Stat3 and Gap Junctions in Normal and Lung Cancer Cells

Abstract
:1. Introduction
2. GJIC Examination
3. Stat3 and GJIC in Cultured, Normal and Src-Transformed Cells
3.1. Cell Density Upregulates GJIC and Connexin-43 Protein Levels
3.2. Stat3 Does Not Transduce Src Signals to GJIC Suppression
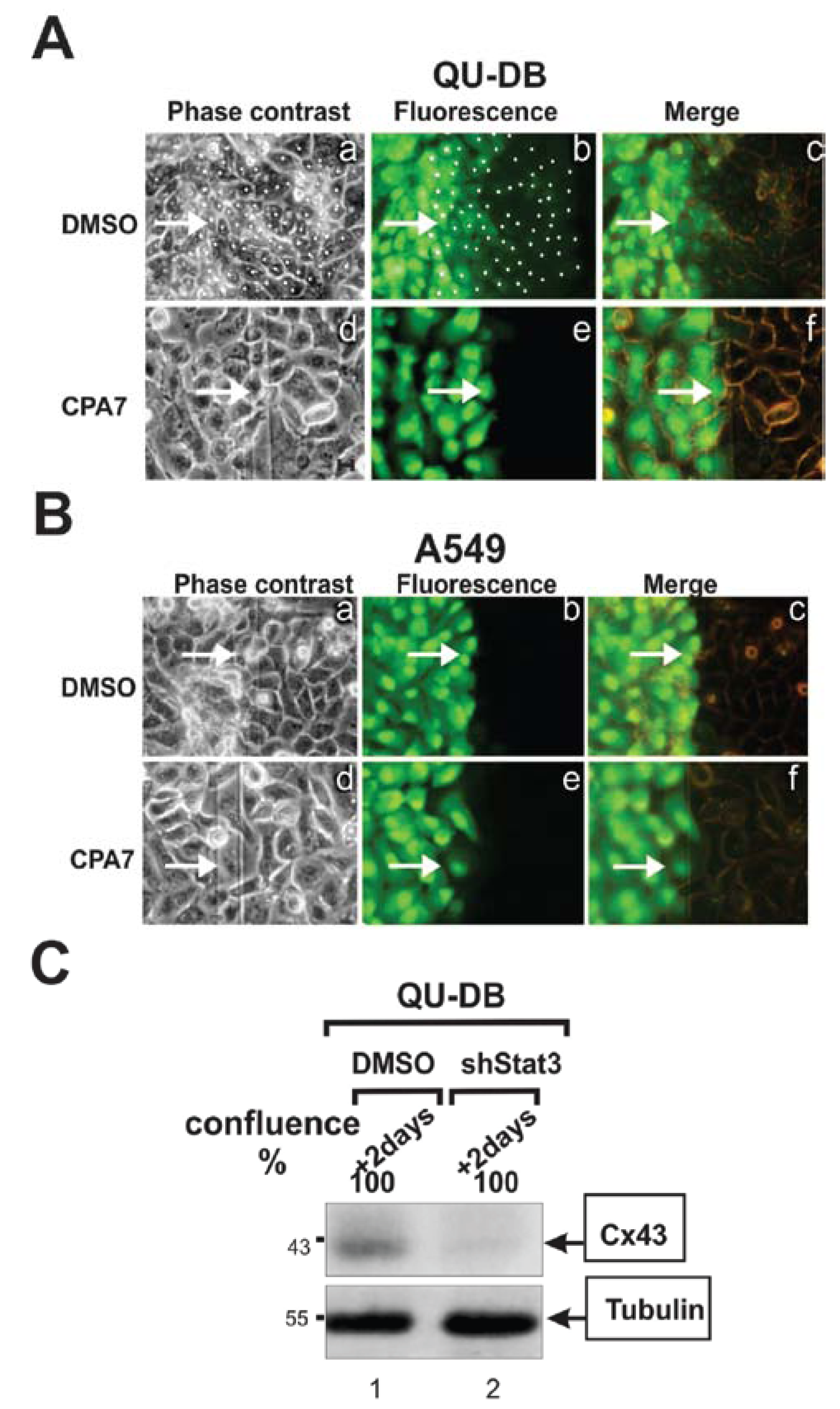
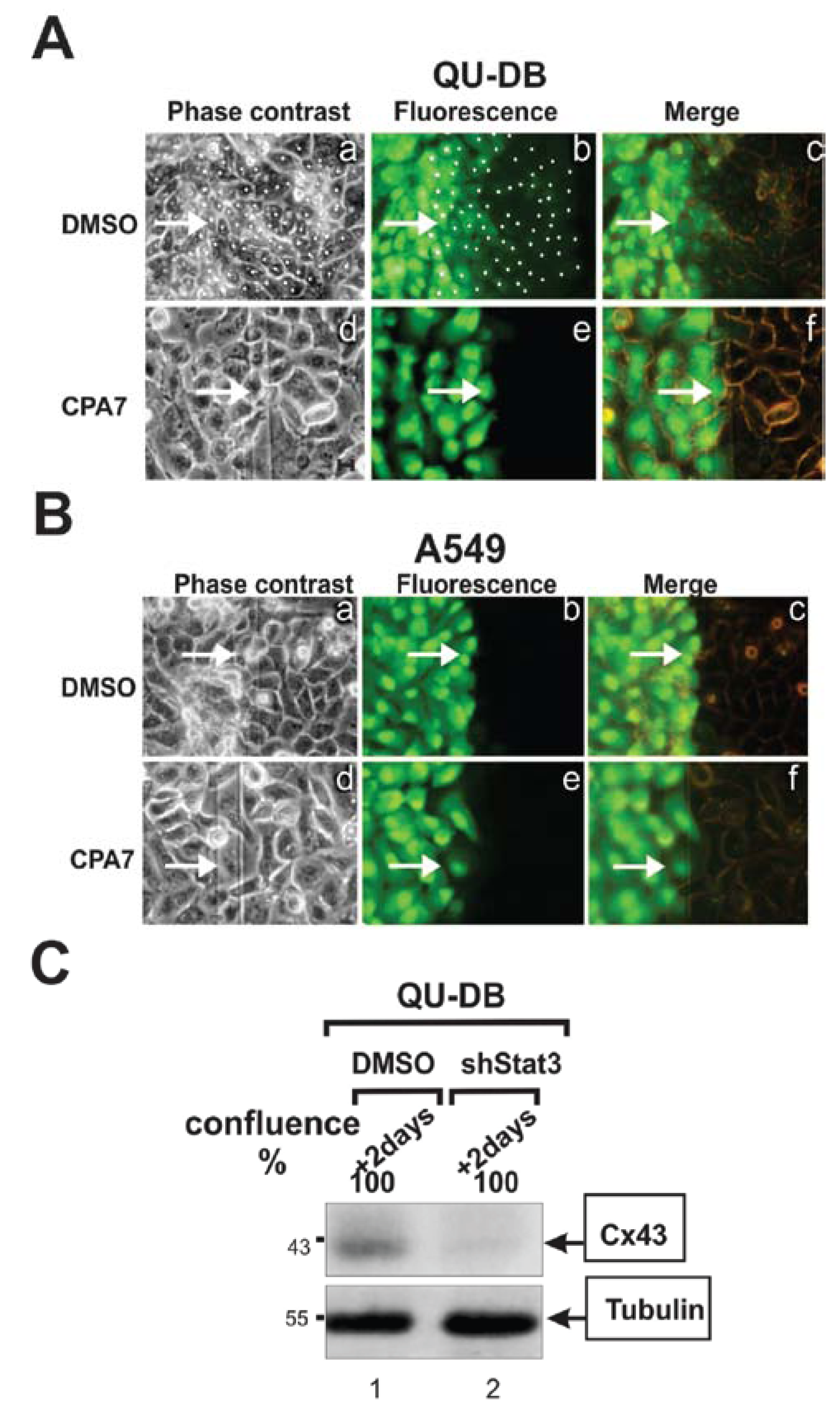
3.3. Stat3 Is a Positive Regulator of GJIC and Cx43 Levels
4. Src as a Stat3 Activator in Non-Small Cell Lung Cancer
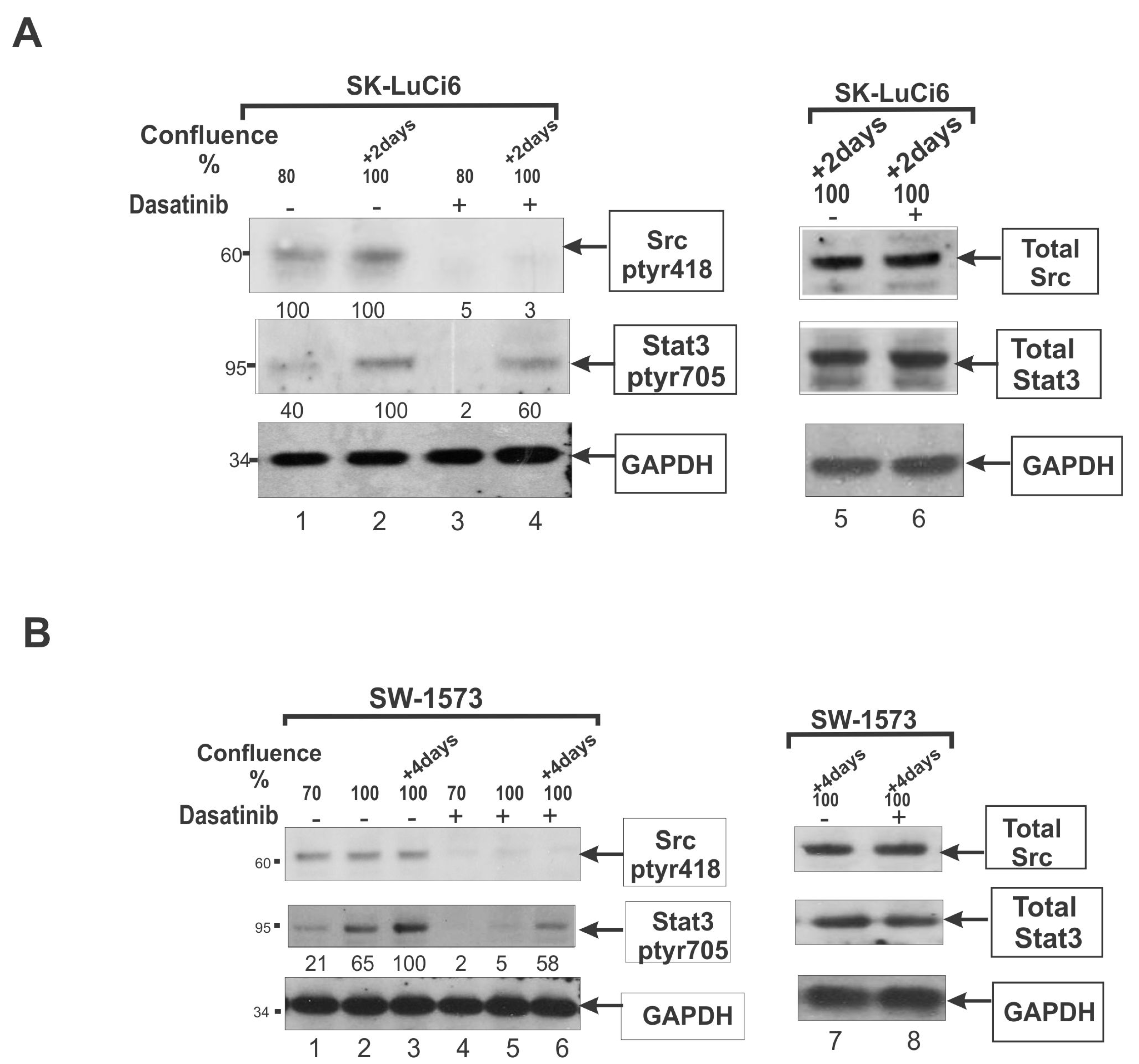
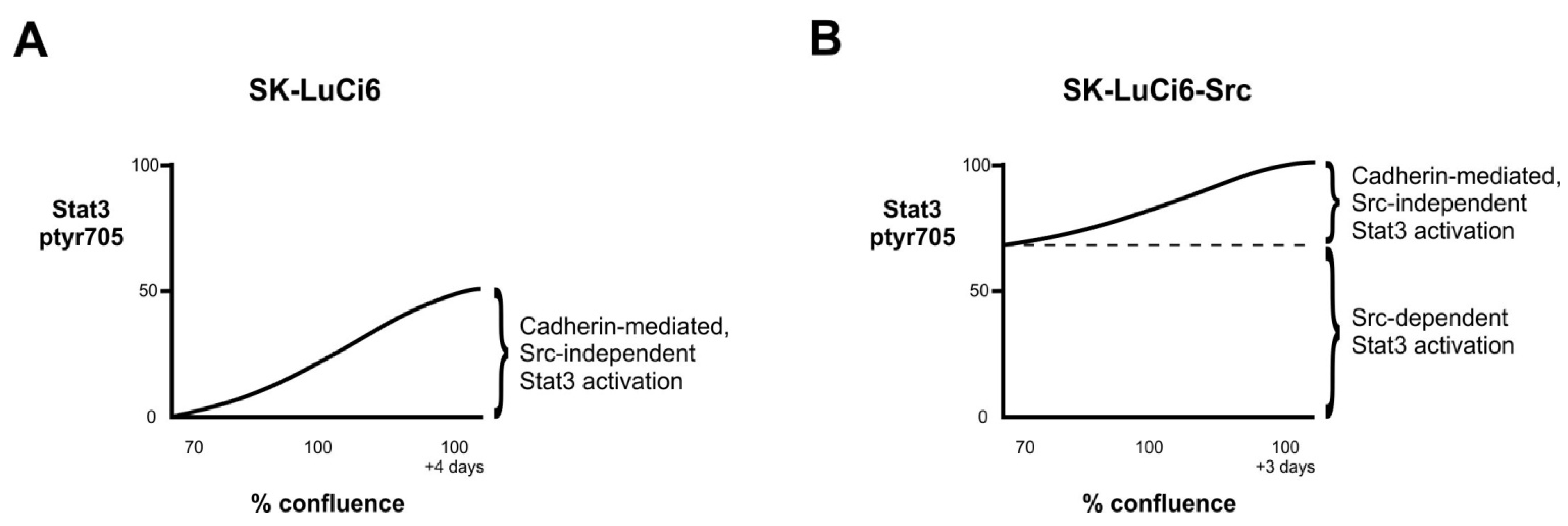
4.1. Cell Density Increases Stat3, in a Src-Independent Manner
4.2. Src Is a Major Stat3 Activator in Certain NSCLC Lines

{kind=link}
{kind=link}
{kind=link}
| Cell line | Srcα (%) | Stat3 α (%) | GJIC β | |
|---|---|---|---|---|
| 50% confluent | 100 + 3 days | |||
| Lines with high Src-418 levels | ||||
| SK-LuCi6-Src | 100 ± 12 | 100 ± 10 | 420 ± 33 | 0.2 ± 0.1 |
| A549 | 95 ± 11 | 93 ± 12 | 320 ± 32 | 0.3 ± 0.1 |
| SK-Lu1 | 85 ± 5 | 90 ± 11 | 311 ± 23 | 1 ± 0.2 |
| Calu-1 | 96 ± 9 | 100 ± 10 | 290 ± 12 | 0.1 ± 0.1 |
| SW-900 | 100 ± 13 | 100 ± 12 | 405 ± 21 | 0.1 ± 0.1 |
| Calu-6 | 95 ± 10 | 90 ± 12 | 300 ± 18 | 0.1 ± 0.1 |
| SW-1573 | 70 ± 9 | 70 ± 8 | 180 ± 12 | 0.2 ± 0.1 |
| WT-E | 60 ± 9 | 30 ± 4 | 80 ± 11 | 0.2 ± 0.1 |
| BEN | 42 ± 5 | 60 ± 8 | 182 ± 25 | 0.2 ± 0.1 |
| H1299 | 30 ± 4 | 30 ± 5 | 60 ± 18 | 0.2 ± 0.1 |
| FRE | 25 ± 3 | 70 ± 4 | 180 ± 25 | 0.5 ± 0.1 |
| SK-MES | 25 ± 2 | 90 ± 6 | 248 ± 22 | 1 ± 0.3 |
| Lines with low Src-418 levels | ||||
| LCT | 2 ± 0.2 | 90 ± 8 | 222 ± 31 | 0.1 ± 0.1 |
| SHP-77 | 5 ± 1 | 2 ± 1 | 12 ± 2 | 0.1 ± 0.1 |
| BHE | 1 ± 0.1 | 1 ± 0.2 | 8 ± 2 | 0.1 ± 0.1 |
| Lines with low Src-418 and high GJIC | ||||
| QUDB | 7 ± 1 | 9 ± 2 | 20 ± 4 | 6.3 ± 1 |
| SK-LuCi6 | 10 ± 1 | 8 ± 2 | 21 ± 4 | 6.5 ± 1 |
| Rat F111 | 0.2 ± 0.1 | 0.2 ± 0.1 | 28 ± 3 | 5.1 ± 1 |
| T51B | 5 ± 1 | 5 ± 3 | 18 ± 2 | 6.0 ± 1 |
| E10 | 6 ± 1 | 6 ± 4 | 26 ± 9 | 6.0 ± 1 |
4.3. Effect of Src and Stat3 upon GJIC in NSCLC Lines
5. Discussion
5.1. Stat3 Does Not Transmit Src Signals to Gap Junction Closure

5.2. Stat3 Plays a Positive Role in Gap Junctional Communication
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sohl, G.; Willecke, K. An update on connexin genes and their nomenclature in mouse and man. Cell Commun. Adhes. 2003, 10, 173–180. [Google Scholar]
- Laird, D.W. The gap junction proteome and its relationship to disease. Trends Cell. Biol. 2010, 20, 92–101. [Google Scholar] [CrossRef]
- Laird, D.W. Life cycle of connexins in health and disease. Biochem. J. 2006, 394, 527–543. [Google Scholar] [CrossRef]
- Sohl, G.; Willecke, K. Gap junctions and the connexin protein family. Cardiovasc. Res. 2004, 62, 228–232. [Google Scholar] [CrossRef]
- Beardslee, M.A.; Laing, J.G.; Beyer, E.C.; Saffitz, J.E. Rapid turnover of connexin43 in the adult rat heart. Circ. Res. 1998, 83, 629–635. [Google Scholar] [CrossRef]
- Vanslyke, J.K.; Naus, C.C.; Musil, L.S. Conformational maturation and post-ER multisubunit assembly of gap junction proteins. Mol. Biol. Cell 2009, 20, 2451–2463. [Google Scholar] [CrossRef]
- Gaietta, G.; Deerinck, T.J.; Adams, S.R.; Bouwer, J.; Tour, O.; Laird, D.W.; Sosinsky, G.E.; Tsien, R.Y.; Ellisman, M.H. Multicolor and electron microscopic imaging of connexin trafficking. Science 2002, 296, 503–507. [Google Scholar] [CrossRef]
- Lin, R.; Warn-Cramer, B.J.; Kurata, W.E.; Lau, A.F. v-Src Phosphorylation of connexin 43 on Tyr247 and Tyr265 disrupts gap junctional communication. J. Cell Biol. 2001, 154, 815–827. [Google Scholar] [CrossRef]
- Raptis, L.; Brownell, H.L.; Firth, K.L.; MacKenzie, L.W. novel technique for the study of intercellular, junctional communication; electroporation of adherent cells on a partly conductive slide. DNA Cell Biol. 1994, 13, 963–975. [Google Scholar] [CrossRef]
- Schaffhausen, B.S.; Roberts, T.M. Lessons from polyoma middle T antigen on signaling and transformation: A DNA tumor virus contribution to the war on cancer. Virology 2009, 384, 304–316. [Google Scholar] [CrossRef]
- Grammatikakis, N.; Vultur, A.; Ramana, C.V.; Siganou, A.; Schweinfest, C.W.; Raptis, L. The role of Hsp90N, a new member of the Hsp90 family, in signal transduction and neoplastic transformation. J.Biol. Chem. 2002, 277, 8312–8320. [Google Scholar]
- Brownell, H.L.; Narsimhan, R.; Corbley, M.J.; Mann, V.M.; Whitfield, J.F.; Raptis, L. Ras is involved in gap junction closure in mouse fibroblasts or preadipocytes but not in differentiated adipocytes. DNA Cell Biol. 1996, 15, 443–451. [Google Scholar] [CrossRef]
- Atkinson, M.M.; Sheridan, J.D. Altered junctional permeability between cells transformed by v-ras, v-mos, or v-src. Am. J. Physiol. 1988, 255, C674–C683. [Google Scholar]
- Solan, J.L.; Lampe, P.D. Connexin43 phosphorylation: Structural changes and biological effects. Biochem. J. 2009, 419, 261–272. [Google Scholar] [CrossRef]
- Geletu, M.; Trotman-Grant, A.; Raptis, L. ind the gap; regulation of gap junctional, intercellular communication by the SRC oncogene product and its effectors. Anticancer Res. 2012, 32, 4245–4250. [Google Scholar]
- Pahujaa, M.; Anikin, M.; Goldberg, G.S. Phosphorylation of connexin43 induced by Src: Regulation of gap junctional communication between transformed cells. Exp. Cell Res. 2007, 313, 4083–4090. [Google Scholar] [CrossRef]
- Ito, S.; Ito, Y.; Senga, T.; Hattori, S.; Matsuo, S.; Hamaguchi, M. v-Src requires Ras signaling for the suppression of gap junctional intercellular communication. Oncogene 2006, 25, 2420–2424. [Google Scholar] [CrossRef]
- Zhang, Y.; Turkson, J.; Carter-Su, C.; Smithgall, T.; Levitzki, A.; Kraker, A.; Krolewski, J.J.; Medveczky, P.; Jove, R. Activation of Stat3 in v-Src transformed fibroblasts requires cooperation of Jak1 kinase activity. J. Biol. Chem. 2000, 275, 24935–24944. [Google Scholar] [CrossRef]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Hung, W.; Elliott, B. Co-operative effect of c-Src tyrosine kinase and Stat3 in activation of hepatocyte growth factor expression in mammary carcinoma cells. J. Biol. Chem. 2001, 276, 12395–12403. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Arulanandam, R.; Vultur, A.; Cao, J.; Carefoot, E.; Truesdell, P.; Elliott, B.; Larue, L.; Feracci, H.; Raptis, L. Cadherin-cadherin engagement promotes survival via Rac/Cdc42 and Stat3. Mol. Cancer Res. 2009, 17, 1310–1327. [Google Scholar]
- Vultur, A.; Cao, J.; Arulanandam, R.; Turkson, J.; Jove, R.; Greer, P.; Craig, A.; Elliott, B.E.; Raptis, L. Cell to cell adhesion modulates Stat3 activity in normal and breast carcinoma cells. Oncogene 2004, 23, 2600–2616. [Google Scholar] [CrossRef]
- Vultur, A.; Arulanandam, R.; Turkson, J.; Niu, G.; Jove, R.; Raptis, L. Stat3 is required for full neoplastic transformation by the Simian Virus 40 large tumor antigen. Mol. Biol. Cell 2005, 16, 3832–3846. [Google Scholar] [CrossRef]
- Raptis, L.; Arulanandam, R.; Vultur, A.; Geletu, M.; Chevalier, S.; Feracci, H. Beyond structure, to survival: Stat3 Activation by cadherin engagement. Biochem. Cell Biol. 2009, 87, 835–843. [Google Scholar] [CrossRef]
- Geletu, M.; Guy, S.; Arulanandam, R.; Feracci, H.; Raptis, L. Engaged for survival: From cadherin ligation to Stat3 activation. JAK-STAT 2013, 2, e27363. [Google Scholar] [CrossRef]
- El-Fouly, M.H.; Trosko, J.E.; Chang, C.C. Scrape-loading and dye transfer: A rapid and simple technique to study gap junctional intercellular communication. Exp. Cell Res. 1987, 168, 442–430. [Google Scholar]
- Goldberg, G.S.; Bechberger, J.F.; Naus, C.C. A pre-loading method of evaluating gap junctional communication by fluorescent dye transfer. Biotechniques 1995, 18, 490–497. [Google Scholar]
- Wade, M.H.; Trosko, J.E.; Schindler, M. A fluorescence photobleaching assay of gap junction-mediated communication between human cells. Science 1986, 232, 525–528. [Google Scholar]
- Raptis, L.; Vultur, A.; Brownell, H.L.; Tomai, E.; Anagnostopoulou, A.; Arulanandam, R.; Cao, J.; Firth, K.L. Electroporation Protocols; Li, S., Ed.; The Humana Press Inc.: Totowa, NJ, USA, 2008; pp. 167–183. [Google Scholar]
- Anagnostopoulou, A.; Cao, J.; Vultur, A.; Firth, K.L.; Raptis, L. Examination of gap junctional, intercellular communication by in situ electroporation on two co-planar indium-tin oxide electrodes. Mol. Oncol. 2007, 1, 226–231. [Google Scholar] [CrossRef]
- Tomai, E.; Brownell, H.L.; Tufescu, T.; Reid, K.; Raptis, S.; Campling, B.G.; Raptis, L. A functional assay for intercellular, junctional communication in cultured human lung carcinoma cells. Lab. Investig. 1998, 78, 639–640. [Google Scholar]
- Geletu, M.; Arulanandam, R.; Greer, S.; Trotman-Grant, A.; Tomai, E.; Raptis, L. Stat3 is a positive regulator of gap junctional intercellular communication in cultured, human lung carcinoma cells. BMC Cancer 2012, 12. [Google Scholar] [CrossRef]
- Brownell, H.L.; Lydon, N.; Schaefer, E.; Roberts, T.M.; Raptis, L. Inhibition of Epidermal Growth Factor-mediated ERK1/2 activation by in situ electroporation of nonpermeant [(alkylamino)methyl]acrylophenone derivatives. DNA Cell Biol. 1998, 17, 265–274. [Google Scholar] [CrossRef]
- Tomai, E.; Brownell, H.L.; Tufescu, T.; Reid, K.; Raptis, L. Gap junctional communication in lung carcinoma cells. Lung Cancer 1999, 23, 223–231. [Google Scholar] [CrossRef]
- Geletu, M.; Chaize, C.; Arulanandam, R.; Vultur, A.; Kowolik, C.; Anagnostopoulou, A.; Jove, R.; Raptis, L. Stat3 activity is required for gap junctional permeability in normal epithelial cells and fibroblasts. DNA Cell Biol. 2009, 28, 319–327. [Google Scholar] [CrossRef]
- Vultur, A.; Tomai, E.; Peebles, K.; Malkinson, A.M.; Grammatikakis, N.; Forkert, P.G.; Raptis, L. Gap junctional, intercellular communication in cells from urethane-induced tumors in A/J mice. DNA Cell Biol. 2003, 22, 33–40. [Google Scholar] [CrossRef]
- Geletu, M.; Guy, S.; Raptis, L. Effects of SRC and STAT3 upon gap junctional, intercellular communication in lung cancer lines. Anticancer Res. 2013, 33, 4401–4410. [Google Scholar]
- Wei, C.J.; Francis, R.; Xu, X.; Lo, C.W. Connexin43 associated with an N-cadherin-containing multiprotein complex is required for gap junction formation in NIH3T3 cells. J. Biol. Chem. 2005, 280, 19925–19936. [Google Scholar] [CrossRef]
- Turkson, J.; Zhang, S.; Palmer, J.; Kay, H.; Stanko, J.; Mora, L.B.; Sebti, S.; Yu, H.; Jove, R. Inhibition of constitutive signal transducer and activator of transcription 3 activation by novel platinum complexes with potent antitumor activity. Mol. Cancer Ther. 2004, 3, 1533–1542. [Google Scholar]
- Littlefield, S.L.; Baird, M.C.; Anagnostopoulou, A.; Raptis, L. Synthesis, characterization and Stat3 inhibitory properties of the prototypical platinum(IV) anticancer drug, [PtCl3(NO2)(NH3)2] (CPA-7). Inorg. Chem. 2008, 47, 2798–2804. [Google Scholar] [CrossRef]
- Siddiquee, K.; Zhang, S.; Guida, W.C.; Blaskovich, M.A.; Greedy, B.; Lawrence, H.R.; Yip, M.L.; Jove, R.; McLaughlin, M.M.; Lawrence, N.J.; et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl. Acad. Sci. USA 2007, 104, 7391–7396. [Google Scholar] [CrossRef]
- Trotman-Grant, A.; Geletu, M.; Raptis, L. Constitutively active, Signal transducer and activator of transcription-3; an oncogene that increases gap junctional communication. Queen’s University: Kingston, ON, Canada, Unpublished work. 2014. [Google Scholar]
- Masaki, T.; Igarashi, K.; Tokuda, M.; Yukimasa, S.; Han, F.; Jin, Y.J.; Li, J.Q.; Yoneyama, H.; Uchida, N.; Fujita, J.; et al. pp60c-src Activation in lung adenocarcinoma. Eur. J. Cancer 2003, 39, 1447–1455. [Google Scholar] [CrossRef]
- Zhang, J.; Kalyankrishna, S.; Wislez, M.; Thilaganathan, N.; Saigal, B.; Wei, W.; Ma, L.; Wistuba, I.I.; Johnson, F.M.; Kurie, J.M. SRC-family kinases are activated in non-small cell lung cancer and promote the survival of epidermal growth factor receptor-dependent cell lines. Am. J. Pathol. 2007, 170, 366–376. [Google Scholar] [CrossRef]
- Song, L.; Turkson, J.; Karras, J.G.; Jove, R.; Haura, E.B. Activation of Stat3 by receptor tyrosine kinases and cytokines regulates survival in human non-small cell carcinoma cells. Oncogene 2003, 22, 4150–4165. [Google Scholar] [CrossRef]
- Byers, L.A.; Sen, B.; Saigal, B.; Diao, L.; Wang, J.; Nanjundan, M.; Cascone, T.; Mills, G.B.; Heymach, J.V.; Johnson, F.M. Reciprocal regulation of c-Src and STAT3 in non-small cell lung cancer. Clin. Cancer Res. 2009, 15, 6852–6861. [Google Scholar] [CrossRef]
- Raptis, L.; Lamfrom, H.; Benjamin, T.L. Regulation of cellular phenotype and expression of polyomavirus middle T antigen in rat fibroblasts. Mol. Cell. Biol. 1985, 5, 2476–2486. [Google Scholar]
- Turkson, J.; Zhang, S.; Mora, L.B.; Burns, A.; Sebti, S.; Jove, R. A novel platinum compound that inhibits constitutive Stat3 signaling and induces cell cycle arrest and apoptosis of malignant cells. J. Biol. Chem. 2005, 280, 32979–32988. [Google Scholar] [CrossRef]
- Azarnia, R.; Loewenstein, W.R. Polyomavirus middle T antigen downregulates junctional cell-to-cell communication. Mol. Cell. Biol. 1987, 7, 946–950. [Google Scholar]
- Brownell, H.L.; Whitfield, J.F.; Raptis, L. Elimination of intercellular junctional communication requires lower Rasleu61 levels than stimulation of anchorage-independent proliferation. Cancer Detect. Prev. 1997, 21, 289–294. [Google Scholar]
- Wadhawan, A.; Smith, C.; Nicholson, R.I.; Barrett-Lee, P.; Hiscox, S. Src-mediated regulation of homotypic cell adhesion: Implications for cancer progression and opportunities for therapeutic intervention. Cancer Treat. Rev. 2011, 37, 234–241. [Google Scholar] [CrossRef]
- Guy, S.; Geletu, M.; Arulanandam, R.; Raptis, L. Cadherin-11 function is required for full neoplastic transfomation by vSrc. Queen’s University: Kingston, ON, Canada, Unpublished work. 2014. [Google Scholar]
- Aleshin, A.; Finn, R.S. SRC: A century of science brought to the clinic. Neoplasia 2010, 12, 599–607. [Google Scholar]
- Brownell, H.L.; Whitfield, J.F.; Raptis, L. Cellular Ras partly mediates gap junction closure by the polyoma virus middle Tumor antigen. Cancer Lett. 1996, 103, 99–106. [Google Scholar] [CrossRef]
- Shen, Y.; Khusial, P.R.; Li, X.; Ichikawa, H.; Moreno, A.P.; Goldberg, G.S. SRC utilizes Cas to block gap junctional communication mediated by connexin43. J. Biol. Chem. 2007, 282, 18914–18921. [Google Scholar]
- Anagnostopoulou, A.; Vultur, A.; Arulanandam, R.; Cao, J.; Turkson, J.; Jove, R.; Kim, J.S.; Glenn, M.; Hamilton, A.D.; Raptis, L. Differential effects of Stat3 inhibition in sparse vs. confluent normal and breast cancer cells. Cancer Lett. 2006, 242, 120–132. [Google Scholar] [CrossRef]
- Gao, X.; Wang, H.; Yang, J.J.; Liu, X.; Liu, Z.R. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol. Cell 2012, 45, 598–609. [Google Scholar] [CrossRef]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef]
- Demaria, M.; Poli, V. From the nucleus to the mitochondria and back: The odyssey of a multitask STAT3. Cell Cycle 2011, 10, 3221–3222. [Google Scholar] [CrossRef]
- Theiss, C.; Mazur, A.; Meller, K.; Mannherz, H.G. Changes in gap junction organization and decreased coupling during induced apoptosis in lens epithelial and NIH-3T3 cells. Exp. Cell Res. 2007, 313, 38–52. [Google Scholar] [CrossRef]
- Anagnostopoulou, A.; Vultur, A.; Arulanandam, R.; Cao, J.; Turkson, J.; Jove, R.; Kim, J.S.; Glenn, M.; Hamilton, A.D.; Raptis, L. Role of Stat3 in normal and SV40 transformed cells. Res. Trends 2006, 2, 93–103. [Google Scholar]
- Geletu, M.; Guy, S.; Greer, S.; Raptis, L. Differential effects of polyoma virus middle tumor antigen mutants upon gap junctional, intercellular communication. Queen’s University: Kingston, ON, Canada, Unpublished work. 2014. [Google Scholar]
- Ozog, M.A.; Bernier, S.M.; Bates, D.C.; Chatterjee, B.; Lo, C.W.; Naus, C.C. The complex of ciliary neurotrophic factor-ciliary neurotrophic factor receptor alpha up-regulates connexin43 and intercellular coupling in astrocytes via the Janus tyrosine kinase/signal transducer and activator of transcription pathway. Mol. Biol. Cell 2004, 15, 4761–4774. [Google Scholar] [CrossRef]
- Rajasingh, J.; Bord, E.; Hamada, H.; Lambers, E.; Qin, G.; Losordo, D.W.; Kishore, R. STAT3-dependent mouse embryonic stem cell differentiation into cardiomyocytes: Analysis of molecular signaling and therapeutic efficacy of cardiomyocyte precommitted mES transplantation in a mouse model of myocardial infarction. Circ. Res. 2007, 101, 910–918. [Google Scholar] [CrossRef]
- Andersson, H.; Brittebo, E. Proangiogenic effects of environmentally relevant levels of bisphenol A in human primary endothelial cells. Arch. Toxicol. 2012, 86, 465–474. [Google Scholar] [CrossRef]
- Li, Q.; Omori, Y.; Nishikawa, Y.; Yoshioka, T.; Yamamoto, Y.; Enomoto, K. Cytoplasmic accumulation of connexin32 protein enhances motility and metastatic ability of human hepatoma cells in vitro and in vivo. Int. J. Cancer 2007, 121, 536–546. [Google Scholar] [CrossRef]
- Ezumi, K.; Yamamoto, H.; Murata, K.; Higashiyama, M.; Damdinsuren, B.; Nakamura, Y.; Kyo, N.; Okami, J.; Ngan, C.Y.; Takemasa, I.; et al. Aberrant expression of connexin 26 is associated with lung metastasis of colorectal cancer. Clin. Cancer Res. 2008, 14, 677–684. [Google Scholar] [CrossRef]
- Elzarrad, M.K.; Haroon, A.; Willecke, K.; Dobrowolski, R.; Gillespie, M.N.; Al Mehdi, A.B. Connexin-43 upregulation in micrometastases and tumor vasculature and its role in tumor cell attachment to pulmonary endothelium. BMC Med. 2008, 6, e20. [Google Scholar] [CrossRef]
- Naus, C.C.; Laird, D.W. Implications and challenges of connexin connections to cancer. Nat. Rev. Cancer 2010, 10, 435–441. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Guy, S.; Geletu, M.; Arulanandam, R.; Raptis, L. Stat3 and Gap Junctions in Normal and Lung Cancer Cells. Cancers 2014, 6, 646-662. https://doi.org/10.3390/cancers6020646
Guy S, Geletu M, Arulanandam R, Raptis L. Stat3 and Gap Junctions in Normal and Lung Cancer Cells. Cancers. 2014; 6(2):646-662. https://doi.org/10.3390/cancers6020646
Chicago/Turabian StyleGuy, Stephanie, Mulu Geletu, Rozanne Arulanandam, and Leda Raptis. 2014. "Stat3 and Gap Junctions in Normal and Lung Cancer Cells" Cancers 6, no. 2: 646-662. https://doi.org/10.3390/cancers6020646