Gene Expression Profiling Identifies Important Genes Affected by R2 Compound Disrupting FAK and P53 Complex

Abstract
:1. Introduction
2. Results
2.1. Global Microarray Gene Profiling in HCT116 Cells Treated with R2 Demonstrated Up- and Down-Regulates Genes in a p53-Dependent Manner
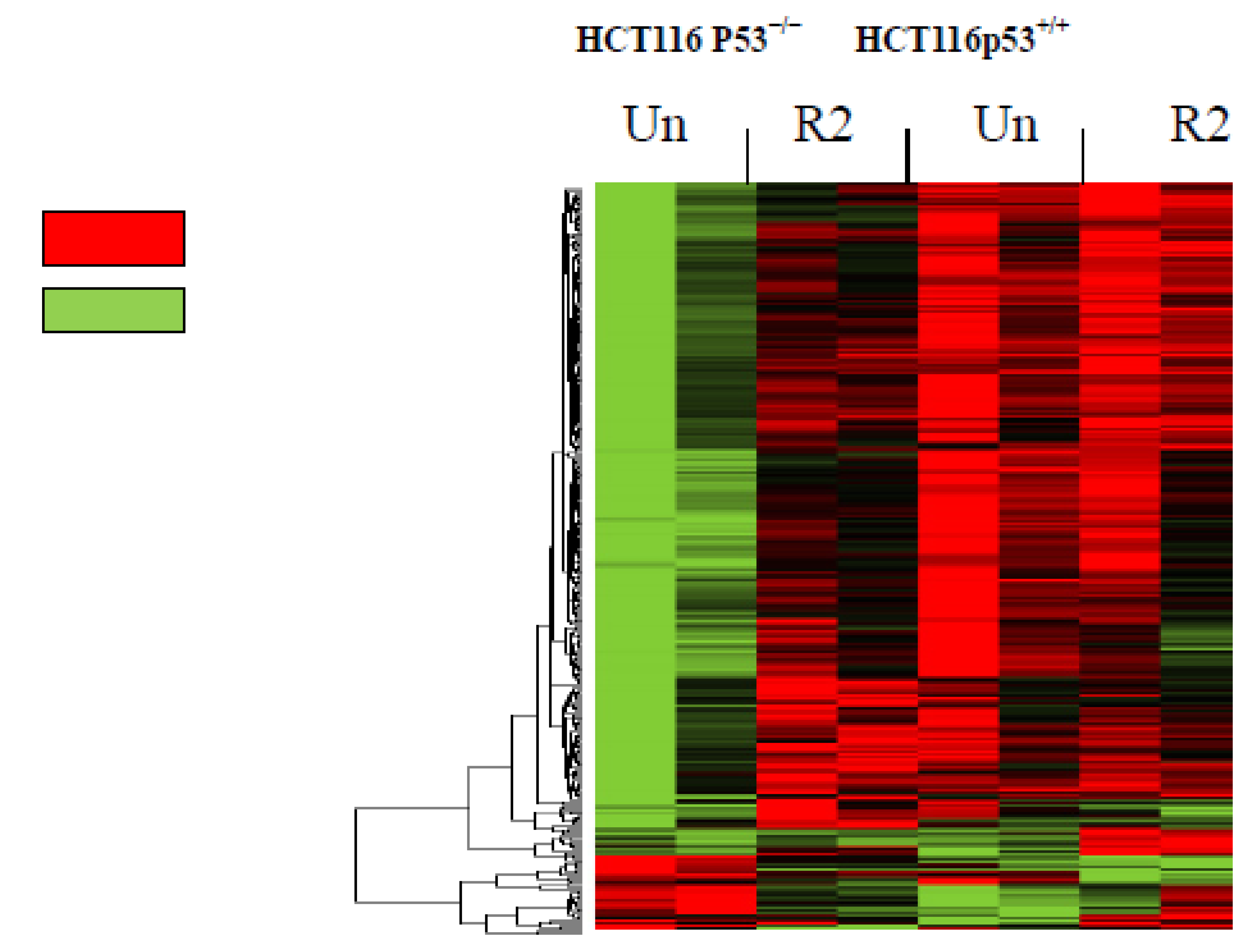

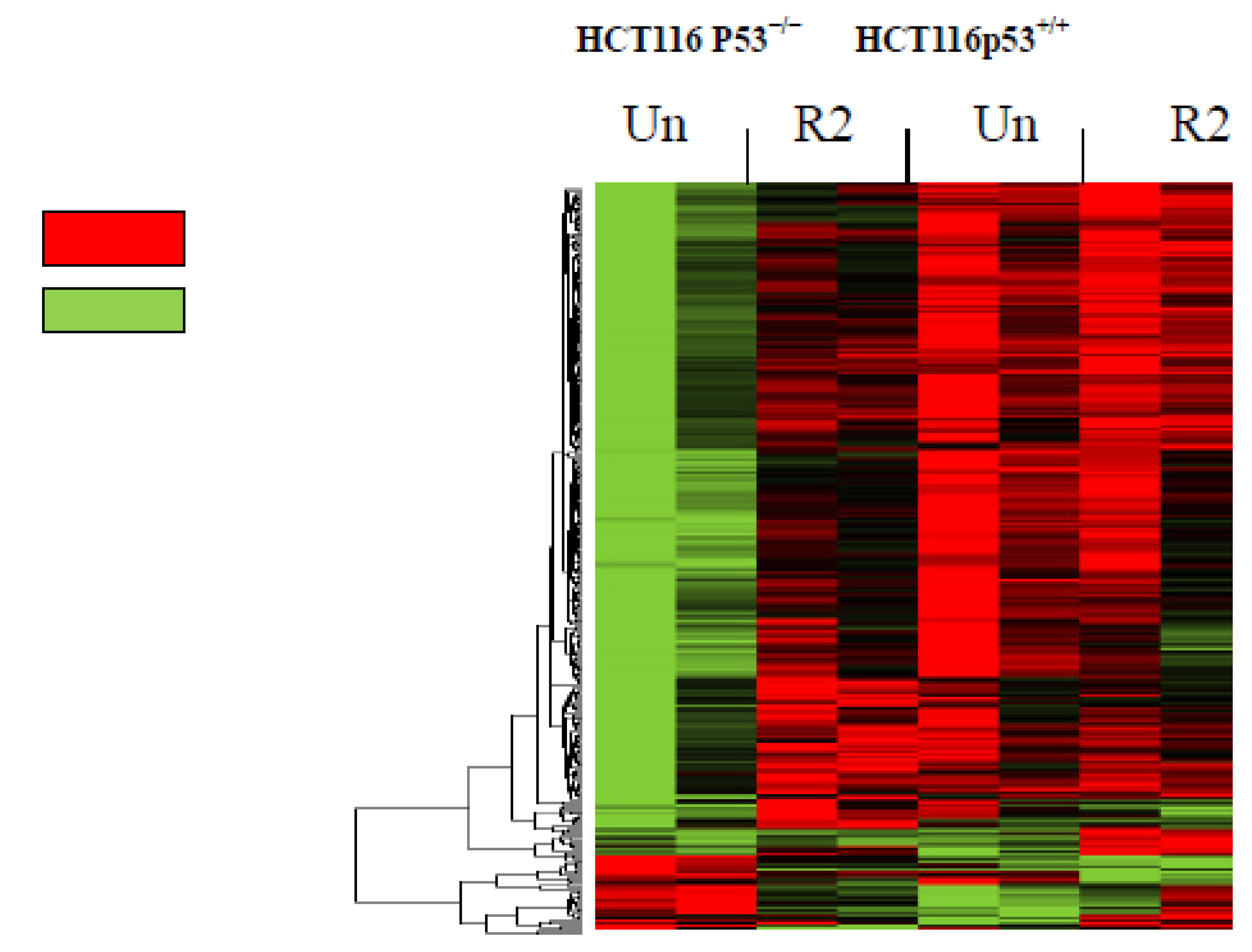{kind=link}
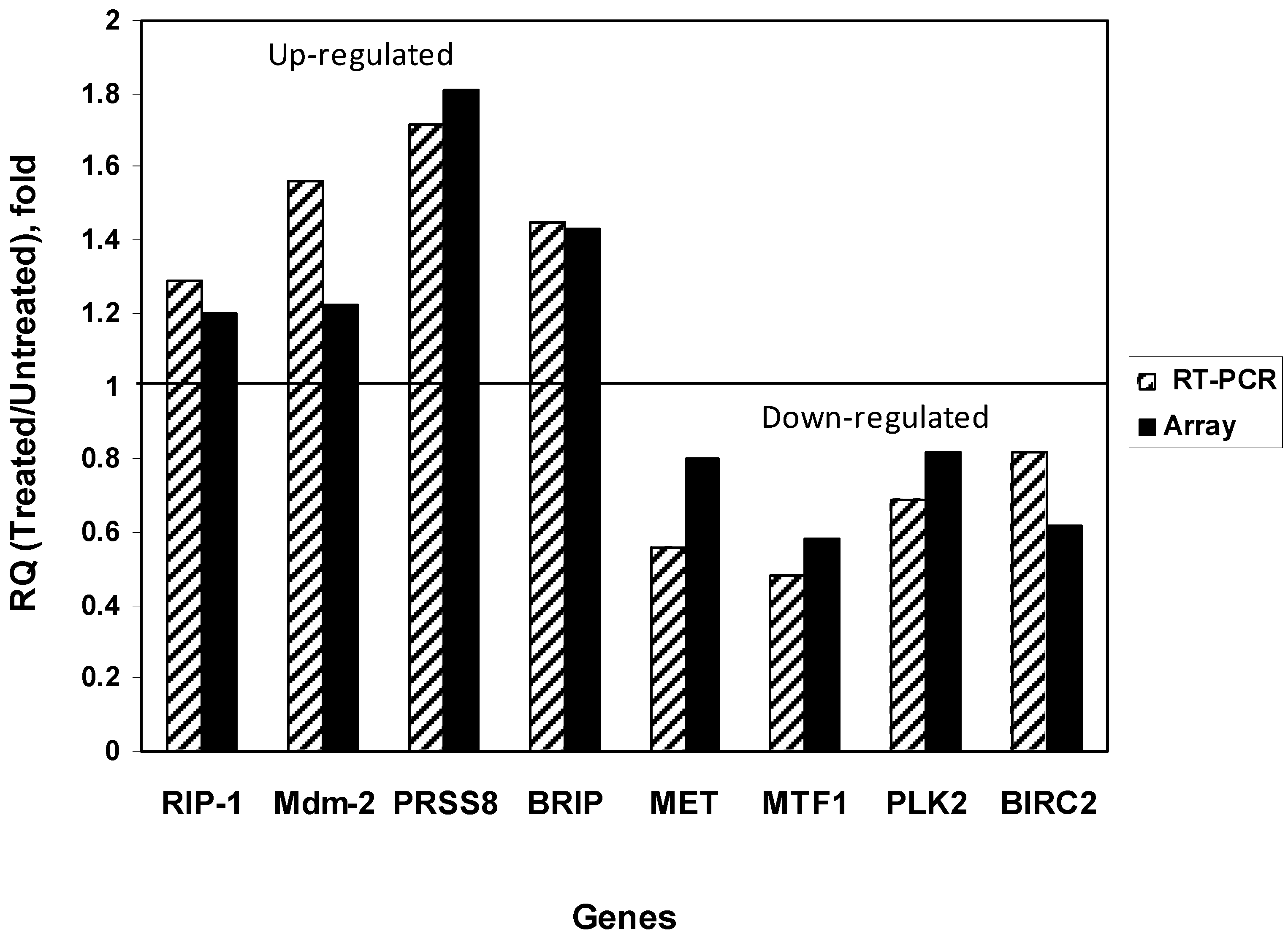{kind=link}
{kind=link}
| Up-regulated genes | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HCT116 p53+/+ cells | HCT116p53−/− cells | ||||||||||||
| Entrez Gene ID | Gene Symbol | Name | Function | Fold Change R2-treated/Untreated | p-value | Fold ChangeR2-treated/Untreated | p-value | ||||||
| 8737 | RIPK1 | Receptor (TNFRSF)-interacting serine-threonine kinase 1 | Apoptosis, regulation of I-kappa B/NF-kappa B cascade | 1.20 | 0.01 | 0.95 | 0.46 | ||||||
| 3725 | JUN | Jun oncogene Serine-threonine kinase 1 | Transcription | 1.21 | 0.07 | 1.01 | 0.83 | ||||||
| 10811 | NOXA1 | NADPH oxidase activator | Superoxide metabolic process | 1.20 | 0.01 | 0.85 | 0.02 | ||||||
| 8739 | HRK | Harakiri, Bcl2 interacting protein | Induction of apoptosis | 1.22 | 0.01 | 1.06 | 0.37 | ||||||
| 4193 | MDM2 | Mdm2 p53 binding protein homolog (mouse) | Ubiquitination, negative regulation of proliferation | 1.22 | 0.03 | 0.92 | 0.29 | ||||||
| 10608 | MXD4 | MAX dimerization protein 4 | Negative regulation of transcription | 1.23 | 0.025 | 1.00 | 0.93 | ||||||
| 5602 | MAPK13 | Mitogen-activated protein kinase 13 | Protein phosphorylation | 1.23 | 0.017 | 0.87 | 0.08 | ||||||
| 3429 | IFI27 | Interferon inducible protein 27 | Interferon-regulated process | 1.25 | 0.013 | 0.99 | 0.93 | ||||||
| 5300 | PIN1 | Peptidylprolil cis/trans isomerase | Cell cycle, protein folding | 1.26 | 0.004 | 0.95 | 0.49 | ||||||
| 7159 | TP53BP2 * | Tumor p53 binding protein 2 | Apoptosis | 1.28 | 0.014 | 0.79 | 0.02 | ||||||
| 9064 | MAP3K6 | Mitogen-activated kinase-kinase 6 | Protein phosphorylation | 1.28 | 0.003 | 1.08 | 0.22 | ||||||
| 1396 | CRIP1 | Cysteine-rich protein 1 | Cell proliferation | 1.31 | 0.026 | 1.17 | 0.046 | ||||||
| 7867 | MAPKAPK3 | Mitogen-activated protein kinase 3 | Protein phosphorylation | 1.36 | 0.0008 | 0.95 | 0.48 | ||||||
| 3675 | ITGA3 | Integrin, alpha 3 | Cell adhesion | 1.40 | 0.0003 | 1.1 | 0.13 | ||||||
| 5296 | PIK3R2 | Phosphoinositide-3-kinase, regulatory subunit | Signal transduction | 1.41 | 0.00019 | 0.88 | 0.084 | ||||||
| 83990 | BRIP1 * | BRCA1 interacting protein C-terminal helicase 1 | DNA damage checkpoijnt metabolic process | 1.43 | 0.0024 | 0.64 | 0.00056 | ||||||
| 90850 | ZNF598 | Zinc finger protein 598 | Protein binding | 1.51 | 0.0029 | 1.07 | 0.41 | ||||||
| 3659 | IRF1 | Interferon regulatory factor 1 | Regulation of interleukin-12 | 1.55 | 9.04E-05 | 1.00 | 0.99 | ||||||
| 85441 | PRIC285 | Peroxisomal proliferator-activated receptor A interacting complex 285 | Regulation of transcription | 1.58 | 4.98E-05 | 0.83 | 0.028 | ||||||
| 22937 | SCAP | SREBF chaperone | Lipid metabolic process | 1.61 | 1.5E-0.5 | 0.96 | 0.51 | ||||||
| 5652 | PRSS8 | Protease, serine, 8 | Proteolysis | 1.81 | 2.21E-06 | 0.96 | 0.59 | ||||||
| 147166 | TRIM16L * | Tripartite motif-containing 16-like | 1.84 | 0.008 | 0.47 | 0.0025 | |||||||
| Down-regulated genes | |||||||||||||
| HCT116 p53+/+ cells | HCT116p53−/− cells | ||||||||||||
| Entrez Gene ID | Gene Symbol | Name | Function | Fold Change R2-treated/Untreated | p-value | Fold Change R2-treated/Untreated | p-value | ||||||
| 7272 | TTK * | TTK protein kinase | Protein amino acid phosphorylation; Mitotic spindle organization | 0.82 | 0.016 | 1.2 | 0.02 | ||||||
| 10769 | PLK2 | Polo-like kinase 2 | Mitotic cell cycle | 0.82 | 0.019 | 1.05 | 0.47 | ||||||
| 4233 | MET | Met proto-oncogene | Protein amino acid phosphorylation; Multicellular organismal development | 0.81 | 0.01 | 0.89 | 0.14 | ||||||
| 27085 | MTBP | Mdm-2, transformed 3T3 cell double minute 2, p53 binding protein | Ubiquitin-dependent protein catabolic process; cell cycle | 0.80 | 0.0066 | 1.13 | 0.1 | ||||||
| 4436 | MSH2 | Mut S homolog 2 | Mismatch repair | 0.8 | 0.017 | 0.88 | 0.13 | ||||||
| 2353 | FOS | FOS oncogene | DNA methylation, transcription | 0.77 | 0.032 | 1.07 | 0.51 | ||||||
| 64282 | PAPD5 * | PAP associated domain 5 | DNA replication; cell cycle | 0.77 | 0.028 | 1.77 | 0.0007 | ||||||
| 9928 | KIF14 | Kinesin 14 | Microtubule-based movement | 0.76 | 0.01 | 1.02 | 0.82 | ||||||
| 9112 | MTA1 | Metastasis associated 1 | Regulation of transcription | 0.75 | 0.026 | 1.26 | 0.06 | ||||||
| 7013 | TERF1 * | Telomeric repeat binding factor 1 | Telomere maintenance via telomerase | 0.74 | 0.001 | 1.26 | 0.0058 | ||||||
| 5099 | PCDH7 | Procadherin 7 | Cell adhesion | 0.73 | 0.002 | 1.16 | 0.09 | ||||||
| 4774 | NFIA | Nuclear factor 1/A | DNA replication; transcription | 0.72 | 0.008 | 1.23 | 0.06 | ||||||
| 54434 | SSH1 * | Slingshot homolog 1 | Cell morphogenesis; cytoskeleton | 0.65 | 0.015 | 1.53 | 0.015 | ||||||
| 329 | BIRC2 | Baculoviaral IAP repeat-containing 2 | Cell surface receptor linked signaling pathway; regulation of apoptosis | 0.62 | 1.43E-05 | 0.93 | 0.36 | ||||||
| 4520 | MTF 1 * | Metal regulatory transcription factor 1 | Regulation of transcription; Response to oxidative stress | 0.58 | 0.003 | 1.66 | 0.004 | ||||||

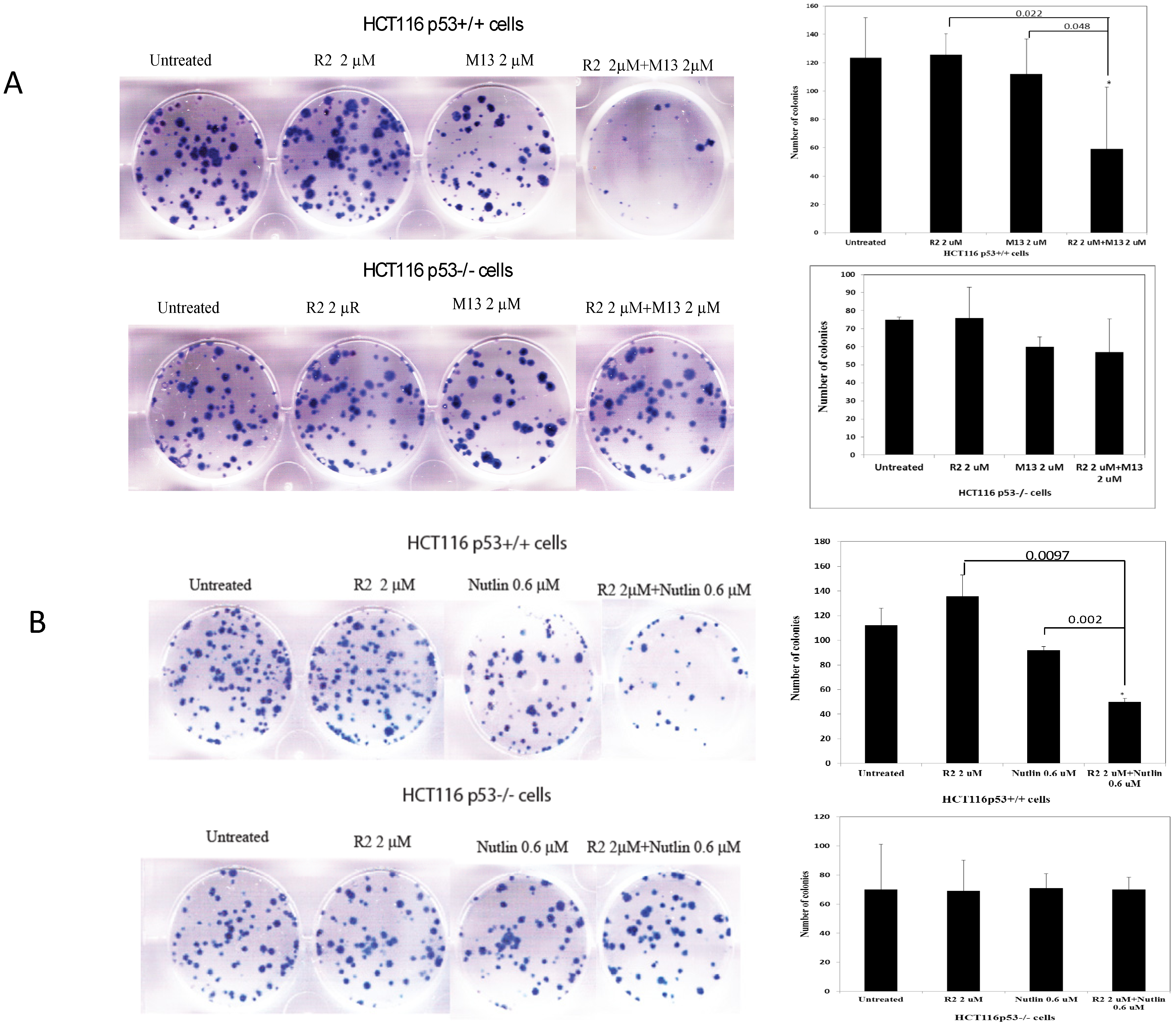
2.2. The R2 Sensitized Cancer Cells to M13 (Disrupting FAK and Mdm-2) and Nutlin-1 (Disrupting p53 and Mdm-2) Treatments
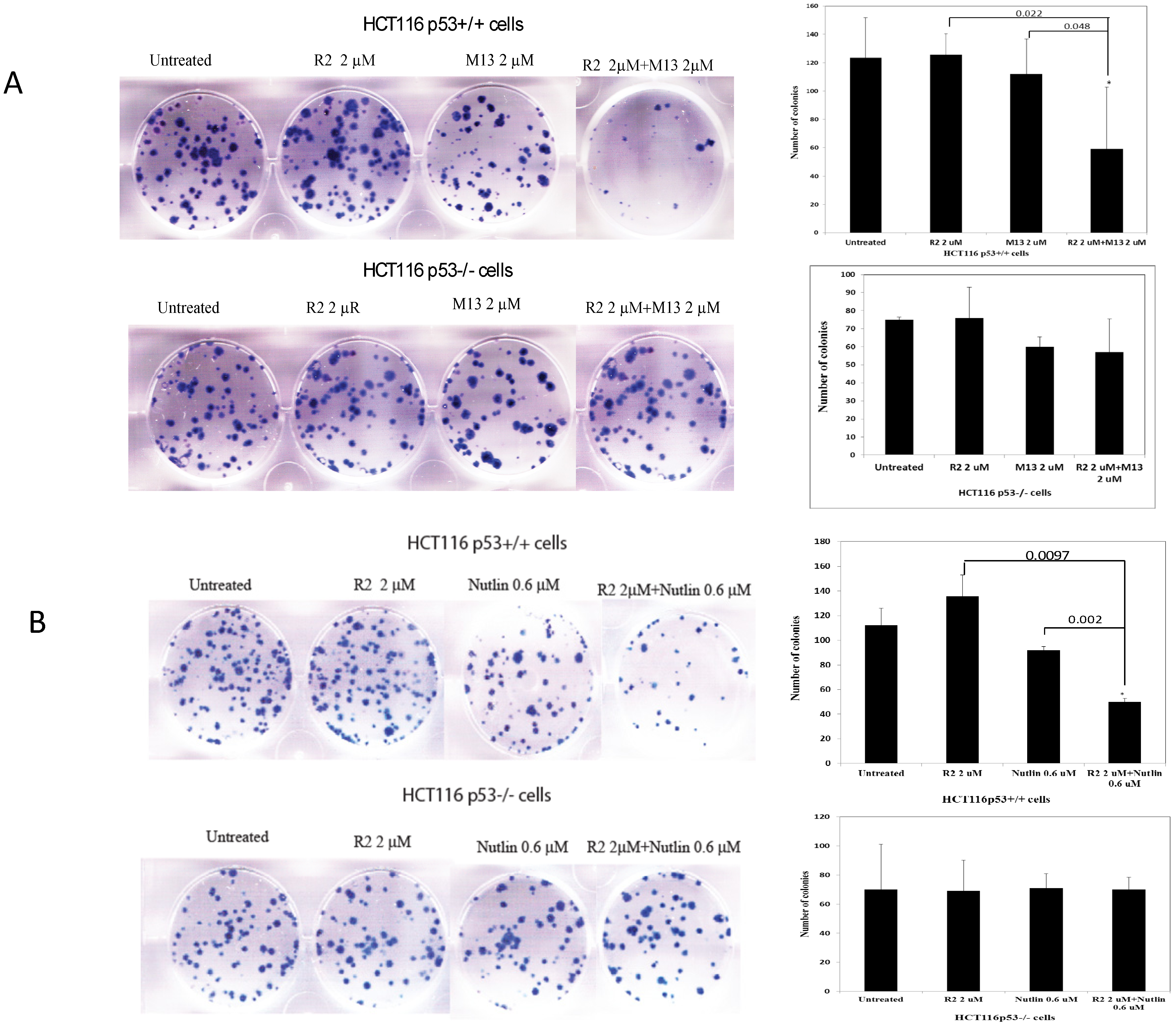
3. Experimental
3.1. Cell Lines and Culture
3.2. Reagents
3.3. Clonogenicity Assay
3.4. RNA Isolation
3.5. Microarray Analysis
3.6. Bioinformatics and Statistical Analyses
3.7. Real-Time PCR
3.8. Statistical Analyses
4. Discussion
5. Conclusions
Acknowledgments
Authors Contributions
Conflicts of Interest
References
- Hanks, S.K.; Polte, T.R. Signaling through focal adhesion kinase. Bioessays 1997, 19, 137–145. [Google Scholar] [CrossRef]
- Mitra, S.K.; Schlaepfer, D.D. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 2006, 18, 516–523. [Google Scholar] [CrossRef]
- McLean, G.W.; Carragher, N.O.; Avizienyte, E.; Evans, J.; Brunton, V.G.; Frame, M.C. The role of focal-adhesion kinase in cancer—A new therapeutic opportunity. Nat. Rev. Cancer 2005, 5, 505–515. [Google Scholar] [CrossRef]
- Golubovskaya, V.M.; Cance, W.G. Focal adhesion kinase and p53 signaling in cancer cells. Int. Rev. Cytol. 2007, 263, 103–153. [Google Scholar]
- Golubovskaya, V.; Kaur, A.; Cance, W. Cloning and characterization of the promoter region of human focal adhesion kinase gene: Nuclear factor kappa B and p53 binding sites*1. Biochim. Biophys. Acta Gene Struct. Expr. 2004, 1678, 111–125. [Google Scholar] [CrossRef]
- Golubovskaya, V.M.; Conway-Dorsey, K.; Edmiston, S.N.; Tse, C.K.; Lark, A.A.; Livasy, C.A.; Moore, D.; Millikan, R.C.; Cance, W.G. FAK overexpression and p53 mutations are highly correlated in human breast cancer. Int. J. Cancer 2009, 125, 1735–1738. [Google Scholar] [CrossRef]
- Cance, W.G.; Golubovskaya, V.M. Focal adhesion kinase versus p53: Apoptosis or survival? Sci. Signal. 2008, 1, pe22. [Google Scholar]
- Anaganti, S.; Fernandez-Cuesta, L.; Langerod, A.; Hainaut, P.; Olivier, M. p53-Dependent repression of focal adhesion kinase in response to estradiol in breast cancer cell-lines. Cancer Lett. 2011, 300, 215–224. [Google Scholar] [CrossRef]
- Golubovskaya, V.M.; Cance, W. Focal adhesion kinase and p53 signal transduction pathways in cancer. Front. Biosci. 2010, 15, 901–912. [Google Scholar] [CrossRef]
- Golubovskaya, V.M.; Finch, R.; Cance, W.G. Direct interaction of the N-terminal domain of focal adhesion kinase with the N-terminal transactivation domain of p53. J. Biol. Chem. 2005, 280, 25008–25021. [Google Scholar] [CrossRef]
- Lim, S.T.; Chen, X.L.; Lim, Y.; Hanson, D.A.; Vo, T.T.; Howerton, K.; Larocque, N.; Fisher, S.J.; Schlaepfer, D.D.; Ilic, D. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol. Cell. 2008, 29, 9–22. [Google Scholar] [CrossRef]
- Golubovskaya, V.M.; Finch, R.; Zheng, M.; Kurenova, E.V.; Cance, W.G. The 7-amino-acid site in the proline-rich region of the N-terminal domain of p53 is involved in the interaction with FAK and is critical for p53 functioning. Biochem. J. 2008, 411, 151–160. [Google Scholar] [CrossRef]
- Golubovskaya, V.M.; Ho, B.; Zheng, M.; Magis, A.; Ostrov, D.; Morrison, C.; Cance, W.G. Disruption of focal adhesion kinase and p53 interaction with small molecule compound R2 reactivated p53 and blocked tumor growth. BMC Cancer 2013, 13, 342–348. [Google Scholar] [CrossRef]
- Golubovskaya, V.; Palma, N.L.; Zheng, M.; Ho, B.; Magis, A.; Ostrov, D.; Cance, W.G. A small-molecule inhibitor, 5'-O-Tritylthymidine, targets FAK And Mdm-2 interaction, and blocks breast and colon tumorigenesis in vivo. Anticancer Agents Med. Chem. 2013, 13, 532–545. [Google Scholar] [CrossRef]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef]
- Vassilev, L.T. p53 Activation by small molecules: Application in oncology. J. Med. Chem. 2005, 48, 4491–4499. [Google Scholar] [CrossRef]
- Kurenova, E.; Xu, L.-H.; Yang, X.; Baldwin, A.S., Jr.; Craven, R.J.; Hanks, S.K.; Liu, Z.-G.; Cance, W.G. Focal adhesion kinase suppresses apoptosis by binding to the death domain of receptor-interacting protein. Mol. Cell. Biol. 2004, 24, 4361–4371. [Google Scholar] [CrossRef]
- Shibue, T.; Takeda, K.; Oda, E.; Tanaka, H.; Murasawa, H.; Takaoka, A.; Morishita, Y.; Akira, S.; Taniguchi, T.; Tanaka, N. Integral role of Noxa in p53-mediated apoptotic response. Genes Dev. 2003, 17, 2233–2238. [Google Scholar] [CrossRef]
- Xie, J.; Litman, R.; Wang, S.; Peng, M.; Guillemette, S.; Rooney, T.; Cantor, S.B. Targeting the FANCJ-BRCA1 interaction promotes a switch from recombination to poleta-dependent bypass. Oncogene 2010, 29, 2499–2508. [Google Scholar] [CrossRef]
- Chen, L.M.; Verity, N.J.; Chai, K.X. Loss of prostasin (PRSS8) in human bladder transitional cell carcinoma cell lines is associated with epithelial-mesenchymal transition (EMT). BMC Cancer 2009, 9, 377. [Google Scholar] [CrossRef]
- Chen, S.-Y.; Chen, H.-C. Direct interaction of Focal Adhesion Kinase (FAK) with Met is required for FAK to promote hepatocyte growth factor-induced cell invasion. Mol. Cell. Biol. 2006, 26, 5155–5167. [Google Scholar] [CrossRef]
- Smith, P.; Syed, N.; Crook, T. Epigenetic inactivation implies a tumor suppressor function in hematologic malignancies for Polo-like kinase 2 but not Polo-like kinase 3. Cell Cycle 2006, 5, 1262–1264. [Google Scholar] [CrossRef]
- Burns, T.F.; Fei, P.; Scata, K.A.; Dicker, D.T.; El-Deiry, W.S. Silencing of the novel p53 target gene Snk/Plk2 leads to mitotic catastrophe in paclitaxel (taxol)-exposed cells. Mol. Cell. Biol. 2003, 23, 5556–5571. [Google Scholar] [CrossRef]
- Matthew, E.M.; Hart, L.S.; Astrinidis, A.; Navaraj, A.; Dolloff, N.G.; Dicker, D.T.; Henske, E.P.; El-Deiry, W.S. The p53 target Plk2 interacts with TSC proteins impacting mTOR signaling, tumor growth and chemosensitivity under hypoxic conditions. Cell Cycle 2009, 8, 4168–4175. [Google Scholar] [CrossRef]
- Boyd, M.T.; Vlatkovic, N.; Haines, D.S. A novel cellular protein (MTBP) binds to MDM2 and induces a G1 arrest that is suppressed by MDM2. J. Biol. Chem. 2000, 275, 31883–31890. [Google Scholar] [CrossRef]
- Issaeva, N.; Bozko, P.; Enge, M.; Protopopova, M.; Verhoef, L.G.; Masucci, M.; Pramanik, A.; Selivanova, G. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat. Med. 2004, 10, 1321–1328. [Google Scholar] [CrossRef]
- Cance, W.G.; Kurenova, E.; Marlowe, T.; Golubovskaya, V. Disrupting the scaffold to improve focal adhesion kinase-targeted cancer therapeutics. Sci. Signal. 2013, 6, pe10. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Golubovskaya, V.M.; Ho, B.; Conroy, J.; Liu, S.; Wang, D.; Cance, W.G. Gene Expression Profiling Identifies Important Genes Affected by R2 Compound Disrupting FAK and P53 Complex. Cancers 2014, 6, 166-178. https://doi.org/10.3390/cancers6010166
Golubovskaya VM, Ho B, Conroy J, Liu S, Wang D, Cance WG. Gene Expression Profiling Identifies Important Genes Affected by R2 Compound Disrupting FAK and P53 Complex. Cancers. 2014; 6(1):166-178. https://doi.org/10.3390/cancers6010166
Chicago/Turabian StyleGolubovskaya, Vita M., Baotran Ho, Jeffrey Conroy, Song Liu, Dan Wang, and William G. Cance. 2014. "Gene Expression Profiling Identifies Important Genes Affected by R2 Compound Disrupting FAK and P53 Complex" Cancers 6, no. 1: 166-178. https://doi.org/10.3390/cancers6010166