Specific Inhibition of the VEGFR-3 Tyrosine Kinase by SAR131675 Reduces Peripheral and Tumor Associated Immunosuppressive Myeloid Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
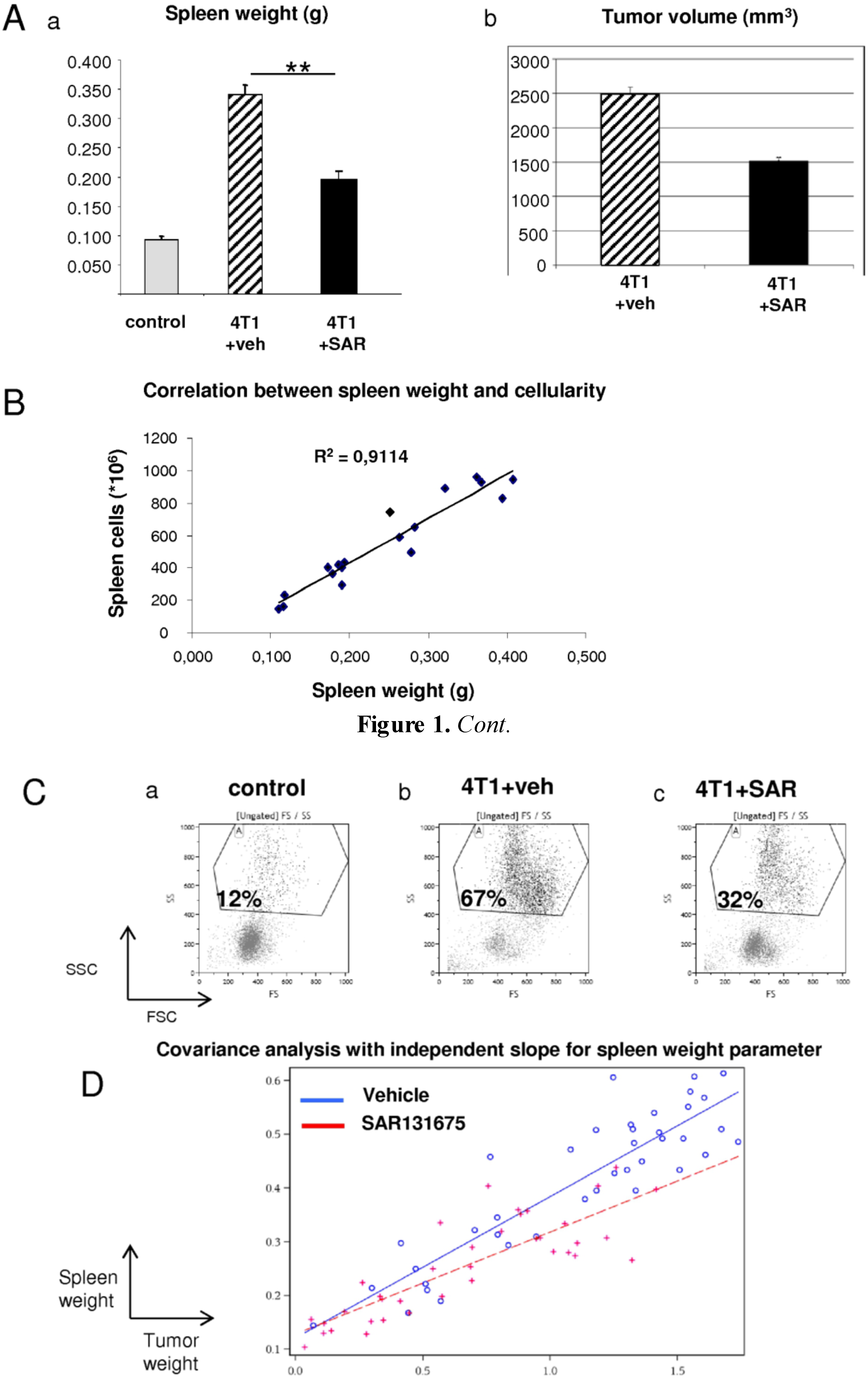
2.1. Effect of SAR131675 on Spleen Weight of 4T1-Bearing Mice

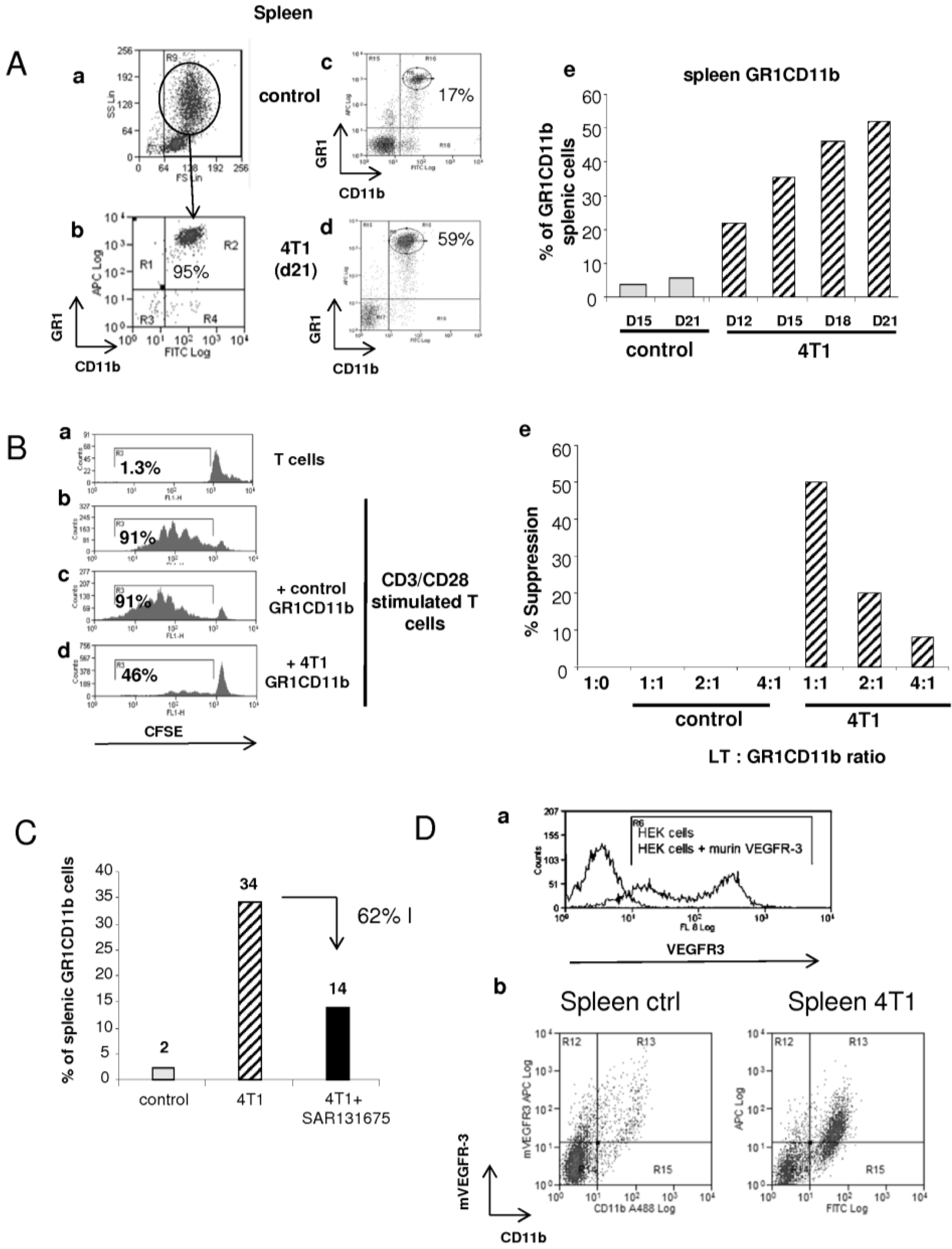
2.2. Effect of SAR131675 on 4T1 Tumor-Induced Splenic and Blood GR1CD11b Cells

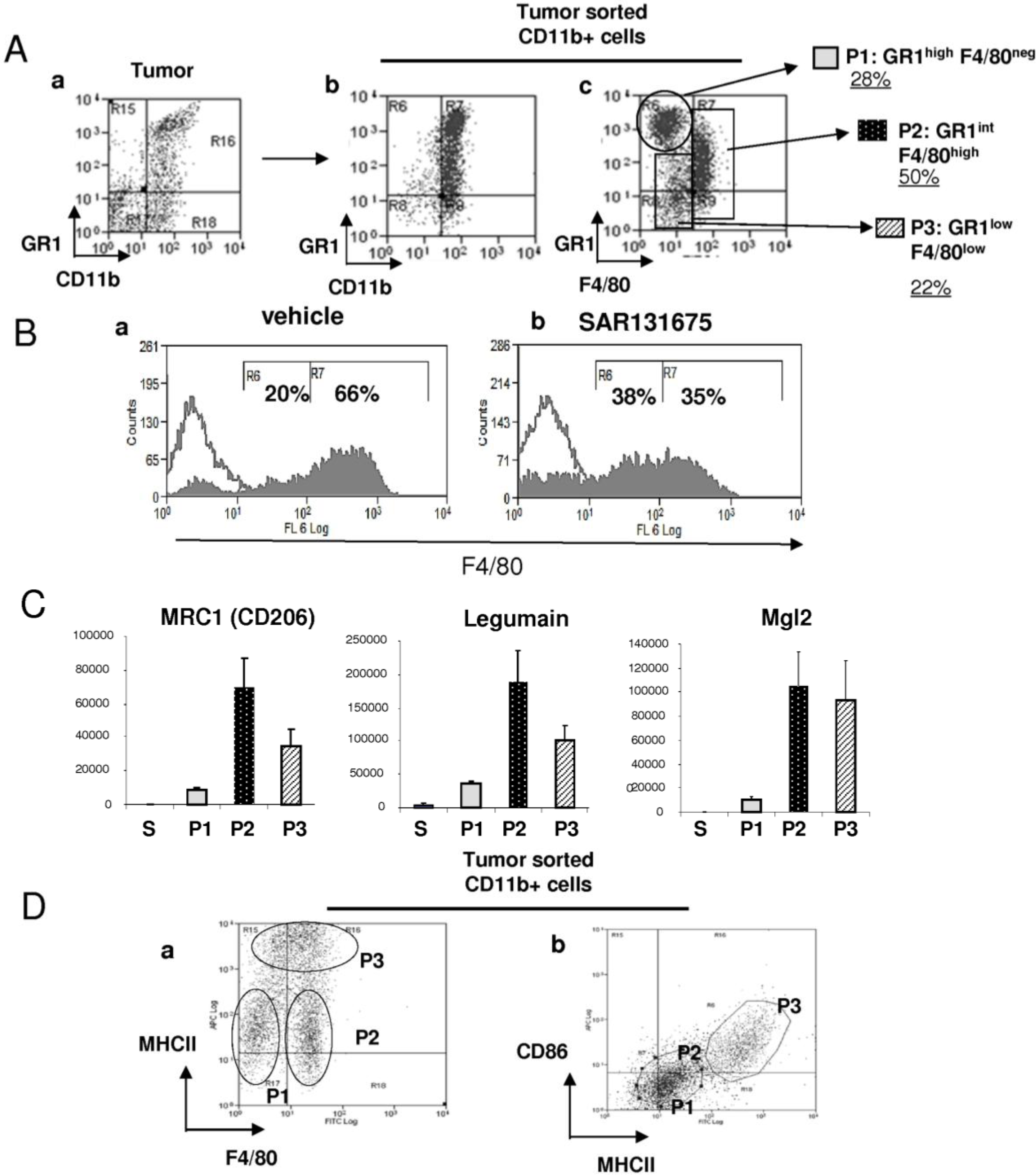
2.3. Characterization of Tumor Infiltrating Myeloid Subpopulations and Effect of SAR131675
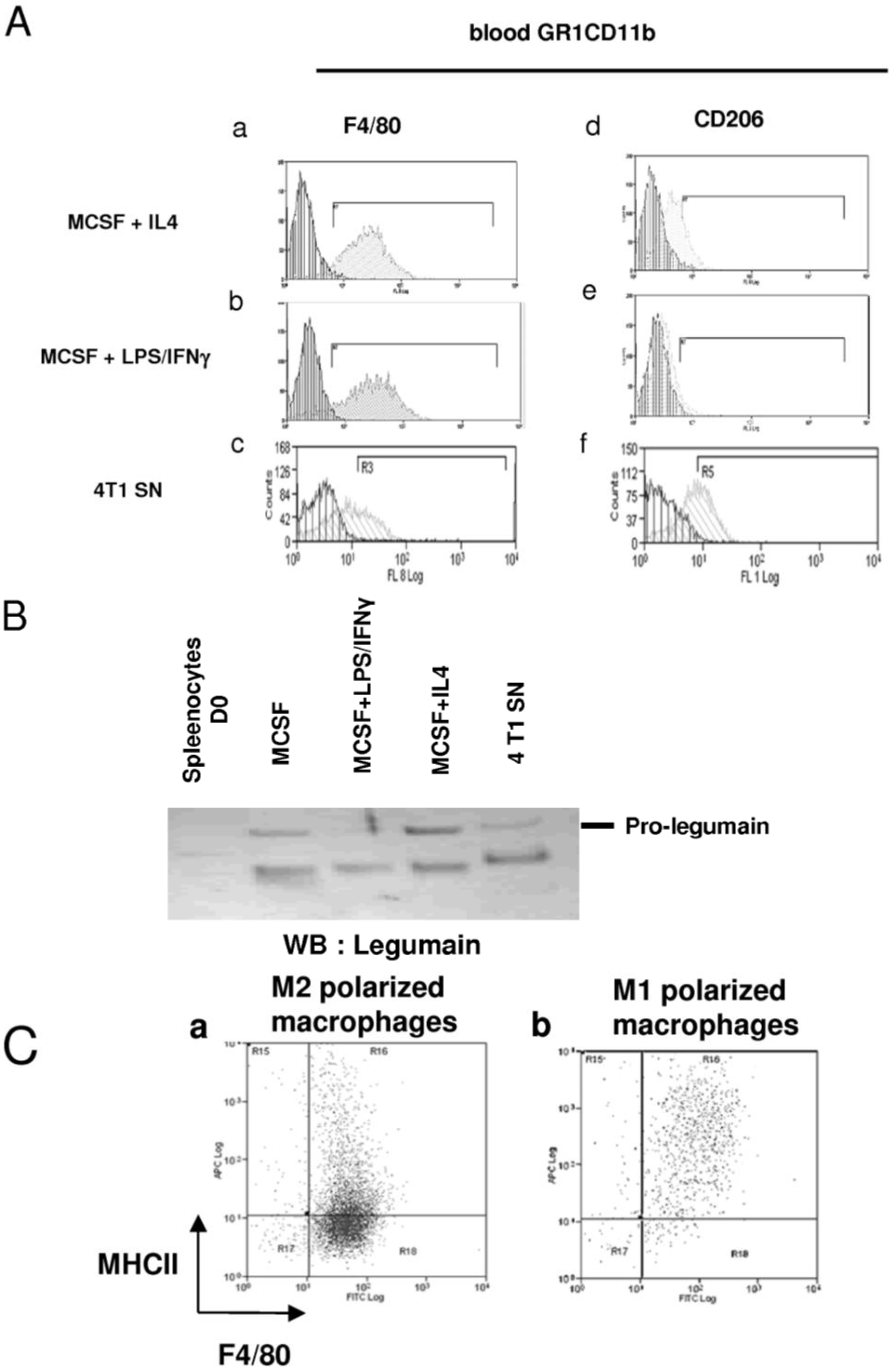
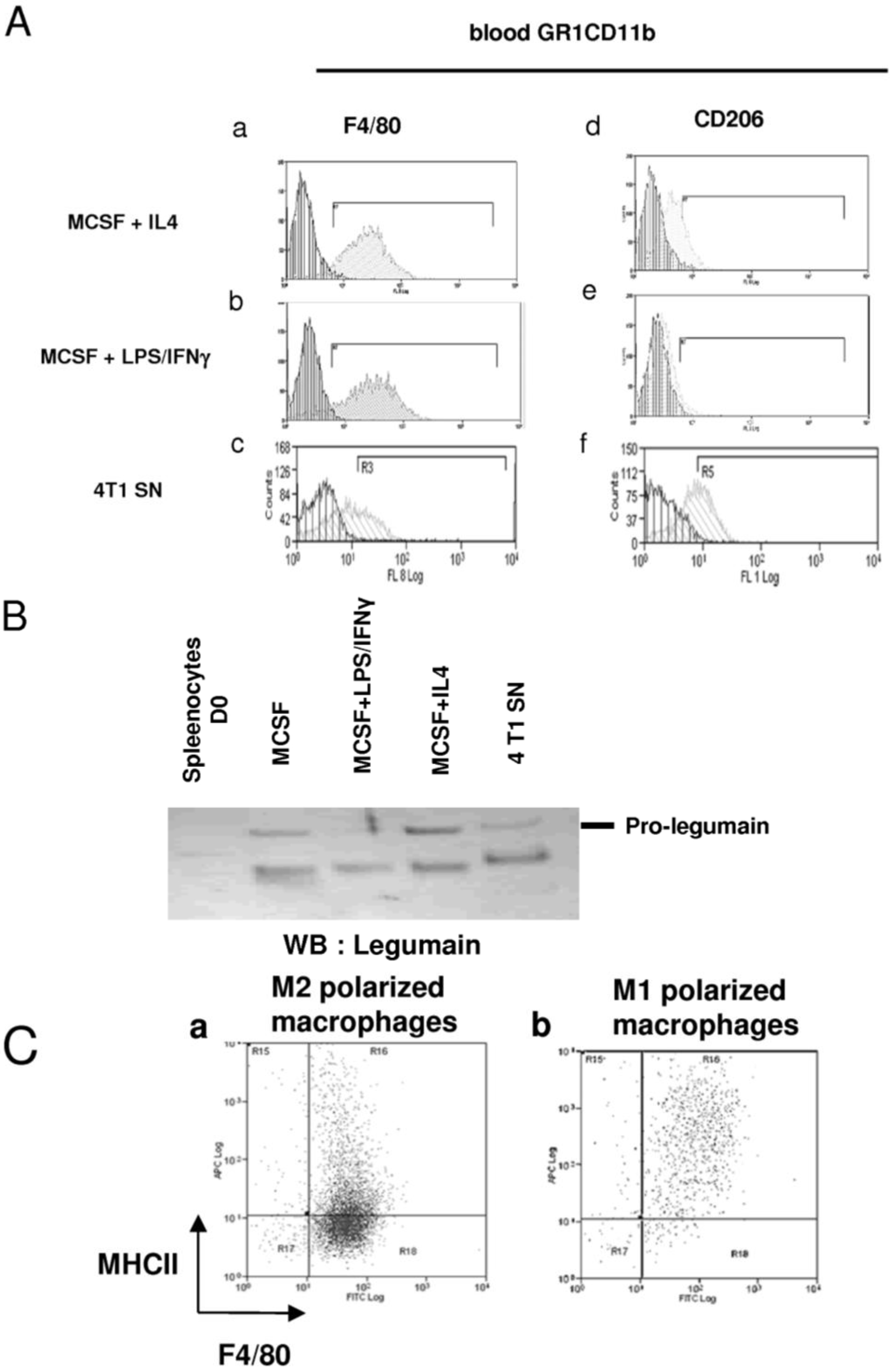
2.4. 4T1-Derived Factors Induce the Differentiation of GR1CD11b Cells in F4/80+ Macrophages

3. Discussion
4. Experimental
4.1. Reagents, Cell Line and Tumor Model
4.2. Isolation of GR1CD11b Cells and TAMs
4.3. Flow Cytometry Analysis
4.4. Immunosuppression Assay
4.5. In Vitro Proliferation and Differentiation of GR1CD11b Cells
4.6. M1/M2 Polarization in a Murine Model
4.7. Transcriptomic Analysis
5. Conclusions
Supplementary Files
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| VEGFR | Vascular Endothelial Growth Factor Receptor |
| TAMs | Tumor Associated Macrophages |
| MDSC | Myeloid Derived Suppressive Cells |
References
- Almand, B.; Clark, J.I.; Nikitina, E.; van Beynen, J.; English, N.R.; Knight, S.C.; Carbone, D.P.; Gabrilovich, D.I. Increased production of immature myeloid cells in cancer patients: A mechanism of immunosuppression in cancer. J. Immunol. 2001, 166, 678–689. [Google Scholar]
- Pak, A.S.; Wright, M.A.; Matthews, J.P.; Collins, S.L.; Petruzzelli, G.J.; Young, M.R. Mechanisms of immune suppression in patients with head and neck cancer: Presence of CD34(+) cells which suppress immune functions within cancers that secrete granulocyte-macrophage colony-stimulating factor. Clin. Cancer Res. 1995, 1, 95–103. [Google Scholar]
- Zea, A.H.; Rodriguez, P.C.; Atkins, M.B.; Hernandez, C.; Signoretti, S.; Zabaleta, J.; McDermott, D.; Quiceno, D.; Youmans, A.; O’Neill, A.; et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: A mechanism of tumor evasion. Cancer Res. 2005, 65, 3044–3048. [Google Scholar]
- Baniyash, M. Chronic inflammation, immunosuppression and cancer: New insights and outlook. Semin. Cancer Biol. 2006, 16, 80–88. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Movahedi, K.; Guilliams, M.; van den Bossche, J.; van den Bergh, R.; Gysemans, C.; Beschin, A.; de Baetselier, P.; van Ginderachter, J.A. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 2008, 111, 4233–4244. [Google Scholar] [CrossRef]
- Youn, J.I.; Nagaraj, S.; Collazo, M.; Gabrilovich, D.I. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J. Immunol. 2008, 181, 5791–5802. [Google Scholar]
- Kusmartsev, S.; Su, Z.; Heiser, A.; Dannull, J.; Eruslanov, E.; Kubler, H.; Yancey, D.; Dahm, P.; Vieweg, J. Reversal of myeloid cell-mediated immunosuppression in patients with metastatic renal cell carcinoma. Clin. Cancer Res. 2008, 14, 8270–8278. [Google Scholar] [CrossRef]
- Diaz-Montero, C.M.; Salem, M.L.; Nishimura, M.I.; Garrett-Mayer, E.; Cole, D.J.; Montero, A.J. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol. Immunother. 2009, 58, 49–59. [Google Scholar] [CrossRef]
- Liu, C.Y.; Wang, Y.M.; Wang, C.L.; Feng, P.H.; Ko, H.W.; Liu, Y.H.; Wu, Y.C.; Chu, Y.; Chung, F.T.; Kuo, C.H.; et al. Population alterations of L-arginase- and inducible nitric oxide synthase-expressed CD11b+/CD14(−)/CD15+/CD33+ myeloid-derived suppressor cells and CD8+ T lymphocytes in patients with advanced-stage non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2010, 136, 35–45. [Google Scholar] [CrossRef]
- Wang, L.; Chang, E.W.; Wong, S.C.; Ong, S.M.; Chong, D.Q.; Ling, K.L. Increased myeloid-derived suppressor cells in gastric cancer correlate with cancer stage and plasma S100A8/A9 proinflammatory proteins. J. Immunol. 2013, 190, 794–804. [Google Scholar] [CrossRef]
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative activation of macrophages: An immunologic functional perspective. Annu. Rev. Immunol. 2009, 27, 451–483. [Google Scholar] [CrossRef]
- Sica, A.; Schioppa, T.; Mantovani, A.; Allavena, P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: Potential targets of anti-cancer therapy. Eur. J. Cancer 2006, 42, 717–727. [Google Scholar] [CrossRef]
- Murdoch, C.; Muthana, M.; Coffelt, S.B.; Lewis, C.E. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef]
- Lin, C.Y.; Lin, C.J.; Chen, K.H.; Wu, J.C.; Huang, S.H.; Wang, S.M. Macrophage activation increases the invasive properties of hepatoma cells by destabilization of the adherens junction. FEBS Lett. 2006, 580, 3042–3050. [Google Scholar] [CrossRef]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef]
- Ryder, M.; Ghossein, R.A.; Ricarte-Filho, J.C.; Knauf, J.A.; Fagin, J.A. Increased density of tumor-associated macrophages is associated with decreased survival in advanced thyroid cancer. Endocr. Relat. Cancer 2008, 15, 1069–1074. [Google Scholar] [CrossRef]
- Shabo, I.; Stal, O.; Olsson, H.; Dore, S.; Svanvik, J. Breast cancer expression of CD163, a macrophage scavenger receptor, is related to early distant recurrence and reduced patient survival. Int. J. Cancer 2008, 123, 780–786. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Mishalian, I.; Singhal, S.; Cheng, G.; Kapoor, V.; Horng, W.; Fridlender, G.; Bayuh, R.; Worthen, G.S.; et al. Transcriptomic analysis comparing tumor-associated neutrophils with granulocytic myeloid-derived suppressor cells and normal neutrophils. PLoS One 2012, 7, e31524. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef]
- Kusmartsev, S.; Gabrilovich, D.I. STAT1 signaling regulates tumor-associated macrophage-mediated T cell deletion. J. Immunol. 2005, 174, 4880–4891. [Google Scholar]
- Alam, A.; Blanc, I.; Gueguen-Dorbes, G.; Duclos, O.; Bonnin, J.; Barron, P.; Laplace, M.C.; Morin, G.; Gaujarengues, F.; Dol, F.; et al. SAR131675, a potent and selective VEGFR-3-TK inhibitor with antilymphangiogenic, antitumoral, and antimetastatic activities. Mol. Cancer Ther. 2012, 11, 1637–1649. [Google Scholar] [CrossRef]
- DuPre, S.A.; Hunter, K.W., Jr. Murine mammary carcinoma 4T1 induces a leukemoid reaction with splenomegaly: Association with tumor-derived growth factors. Exp. Mol. Pathol. 2007, 82, 12–24. [Google Scholar] [CrossRef]
- Pulaski, B.A.; Ostrand-Rosenberg, S. Mouse 4T1 breast tumor model. Curr. Protoc. Immunol. 2001. [Google Scholar] [CrossRef]
- Koebel, C.M.; Vermi, W.; Swann, J.B.; Zerafa, N.; Rodig, S.J.; Old, L.J.; Smyth, M.J.; Schreiber, R.D. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 2007, 450, 903–907. [Google Scholar]
- Sinha, P.; Clements, V.K.; Ostrand-Rosenberg, S. Interleukin-13-regulated M2 macrophages in combination with myeloid suppressor cells block immune surveillance against metastasis. Cancer Res. 2005, 65, 11743–11751. [Google Scholar] [CrossRef]
- Shojaei, F.; Wu, X.; Malik, A.K.; Zhong, C.; Baldwin, M.E.; Schanz, S.; Fuh, G.; Gerber, H.P.; Ferrara, N. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat. Biotechnol. 2007, 25, 911–920. [Google Scholar] [CrossRef]
- Movahedi, K.; Laoui, D.; Gysemans, C.; Baeten, M.; Stange, G.; van den Bossche, J.; Mack, M.; Pipeleers, D.; In’t Veld, P.; de Baetselier, P.; et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010, 70, 5728–5739. [Google Scholar] [CrossRef]
- Loercher, A.E.; Nash, M.A.; Kavanagh, J.J.; Platsoucas, C.D.; Freedman, R.S. Identification of an IL-10-producing HLA-DR-negative monocyte subset in the malignant ascites of patients with ovarian carcinoma that inhibits cytokine protein expression and proliferation of autologous T cells. J. Immunol. 1999, 163, 6251–6260. [Google Scholar]
- Schartner, J.M.; Hagar, A.R.; van Handel, M.; Zhang, L.; Nadkarni, N.; Badie, B. Impaired capacity for upregulation of MHC class II in tumor-associated microglia. Glia 2005, 51, 279–285. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Albelda, S.M. Tumor-associated neutrophils: Friend or foe? Carcinogenesis 2012, 33, 949–955. [Google Scholar] [CrossRef]
- Gu, Y.; Zhao, W.; Meng, F.; Qu, B.; Zhu, X.; Sun, Y.; Shu, Y.; Xu, Q. Sunitinib impairs the proliferation and function of human peripheral T cell and prevents T-cell-mediated immune response in mice. Clin. Immunol. 2010, 135, 55–62. [Google Scholar] [CrossRef]
- Zhao, W.; Gu, Y.H.; Song, R.; Qu, B.Q.; Xu, Q. Sorafenib inhibits activation of human peripheral blood T cells by targeting LCK phosphorylation. Leukemia 2008, 22, 1226–1233. [Google Scholar] [CrossRef]
- Van Dongen, M.; Savage, N.D.; Jordanova, E.S.; Briaire-de Bruijn, I.H.; Walburg, K.V.; Ottenhoff, T.H.; Hogendoorn, P.C.; van der Burg, S.H.; Gelderblom, H.; van Hall, T. Anti-inflammatory M2 type macrophages characterize metastasized and tyrosine kinase inhibitor-treated gastrointestinal stromal tumors. Int. J. Cancer 2010, 127, 899–909. [Google Scholar]
- Zhang, W.; Zhu, X.D.; Sun, H.C.; Xiong, Y.Q.; Zhuang, P.Y.; Xu, H.X.; Kong, L.Q.; Wang, L.; Wu, W.Z.; Tang, Z.Y. Depletion of tumor-associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clin. Cancer Res. 2010, 16, 3420–3430. [Google Scholar] [CrossRef]
- Baluk, P.; Tammela, T.; Ator, E.; Lyubynska, N.; Achen, M.G.; Hicklin, D.J.; Jeltsch, M.; Petrova, T.V.; Pytowski, B.; Stacker, S.A.; et al. Pathogenesis of persistent lymphatic vessel hyperplasia in chronic airway inflammation. J. Clin. Investig. 2005, 115, 247–257. [Google Scholar] [CrossRef]
- Cursiefen, C.; Chen, L.; Borges, L.P.; Jackson, D.; Cao, J.; Radziejewski, C.; D’Amore, P.A.; Dana, M.R.; Wiegand, S.J.; Streilein, J.W. VEGF-A stimulates lymphangiogenesis and hemangiogenesis in inflammatory neovascularization via macrophage recruitment. J. Clin. Investig. 2004, 113, 1040–1050. [Google Scholar] [CrossRef]
- Hamrah, P.; Chen, L.; Zhang, Q.; Dana, M.R. Novel expression of vascular endothelial growth factor receptor (VEGFR)-3 and VEGF-C on corneal dendritic cells. Am. J. Pathol. 2003, 163, 57–68. [Google Scholar] [CrossRef]
- Maruyama, K.; Ii, M.; Cursiefen, C.; Jackson, D.G.; Keino, H.; Tomita, M.; van Rooijen, N.; Takenaka, H.; D’Amore, P.A.; Stein-Streilein, J.; et al. Inflammation-induced lymphangiogenesis in the cornea arises from CD11b-positive macrophages. J. Clin. Investig. 2005, 115, 2363–2372. [Google Scholar] [CrossRef]
- Shin, Y.J.; Choi, J.S.; Choi, J.Y.; Hou, Y.; Cha, J.H.; Chun, M.H.; Lee, M.Y. Induction of vascular endothelial growth factor receptor-3 mRNA in glial cells following focal cerebral ischemia in rats. J. Neuroimmunol. 2010, 229, 81–90. [Google Scholar] [CrossRef]
- Schoppmann, S.F.; Birner, P.; Stockl, J.; Kalt, R.; Ullrich, R.; Caucig, C.; Kriehuber, E.; Nagy, K.; Alitalo, K.; Kerjaschki, D. Tumor-associated macrophages express lymphatic endothelial growth factors and are related to peritumoral lymphangiogenesis. Am. J. Pathol. 2002, 161, 947–956. [Google Scholar] [CrossRef]
- Yang, H.; Kim, C.; Kim, M.J.; Schwendener, R.A.; Alitalo, K.; Heston, W.; Kim, I.; Kim, W.J.; Koh, G.Y. Soluble vascular endothelial growth factor receptor-3 suppresses lymphangiogenesis and lymphatic metastasis in bladder cancer. Mol. Cancer 2011, 10, 36. [Google Scholar] [CrossRef] [Green Version]
- Alitalo, A.K.; Proulx, S.T.; Karaman, S.; Aebischer, D.; Martino, S.; Jost, M.; Schneider, N.; Bry, M.; Detmar, M. VEGF-C and VEGF-D blockade inhibits inflammatory skin carcinogenesis. Cancer Res. 2013, 73, 4212–4221. [Google Scholar] [CrossRef]
- Bogos, K.; Renyi-Vamos, F.; Dobos, J.; Kenessey, I.; Tovari, J.; Timar, J.; Strausz, J.; Ostoros, G.; Klepetko, W.; Ankersmit, H.J.; et al. High VEGFR-3-positive circulating lymphatic/vascular endothelial progenitor cell level is associated with poor prognosis in human small cell lung cancer. Clin. Cancer Res. 2009, 15, 1741–1746. [Google Scholar] [CrossRef]
- Favier, B.; Alam, A.; Barron, P.; Bonnin, J.; Laboudie, P.; Fons, P.; Mandron, M.; Herault, J.P.; Neufeld, G.; Savi, P.; et al. Neuropilin-2 interacts with VEGFR-2 and VEGFR-3 and promotes human endothelial cell survival and migration. Blood 2006, 108, 1243–1250. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Espagnolle, N.; Barron, P.; Mandron, M.; Blanc, I.; Bonnin, J.; Agnel, M.; Kerbelec, E.; Herault, J.P.; Savi, P.; Bono, F.; et al. Specific Inhibition of the VEGFR-3 Tyrosine Kinase by SAR131675 Reduces Peripheral and Tumor Associated Immunosuppressive Myeloid Cells. Cancers 2014, 6, 472-490. https://doi.org/10.3390/cancers6010472
Espagnolle N, Barron P, Mandron M, Blanc I, Bonnin J, Agnel M, Kerbelec E, Herault JP, Savi P, Bono F, et al. Specific Inhibition of the VEGFR-3 Tyrosine Kinase by SAR131675 Reduces Peripheral and Tumor Associated Immunosuppressive Myeloid Cells. Cancers. 2014; 6(1):472-490. https://doi.org/10.3390/cancers6010472
Chicago/Turabian StyleEspagnolle, Nicolas, Pauline Barron, Marie Mandron, Isabelle Blanc, Jacques Bonnin, Magali Agnel, Erwan Kerbelec, Jean Pascal Herault, Pierre Savi, Françoise Bono, and et al. 2014. "Specific Inhibition of the VEGFR-3 Tyrosine Kinase by SAR131675 Reduces Peripheral and Tumor Associated Immunosuppressive Myeloid Cells" Cancers 6, no. 1: 472-490. https://doi.org/10.3390/cancers6010472