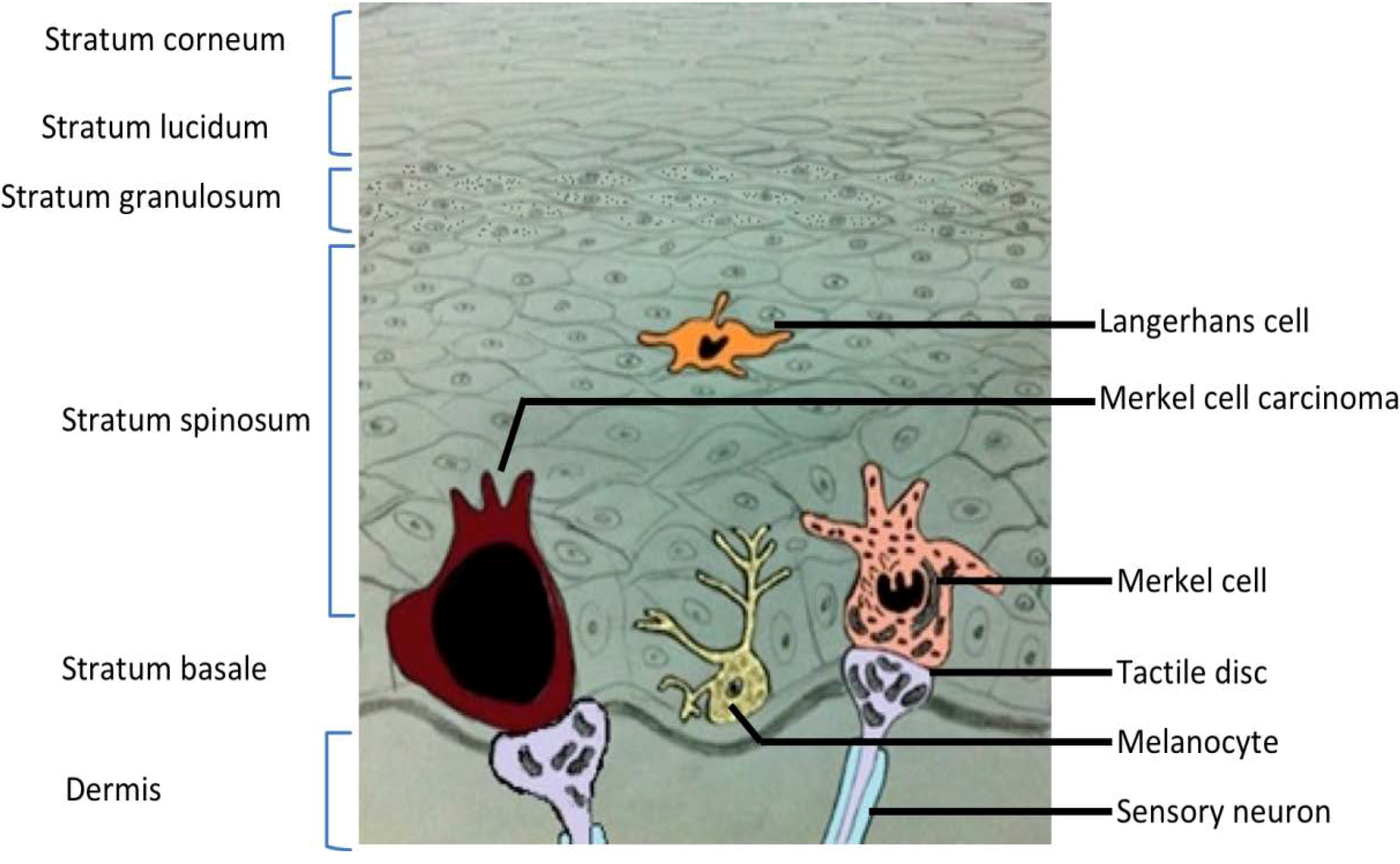
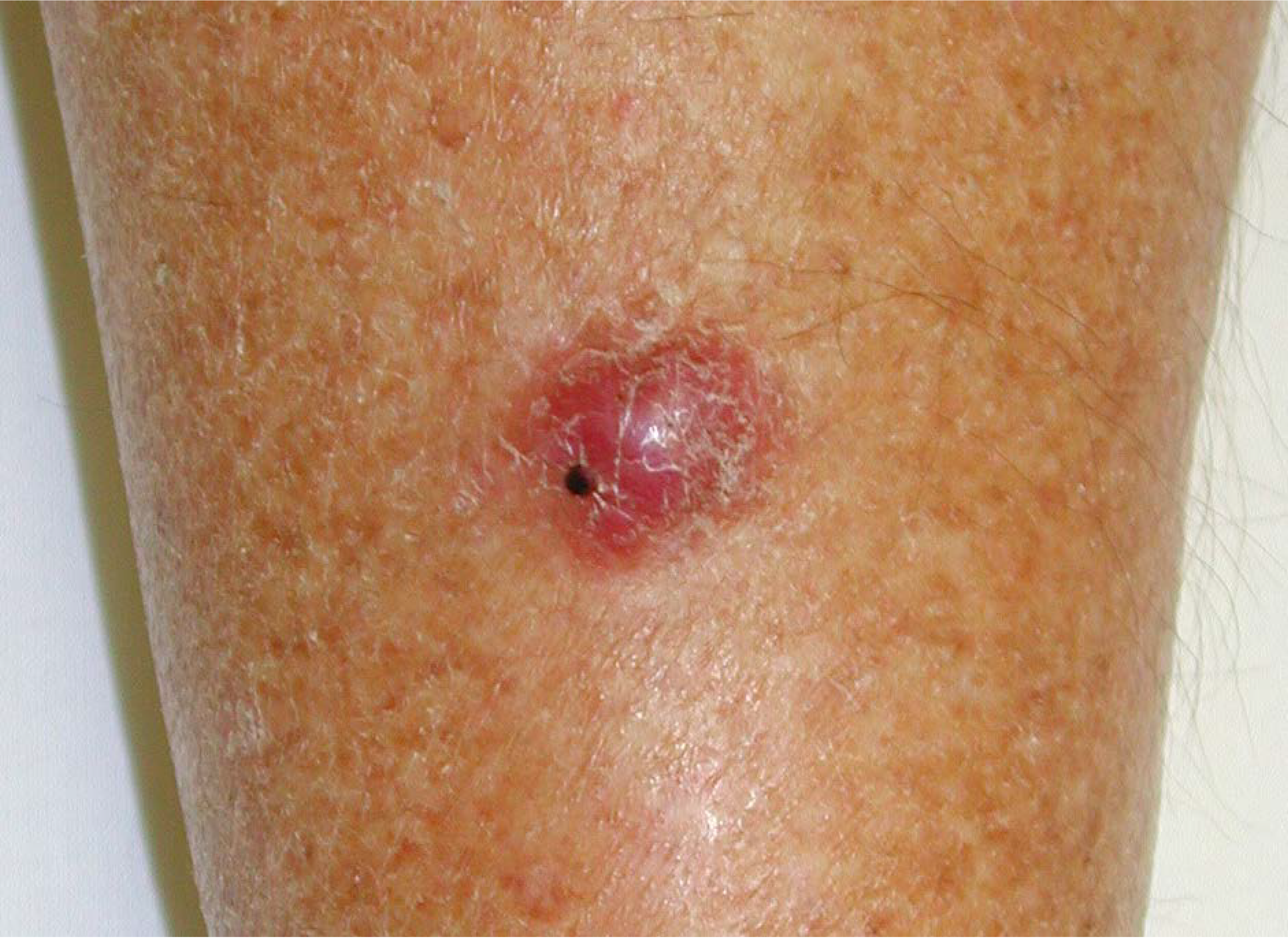
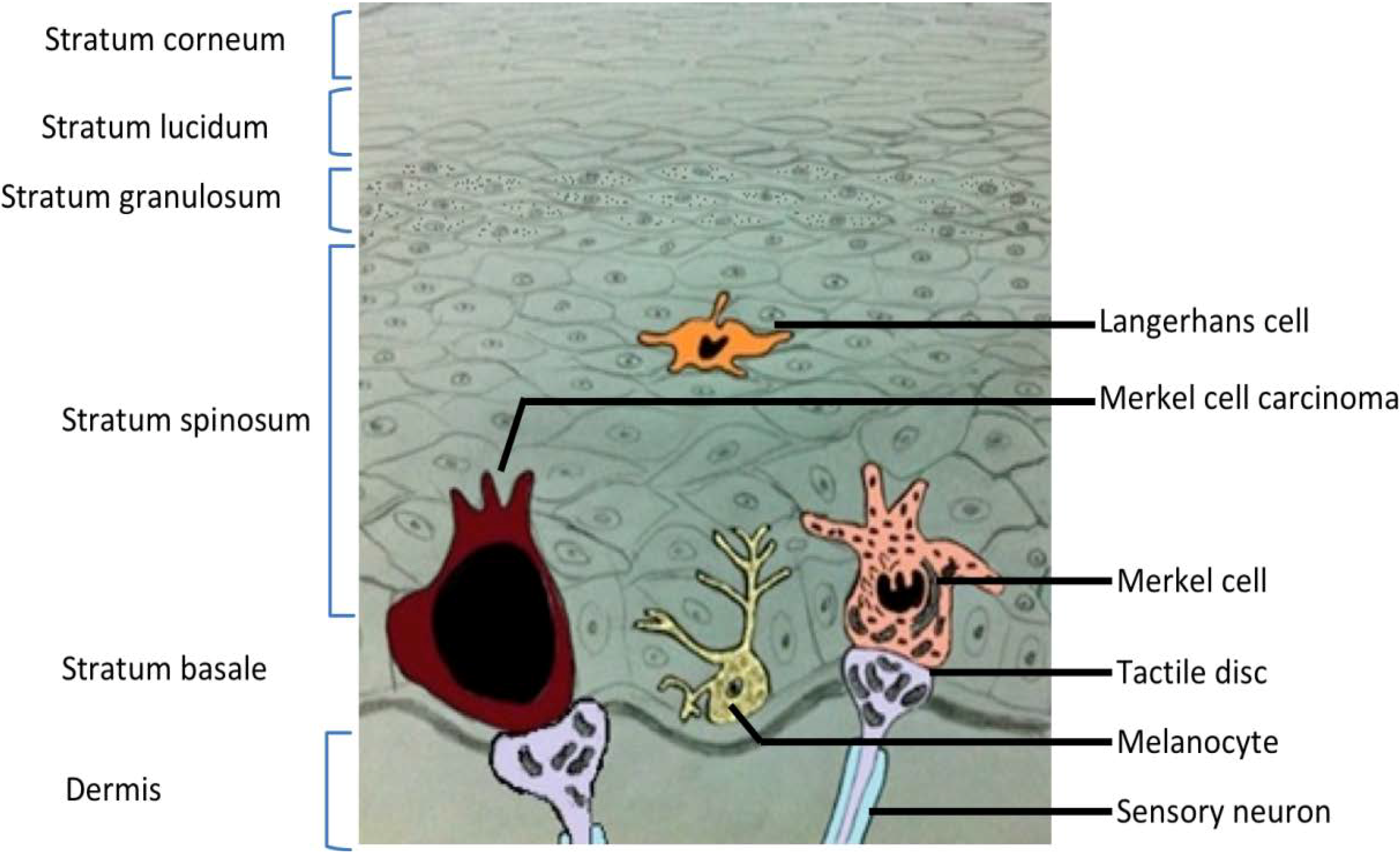
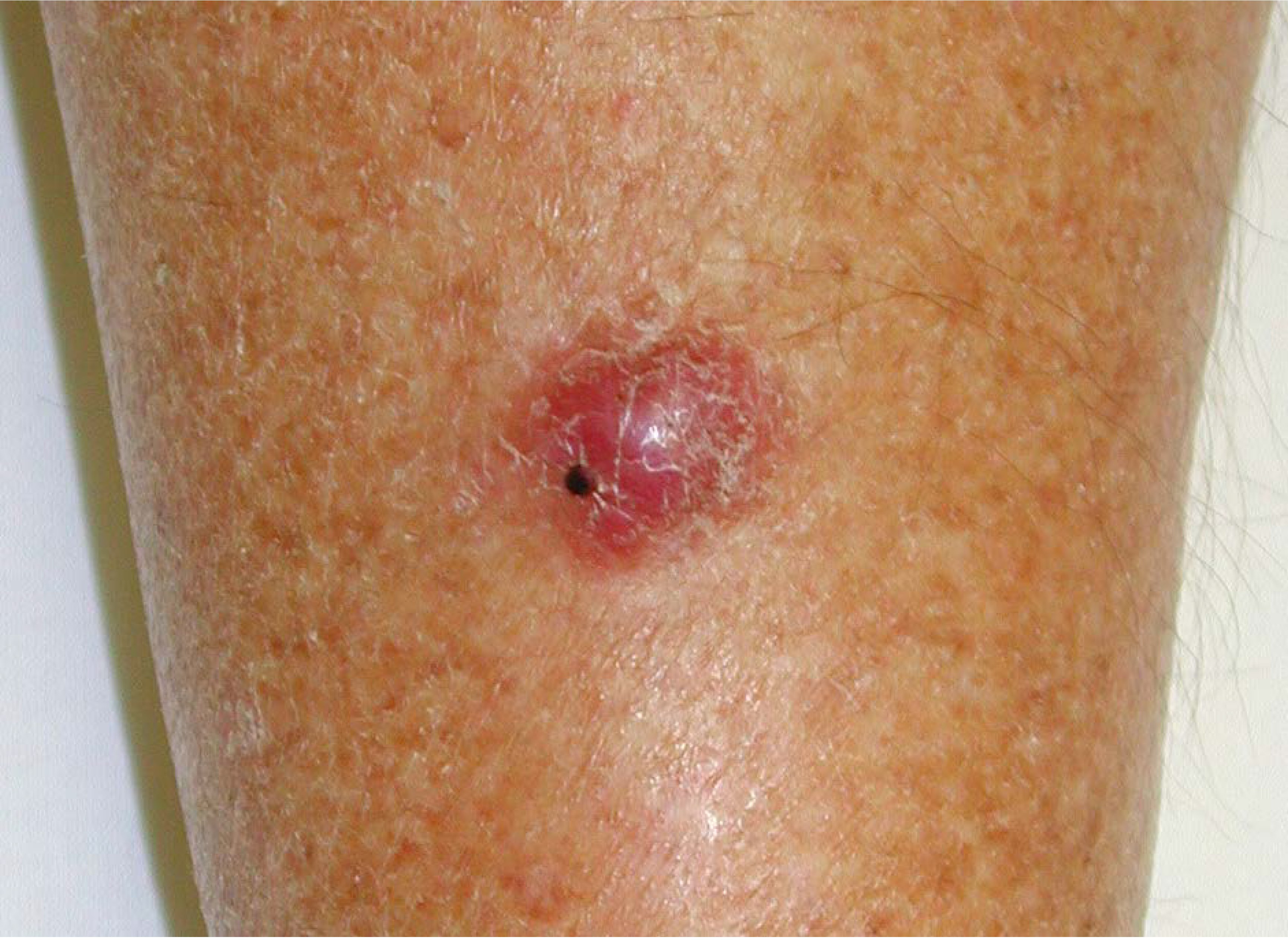
2. Merkel Cell Carcinoma
3. Merkel Cell Polyomavirus
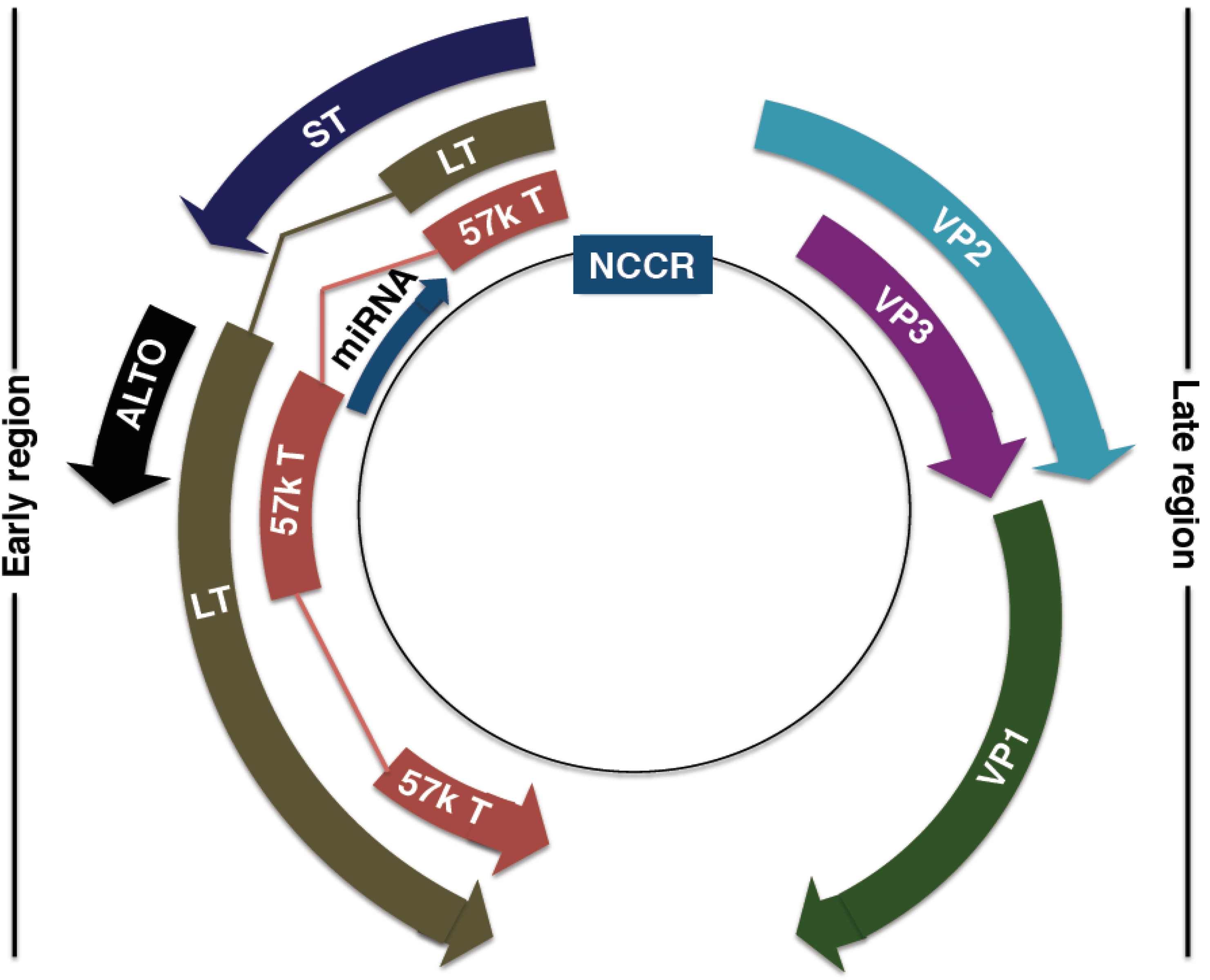
4. MCPyV Genome
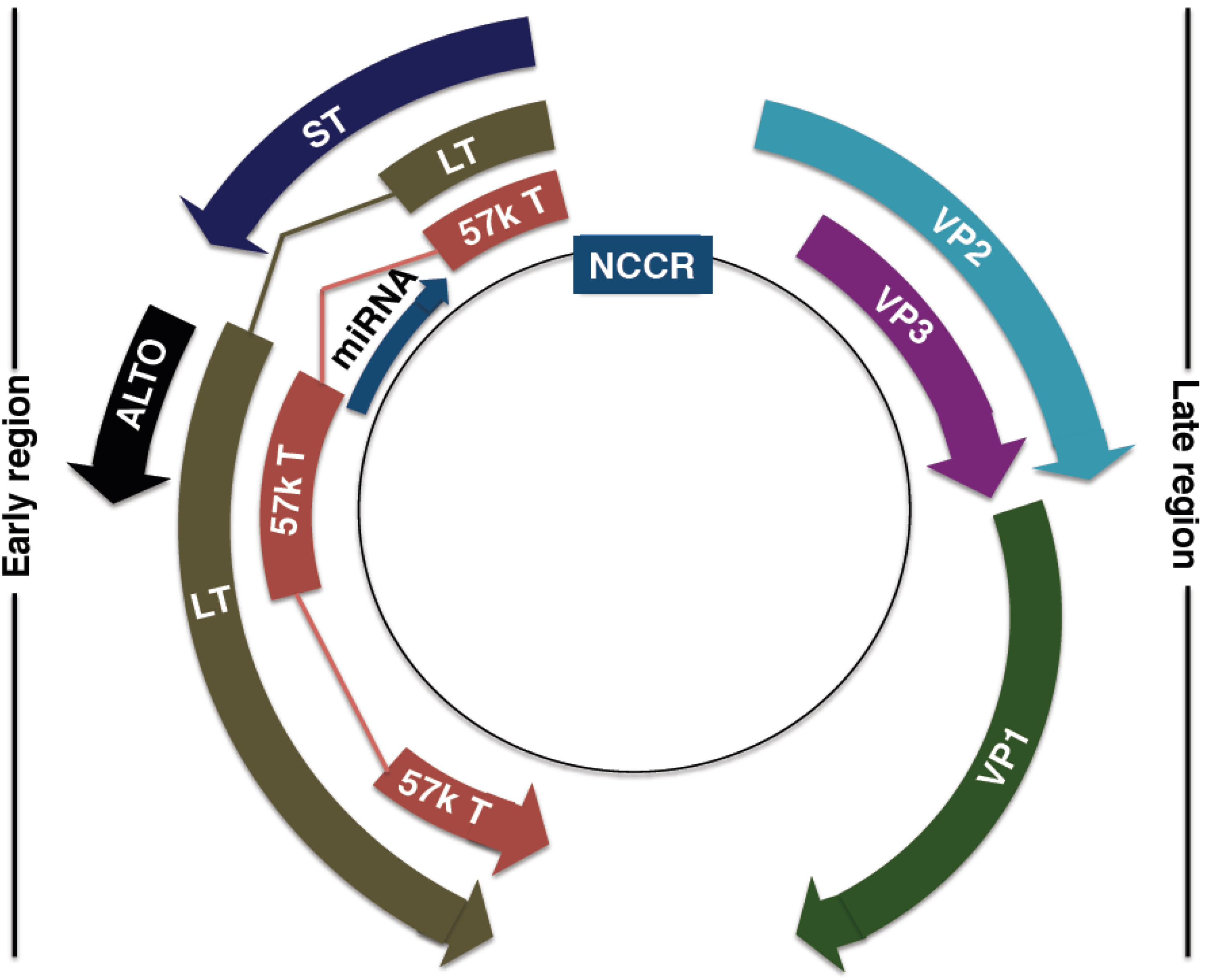
4.1. MCPyV Origin of Replication
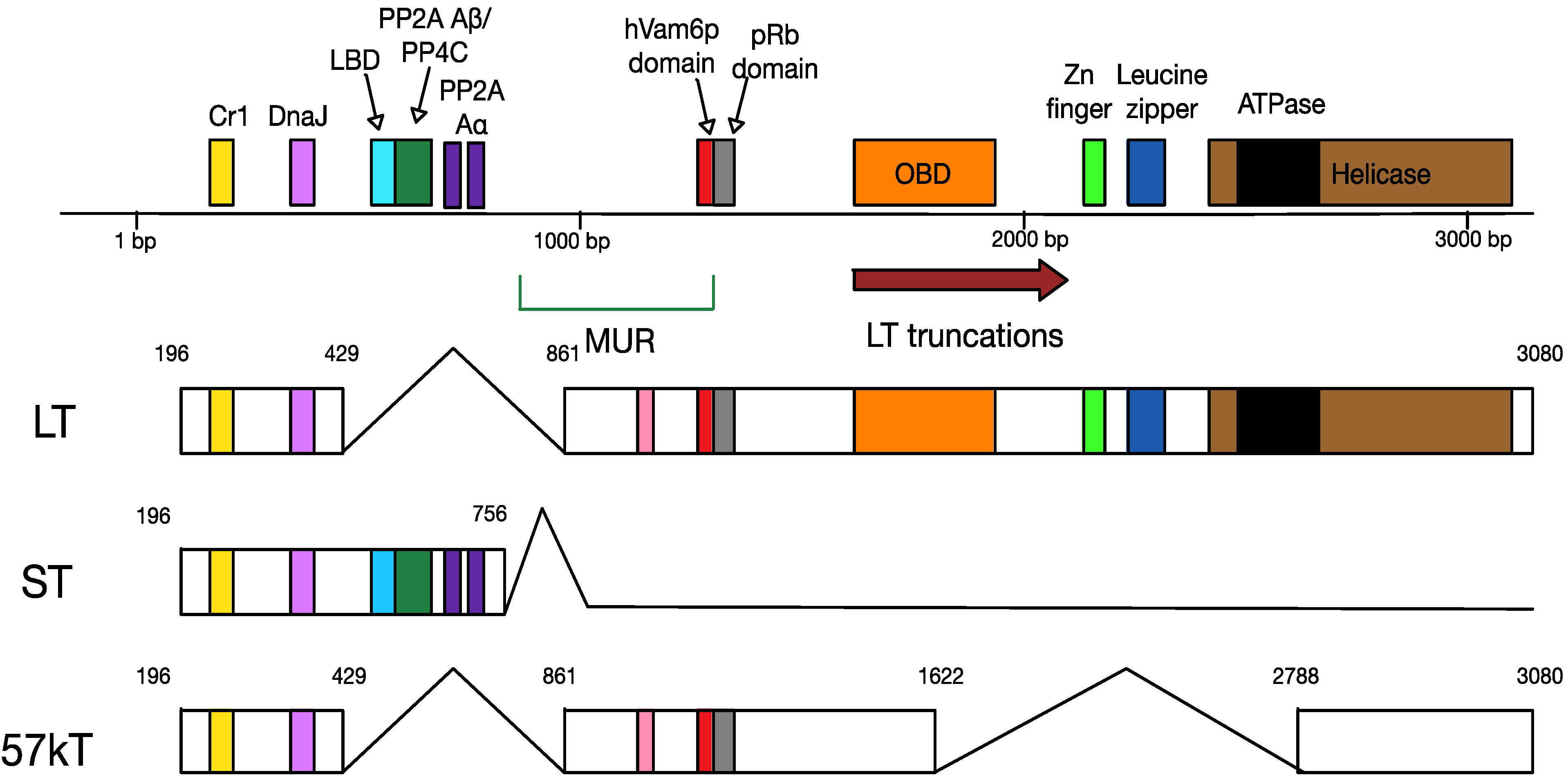
4.2. MCPyV T Antigen Locus
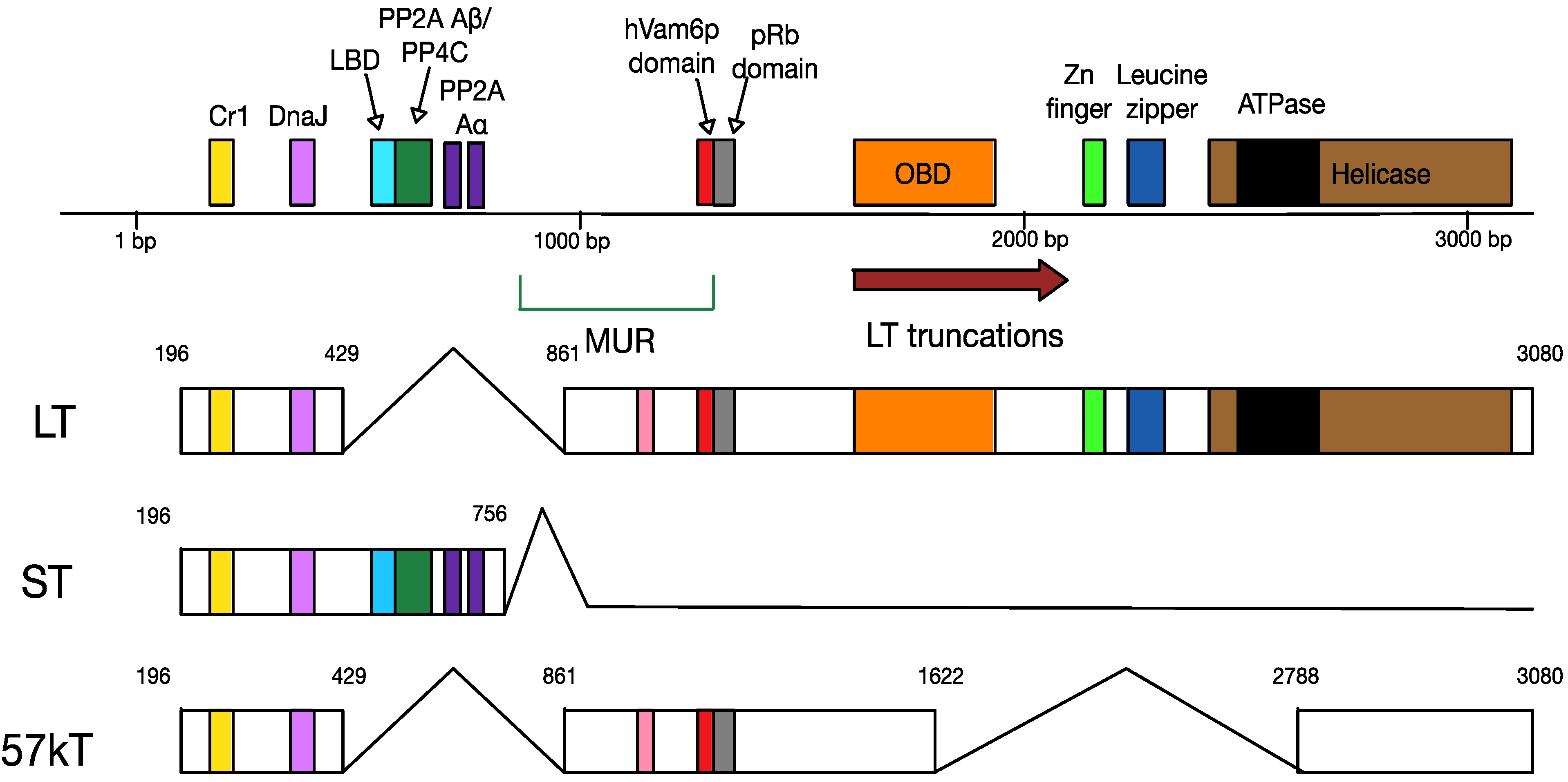
4.3. MCPyV Late Proteins
4.4. MCPyV MicroRNA
5. The Lifecycle of MCPyV
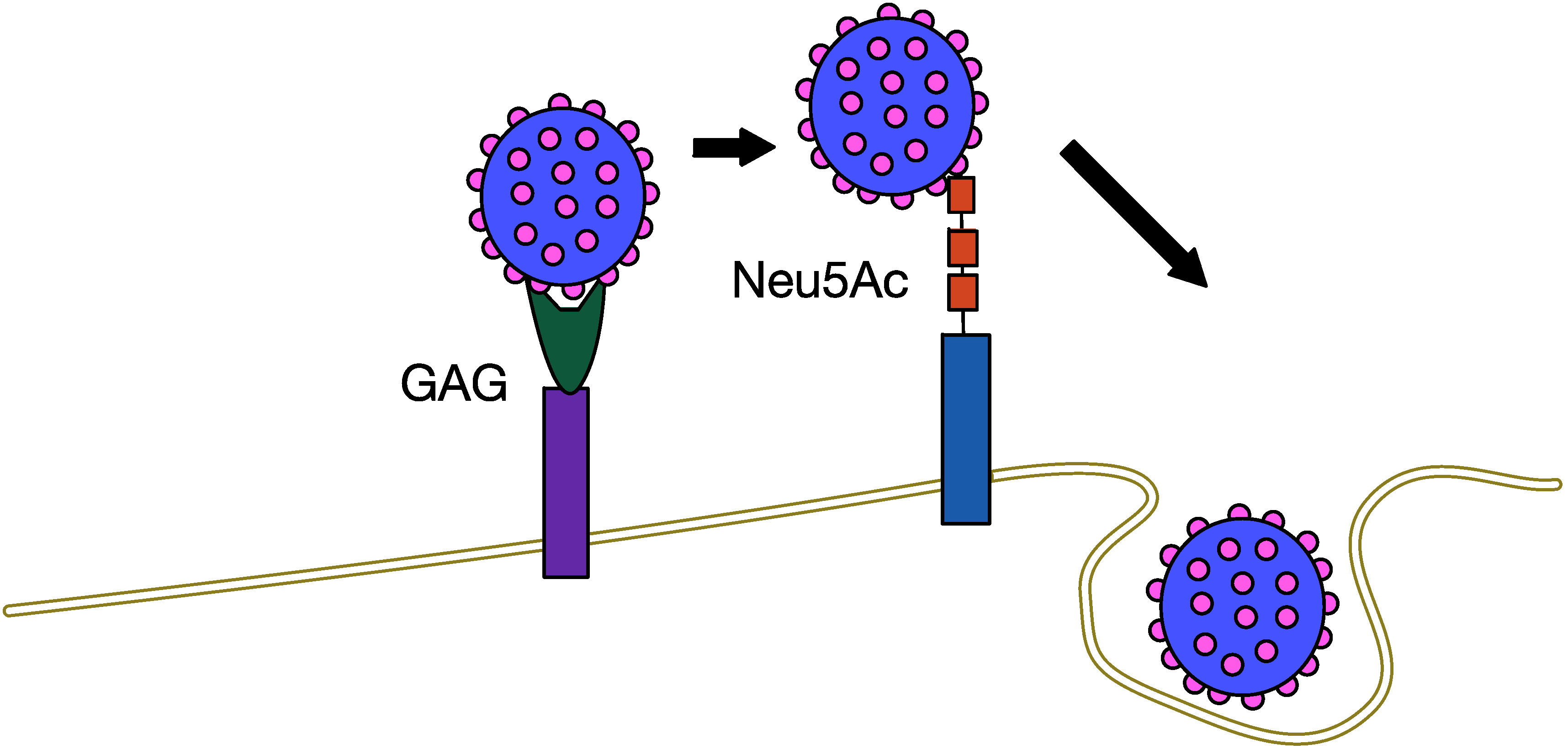
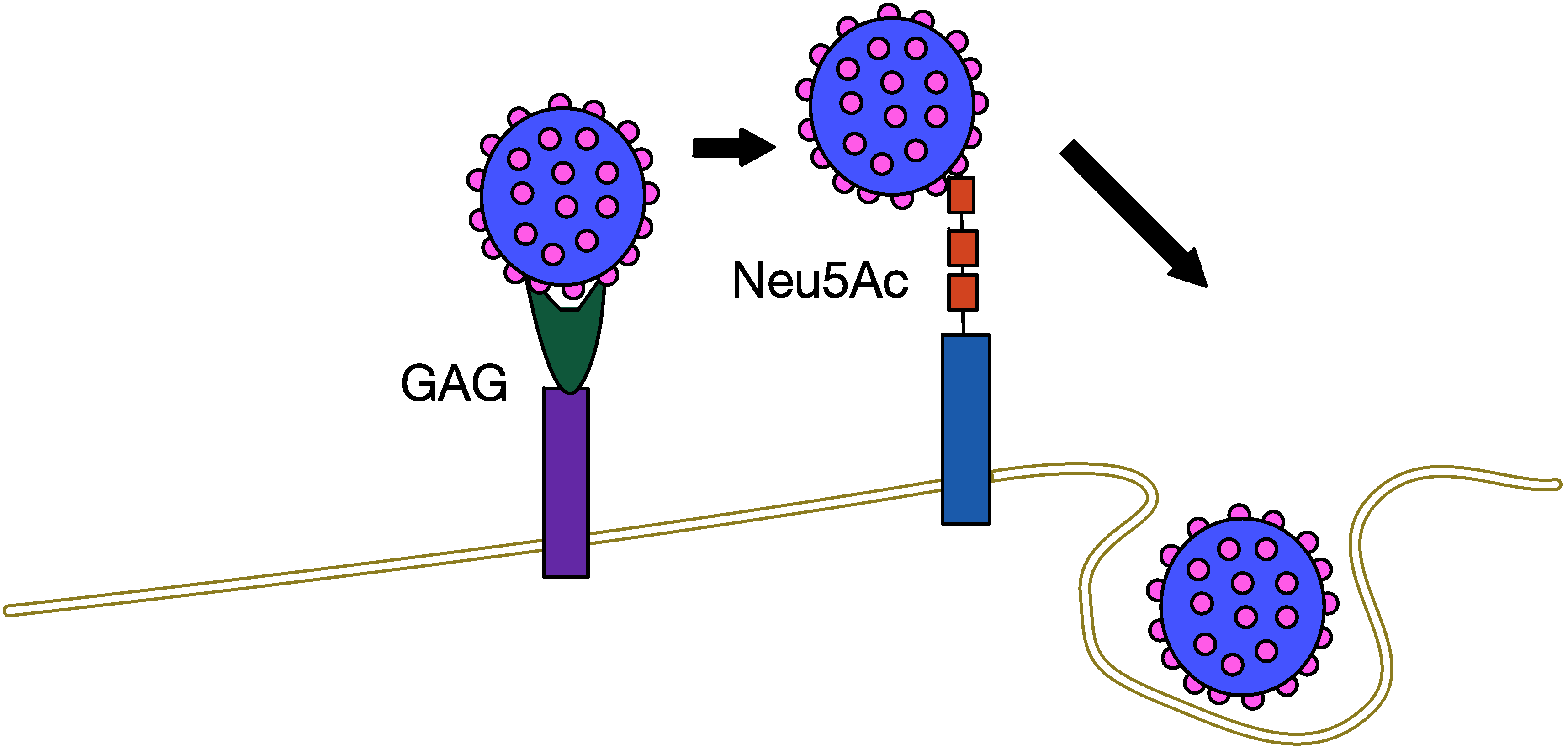
5.1. MCPyV Attachment and Entry
5.2. MCPyV Replication
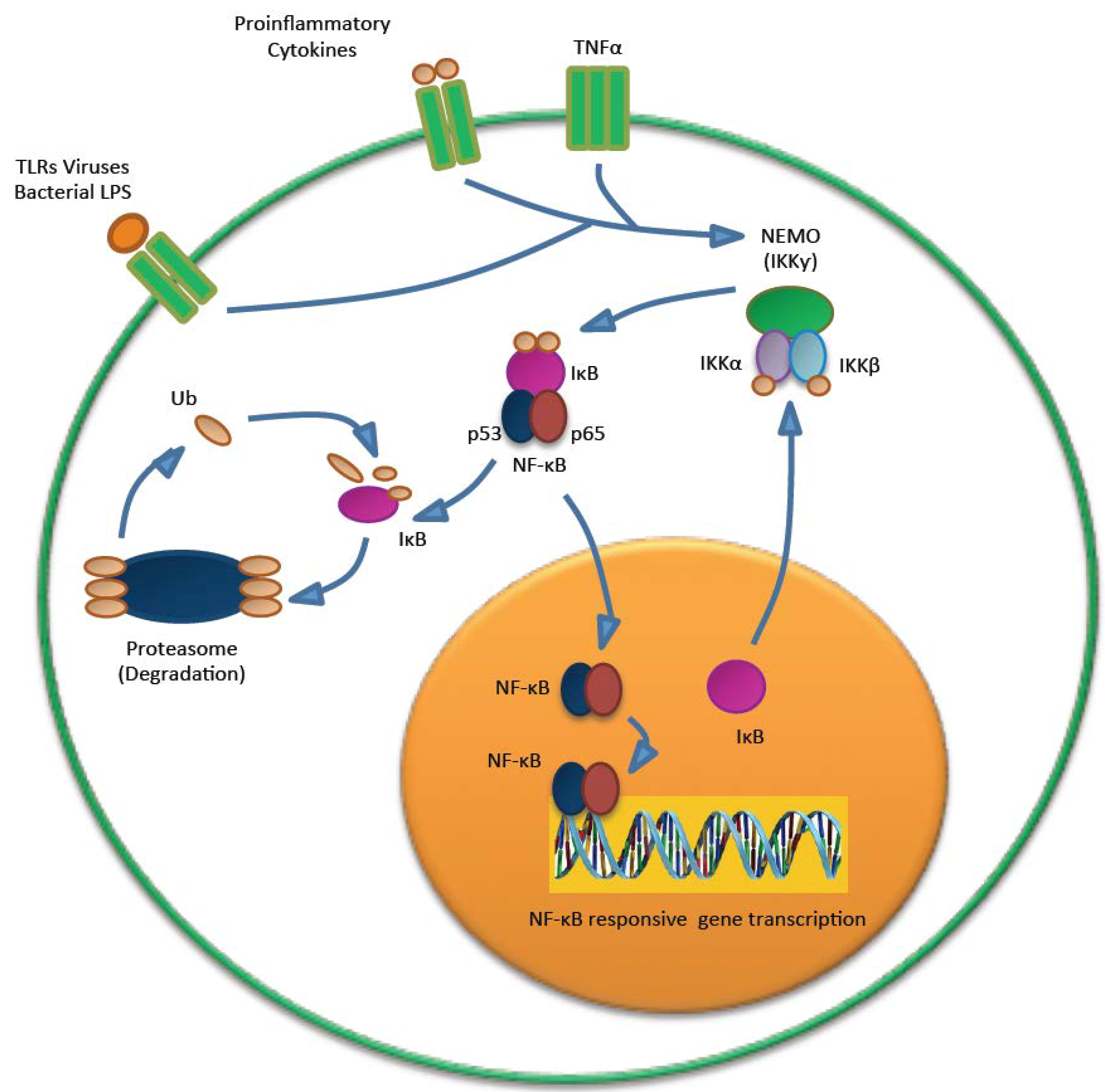
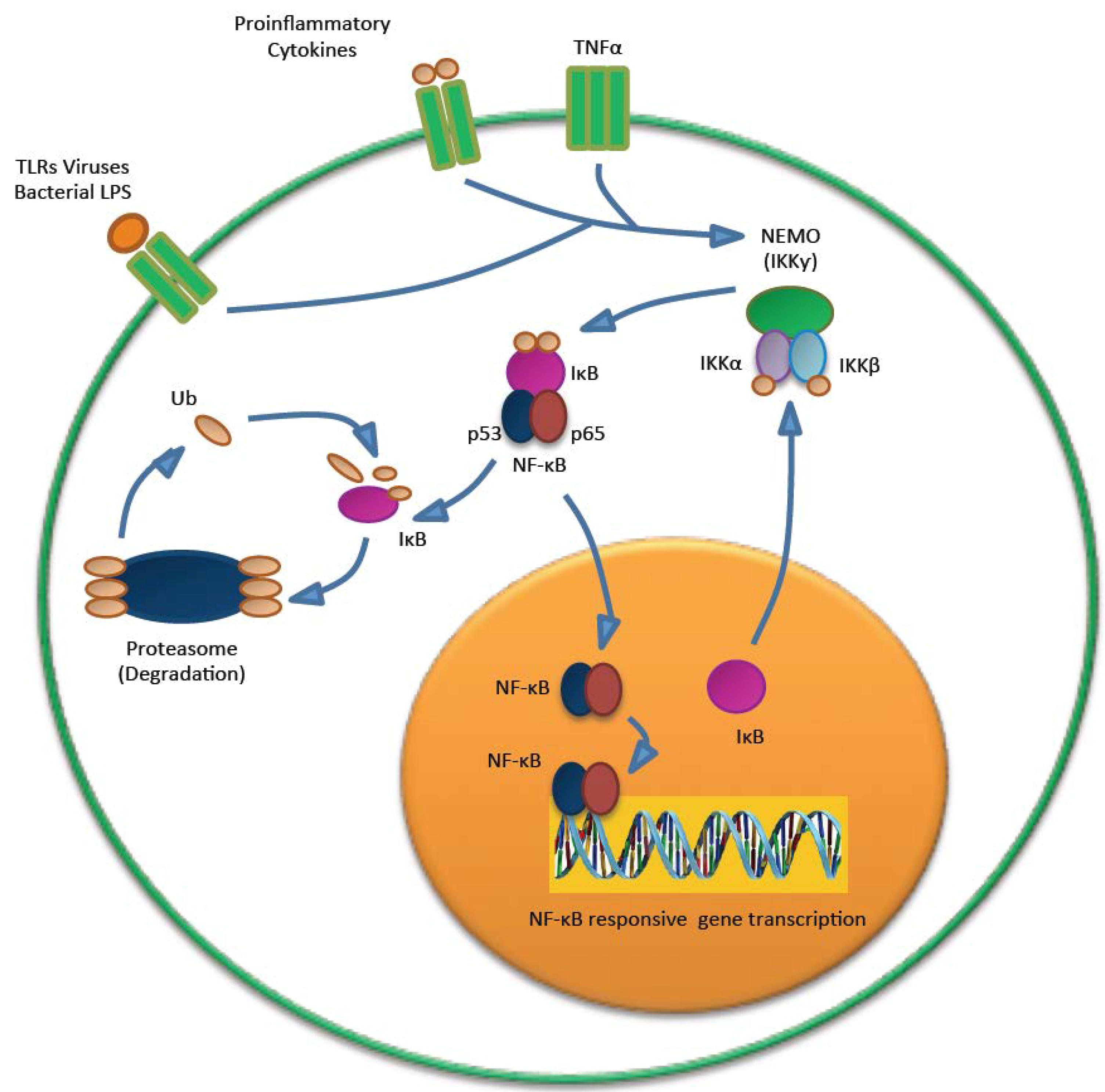
5.3. MCPyV and the Immune Response
5.4. MCPyV Assembly and Egress
6. MCPyV and Tumourigenesis
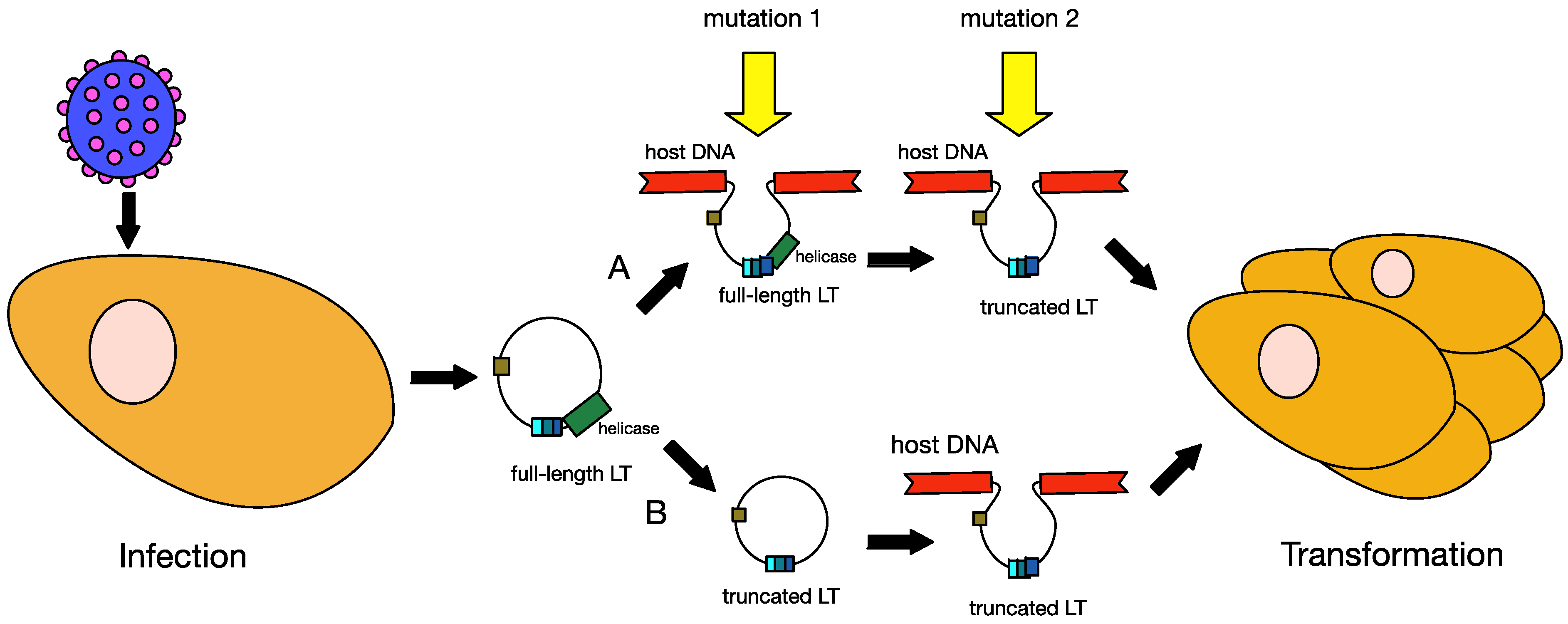
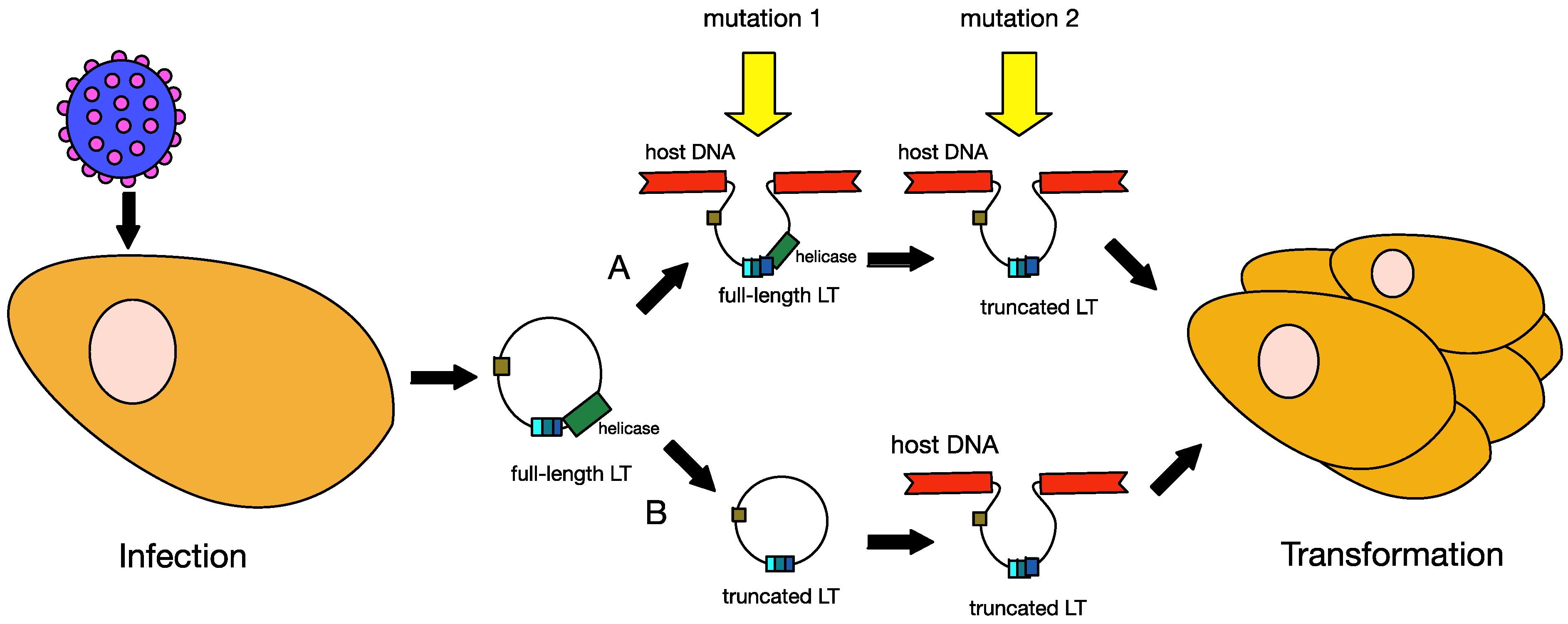
6.1. Truncation of MCPyV Large T Antigen
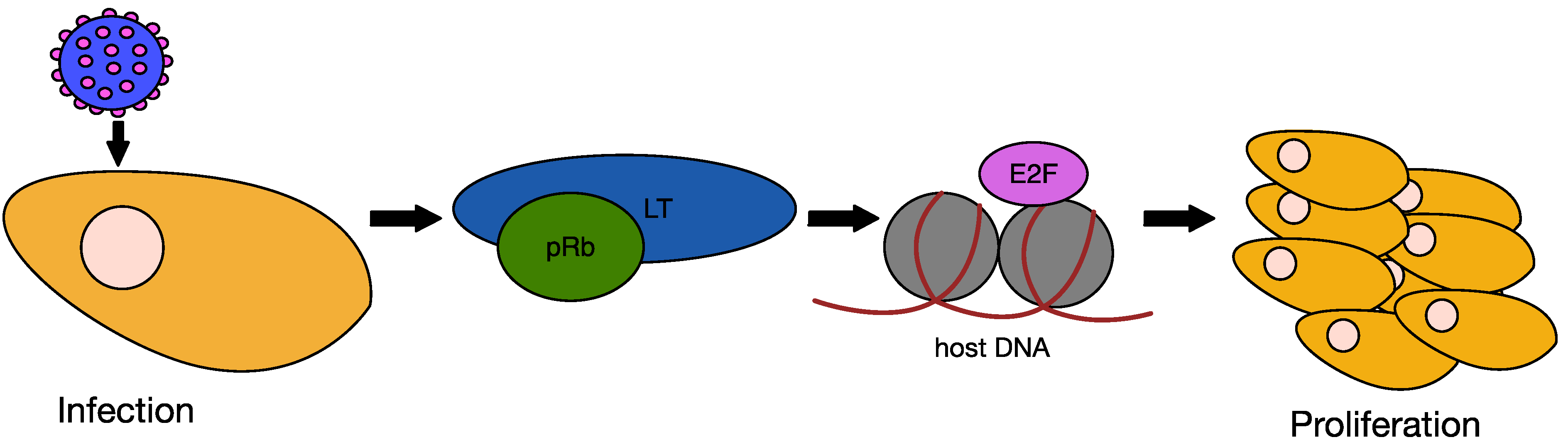
6.2. MCPyV Large T Antigen as an Oncogene
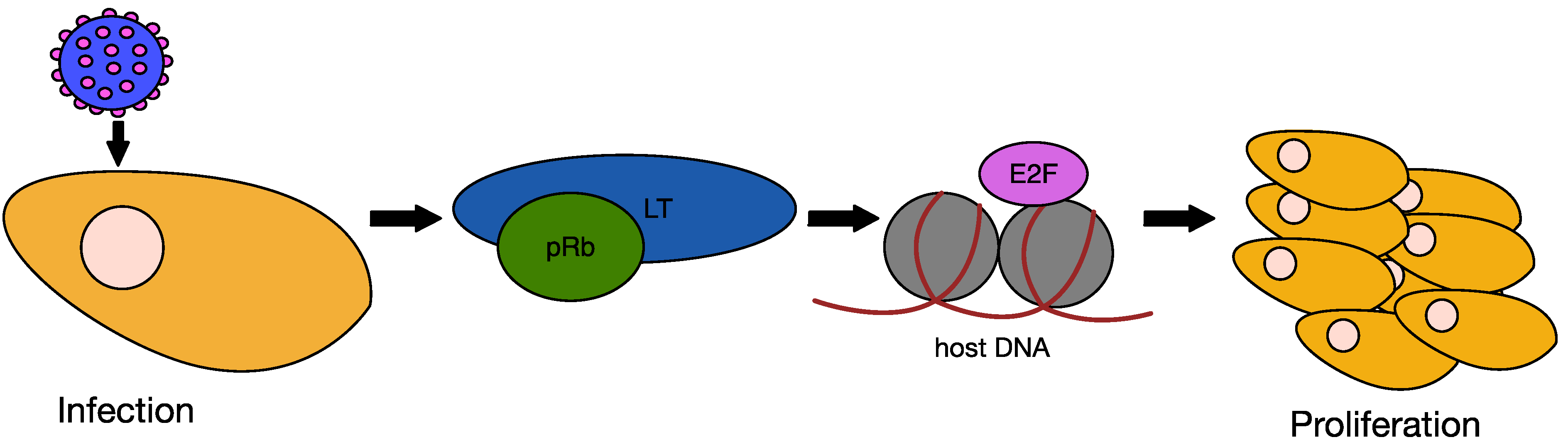
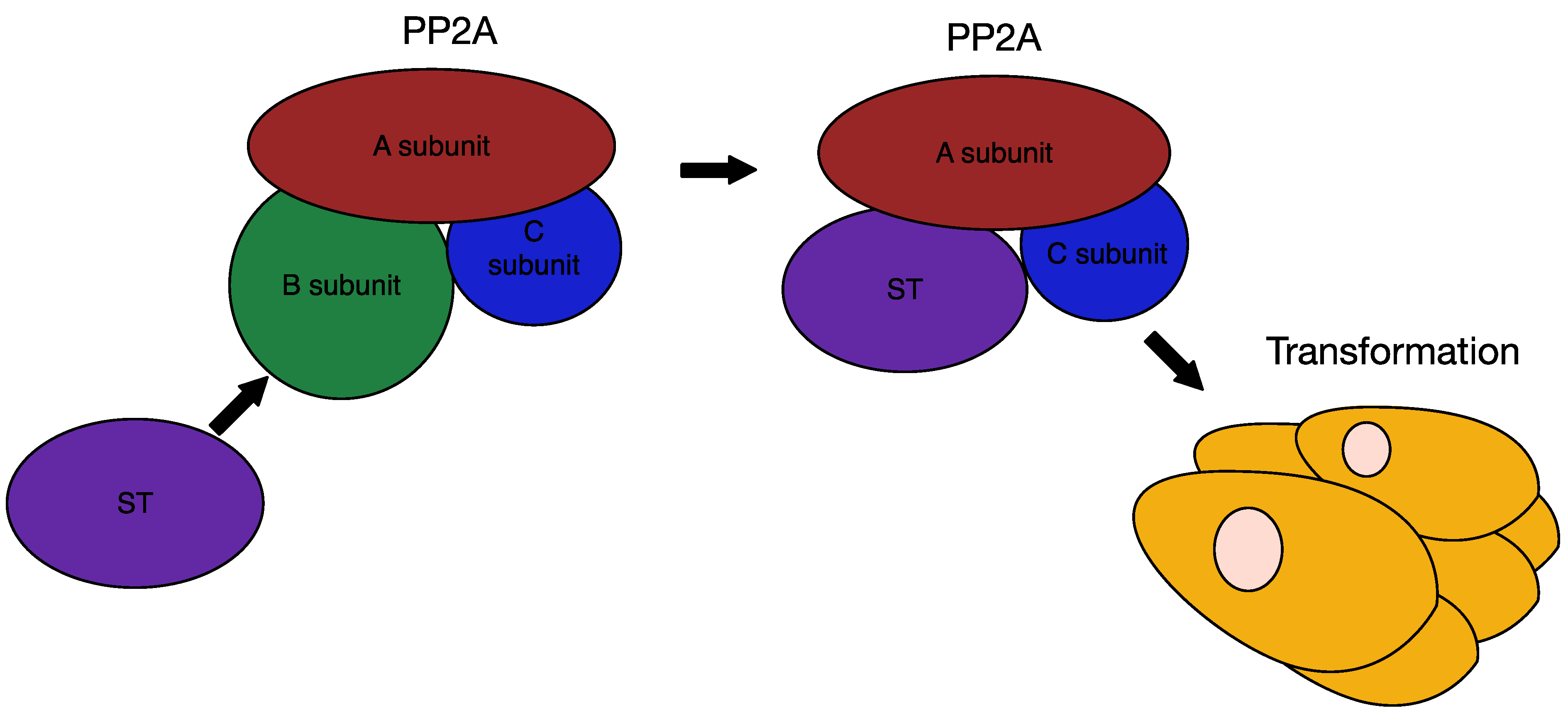
6.3. MCPyV Small T Antigen as an Oncogene
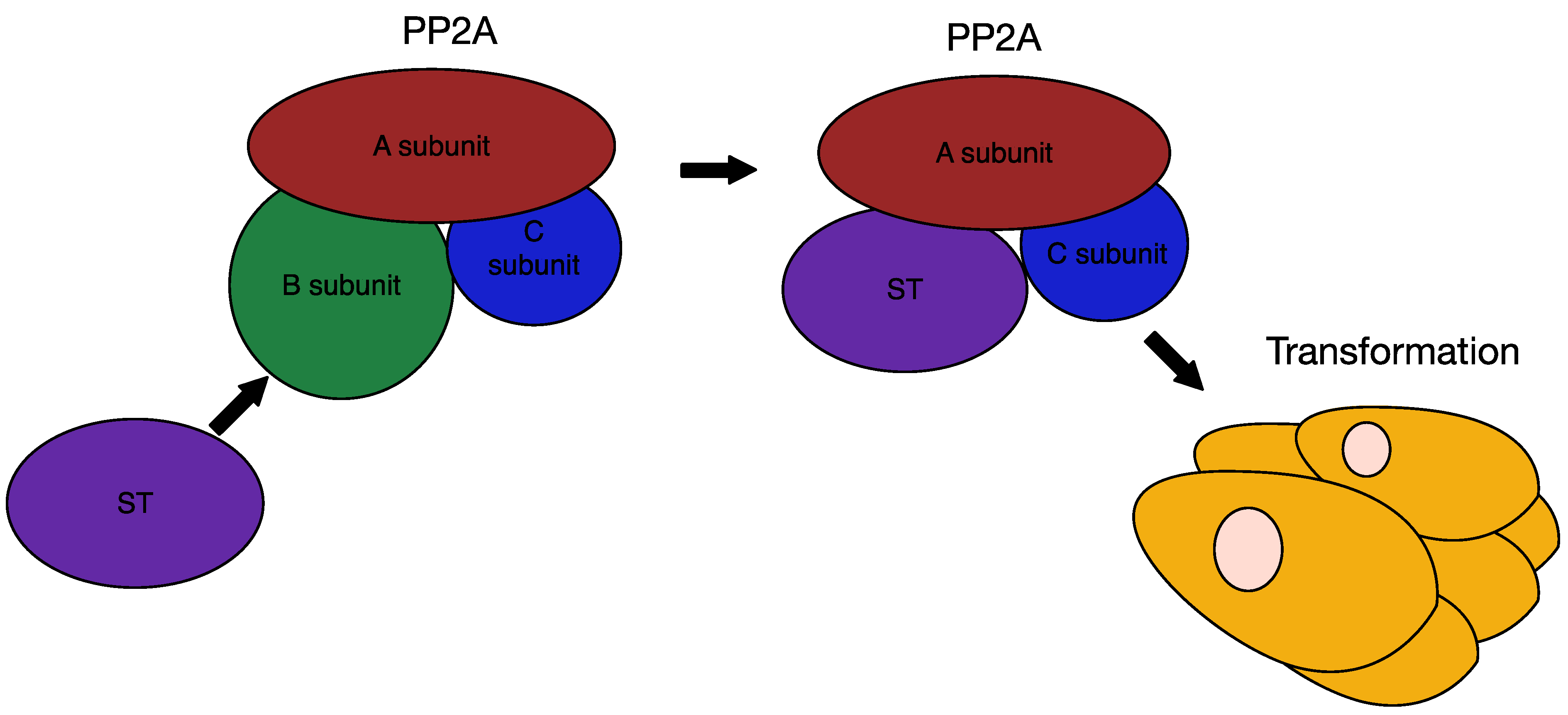
7. Therapies for MCC
8. Conclusions
Acknowledgements
Conflicts of Interest
References
- Parkin, D.M. The global health burden of infection-associated cancers in the year 2002. Int. J.Cancer 2006, 118, 3030–3044. [Google Scholar] [CrossRef]
- Javier, R.T.; Butel, J.S. The History of tumor virology. Cancer Res. 2008, 68, 7693–7706. [Google Scholar] [CrossRef]
- Moore, P.S.; Chang, Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 2010, 10, 878–889. [Google Scholar]
- Stewart, S.E.; Eddy, B.E.; Borgese, N. Neoplasms in mice inoculated with a tumor agent carried in tissue culture. J. Natl. Cancer Inst. 1958, 20, 1223–1243. [Google Scholar]
- Sweet, B.H.; Hilleman, M.R. The Vacuolating Virus, SV40. Exp. Biol. Med. 1960, 105, 420–427. [Google Scholar] [CrossRef]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef]
- Kaae, J.; Hansen, A.V.; Biggar, R.J.; Boyd, H.A.; Moore, P.S.; Wohlfahrt, J.; Melbye, M. Merkel cell carcinoma: Incidence, mortality, and risk of other cancers. J. Natl. Cancer Inst. 2010, 102, 793–801. [Google Scholar] [CrossRef]
- Lanoy, E.; Costagliola, D.; Engels, E.A. Skin cancers associated with HIV infection and solid-organ transplantation among elderly adults. Int. J. Cancer 2010, 126, 1724–1731. [Google Scholar]
- Toker, C. Trabecular Carcinoma of the Skin. Arch. Dermatol. 1972, 105, 107–110. [Google Scholar] [CrossRef]
- Merkel, F. Tastzellen and Tastkoerperchenbei den Hausthieren und beim Menschen. Arch. Mikr. Anat. 1875, 11, 636–652. [Google Scholar] [CrossRef]
- Lacour, J.P.; Dubois, D.; Pisani, A.; Ortonne, J.P. Anatomical mapping of Merkel cells in normal human adult epidermis. Br.J. Dermatol. 1991, 125, 535–542. [Google Scholar] [CrossRef]
- May, C.A.; Osterland, I. Merkel cell distribution in the human eyelid. Eur. J. Histochem. 2013, 57, 224–226. [Google Scholar]
- Koljonen, V.; Kluger, N.; Sihto, H.; Bohling, T. Lateral distribution of merkel cell carcinoma in a nationwide cohort. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 884–888. [Google Scholar] [CrossRef]
- Hodgson, N.C. Merkel Cell Carcinoma: Changing Incidence Trends. J. Surg. Oncol. 2005, 89, 1–4. [Google Scholar] [CrossRef]
- Bzhalava, D.; Bray, F.; Storm, H.; Dillner, J. Risk of second cancers after the diagnosis of Merkel cell carcinoma in Scandinavia. Br. J. Cancer 2011, 104, 178–180. [Google Scholar] [CrossRef]
- Gupta, S.G.; Wang, L.C.; Penas, P.F.; Gellenthin, M.; Lee, S.J.; Nghiem, P. Sentinel lymph node biopsy for evaluation and treatment of patients with Merkel cell carcinoma: The dana-farber experience and meta-analysis of the literature. Arch. Dermatol. 2006, 142, 685–690. [Google Scholar]
- Vernadakis, S.; Moris, D.; Bankfalvi, A.; Makris, N.; Sotiropoulos, G.C. Metastatic Merkel cell carcinoma (MCC) of pancreas and breast: A unique case. World J. Surg. Oncol. 2013, 11, 261. [Google Scholar] [CrossRef]
- Suttie, C.F.; Hruby, G.; Horvath, L.; Thompson, J. Cardiac metastasis in Merkel cell carcinoma. J. Clin. Oncol. 2014, 32, 1–2. [Google Scholar] [CrossRef]
- Vasileiadis, I.; Sofopoulos, M.; Arnogiannaki, N.; Georgopoulos, S. A Merkel cell carcinoma metastatic to the tonsil: A case report and review of the literature. J. Oral Maxillofac. Surg. 2013, 71, 1812. [Google Scholar]
- Laude, H.C.; Jonchère, B.; Maubec, E.; Carlotti, A.; Marinho, E.; Couturaud, B.; Peter, M.; Sastre-Garau, X.; Avril, M.-F.; Dupin, N.; et al. Distinct Merkel cell polyomavirus molecular features in tumour and non tumour specimens from patients with merkel cell carcinoma. PLoS Pathog. 2010, 6, e1001076. [Google Scholar] [CrossRef]
- Martel-Jantin, C.; Filippone, C.; Cassar, O.; Peter, M.; Tomasic, G.; Vielh, P.; Briere, J.; Petrella, T.; Aubriot-Lorton, M.H.; Mortier, L.; et al. Genetic Variability and integration of Merkel cell polyomavirus in Merkel cell carcinoma. Virology 2012, 426, 134–142. [Google Scholar] [CrossRef]
- Sastre-Garau, X.; Peter, M.; Avril, M.-F.; Laude, H.; Couturier, J.; Rozenberg, F.; Almeida, A.; Boitier, F.; Carlotti, A.; Couturaud, B.; et al. Merkel cell carcinoma of the skin: Pathological and molecular evidence for a causative role of MCV in oncogenesis. J. Pathol. 2009, 218, 48–56. [Google Scholar]
- Rodig, S.J.; Cheng, J.; Wardzala, J.; DoRosario, A.; Scanlon, J.J.; Laga, A.C.; Martinez-Fernandez, A.; Barletta, J.A.; Bellizzi, A.M.; Sadasivam, S.; et al. Improved detection suggests all Merkel cell carcinomas harbor merkel polyomavirus. J. Clin. Invest. 2012, 122, 4645–4653. [Google Scholar] [CrossRef]
- Donepudi, S.; DeConti, R.C.; Samlowski, W.E. Recent Advances in the understanding of the genetics, etiology, and treatment of Merkel cell carcinoma. Semin.Oncol. 2012, 39, 163–172. [Google Scholar] [CrossRef]
- Shuda, M.; Arora, R.; Kwun, H.J.; Feng, H.; Sarid, R.; Fernandez-Figueras, M.-T.; Tolstov, Y.; Gjoerup, O.; Mansukhani, M.M.; Swerdlow, S.H.; et al. Human Merkel cell polyomavirus infection I. MCV T antigen expression in Merkel cell carcinoma, lymphoid tissues and lymphoid tumors. Int. J. Cancer 2009, 125, 1243–1249. [Google Scholar] [CrossRef]
- Viscidi, R.P.; Rollison, D.E.; Sondak, V.K.; Silver, B.; Messina, J.L.; Giuliano, A.R.; Fulp, W.; Ajidahun, A.; Rivanera, D. Age-specific seroprevalence of Merkel cell polyomavirus, BK virus, and JC virus. Clin.Vaccine Immunol. 2011, 18, 1737–1743. [Google Scholar] [CrossRef]
- Tolstov, Y.L.; Pastrana, D.V.; Feng, H.; Becker, J.C.; Jenkins, J.J.; Moschos, S.; Chang, Y.; Buck, C.B.; Moore, P.S. Human Merkel cell polyomavirus infection II. MCV is a common human infection that can be detected by conformational capsid epitope immunoassays. Int. J. Cancer 2009, 125, 1250–1256. [Google Scholar] [CrossRef]
- Becker, J.C.; Houben, R.; Ugurel, S.; Trefzer, U.; Pfohler, C.; Schrama, D. MC polyomavirus is frequently present in Merkel cell carcinoma of European patients. J. Invest. Dermatol. 2009, 129, 248–250. [Google Scholar] [CrossRef]
- Matsushita, M.; Kuwamoto, S.; Iwasaki, T.; Higaki-Mori, H.; Yashima, S.; Kato, M.; Murakami, I.; Horie, Y.; Kitamura, Y.; Hayashi, K. Detection of Merkel cell polyomavirus in the human tissues from 41 japanese autopsy cases using polymerase chain reaction. Intervirology 2013, 56, 1–5. [Google Scholar] [CrossRef]
- Martel-Jantin, C.; Pedergnana, V.; Nicol, J.T.J.; Leblond, V.; Trégouët, D.-A.; Tortevoye, P.; Plancoulaine, S.; Coursaget, P.; Touzé, A.; Abel, L.; et al. Merkel Cell polyomavirus infection occurs during early childhood and is transmitted between siblings. J. Clin. Virol. 2013, 58, 288–291. [Google Scholar] [CrossRef]
- van der Meijden, E.; Bialasiewicz, S.; Rockett, R.J.; Tozer, S.J.; Sloots, T.P.; Feltkamp, M.C.W. Different serologic behavior of MCPyV, TSPyV, HPyV6, HPyV7 and HPyV9 polyomaviruses found on the skin. PLoS One 2013, 8, e81078:1–e81078:9. [Google Scholar]
- Hashida, Y.; Imajoh, M.; Kamioka, M.; Taniguchi, A.; Kuroda, N.; Hayashi, K.; Nakajima, H.; Sano, S.; Daibata, M. Phylogenetic analysis of Merkel cell polyomavirus based on full-length LT and VP1 gene sequences derived from neoplastic tumours in Japanese patients. J. Gen. Virol. 2014, 95, 135–141. [Google Scholar] [CrossRef]
- Matsushita, M.; Iwasaki, T.; Kuwamoto, S.; Kato, M.; Nagata, K.; Murakami, I.; Kitamura, Y.; Hayashi, K. Merkel cell polyomavirus (MCPyV) strains in Japanese Merkel cell carcinomas (MCC) are distinct from caucasian type MCPyVs: Genetic variability and phylogeny of MCPyV genomes obtained from Japanese MCPyV-infected MCCs. Virus Genes 2014, 48, 233–242. [Google Scholar] [CrossRef]
- Martel-Jantin, C.; Filippone, C.; Tortevoye, P.; Afonso, P.V.; Betsem, E.; Descorps-Declere, S.; Nicol, J.T.J.; Touzé, A.; Coursaget, P.; Crouzat, M.; et al. Molecular epidemiology of Merkel cell polyomavirus: Evidence for geographically related variant genotypes. J. Clin. Microbiol. 2014, 52, 1687–1690. [Google Scholar] [CrossRef]
- Dalianis, T.; Ramqvist, T.; Andreasson, K.; Kean, J.M.; Garcea, R.L. KI, WU and Merkel cell polyomaviruses: A new era for human polyomavirus research. Semin. Cancer Biol. 2009, 19, 270–275. [Google Scholar] [CrossRef]
- Johne, R.; Buck, C.B.; Allander, T.; Atwood, W.J.; Garcea, R.L.; Imperiale, M.J.; Major, E.O.; Ramqvist, T.; Norkin, L.C. Taxonomical developments in the family polyomaviridae. Arch. Virol. 2011, 156, 1627–1634. [Google Scholar] [CrossRef]
- Kwun, H.J.; Guastafierro, A.; Shuda, M.; Meinke, G.; Bohm, A.; Moore, P.S.; Chang, Y. The minimum replication origin of Merkel cell polyomavirus has a unique large T-antigen loading architecture and requires small T-antigen expression for optimal replication. J. Virol. 2009, 83, 12118–12128. [Google Scholar] [CrossRef]
- Peden, K.W.C.; Srinivasan, A.; Farber, J.M.; Pipas, J.M. Mutants with changes within or near a hydrophobic region of simian virus-40 Large Tumor Antigen are Defective for Binding Cellular Protein p53. Virology 1989, 168, 13–21. [Google Scholar] [CrossRef]
- Kierstead, T.D.; Tevethia, M.J. Association of p53 binding and immortalization of primary C57bl/6 mouse embryo fibroblasts by using simian-virus 40 T-antigen mutants bearing internal overlapping deletion mutations. J. Virol. 1993, 67, 1817–1829. [Google Scholar]
- Topalis, D.; Andrei, G.; Snoeck, R. The large tumor antigen: A “Swiss Army Knife” protein possessing the functions required for the polyomavirus life cycle. Antiviral Res. 2013, 97, 122–136. [Google Scholar] [CrossRef]
- Nakamura, T.; Sato, Y.; Watanabe, D.; Ito, H.; Shimonohara, N.; Tsuji, T.; Nakajima, N.; Suzuki, Y.; Matsuo, K.; Nakagawa, H.; et al. Nuclear localization of Merkel cell polyomavirus large T antigen in Merkel cell carcinoma. Virology 2010, 398, 273–279. [Google Scholar] [CrossRef]
- Liu, X.; Hein, J.; Richardson, S.C.W.; Basse, P.H.; Toptan, T.; Moore, P.S.; Gjoerup, O.V.; Chang, Y. Merkel cell polyomavirus large T antigen disrupts lysosome clustering by translocating human vam6p from the cytoplasm to the nucleus. J. Biol. Chem. 2011, 286, 17079–17090. [Google Scholar] [CrossRef]
- DeCaprio, J.A.; Ludlow, J.W.; Figge, J.; Shew, J.Y.; Huang, C.M.; Lee, W.H.; Marsilio, E.; Paucha, E.; Livingston, D.M. SV40 large tumor-antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell 1988, 54, 275–283. [Google Scholar] [CrossRef]
- Srinivasan, A.; McClellan, A.J.; Vartikar, J.; Marks, I.; Cantalupo, P.; Li, Y.; Whyte, P.; Rundell, K.; Brodsky, J.L.; Pipas, J.M. The amino-terminal transforming region of simian virus 40 large T and small T antigens functions as a J domain. Mol. Cell. Biol. 1997, 17, 4761–4773. [Google Scholar]
- Ali, S.H.; Kasper, J.S.; Arai, T.; DeCaprio, J.A. Cu17/p185/p193 binding to simian virus 40 large T antigen has a role in cellular transformation. J. Virol. 2004, 78, 2749–2757. [Google Scholar] [CrossRef]
- Cotsiki, M.; Lock, R.L.; Cheng, Y.; Williams, G.L.; Zhao, J.; Perera, D.; Freire, R.; Entwistle, A.; Golemis, E.A.; Roberts, T.M.; et al. Simian virus 40 large T antigen targets the spindle assembly checkpoint protein Bub1. Proc. Natl. Acad. Sci. USA 2004, 101, 947–952. [Google Scholar] [CrossRef]
- Lane, D.P.; Crawford, L.V. T-antigen is bound to a host protein in SV40-transformed cells. Nature 1979, 278, 261–263. [Google Scholar] [CrossRef]
- Poulin, D.L.; Kung, A.L.; DeCaprio, J.A. p53 Targets Simian virus 40 large T antigen for acetylation by CBP. J. Virol. 2004, 78, 8245–8253. [Google Scholar] [CrossRef]
- Seo, G.J.; Chen, C.J.; Sullivan, C.S. Merkel cell polyomavirus encodes a MicroRNA with the ability to autoregulate viral gene expression. Virology 2009, 383, 183–187. [Google Scholar] [CrossRef]
- Zerrahn, J.; Knippschild, U.; Winkler, T.; Deppert, W. Independent expression of the transforming amino-terminal domain of SV40 large T-antigen from an alternatively spliced 3rd SV40 early messenger-RNA. EMBO J. 1993, 12, 4739–4746. [Google Scholar]
- Comerford, S.A.; Schultz, N.; Hinnant, E.A.; Klapproth, S.; Hammer, R.E. Comparative analysis of SV40 17kT and LT function in vivo demonstrates that it’s C-terminus reprograms hepatic gene expression and is necessary for tumorigenesis in the liver. Oncogenesis 2012, 1, e28. [Google Scholar] [CrossRef]
- Moens, U.; van Ghelue, M.; Johannessen, M. Oncogenic potentials of the human polyomavirus regulatory proteins. Cell. Mol. Life. Sci. 2007, 64, 1656–1678. [Google Scholar] [CrossRef]
- Pallas, D.C.; Shahrik, L.K.; Martin, B.L.; Jaspers, S.; Miller, T.B.; Brautigan, D.L.; Roberts, T.M. Polyoma small and middle T-antigens and SV40 small T-antigen form stable complexes with protein phosphatase-2A. Cell 1990, 60, 167–176. [Google Scholar] [CrossRef]
- Griffiths, D.A.; Abdul-Sada, H.; Knight, L.M.; Jackson, B.R.; Richards, K.; Prescott, E.L.; Peach, A.H.; Blair, G.E.; Macdonald, A.; Whitehouse, A. Merkel cell polyomavirus small T antigen targets the NEMO adaptor protein to disrupt inflammatory signaling. J. Virol. 2013, 87, 13853–13867. [Google Scholar] [CrossRef]
- Kwun, H.J.; Shuda, M.; Feng, H.; Camacho, C.J.; Moore, P.S.; Chang, Y. Merkel cell polyomavirus small T antigen controls viral replication and oncoprotein expression by Targeting the cellular ubiquitin ligase SCFFbw7. Cell Host Microbe 2013, 14, 125–135. [Google Scholar] [CrossRef]
- Carter, J.J.; Daugherty, M.D.; Qi, X.; Bheda-Malge, A.; Wipf, G.C.; Robinson, K.; Roman, A.; Malik, H.S.; Galloway, D.A. Identification of an overprinting gene in Merkel cell polyomavirus provides evolutionary insight into the birth of viral genes. Proc. Natl. Acad. Sci. USA 2013, 110, 12744–12749. [Google Scholar] [CrossRef]
- Schowalter, R.M.; Buck, C.B. The Merkel cell polyomavirus minor capsid protein. PLoS Pathog. 2013, 9, e1003558. [Google Scholar] [CrossRef]
- Neu, U.; Hengel, H.; Blaum, B.S.; Schowalter, R.M.; Macejak, D.; Gilbert, M.; Wakarchuk, W.W.; Imamura, A.; Ando, H.; Kiso, M.; et al. Structures of Merkel cell polyomavirus VP1 complexes define a sialic acid binding site required for infection. PLoS Pathog. 2012, 8, e1002738. [Google Scholar]
- Sullivan, C.S.; Grundhoff, A.T.; Tevethia, S.; Pipas, J.M.; Ganem, D. SV40-encoded MicroRNAs regulate viral gene expression and reduce susceptibility to cytotoxic T cells. Nature 2005, 435, 682–686. [Google Scholar] [CrossRef]
- Seo, G.J.; Fink, L.H.L.; O’Hara, B.; Atwood, W.J.; Sullivan, C.S. Evolutionarily conserved function of a viral microRNA. J. Virol. 2008, 82, 9823–9828. [Google Scholar] [CrossRef]
- Lee, S.; Paulson, K.G.; Murchison, E.P.; Afanasiev, O.K.; Alkan, C.; Leonard, J.H.; Byrd, D.R.; Hannon, G.J.; Nghiem, P. Identification and validation of a novel mature microRNA encoded by the Merkel cell polyomavirus in human Merkel cell carcinomas. J. Clin. Virol. 2011, 52, 272–275. [Google Scholar] [CrossRef]
- Schowalter, R.M.; Pastrana, D.V.; Pumphrey, K.A.; Moyer, A.L.; Buck, C.B. Merkel cell polyomavirus and two previously unknown polyomaviruses are chronically shed from human skin. Cell Host Microbe 2010, 7, 509–515. [Google Scholar] [CrossRef]
- Schowalter, R.M.; Reinhold, W.C.; Buck, C.B. Entry tropism of BK and Merkel cell polyomaviruses in cell culture. PLoS One 2012, 7, e42181. [Google Scholar] [CrossRef]
- Schowalter, R.M.; Pastrana, D.V.; Buck, C.B. Glycosaminoglycans and sialylated glycans sequentially facilitate Merkel cell polyomavirus infectious entry. PLoS Pathog. 2011, 7, e1002161. [Google Scholar] [CrossRef]
- Bauer, P.H.; Cui, C.; Stehle, T.; Harrison, S.C.; DeCaprio, J.A.; Benjamin, T.L. Discrimination between sialic acid-containing receptors and pseudoreceptors regulates polyomavirus spread in the mouse. J. Virol. 1999, 7, 5826–5832. [Google Scholar]
- Sapp, M.; Day, P.M. Structure, attachment and entry of polyoma- and papillomaviruses. Virology 2009, 384, 400–409. [Google Scholar] [CrossRef]
- Wessel, R.; Schweizer, J.; Stahl, H. Simian virus-40 T-antigen DNA helicase is a hexamer which forms a binary complex during bidirectional unwinding from the viral origin of DNA-replication. J. Virol. 1992, 66, 804–815. [Google Scholar]
- Harrison, C.J.; Meinke, G.; Kwun, H.J.; Rogalin, H.; Phelan, P.J.; Bullock, P.A.; Chang, Y.; Moore, P.S.; Bohm, A. Asymmetric assembly of Merkel cell polyomavirus large T-antigen origin binding domains at the viral origin. J. Molec. Biol. 2011, 409, 529–542. [Google Scholar] [CrossRef]
- Feng, H.; Kwun, H.J.; Liu, X.; Gjoerup, O.; Stolz, D.B.; Chang, Y.; Moore, P.S. Cellular and viral factors regulating Merkel cell polyomavirus replication. PLoS One 2011, 6, e22468. [Google Scholar]
- Wang, X.; Li, J.; Schowalter, R.M.; Jiao, J.; Buck, C.B.; You, J. Bromodomain protein Brd4 plays a key role in Merkel cell polyomavirus DNA replication. PLoS Pathog. 2012, 8, e1003021. [Google Scholar] [CrossRef]
- Tsang, S.H.; Wang, X.; Li, J.; Buck, C.B.; You, J. Host DNA damage response factors localize to Merkel cell polyomavirus DNA replication sites to support efficient viral DNA replication. J. Virol. 2014, 88, 3285–3297. [Google Scholar] [CrossRef]
- Gillespie, K.A.; Mehta, K.P.; Laimins, L.A.; Moody, C.A. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J. Virol. 2012, 86, 9520–9526. [Google Scholar] [CrossRef]
- Wang, X.; Helfer, C.M.; Pancholi, N.; Bradner, J.E.; You, J. Recruitment of Brd4 to the human papillomavirus type 16 DNA replication complex is essential for replication of viral DNA. J. Virol. 2013, 87, 3871–3884. [Google Scholar] [CrossRef]
- Shuda, M.; Kwun, H.J.; Feng, H.; Chang, Y.; Moore, P.S. Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J. Clin. Invest. 2011, 121, 3623–3634. [Google Scholar] [CrossRef]
- Welcker, M.; Clurman, B.E. FBW7 ubiquitin ligase: A tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer 2008, 8, 83–93. [Google Scholar] [CrossRef]
- Maser, R.S.; Choudhury, B.; Campbell, P.J.; Feng, B.; Wong, K.K.; Protopopov, A.; O’Neil, J.; Gutierrez, A.; Ivanova, E.; Perna, I.; et al. Chromosomally Unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature 2007, 447, 966–971. [Google Scholar] [CrossRef]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjöblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef]
- Mao, J.H.; Perez-Losada, J.; Wu, D.; Delrosario, R.; Tsunematsu, R.; Nakayama, K.I.; Brown, K.; Bryson, S.; Balmain, A. Fbxw7/Cdc4 Is a p53-dependent, haploinsufficient tumour suppressor gene. Nature 2004, 432, 775–779. [Google Scholar] [CrossRef]
- Rajagopalan, H.; Jallepalli, P.V.; Rago, C.; Velculescu, V.E.; Kinzler, K.W.; Vogelstein, B.; Lengauer, C. Inactivation of hCDC4 can cause chromosomal instability. Nature 2004, 428, 77–81. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Signaling to NF-kappa B. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef]
- Le Negrate, G. Viral interference with innate immunity by preventing NF-kappa B activity. Cell. Microbiol. 2012, 14, 168–181. [Google Scholar] [CrossRef]
- Joo, M.S.; Hahn, Y.S.; Kwon, M.; Sadikot, R.T.; Blackwell, T.S.; Christman, J.W. Hepatitis C virus core protein suppresses NF-kappa B activation and cyclooxygenase-2 expression by direct interaction with I kappa B kinase beta. J. Virol. 2005, 79, 7648–7657. [Google Scholar] [CrossRef]
- Spitkovsky, D.; Hehner, S.P.; Hofmann, T.G.; Moller, A.; Schmitz, M.L. The Human papillomavirus oncoprotein E7 attenuates NF-kappa B activation by targeting the I kappa B kinase complex. J. Biol. Chem. 2002, 277, 25576–25582. [Google Scholar]
- Randall, C.M.H.; Jokela, J.A.; Shisler, J.L. The MC159 Protein from the molluscum contagiosum poxvirus inhibits NF-kappa B activation by interacting with the I kappa B kinase complex. J. Immunol. 2012, 188, 2371–2379. [Google Scholar] [CrossRef]
- Fliss, P.M.; Jowers, T.P.; Brinkmann, M.M.; Holstermann, B.; Mack, C.; Dickinson, P.; Hohenberg, H.; Ghazal, P.; Brune, W. Viral mediated redirection of NEMO/IKK gamma to autophagosomes curtails the inflammatory cascade. PLoS Pathog. 2012, 8, e1002517. [Google Scholar] [CrossRef]
- Moreno, C.S.; Ramachandran, S.; Ashby, D.G.; Laycock, N.; Plattner, C.A.; Chen, W.; Hahn, W.C.; Pallas, D.C. Signaling and transcriptional changes critical for transformation of human cells by simian virus 40 small tumor antigen or protein phosphatase 2A B56 gamma knockdown. Cancer Res. 2004, 64, 6978–6988. [Google Scholar] [CrossRef]
- Shahzad, N.; Shuda, M.; Gheit, T.; Kwun, H.J.; Cornet, I.; Saidj, D.; Zannetti, C.; Hasan, U.; Chang, Y.; Moore, P.S.; et al. The T antigen locus of Merkel Cell polyomavirus downregulates human toll-like receptor 9 expression. J. Virol. 2013, 87, 13009–13019. [Google Scholar] [CrossRef]
- Takeda, K.; Akira, S. Toll-like receptors in innate immunity. Int.Immunol. 2005, 17, 1–14. [Google Scholar]
- Beutler, B. Inferences, questions and possibilities in toll-like receptor signalling. Nature 2004, 430, 257–263. [Google Scholar] [CrossRef]
- Tsujimura, H.; Tamura, T.; Kong, H.J.; Nishiyama, A.; Ishii, K.J.; Klinman, D.M.; Ozato, K. Toll-like receptor 9 signaling activates NF-κB through IFN regulatory factor-8/IFN consensus sequence binding protein in dendritic cells. J. Immunol. 2004, 172, 6820–6827. [Google Scholar] [CrossRef]
- Fathallah, I.; Parroche, P.; Gruffat, H.; Zannetti, C.; Johansson, H.; Yue, J.; Manet, E.; Tommasino, M.; Sylla, B.S.; Hasan, U.A. EBV latent membrane protein 1 is a negative regulator of TLR9. J. Immunol. 2010, 185, 6439–6447. [Google Scholar] [CrossRef]
- Hasan, U.A.; Bates, E.; Takeshita, F.; Biliato, A.; Accardi, R.; Bouvard, V.; Mansour, M.; Vincent, I.; Gissmann, L.; Iftner, T.; et al. TLR9 Expression and Function is abolished by the cervical cancer-associated human papillomavirus type 16. J. Immunol. 2007, 178, 3186–3197. [Google Scholar] [CrossRef]
- Vincent, I.E.; Zannetti, C.; Lucifora, J.; Norder, H.; Protzer, U.; Hainaut, P.; Zoulim, F.; Tommasino, M.; Trépo, C.; Hasan, U.; et al. Hepatitis B virus impairs TLR9 expression and function in plasmacytoid dendritic cells. PLoS One 2011, 6, e26315. [Google Scholar]
- Neumann, F.; Borchert, S.; Schmidt, C.; Reimer, R.; Hohenberg, H.; Fischer, N.; Grundhoff, A. Replication, gene expression and particle production by a consensus Merkel cell polyomavirus (MCPyV) genome. PLoS One 2011, 6, e29112. [Google Scholar]
- Khalili, K.; White, M.K.; Sawa, H.; Nagashima, K.; Safak, M. The agnoprotein of polyomaviruses: A multifunctional auxiliary protein. J. Cell Physiol. 2005, 204, 1–7. [Google Scholar] [CrossRef]
- Daniels, R.; Sadowicz, D.; Hebert, D.N. A Very late viral protein triggers the lytic release of SV40. PLoS Pathog. 2007, 3, 0929–0938. [Google Scholar]
- Clayson, E.T.; Brando, L.V.; Compans, R.W. Release of simian virus 40 virions from epithelial cells is polarized and occurs without cell lysis. J. Virol. 1989, 63, 2278–2288. [Google Scholar]
- Hahn, W.C.; Dessain, S.K.; Brooks, M.W.; King, J.E.; Elenbaas, B.; Sabatini, D.M.; DeCaprio, J.A.; Weinberg, R.A. Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol. Cell. Biol. 2002, 22, 2111–2123. [Google Scholar]
- Bikel, I.; Montano, X.; Agha, M.E.; Brown, M.; McCormack, M.; Boltax, J.; Livingston, D.M. SV40 small T-antigen enhances the transformation activity of limiting concentrations of SV40 Large T-antigen. Cell 1987, 48, 321–330. [Google Scholar]
- Houben, R.; Shuda, M.; Weinkam, R.; Schrama, D.; Feng, H.; Chang, Y.; Moore, P.S.; Becker, J.C. Merkel cell polyomavirus-infected merkel cell carcinoma cells require expression of viral T antigens. J. Virol. 2010, 84, 7064–7072. [Google Scholar] [CrossRef]
- Fischer, N.; Brandner, J.; Fuchs, F.; Moll, I.; Grundhoff, A. Detection of merkel cell polyomavirus (MCPyV) in Merkel cell carcinoma cell lines: Cell morphology and growth phenotype do not reflect presence of the virus. Int. J. Cancer 2010, 126, 2133–2142. [Google Scholar]
- Katano, H.; Ito, H.; Suzuki, Y.; Nakamura, T.; Sato, Y.; Tsuji, T.; Matsuo, K.; Nakagawa, H.; Sata, T. Detection of Merkel cell polyomavirus in Merkel cell carcinoma and Kaposi’s sarcoma. J. Med. Virol. 2009, 81, 1951–1958. [Google Scholar] [CrossRef]
- Shuda, M.; Feng, H.; Kwun, H.J.; Rosen, S.T.; Gjoerup, O.; Moore, P.S.; Chang, Y. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc. Natl. Acad. Sci. USA 2008, 105, 16272–16277. [Google Scholar]
- Kassem, A.; Schopflin, A.; Diaz, C.; Weyers, W.; Stickeler, E.; Werner, M.; Zur Hausen, A. Frequent detection of Merkel cell polyomavirus in human Merkel cell carcinomas and identification of a unique deletion in the VP1 gene. Cancer Res. 2008, 68, 5009–5013. [Google Scholar]
- DeCaprio, J.A.; Garcea, R.L. A cornucopia of human polyomaviruses. Nat. Rev. Microbiol. 2013, 11, 264–276. [Google Scholar] [CrossRef]
- Demetriou, S.K.; Ona-Vu, K.; Sullivan, E.M.; Dong, T.K.; Hsu, S.-W.; Oh, D.H. Defective DNA repair and cell cycle arrest in cells expressing Merkel cell polyomavirus T antigen. Int. J. Cancer 2012, 131, 1818–1827. [Google Scholar] [CrossRef]
- Busam, K.J.; Jungbluth, A.A.; Rekthman, N.; Coit, D.; Pulitzer, M.; Bini, J.; Arora, R.; Hanson, N.C.; Tassello, J.A.; Frosina, D.; et al. Merkel cell polyomavirus expression in Merkel cell carcinomas and its absence in combined tumors and pulmonary neuroendocrine carcinomas. Am. J. Surg. Pathol. 2009, 33, 1378–1385. [Google Scholar] [CrossRef]
- Shuda, M.; Arora, R.; Kwun, H.J.; Feng, H.; Sarid, R.; Fernandez-Figueras, M.-T.; Tolstov, Y.; Gjoerup, O.; Mansukhani, M.M.; Swerdlow, S.H.; et al. Human Merkel cell polyomavirus infection I. MCV T antigen expression in Merkel cell carcinoma, lymphoid tissues and lymphoid tumors. Int. J. Cancer 2009, 125, 1243–1249. [Google Scholar] [CrossRef]
- Brown, M.; McCormack, M.; Zinn, K.G.; Farrell, M.P.; Bikel, I.; Livingston, D.M. A recombinant murine retrovirus for simian virus-40 large T-cDNA transforms mouse fibroblasts to anchorage-independent growth. J. Virol. 1986, 60, 290–293. [Google Scholar]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell. Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef]
- Sullivan, C.S.; Cantalupo, P.; Pipas, J.M. The molecular chaperone activity of simian virus 40 large T antigen is required to Disrupt Rb-E2F family complexes by an ATP-dependent mechanism. Mol. Cell. Biol. 2000, 20, 6233–6243. [Google Scholar]
- Cheng, J.; Rozenblatt-Rosen, O.; Paulson, K.G.; Nghiem, P.; DeCaprio, J.A. Merkel cell polyomavirus large T antigen has growth-promoting and inhibitory activities. J. Virol. 2013, 87, 6118–6126. [Google Scholar] [CrossRef]
- Li, J.; Wang, X.; Diaz, J.; Tsang, S.H.; Buck, C.B.; You, J. Merkel cell polyomavirus large T antigen disrupts host genomic integrity and inhibits cellular proliferation. J. Virol. 2013, 87, 9173–9188. [Google Scholar] [CrossRef]
- Shi, Y.L.; Dodson, G.E.; Rundell, K.; Tibbetts, R.S. Ataxia-telangiectasia-mutated (ATM) Is a T-antigen kinase that controls SV40 viral replication in vivo. J. Biol. Chem. 2005, 280, 40195–40200. [Google Scholar]
- Arora, R.; Shuda, M.; Guastafierro, A.; Feng, H.; Toptan, T.; Tolstov, Y.; Normolle, D.; Vollmer, L.L.; Vogt, A.; Doemling, A.; et al. Survivin is a therapeutic target in Merkel cell carcinoma. Sci. Transl. Med. 2012, 4, 133ra56. [Google Scholar]
- Coumar, M.S.; Tsai, F.Y.; Kanwar, J.R.; Sarvagalla, S.; Cheung, C.H.A. Treat cancers by targeting survivin: Just a dream or future reality? Cancer Treat. Rev. 2013, 39, 802–811. [Google Scholar] [CrossRef]
- Angermeyer, S.; Hesbacher, S.; Becker, J.C.; Schrama, D.; Houben, R. Merkel cell polyomavirus-positive Merkel cell carcinoma cells do not require expression of the viral small T antigen. J. Invest. Dermatol. 2013, 133, 2059–2064. [Google Scholar] [CrossRef]
- Sontag, E.; Fedorov, S.; Kamibayashi, C.; Robbins, D.; Cobb, M.; Mumby, M. The interaction of SV40 small tumor-antigen with protein phosphatase-2a stimulates the MAP kinase pathway and induces cell-proliferation. Cell 1993, 75, 887–897. [Google Scholar] [CrossRef]
- Ruediger, R.; Hentz, M.; Fait, J.; Mumby, M.; Walter, G. Molecular Model of the a subunit of protein phosphatase 2A: Interaction with other subunits and tumor antigens. J. Virol. 1994, 68, 123–129. [Google Scholar]
- Yang, S.I.; Lickteig, R.L.; Estes, R.; Rundell, K.; Walter, G.; Mumby, M.C. Control of protein phosphatase-2A by simian virus-40 small-T antigen. Mol. Cell. Biol. 1991, 11, 1988–1995. [Google Scholar]
- Yu, J.; Boyapati, A.; Rundell, K. Critical role for SV40 small-T antigen in human cell transformation. Virology 2001, 290, 192–198. [Google Scholar] [CrossRef]
- Zhao, J.J.; Gjoerup, O.V.; Subramanian, R.R.; Cheng, Y.; Chen, W.; Roberts, T.M.; Hahn, W.C. Human Mammary epithelial cell transformation through the activation of phosphatidylinositol 3-kinase. Cancer Cell 2003, 3, 483–495. [Google Scholar] [CrossRef]
- Rodriguez-Viciana, P.; Collins, C.; Fried, M. Polyoma and SV40 proteins differentially regulate PP2A to activate distinct cellular signaling pathways involved in growth control. Proc. Natl. Acad. Sci. USA 2006, 103, 19290–19295. [Google Scholar] [CrossRef]
- Buchkovich, N.J.; Yu, Y.; Zampieri, C.A.; Alwine, J.C. The TORrid affairs of viruses: Effects of mammalian DNA viruses on the PI3K-Akt-mTOR signalling pathway. Nat. Rev. Microbiol. 2008, 6, 266–275. [Google Scholar] [CrossRef]
- Dowling, R.J.O.; Topisirovic, I.; Alain, T.; Bidinosti, M.; Fonseca, B.D.; Petroulakis, E.; Wang, X.; Larsson, O.; Selvaraj, A.; Liu, Y.; et al. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science 2010, 328, 1172–1176. [Google Scholar] [CrossRef]
- She, Q.-B.; Halilovic, E.; Ye, Q.; Zhen, W.; Shirasawa, S.; Sasazuki, T.; Solit, D.B.; Rosen, N. 4E-BP1 is a key effector of the oncogenic activation of the AKT and ERK signaling pathways that integrates their function in tumors. Cancer Cell 2010, 18, 39–51. [Google Scholar] [CrossRef]
- Richter, J.D.; Sonenberg, N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature 2005, 433, 477–480. [Google Scholar] [CrossRef]
- Pause, A.; Belsham, G.J.; Gingras, A.C.; Donzé, O.; Lin, T.A.; Lawrence, J.C., Jr.; Sonenberg, N. Insulin-dependent stimulation of protein-synthesis by phosphorylation of a regulator of 5'-Cap function. Nature 1994, 371, 762–767. [Google Scholar] [CrossRef]
- Lazariskaratzas, A.; Montine, K.S.; Sonenberg, N. Malignant transformation by a eukaryotic initiation-factor subunit that binds to messenger-RNA 5' cap. Nature 1990, 345, 544–547. [Google Scholar] [CrossRef]
- Avdulov, S.; Li, S.; Michalek, V.; Burrichter, D.; Peterson, M.; Perlman, D.M.; Manivel, J.C.; Sonenberg, N.; Yee, D.; Bitterman, P.B.; et al. Activation of translation complex eIF4F is essential for the genesis and maintenance of the malignant phenotype in human mammary epithelial cells. Cancer Cell 2004, 5, 553–563. [Google Scholar] [CrossRef]
- Beretta, L.; Gingras, A.C.; Svitkin, Y.V.; Hall, M.N.; Sonenberg, N. Rapamycin blocks the phosphorylation of 4E-BP1 and inhibits cap-dependent initiation of translation. EMBO J. 1996, 15, 658–664. [Google Scholar]
- Yu, J.; Boyapati, A.; Rundell, K. Critical role for SV40 small-T antigen in human cell transformation. Virology 2001, 290, 192–198. [Google Scholar] [CrossRef]
- Jouary, T.; Leyral, C.; Dreno, B.; Doussau, A.; Sassolas, B.; Beylot-Barry, M.; Renaud-Vilmer, C.; Guillot, B.; Bernard, P.; Lok, C.; et al. Adjuvant prophylactic regional radiotherapy versus observation in stage I Merkel cell carcinoma: A multicentric prospective randomized study. Ann. Oncol. 2012, 23, 1074–1080. [Google Scholar] [CrossRef]
- Pape, E.; Rezvoy, N.; Penel, N.; Salleron, J.; Martinot, V.; Guerreschi, P.; Dziwniel, V.; Darras, S.; Mirabel, X.; Mortier, L. Radiotherapy alone for Merkel cell carcinoma: A comparative and retrospective study of 25 patients. J. Am. Acad. Dermatol. 2011, 65, 983–990. [Google Scholar] [CrossRef]
- Tai, P.T.; Yu, E.; Winquist, E.; Hammond, A.; Stitt, L.; Tonita, J.; Gilchrist, J. Chemotherapy in neuroendocrine/Merkel cell carcinoma of the skin: Case series and review of 204 cases. J. Clin. Oncol. 2000, 18, 2493–2499. [Google Scholar]
- Willmes, C.; Adam, C.; Alb, M.; Völkert, L.; Houben, R.; Becker, J.C.; Schrama, D. Type I and II IFNs inhibit Merkel cell carcinoma via modulation of the Merkel Cell polyomavirus T antigens. Cancer Res. 2012, 72, 2120–2128. [Google Scholar] [CrossRef]
- Biver-Dalle, C.; Nguyen, T.; Touzé, A.; Saccomani, C.; Penz, S.; Cunat-Peultier, S.; Riou-Gotta, M.-O.; Humbert, P.; Coursaget, P.; Aubin, F. Use of interferon-alpha in two patients with Merkel cell carcinoma positive for Merkel cell polyomavirus. Acta Oncol. 2010, 50, 479–480. [Google Scholar]
- Dresang, L.R.; Guastafierro, A.; Arora, R.; Normolle, D.; Chang, Y.; Moore, P.S. Response of Merkel cell polyomavirus-positive Merkel cell carcinoma xenografts to a survivin inhibitor. PLoS One 2013, 11, e80543. [Google Scholar]
- Davids, M.; Charlton, A.; Ng, S.S.; Chong, M.L.; Laubscher, K.; Dar, M.; Hodge, J.; Soong, R.; Goh, B.C. Response to a novel multitargeted tyrosine kinase inhibitor pazopanib in metastatic Merkel cell carcinoma. J. Clin. Oncol. 2009, 27, e97–e100. [Google Scholar] [CrossRef]
- Sugamata, A.; Goya, K.; Yoshizawa, N. A Case of complete spontaneous regression of extremely advanced Merkel cell carcinoma. J. Surg. Case Rep. 2011, 10, 7. [Google Scholar]
- Iyer, J.G.; Afanasiev, O.K.; McClurkan, C.; Paulson, K.; Nagase, K.; Jing, L.; Marshak, J.O.; Dong, L.; Carter, J.; Lai, I.; et al. Merkel cell polyomavirus-specific CD8+ and CD4+ T-cell responses identified in Merkel cell carcinomas and blood. Clin. Cancer Res. 2011, 17, 6671–6680. [Google Scholar] [CrossRef]
- Lyngaa, R.; Pedersen, N.W.; Schrama, D.; Thrue, C.A.; Ibrani, D.; Met, Ö.; Thor Straten, P.; Nghiem, P.; Becker, J.C.; Hadrup, S.R. T-cell responses to oncogenic Merkel cell polyomavirus proteins distinguish Merkel cell carcinoma patients from healthy donors. Clin. Cancer Res. 2014, 20, 1768–1778. [Google Scholar] [CrossRef]
- Gomez, B.; He, L.; Tsai, Y.C.; Wu, T.-C.; Viscidi, R.P.; Hung, C.-F. Creation of a Merkel cell polyomavirus small T antigen-expressing murine tumor model and a DNA vaccine targeting small T antigen. Cell Biosci. 2013, 3, 29. [Google Scholar] [CrossRef]
- Zeng, Q.; Gomez, B.P.; Viscidi, R.P.; Peng, S.; He, L.; Ma, B.; Wu, T.-C.; Hung, C.-F. Development of a DNA vaccine targeting Merkel cell polyomavirus. Vaccine 2012, 30, 1322–1329. [Google Scholar]
- Afanasiev, O.K.; Yelistratova, L.; Miller, N.; Nagase, K.; Paulson, K.; Iyer, J.G.; Ibrani, D.; Koelle, D.M.; Nghiem, P. Merkel polyomavirus-specific T cells fluctuate with Merkel cell carcinoma burden and express therapeutically targetable PD-1 and Tim-3 exhaustion markers. Clin. Cancer Res. 2013, 19, 5351–5360. [Google Scholar] [CrossRef]
- Pantulu, N.D.; Pallasch, C.P.; Kurz, A.K.; Kassem, A.; Frenzel, L.; Sodenkamp, S.; Kvasnicka, H.M.; Wendtner, C.M.; Zur Hausen, A. Detection of a novel truncating Merkel cell polyomavirus large T antigen deletion in chronic lymphocytic leukemia cells. Blood 2010, 116, 5280–5284. [Google Scholar]
- Koljonen, V.; Kukko, H.; Pukkala, E.; Sankila, R.; Bohling, T.; Tukiainen, E.; Sihto, H.; Joensuu, H. Chronic lymphocytic leukaemia patients have a high risk of Merkel-Cell polyomavirus DNA-positive Merkel cell carcinoma. Br. J. Cancer 2009, 101, 1444–1447. [Google Scholar] [CrossRef]
- Teman, C.J.; Tripp, S.R.; Perkins, S.L.; Duncavage, E.J. Merkel cell polyomavirus (MCPyV) in chronic lymphocytic leukemia/small lymphocytic lymphoma. Leuk Res. 2011, 35, 689–692. [Google Scholar] [CrossRef]
- Andres, C.; Belloni, B.; Puchta, U.; Sander, C.A.; Flaig, M.J. Prevalence of MCPyV in Merkel cell carcinoma and non-MCC tumors. J. Cutan. Pathol. 2010, 37, 28–34. [Google Scholar] [CrossRef]
- Reisinger, D.M.; Shiffer, J.D.; Cognetta, A.B., Jr.; Chang, Y.; Moore, P.S. Lack of evidence for basal or squamous cell carcinoma infection with Merkel cell polyomavirus in immunocompetent patients with Merkel cell carcinoma. J. Am. Acad. Dermatol. 2010, 63, 400–403. [Google Scholar] [CrossRef]
- Rollison, D.E.; Giuliano, A.R.; Messina, J.L.; Fenske, N.A.; Cherpelis, B.S.; Sondak, V.K.; Roetzheim, R.G.; Iannacone, M.R.; Michael, K.M.; Gheit, T.; et al. Case-control study of Merkel cell polyomavirus infection and cutaneous squamous cell carcinoma. Cancer Epidemiol. Biomarkers Prev. 2012, 21, 74–81. [Google Scholar] [CrossRef]
- Dworkin, A.M.; Tseng, S.Y.; Allain, D.C.; Iwenofu, O.H.; Peters, S.B.; Toland, A.E. Merkel cell polyomavirus in cutaneous squamous cell carcinoma of immunocompetent individuals. J. Invest. Dermatol. 2009, 129, 2868–2874. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Stakaitytė, G.; Wood, J.J.; Knight, L.M.; Abdul-Sada, H.; Adzahar, N.S.; Nwogu, N.; Macdonald, A.; Whitehouse, A. Merkel Cell Polyomavirus: Molecular Insights into the Most Recently Discovered Human Tumour Virus. Cancers 2014, 6, 1267-1297. https://doi.org/10.3390/cancers6031267
Stakaitytė G, Wood JJ, Knight LM, Abdul-Sada H, Adzahar NS, Nwogu N, Macdonald A, Whitehouse A. Merkel Cell Polyomavirus: Molecular Insights into the Most Recently Discovered Human Tumour Virus. Cancers. 2014; 6(3):1267-1297. https://doi.org/10.3390/cancers6031267
Chicago/Turabian StyleStakaitytė, Gabrielė, Jennifer J. Wood, Laura M. Knight, Hussein Abdul-Sada, Noor Suhana Adzahar, Nnenna Nwogu, Andrew Macdonald, and Adrian Whitehouse. 2014. "Merkel Cell Polyomavirus: Molecular Insights into the Most Recently Discovered Human Tumour Virus" Cancers 6, no. 3: 1267-1297. https://doi.org/10.3390/cancers6031267
APA StyleStakaitytė, G., Wood, J. J., Knight, L. M., Abdul-Sada, H., Adzahar, N. S., Nwogu, N., Macdonald, A., & Whitehouse, A. (2014). Merkel Cell Polyomavirus: Molecular Insights into the Most Recently Discovered Human Tumour Virus. Cancers, 6(3), 1267-1297. https://doi.org/10.3390/cancers6031267