Proton Radiotherapy for Pediatric Sarcoma

{kind=link}
{kind=link}
Abstract
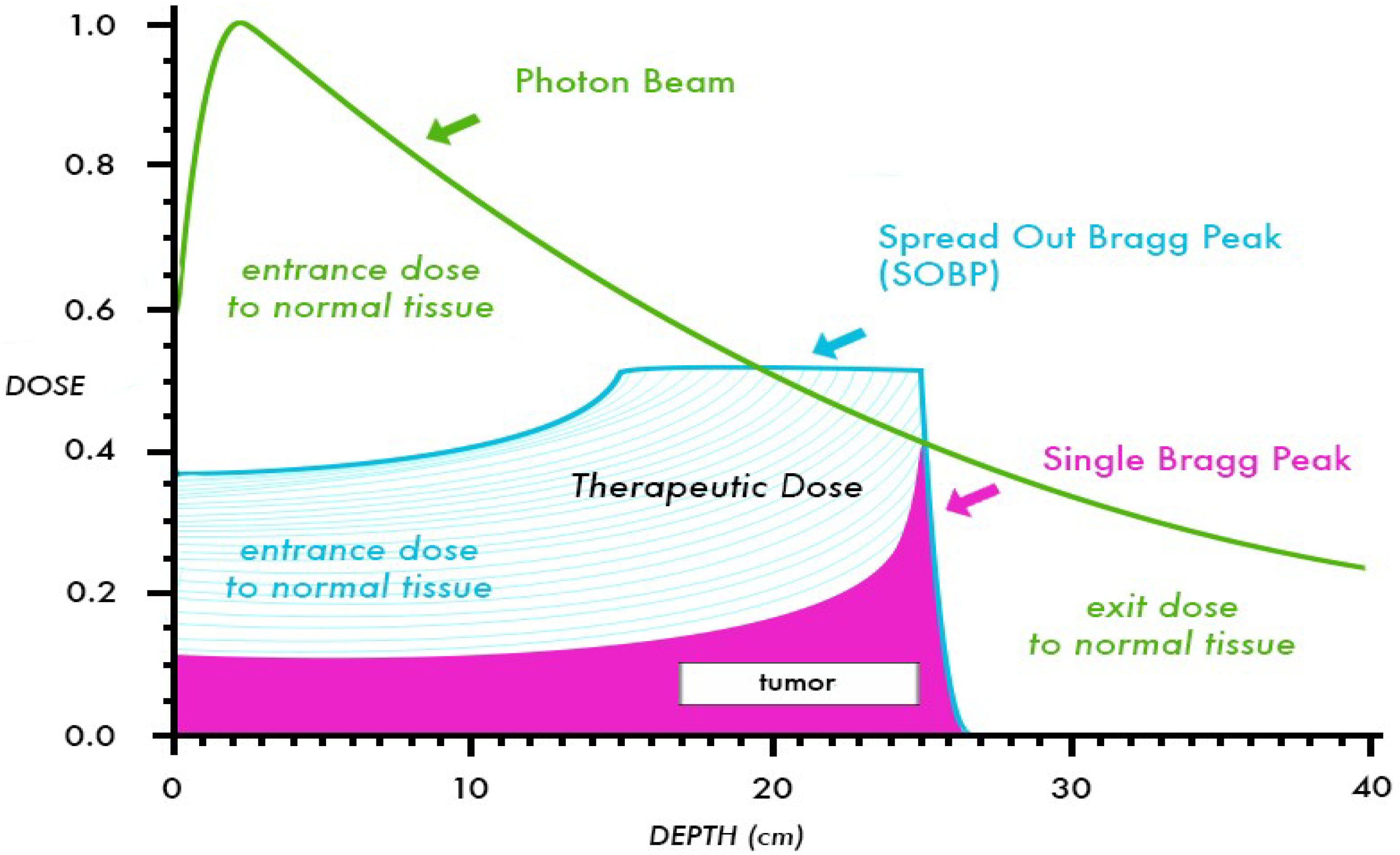
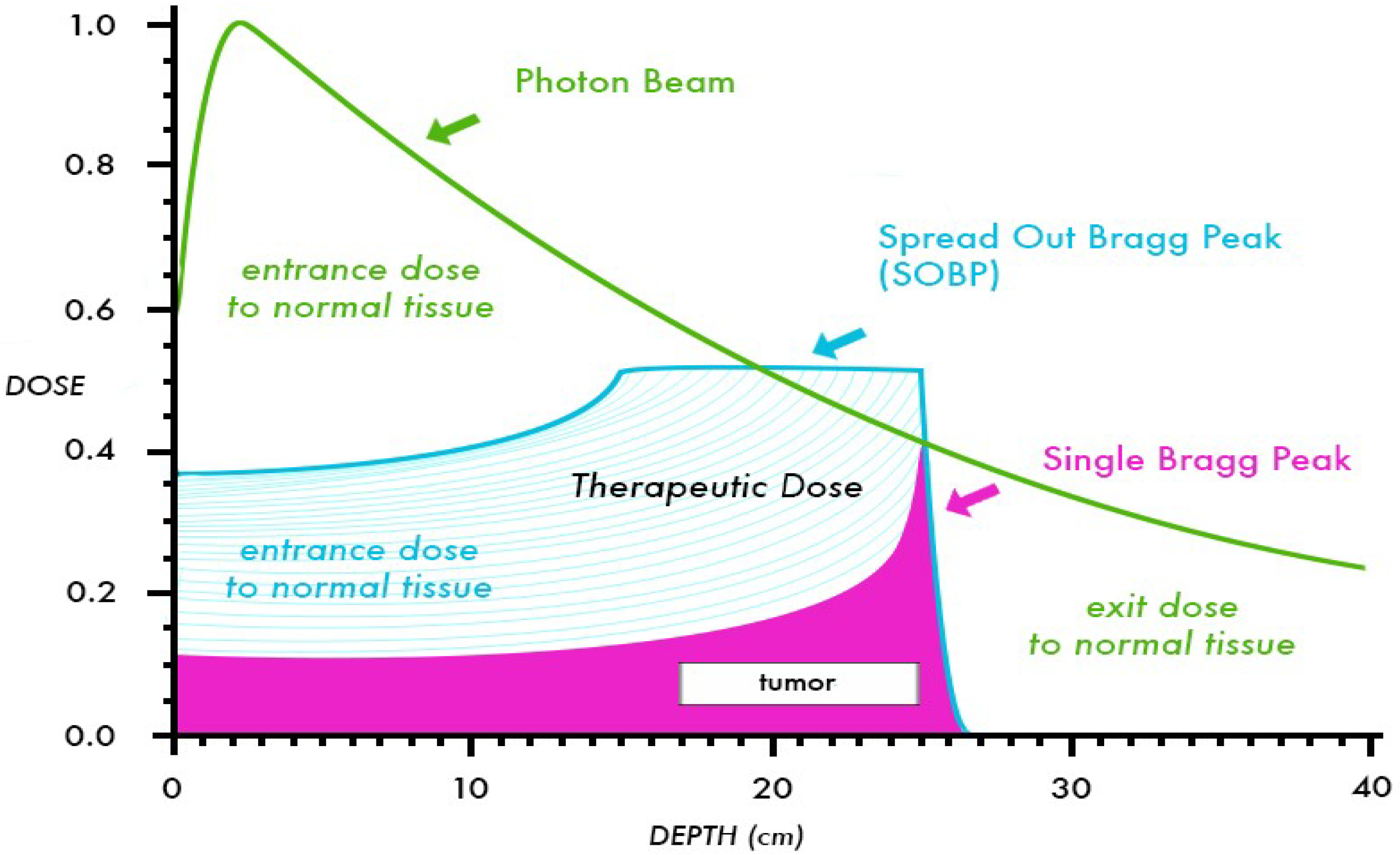
:1. Introduction
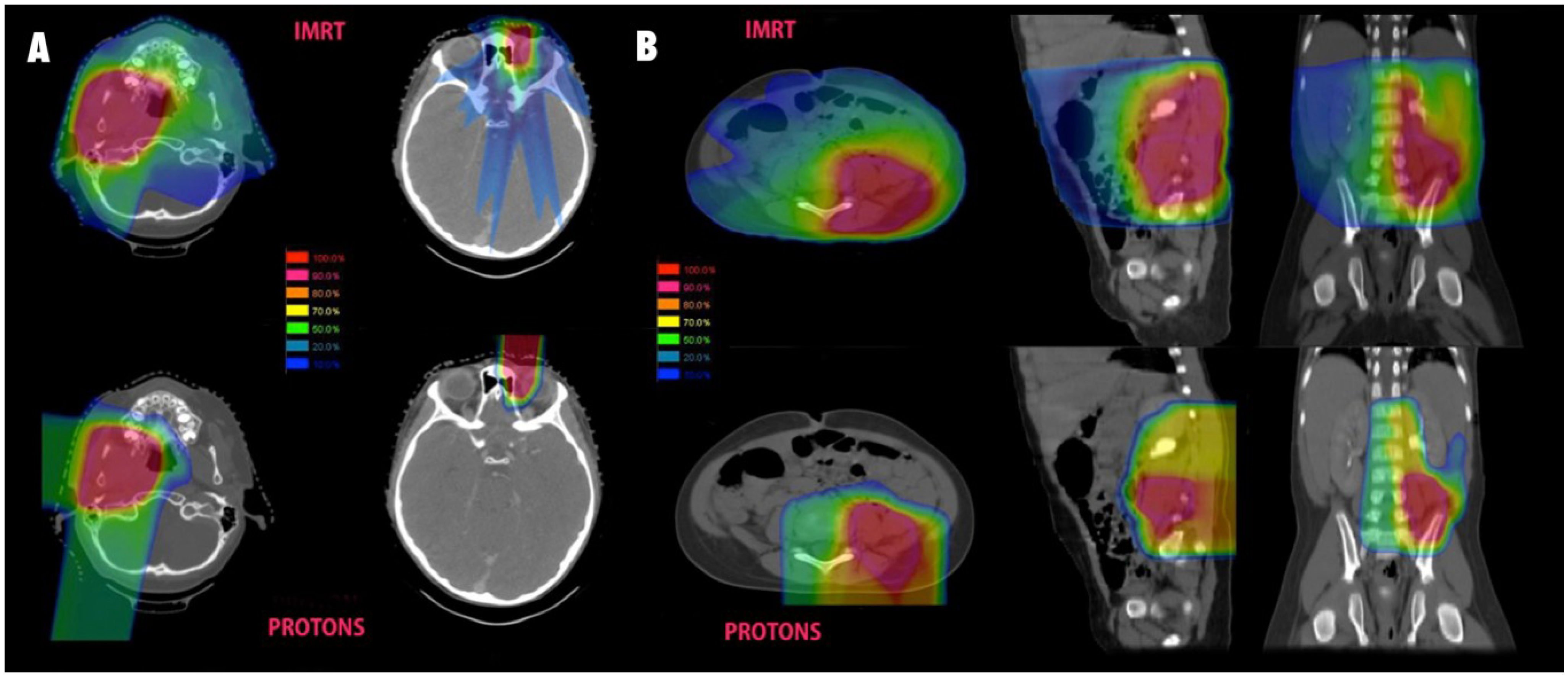
2. Rhabdomyosarcoma and Soft Tissue Sarcoma
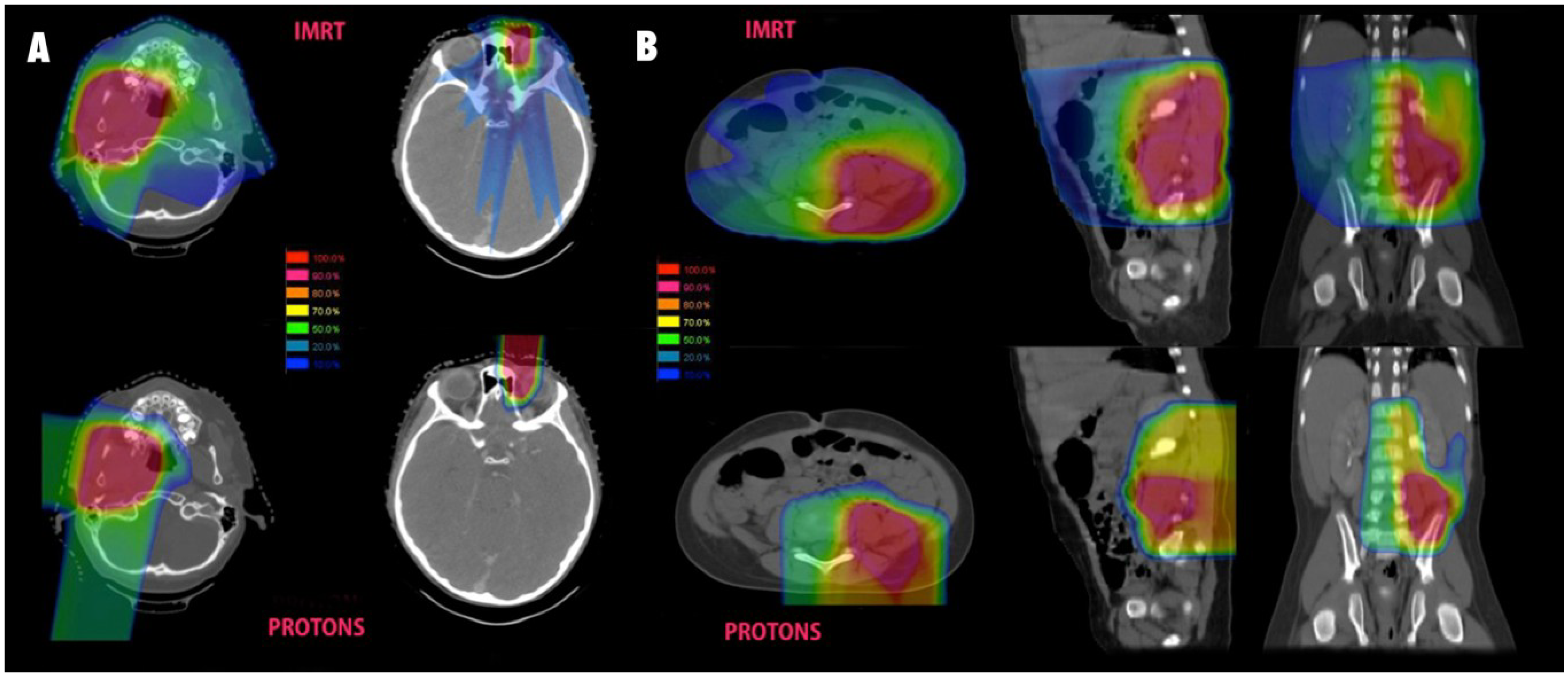
3. Non-Rhabdomyosarcoma Soft Tissue Sarcoma
4. Osteosarcoma
5. Ewing Sarcoma
6. Chordoma and Chondrosarcoma
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Schwartz, C.L. Long-term survivors of childhood cancer: The late effects of therapy. Oncologist 1999, 4, 45–54. [Google Scholar]
- Goitein, M.; Jermann, M. The relative costs of proton and x-ray radiation therapy. Clin. Oncol. 2003, 15, S37–S50. [Google Scholar] [CrossRef]
- Pagnetti, H.; Niemierko, A.; Ancukiewitz, M.; Gerweck, L.E.; Goitein, M.; Loeffler, G.S.; Suit, H.D. Relative biological effectiveness (RBE) values for proton beam radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2002, 53, 407–421. [Google Scholar] [CrossRef]
- Lomax, A.J.; Bortfeld, T.; Goitein, G.; Debus, J.; Dykstra, C.; Tercier, P.A.; Coucke, P.A.; Mirimanoff, R.O. A treatment planning inter-comparison of proton and intensity modulated photon radiotherapy. Radiother. Oncol. 1999, 51, 257–271. [Google Scholar] [CrossRef]
- Miralbell, R.; Lomax, A.; Cell, C.; Schneider, U. Potential reduction of the incidence of radiation induced second cancers by using proton beams in the treatment of pediatric tumors. Int. J. Radiat. Oncol. Biol. Phys. 2002, 54, 824–829. [Google Scholar] [CrossRef]
- Cancer Incidence and Survival among Children and Adolescents: United States SEER Program 1975–1995; Ries, L.A.; Smith, M.A.; Gurney, J.G.; Linet, M.; Tamra, T.; Young, J.L.; Bunin, G.R. (Eds.) National Cancer Institute, SEER Program; NIH Publication: Bethesda, MD, USA, 1999; No. 99–4649.
- Pappo, A.S.; Shapiro, D.N.; Crist, W.M.; Maurer, H.M. Biology and therapy of pediatric rhabdomyosarcoma. J. Clin. Oncol. 1995, 13, 2123–2139. [Google Scholar]
- Raney, R.B.; Anderson, J.R.; Barr, F.G.; Donaldson, S.S.; Pappo, A.S.; Qualman, S.J.; Wiener, E.S.; Maurer, H.M.; Crist, W.M. Rhabdomyosarcoma and undifferentiated sarcoma in the first two decades of life: A selective review of intergroup rhabdomyosarcoma study group experience and rationale for Intergroup Rhabdomyosarcoma Study V. J. Pediatr. Hematol. Oncol. 2001, 23, 215–220. [Google Scholar] [CrossRef]
- Ladra, M.; Szymonifka, J.; MacDonald, S.H.; Yong Yeap, B.; Friedmann, A.M.; Kasper, H.; Mahajan, A.; Grosshans, D.; Tarbell, N.; Yock, T.I. Proton Radiation Therapy for Rhabdomyosarcoma: Preliminary Results From a Multicenter Prospective Study. Int. J. Radiat. Oncol. Biol. Phys. 2013, 87, S597–S598. [Google Scholar]
- Crist, W.M.; Anderson, J.R.; Meza, J.L.; Fryer, C.; Raney, R.B.; Ruymann, F.B.; Breneman, J.; Qualman, S.J.; Wiener, E.; Wharam, M.; et al. Intergroup rhabdomyosarcoma study-IV: Results for patients with nonmetastatic disease. J. Clin. Oncol. 2001, 19, 3091–3102. [Google Scholar]
- Arndt, C.A.; Stoner, J.A.; Hawkins, D.S.; Rodeberg, D.A.; Hayes-Jordan, A.A.; Paidas, C.N.; Parham, D.M.; Teot, L.A.; Wharam, M.D.; Breneman, J.C.; et al. Vincristine, actinomycin, and cyclophosphamide compared with vincristine, actinomycin, and cyclophosphamide alternating with vincristine, topotecan, and cyclophosphamide for intermediate-risk rhabdomyosarcoma: Children’s oncology group study D9803. J. Clin. Oncol. 2009, 27, 5182–5188. [Google Scholar] [CrossRef]
- Pappo, A.S.; Lyden, E.; Breitfeld, P.; Donaldson, S.S.; Wiener, E.; Parham, D.; Crews, K.R.; Houghton, P.; Meyer, W.H. Two consecutive phase II window trials of irinotecan alone or in combination with vincristine for the treatment of metastatic rhabdomyosarcoma: The Children’s Oncology Group. J. Clin. Oncol. 2007, 25, 362–369. [Google Scholar] [CrossRef]
- Childs, S.K.; Kozak, K.R.; Friedmann, A.M.; Yeap, B.Y.; Adams, J.; Macdonald, S.M.; Liebsch, N.J.; Tarbell, N.J.; Yock, T.I. Proton radiotherapy for parameningeal rhabdomyosarcoma: Clinical outcomes and late effects. Int. J. Radiat. Oncol. Biol. Phys. 2011, 82, 635–642. [Google Scholar]
- Paulino, A.C.; Simon, J.H.; Zhen, W.; Wen, B.C. Long-term effects in children treated with radiotherapy for head and neck rhabdomyosarcoma. Int. J. Radiat. Oncol. Biol. Phys. 2000, 48, 1489–1495. [Google Scholar] [CrossRef]
- Raney, R.B.; Asmar, L.; Vassilopoulou-Sellin, R.; Klein, M.J.; Donaldson, S.S.; Green, J.; Heyn, R.; Wharam, M.; Glicksman, A.S.; Gehan, E.A.; et al. Late complications of therapy in 213 children with localized, nonorbital soft-tissue sarcoma of the head and neck: A descriptive report from the Intergroup Rhabdomyosarcoma Studies (IRS)-II and -III. Med. Pediatr. Oncol. 1999, 33, 362–371. [Google Scholar] [CrossRef]
- Kozak, K.R.; Adams, J.; Krejcarek, S.J.; Tarbell, N.J.; Yock, T.I. A dosimetric comparison of proton and intensity-modulated photon radiotherapy for pediatric parameningeal rhabdomyosarcomas. Int. J. Radiat. Oncol. Biol. Phys. 2009, 74, 179–186. [Google Scholar] [CrossRef]
- Merchant, T.E.; Rose, S.R.; Bosley, C.; Wu, S.; Xiong, X.; Lustig, R.H. Growth hormone secretion after conformal radiation therapy in pediatric patients with localized brain tumors. J. Clin. Oncol. 2011, 29, 4776–4780. [Google Scholar] [CrossRef]
- Yock, T.; Schneider, R.; Friedmann, A.; Adams, J.; Fullerton, B.; Tarbell, N. Proton radiotherapy for orbital rhabdomyosarcoma: Clinical outcome and a dosimetric comparison with photons. Int. J. Radiat. Oncol. Biol. Phys. 2005, 63, 1161–1168. [Google Scholar] [CrossRef]
- Heyn, R.; Ragab, A.; Raney, R.B., Jr.; Ruymann, F.; Tefft, M.; Lawrence, W., Jr.; Soule, E.; Maurer, H.M. Late effects of therapy in orbital rhabdomyosarcoma in children: A report from the Intergroup Rhabdomyosarcoma Study. Cancer 1986, 57, 1738–1743. [Google Scholar] [CrossRef]
- Wolden, S.L.; Wexler, L.H.; Kraus, D.H.; Laquaglia, M.P.; Lis, E.; Meyers, P.A. Intensity-modulated radiotherapy for head-and-neck rhabdomyosarcoma. Int. J. Radiat. Oncol. Biol. Phys. 2005, 61, 1432–1438. [Google Scholar] [CrossRef]
- Cotter, S.E.; Herrup, D.A.; Friedmann, A.; Macdonald, S.M.; Pieretti, R.V.; Robinson, G.; Adams, J.; Tarbell, N.J.; Yock, T.I. Proton radiotherapy for pediatric bladder/prostate rhabdomyosarcoma: Clinical outcomes and dosimetry compared to intensity-modulated radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 1367–1373. [Google Scholar] [CrossRef]
- Lee, C.T.; Bilton, S.D.; Famiglietti, R.M.; Riley, B.A.; Mahajan, A.; Chang, E.L.; Maor, M.H.; Woo, S.Y.; Cox, J.D.; Smith, A.R. Treatment planning with protons for pediatric retinoblastoma, medulloblastoma, and pelvic sarcoma: How do protons compare with other conformal techniques? Int. J. Radiat. Oncol. Biol. Phys. 2005, 63, 362–372. [Google Scholar] [CrossRef]
- Spunt, S.L.; Hill, D.A.; Motosue, A.M.; Billups, C.A.; Cain, A.M.; Rao, B.N.; Pratt, C.B.; Merchant, T.E.; Pappo, A.S. Clinical features and outcome of initially unresectednonmetastatic pediatric nonrhabdomyosarcoma soft tissue sarcoma. J. Clin. Oncol. 2002, 20, 3225–3235. [Google Scholar] [CrossRef]
- Spunt, S.L.; Poquette, C.A.; Hurt, Y.S.; Cain, A.M.; Rao, B.N.; Merchant, T.E.; Jenkins, J.J.; Santana, V.M.; Pratt, C.B.; Pappo, A.S. Prognostic factors for children and adolescents with surgically resected nonrhabdomyosarcoma soft tissue sarcoma: An analysis of 121 patients treated at St Jude Children’s Research Hospital. J. Clin. Oncol. 1999, 17, 3697–3705. [Google Scholar]
- Pappo, A.S.; Rao, B.N.; Jenkins, J.J.; Merchant, T.; Poquette, C.A.; Cain, A.; Pratt, C.B. Metastatic nonrhabdomyosarcomatous soft-tissue sarcomas in children and adolescents: The St. Jude Children’s Research Hospital experience. Med. Pediatr. Oncol. 1999, 33, 76–82. [Google Scholar] [CrossRef]
- Ferrari, A.; Miceli, R.; Rey, A.; Oberlin, O.; Orbach, D.; Brennan, B.; Mariani, L.; Carli, M.; Bisogno, G.; Cecchetto, G.; et al. Non-metastatic unresectedpaediatric non-rhabdomyosarcoma soft tissue sarcomas: Results of a pooled analysis from United States and European groups. Eur. J. Cancer 2011, 47, 724–731. [Google Scholar] [CrossRef]
- Weber, D.C.; Trofimov, A.V.; Delaney, T.F.; Bortfeld, T. A treatment planning comparison of intensity modulated photon and proton therapy for paraspinal sarcomas. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 1596–1606. [Google Scholar] [CrossRef]
- Weber, D.C.; Rutz, H.P.; Bolsi, A.; Pedroni, E.; Coray, A.; Jermann, M.; Lomax, A.J.; Hug, E.B.; Goitein, G. Spot scanning proton therapy in the curative treatment of adult patients with sarcoma: The Paul Scherrer institute experience. Int. J. Radiat. Oncol. Biol. Phys. 2007, 69, 865–871. [Google Scholar] [CrossRef]
- SEER Pediatric Monograph on Malignant Bone Tumors. Available online: http://seer.cancer.gov/publications/childhood/bone.pdf (accessed on 17 December 2013).
- Ogawa, Y.; Takahashi, T.; Kobayashi, T.; Kariya, S.; Nishioka, A.; Hamasato, S.; Moriki, T.; Seguchi, H.; Yoshida, S.; Sonobe, H. Immunocytochemical characteristics of human osteosarcoma cell line HS-Os-1: Possible implication in apoptotic resistance against irradiation. Int. J. Mol. Med. 2004, 14, 397–403. [Google Scholar]
- Mahajan, A.; Woo, S.Y.; Kornguth, D.G.; Hughes, D.; Huh, W.; Chang, E.L.; Herzog, C.E.; Pelloski, C.E.; Anderson, P. Multimodality treatment of osteosarcoma: Radiation in a high-risk cohort. Pediatr. Blood Cancer 2008, 50, 976–982. [Google Scholar] [CrossRef]
- Guadagnolo, B.A.; Zagars, G.K.; Raymond, A.K.; Benjamin, R.S.; Sturgis, E.M. Osteosarcoma of the jaw/craniofacial region: Outcomes after multimodality treatment. Cancer 2009, 115, 3262–3270. [Google Scholar] [CrossRef]
- Ciernik, I.F.; Niemierko, A.; Harmon, D.C.; Kobayashi, W.; Chen, Y.L.; Yock, T.I.; Ebb, D.H.; Choy, E.; Raskin, K.A.; Liebsch, N.; et al. Proton-based radiotherapy for unresectable or incompletely resected osteosarcoma. Cancer 2011, 117, 4522–4530. [Google Scholar] [CrossRef]
- DeLaney, T.F.; Park, L.; Goldberg, S.I.; Hug, E.B.; Liebsch, N.J.; Munzenrider, J.E.; Suit, H.D. Radiotherapy for local control of osteosarcoma. Int. J. Radiat. Oncol. Biol. Phys. 2005, 61, 492–498. [Google Scholar] [CrossRef]
- Cotterill, S.J.; Ahrens, S.; Paulussen, M.; Jürgens, H.F.; Voûte, P.A.; Gadner, H.; Craft, A.W. Prognostic factors in Ewing’s tumor of bone: Analysis of 975 patients from the European Intergroup Cooperative Ewing’s Sarcoma Study Group. J. Clin. Oncol. 2000, 18, 3108–3114. [Google Scholar]
- Donaldson, S.S.; Torrey, M.; Link, M.P.; Glicksman, A.; Gilula, L.; Laurie, F.; Manning, J.; Neff, J.; Reinus, W.; Thompson, E.; et al. A multidisciplinary study investigating radiotherapy in Ewing’s sarcoma: End results of POG #8346. Pediatric Oncology Group. Int. J. Radiat. Oncol. Biol. Phys. 1998, 42, 125–135. [Google Scholar]
- Shankar, A.G.; Pinkerton, C.R.; Atra, A.; Ashley, S.; Ashley, S.; Lewis, I.; Spooner, D.; Cannon, S.; Grimer, R.; Cotterill, S.J.; Craft, A.W.; et al. Local therapy and other factors influencing site of relapse in patients with localised Ewing’s sarcoma. United Kingdom Children’s Cancer Study Group (UKCCSG). Eur. J. Cancer 1999, 35, 1698–1704. [Google Scholar] [CrossRef]
- Elomaa, I.; Blomqvist, C.P.; Saeter, G.; Akerman, M.; Stenwig, E.; Wiebe, T.; Björk, O.; Alvegård, T.A. Five-year results in Ewing’s sarcoma. The Scandinavian Sarcoma Group experience with the SSG IX protocol. Eur. J. Cancer 2000, 36, 875–880. [Google Scholar] [CrossRef]
- Schuck, A.; Ahrens, S.; Paulussen, M.; Kuhlen, M.; Könemann, S.; Rübe, C.; Winkelmann, W.; Kotz, R.; Dunst, J.; Willich, N.; et al. Local therapy in localized Ewing tumors: Results of 1058 patients treated in the CESS 81, CESS 86, and EICESS 92 trials. Int. J. Radiat. Oncol. Biol. Phys. 2003, 55, 168–177. [Google Scholar] [CrossRef]
- Bacci, G.; Toni, A.; Avella, M.; Manfrini, M.; Sudanese, A.; Ciaroni, D.; Boriani, S.; Emiliani, E.; Campanacci, M. Long-term results in 144 localized Ewing’s sarcoma patients treated with combined therapy. Cancer 1989, 63, 1477–1486. [Google Scholar] [CrossRef]
- Wilkins, R.M.; Pritchard, D.J.; Burgert, E.O., Jr.; Unni, K.K. Ewing’s sarcoma of bone. Experiences with 140 patients. Cancer 1986, 58, 2551–2555. [Google Scholar] [CrossRef]
- Burgert, E.O., Jr.; Unni, K.K. The role of radiation therapy in the management of nonmetastatic Ewing’s sarcoma of bone. Report of the Intergroup Ewing’s sarcoma study. Int. J. Radiat. Oncol. Biol. Phys. 1981, 7, 141–145. [Google Scholar] [CrossRef]
- Jurgens, H.; Bier, V.; Dunst, J.; Harms, D.; Jobke, A.; Kotz, R.; Kühl, J.; Müller-Weihrich, S.; Ritter, J.; Salzer-Kuntschik, M.; et al. The German Society of Pediatric Oncology Cooperative Ewing Sarcoma Studies CESS 81/86: Report after 6 1/2 years. Klin. Padiatr. 1988, 200, 243–252. [Google Scholar] [CrossRef]
- Dunst, J.; Sauer, R.; Burgers, J.M.; Hawliczek, R.; Kürten, R.; Müller, R.P.; Wannenmacher, M.; Jürgens, H. Radiotherapy in ewing sarcoma: Current results of the german society of pediatric oncology studies CESS 81 and CESS 86. Klin. Padiatr. 1988, 200, 261–266. [Google Scholar] [CrossRef]
- Jurgens, H.; Exner, U.; Gardner, H.; Harms, D.; Michaelis, J.; Sauer, R.; Treuner, J.; Voûte, T.; Winkelmann, W.; Winkler, K.; et al. Multidisciplinary treatment of primary Ewing’s sarcoma of bone. A 6-year experience of a European cooperative trial. Cancer 1988, 61, 23–32. [Google Scholar] [CrossRef]
- Rombi, B.; Delaney, T.F.; Macdonald, S.M.; Huang, M.S.; Ebb, D.H.; Liebsch, N.J.; Raskin, K.A.; Yeap, B.Y.; Marcus, K.J.; Tarbell, N.J.; et al. Proton radiotherapy for pediatric Ewing’s sarcoma: Initial clinical outcomes. Int. J. Radiat. Oncol. Biol. Phys. 2011, 82, 1142–1148. [Google Scholar]
- Gray, S.T.; Chen, Y.L.; Lin, D.T. Efficacy of proton beam therapy in the treatment of Ewing’s sarcoma of the paranasal sinuses and anterior skull base. Skull Base 2009, 19, 409–416. [Google Scholar] [CrossRef]
- Cummings, B.J.; Hodson, D.I.; Bush, R.S. Chordoma: The results of megavoltage radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 1983, 9, 633–642. [Google Scholar] [CrossRef]
- Heffelfinger, M.J.; Dahlin, D.C.; MacCarty, C.S.; Beabout, J.W. Chordomas and cartilaginous tumors at the skull base. Cancer 1973, 32, 410–420. [Google Scholar] [CrossRef]
- Wold, L.E.; Laws, E.R., Jr. Cranial chordomas in children and young adults. J. Neurosurg. 1983, 59, 1043–1047. [Google Scholar] [CrossRef]
- Borba, L.A.B.; Al-Mefty, O.; Mrak, R.E.; Suen, J. Cranialchordomas in children and adolescents. J. Neurosurg. 1996, 84, 584–591. [Google Scholar] [CrossRef]
- Hoch, B.L.; Nielsen, G.P.; Liebsch, N.J.; Rosenberg, A.E. Base of skull chordomas in children and adolescents: A clinicopathologic study of 73 cases. Am. J. Surg. Pathol. 2006, 30, 811–818. [Google Scholar] [CrossRef]
- Noel, G.; Harbrand, J.L.; Jauffret, E. Radiation therapy for chordoma and chondrosarcoma of the skull base and the cervical spine. Strahlenther. Onkol. 2003, 179, 241–247. [Google Scholar] [CrossRef]
- Hug, E.B.; Slater, J.D. Proton radiotherapy for chordomas and chondrosarcomas of the skull base. Neurosurg. Clin. N. Am. 2000, 11, 627–634. [Google Scholar]
- Schulz-Ertner, D.; Nikoghosyan, A.; Thilmann, C.; Haberer, T.; Jäkel, O.; Karger, C.; Kraft, G.; Wannenmacher, M.; Debus, J. Results of carbon ion radiotherapy in 152 patients. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 631–640. [Google Scholar] [CrossRef]
- Tzortzidis, F.; Elahi, F.; Wright, D.; Natarajan, S.K.; Sekhar, L.N. Patient outcome at long-term follow-up after aggressive microsurgical resection of cranial base chordomas. Neurosurgery 2006, 59, 230–237. [Google Scholar] [CrossRef]
- Boriani, S.; Bandiera, S.; Biagini, R.; Bacchini, P.; Boriani, L.; Cappuccio, M.; Chevalley, F.; Gasbarrini, A.; Picci, P.; Weinstein, J.N. Chordoma of the mobile spine: Fifty years of experience. Spine 2006, 31, 493–503. [Google Scholar] [CrossRef]
- Fuchs, B.; Dickey, I.D.; Yaszemski, M.J.; Inwards, C.Y.; Sim, F.H. Operative management of sacral chordoma. J. Bone Joint Surg. Am. 2005, 87, 2211–2216. [Google Scholar] [CrossRef]
- Munzenrider, J.E.; Liebsch, N.J. Proton therapy for tumors of the skull base. Strahlenther. Onkol. 1999, 175, 57–63. [Google Scholar] [CrossRef]
- Hug, E.B.; Loredo, L.N.; Slater, J.D.; DeVries, A.; Grove, R.I.; Schaefer, R.A.; Rosenberg, A.E.; Slater, J.M. Proton radiation therapy for chordomas and chondrosarcomas of the skull base. J. Neurosurg. 1999, 91, 432–439. [Google Scholar] [CrossRef]
- Montgomery, A.H.; Wolman, I.J. Sacrococcy gealchordomas in children. Am. J. Dis. Child. 1973, 46, 1263–1281. [Google Scholar]
- Habrand, J.L.; Schneider, R.; Alapetite, C.; Feuvret, L.; Petras, S.; Datchary, J.; Grill, J.; Noel, G.; Helfre, S.; Ferrand, R.; et al. Proton therapy in pediatric skull base and cervical canal low-grade bone malignancies. Int. J. Radiat. Oncol. Biol. Phys. 2008, 71, 672–675. [Google Scholar] [CrossRef]
- Hug, E.B.; Sweeney, R.A.; Nurre, P.M.; Holloway, K.C.; Slater, J.D.; Munzenrider, J.E. Proton radiotherapy in management of pediatric base of skull tumors. Int. J. Radiat. Oncol. Biol. Phys. 2002, 52, 1017–1024. [Google Scholar] [CrossRef]
- Debus, J.; Hug, E.B.; Liebsch, N.J.; O’Farrel, D.; Finkelstein, D.; Efird, J.; Munzenrider, J.E. Brainstem tolerance to conformal radiotherapy of skull base tumors. Int. J. Radiat. Oncol. Biol. Phys. 1997, 39, 967–975. [Google Scholar] [CrossRef]
- Nielsen, G.P.; Rosenberg, A.E.; Liebsch, N.J. Chordomas of the base of skull in children and adolescents: A clinicopathologic study of 35 cases. Mod. Pathol. 1996, 9, 11. [Google Scholar]
- Yasuda, M.; Bresson, D.; Chibbaro, S.; Cornelius, J.F.; Polivka, M.; Feuvret, L.; Takayasu, M.; George, B. Chordomas of the skull base and cervical spine: Clinical outcomes associated with a multimodal surgical resection combined with proton-beam radiation in 40 patients. Neurosurg. Rev. 2012, 35, 171–182. [Google Scholar] [CrossRef]
- Rutz, H.P.; Weber, D.C.; Sugahara, S.; Timmermann, B.; Lomax, A.J.; Bolsi, A.; Pedroni, E.; Coray, A.; Jermann, M.; Goitein, G. Extracranialchordoma: Outcome in patients treated with function-preserving surgery followed by spot-scanning proton beam irradiation. Int. J. Radiat. Oncol. Biol. Phys. 2007, 67, 512–520. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ladra, M.M.; Yock, T.I. Proton Radiotherapy for Pediatric Sarcoma. Cancers 2014, 6, 112-127. https://doi.org/10.3390/cancers6010112
Ladra MM, Yock TI. Proton Radiotherapy for Pediatric Sarcoma. Cancers. 2014; 6(1):112-127. https://doi.org/10.3390/cancers6010112
Chicago/Turabian StyleLadra, Matthew M., and Torunn I. Yock. 2014. "Proton Radiotherapy for Pediatric Sarcoma" Cancers 6, no. 1: 112-127. https://doi.org/10.3390/cancers6010112