The Molecular Crosstalk between the MET Receptor Tyrosine Kinase and the DNA Damage Response—Biological and Clinical Aspects

Abstract
:1. Introduction
2. Results and Discussion
2.1. Radiotherapy
2.2. The DNA Damage Response
2.3. Growth Factor RTK Systems Involved in Responses to DNA Damage
2.3.1. EGFR
2.3.2. IGF-1R
2.3.3. VEGFR, PDGFR, FGFR

{kind=link}
{kind=link}
| DDR-related protein | RTK | Mode of modulation |
|---|---|---|
| DNA-PK (dsDNA breaks repair by non-homologous end-joining) | EGFR | physical interaction [63], lower DNA-PK nuclear activity [63] and redistribution of DNA-PK from nucleus to cytosol upon EGFRi [64], EGFR in complex with DNA-PK [67], IR-dependent nuclear translocation of EGFR [67], defective interaction with EGFR mutants [78] |
| ERCC1 (ssDNA breaks repair by nucleotide excision) | EGFR | MAPK-dependent upregulation [65] |
| XRCC1 (ssDNA breaks repair by base excision) | EGFR | MAPK-dependent upregulation [65] |
| BRCA1 (dsDNA breaks repair by homologous recombination) | EGFR | cytoplasmic sequestration upon EGFRi [68] |
| ATM (dsDNA damage breaks sensing) | IGF-1R, MET | lower levels in IGF-1R antisense-expressing cells [85], A-T cells display low levels of IGF-1R [86], MET-dependent ATM activation upon IR [98], increase in ATM autophosphorylation upon METi [99] |
| RAD51 (dsDNA breaks repair by homologous recombination) | IGF-1R, MET | intracellular trafficking controlled by IGF-1R [88], decrease in phosphorylation and nuclear translocation upon METi [100], interaction with BRCA2 hindered upon METi [101], reduced levels upon METi combined with IR [102] |
| BRCA2 (dsDNA breaks repair by homologous recombination) | MET | interaction with RAD51 hindered by METi [101] |
| γH2AX (dsDNA damage breaks sensing) | IGF-1R, MET | sustained γH2AX expression upon IGF-1R [89] and MET [99] inhibition, sustained γH2AX levels upon METi in combination with IR [99] |
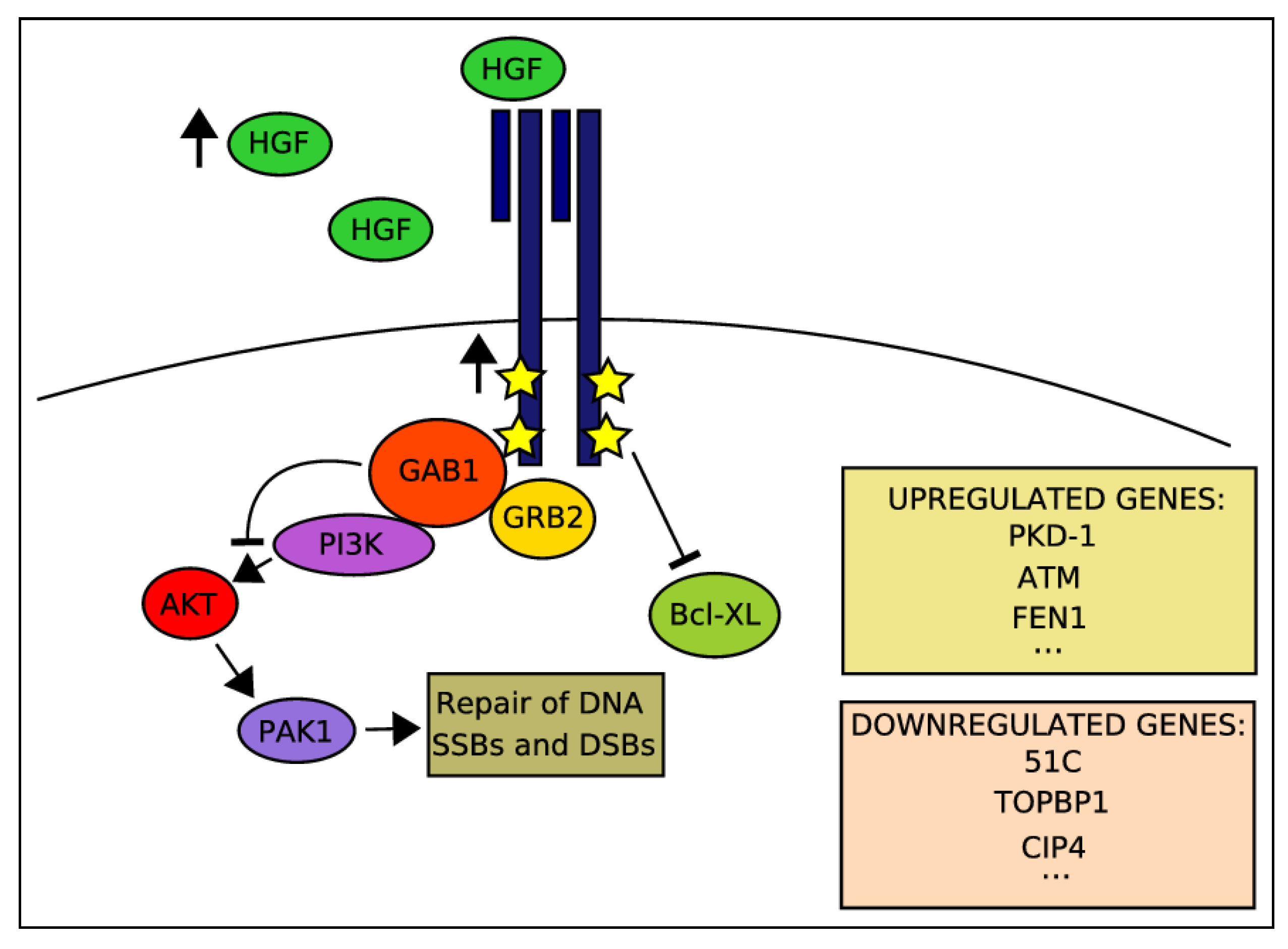
2.4. Activation of HGF-MET Axis Protects Cells from DDAs-Related Cytotoxicity
2.5. Clinical Observations Correlating MET Expression with Responses to RT
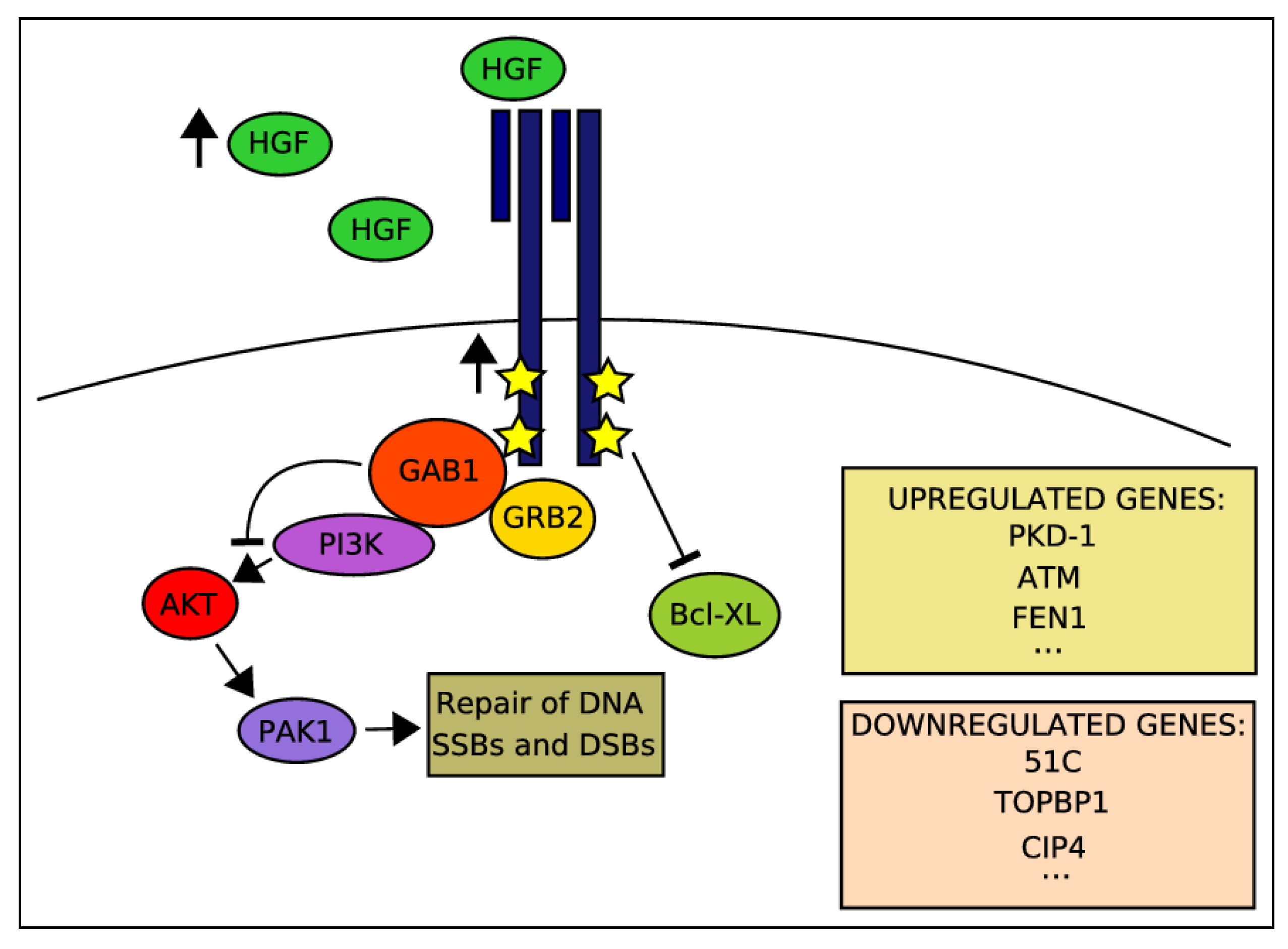
2.6. Potentiation of IR-Induced Cytotoxicity by Targeting MET Signaling
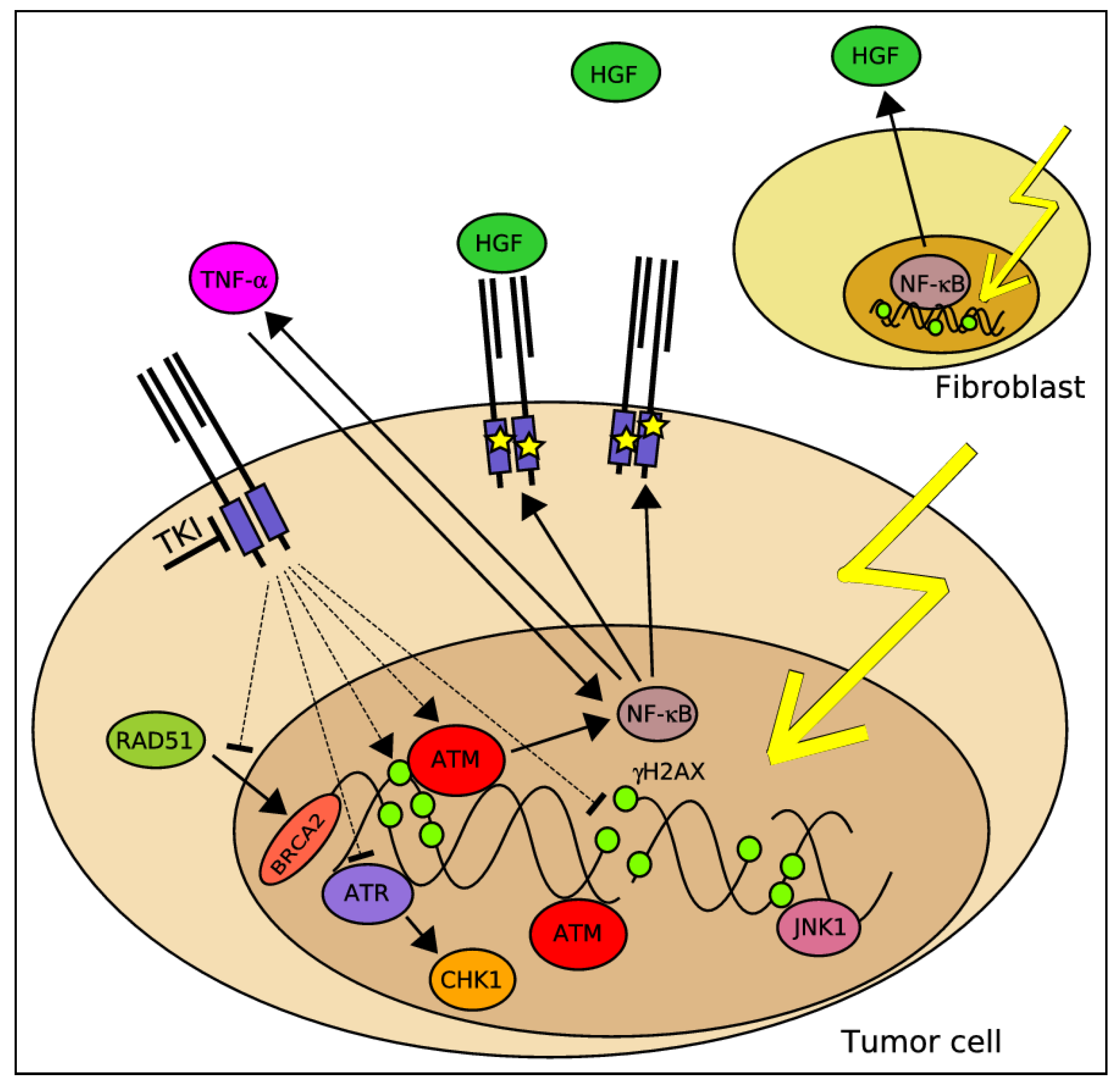
2.7. A Potential Molecular Crosstalk between MET and the DDR
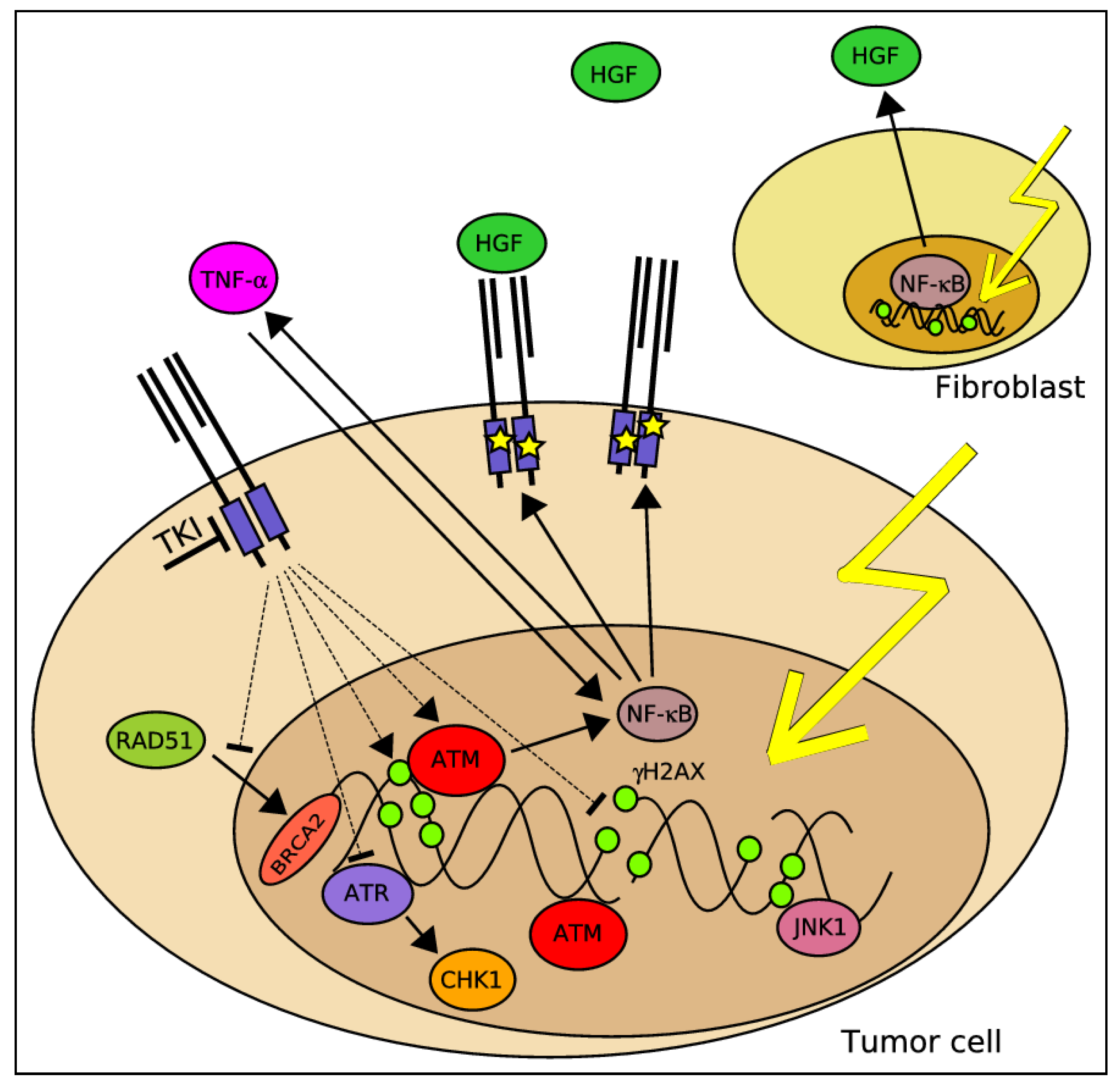
2.8. The Relevance of MET Signaling to Tumor Resistance towards RT under Hypoxic Conditions
3. Conclusions
Acknowledgments
Conflicts of Interest
References
- Cooper, C.S.; Park, M.; Blair, D.G.; Tainsky, M.A.; Huebner, K.; Croce, C.M.; vande Woude, G.F. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 1984, 311, 29–33. [Google Scholar] [CrossRef]
- Bottaro, D.P.; Rubin, J.S.; Faletto, D.L.; Chan, A.M.; Kmiecik, T.E.; vande Woude, G.F.; Aaronson, S.A. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991, 251, 802–804. [Google Scholar]
- Iyer, A.; Kmiecik, T.E.; Park, M.; Daar, I.; Blair, D.; Dunn, K.J.; Sutrave, P.; Ihle, J.N.; Bodescot, M.; vande Woude, G.F. Structure, tissue-specific expression, and transforming activity of the mouse met protooncogene. Cell Growth Differ. 1990, 1, 87–95. [Google Scholar]
- Zhang, Y.W.; Su, Y.; Volpert, O.V.; vande Woude, G.F. Hepatocyte growth factor/scatter factor mediates angiogenesis through positive VEGF and negative thrombospondin 1 regulation. Proc. Natl. Acad. Sci. USA 2003, 100, 12718–12723. [Google Scholar] [CrossRef]
- Knowles, L.M.; Stabile, L.P.; Egloff, A.M.; Rothstein, M.E.; Thomas, S.M.; Gubish, C.T.; Lerner, E.C.; Seethala, R.R.; Suzuki, S.; Quesnelle, K.M.; et al. HGF and c-MET participate in paracrine tumorigenic pathways in head and neck squamous cell cancer. Clin. Cancer Res. 2009, 15, 3740–3750. [Google Scholar]
- Lengyel, E.; Prechtel, D.; Resau, J.H.; Gauger, K.; Welk, A.; Lindemann, K.; Salanti, G.; Richter, T.; Knudsen, B.; vande Woude, G.F.; et al. C-Met overexpression in node-positive breast cancer identifies patients with poor clinical outcome independent of HER2/NEU. Int. J. Cancer 2005, 113, 678–682. [Google Scholar] [CrossRef]
- Tokunou, M.; Niki, T.; Eguchi, K.; Iba, S.; Tsuda, H.; Yamada, T.; Matsuno, Y.; Kondo, H.; Saitoh, Y.; Imamura, H.; et al. c-MET expression in myofibroblasts: Role in autocrine activation and prognostic significance in lung adenocarcinoma. Am. J. Pathol. 2001, 158, 1451–1463. [Google Scholar] [CrossRef]
- Ramirez, R.; Hsu, D.; Patel, A.; Fenton, C.; Dinauer, C.; Tuttle, R.M.; Francis, G.L. Over-expression of hepatocyte growth factor/scatter factor (HGF/SF) and the HGF/SF receptor (cMET) are associated with a high risk of metastasis and recurrence for children and young adults with papillary thyroid carcinoma. Clin. Endocrinol. (Oxf.) 2000, 53, 635–644. [Google Scholar] [CrossRef]
- Koochekpour, S.; Jeffers, M.; Rulong, S.; Taylor, G.; Klineberg, E.; Hudson, E.A.; Resau, J.H.; vande Woude, G.F. MET and hepatocyte growth factor/scatter factor expression in human gliomas. Cancer Res. 1997, 57, 5391–5398. [Google Scholar]
- Di Renzo, M.F.; Poulsom, R.; Olivero, M.; Comoglio, P.M.; Lemoine, N.R. Expression of the MET/hepatocyte growth factor receptor in human pancreatic cancer. Cancer Res. 1995, 55, 1129–1138. [Google Scholar]
- Liu, C.; Park, M.; Tsao, M.S. Overexpression of c-MET proto-oncogene but not epidermal growth factor receptor or c-ERBB-2 in primary human colorectal carcinomas. Oncogene 1992, 7, 181–185. [Google Scholar]
- Kelsen, D.P.; Minsky, B.; Smith, M.; Beitler, J.; Niedzwiecki, D.; Chapman, D.; Bains, M.; Burt, M.; Heelan, R.; Hilaris, B. Preoperative therapy for esophageal cancer: A randomized comparison of chemotherapy versus radiation therapy. J. Clin. Oncol. 1990, 8, 1352–1361. [Google Scholar]
- Pennacchietti, S.; Michieli, P.; Galluzzo, M.; Mazzone, M.; Giordano, S.; Comoglio, P.M. Hypoxia promotes invasive growth by transcriptional activation of the MET protooncogene. Cancer Cell 2003, 3, 347–361. [Google Scholar] [CrossRef]
- Cappuzzo, F.; Marchetti, A.; Skokan, M.; Rossi, E.; Gajapathy, S.; Felicioni, L.; del Grammastro, M.; Sciarrotta, M.G.; Buttitta, F.; Incarbone, M.; et al. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J. Clin. Oncol. 2009, 27, 1667–1674. [Google Scholar] [CrossRef]
- Beau-Faller, M.; Ruppert, A.M.; Voegeli, A.C.; Neuville, A.; Meyer, N.; Guerin, E.; Legrain, M.; Mennecier, B.; Wihlm, J.M.; Massard, G.; et al. MET gene copy number in non-small cell lung cancer: Molecular analysis in a targeted tyrosine kinase inhibitor naive cohort. J. Thorac. Oncol. 2008, 3, 331–339. [Google Scholar] [CrossRef]
- Miller, C.T.; Lin, L.; Casper, A.M.; Lim, J.; Thomas, D.G.; Orringer, M.B.; Chang, A.C.; Chambers, A.F.; Giordano, T.J.; Glover, T.W.; et al. Genomic amplification of MET with boundaries within fragile site FRA7G and upregulation of MET pathways in esophageal adenocarcinoma. Oncogene 2006, 25, 409–418. [Google Scholar]
- Schmidt, L.; Duh, F.M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; Scherer, S.W.; Zhuang, Z.; Lubensky, I.; Dean, M.; et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 1997, 16, 68–73. [Google Scholar] [CrossRef]
- Comoglio, P.M.; Boccaccio, C. Scatter factors and invasive growth. Semin. Cancer Biol. 2001, 11, 153–165. [Google Scholar] [CrossRef]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; vande Woude, G.F. MET, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003, 4, 915–925. [Google Scholar] [CrossRef]
- Gentile, A.; Trusolino, L.; Comoglio, P.M. The MET tyrosine kinase receptor in development and cancer. Cancer Metastasis Rev. 2008, 27, 85–94. [Google Scholar] [CrossRef]
- Ma, P.C.; Tretiakova, M.S.; MacKinnon, A.C.; Ramnath, N.; Johnson, C.; Dietrich, S.; Seiwert, T.; Christensen, J.G.; Jagadeeswaran, R.; Krausz, T.; et al. Expression and mutational analysis of MET in human solid cancers. Genes Chromosom. Cancer 2008, 47, 1025–1037. [Google Scholar] [CrossRef]
- Ferracini, R.; di Renzo, M.F.; Scotlandi, K.; Baldini, N.; Olivero, M.; Lollini, P.; Cremona, O.; Campanacci, M.; Comoglio, P.M. The MET/HGF receptor is over-expressed in human osteosarcomas and is activated by either a paracrine or an autocrine circuit. Oncogene 1995, 10, 739–749. [Google Scholar]
- Rong, S.; Segal, S.; Anver, M.; Resau, J.H.; vande Woude, G.F. Invasiveness and metastasis of NIH 3T3 cells induced by MET-hepatocyte growth factor/scatter factor autocrine stimulation. Proc. Natl. Acad. Sci. USA 1994, 91, 4731–4735. [Google Scholar] [CrossRef]
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; vande Woude, G. Targeting MET in cancer: Rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [Google Scholar] [CrossRef]
- Arteaga, C.L. HER3 and mutant EGFR meet MET. Nat. Med. 2007, 13, 675–677. [Google Scholar] [CrossRef]
- Moding, E.J.; Kastan, M.B.; Kirsch, D.G. Strategies for optimizing the response of cancer and normal tissues to radiation. Nat. Rev. Drug Discov. 2013, 12, 526–542. [Google Scholar] [CrossRef]
- Meyer, J.L.; Leibel, S.; Roach, M.; Vijayakumar, S. New technologies for the radiotherapy of prostate cancer. A discussion of clinical treatment programs. Front. Radiat. Ther. Oncol. 2007, 40, 315–337. [Google Scholar]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef]
- Baumann, M. Keynote comment: Radiotherapy in the age of molecular oncology. Lancet Oncol. 2006, 7, 786–787. [Google Scholar] [CrossRef]
- Rodrigues, G.; Sanatani, M. Age and comorbidity considerations related to radiotherapy and chemotherapy administration. Semin. Radiat. Oncol. 2012, 22, 277–283. [Google Scholar]
- Plataniotis, G.A.; Dale, R.G. Radio-chemotherapy for bladder cancer: Contribution of chemotherapy on local control. World J. Radiol. 2013, 5, 267–274. [Google Scholar] [CrossRef]
- Zaorsky, N.G.; Trabulsi, E.J.; Lin, J.; Den, R.B. Multimodality therapy for patients with high-risk prostate cancer: Current status and future directions. Semin. Oncol. 2013, 40, 308–321. [Google Scholar] [CrossRef]
- Zhivotovsky, B.; Joseph, B.; Orrenius, S. Tumor radiosensitivity and apoptosis. Exp. Cell Res. 1999, 248, 10–17. [Google Scholar] [CrossRef]
- Hockel, M.; Schlenger, K.; Mitze, M.; Schaffer, U.; Vaupel, P. Hypoxia and radiation response in human tumors. Semin. Radiat. Oncol. 1996, 6, 3–9. [Google Scholar] [CrossRef]
- Baumann, M.; Krause, M.; Dikomey, E.; Dittmann, K.; Dorr, W.; Kasten-Pisula, U.; Rodemann, H.P. EGFR-targeted anti-cancer drugs in radiotherapy: Preclinical evaluation of mechanisms. Radiother. Oncol. 2007, 83, 238–248. [Google Scholar] [CrossRef]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef]
- Abraham, R.T. PI 3-kinase related kinases: ‘Big’ players in stress-induced signaling pathways. DNA Repair (Amst.) 2004, 3, 883–887. [Google Scholar] [CrossRef]
- Shiloh, Y. ATM and ATR: Networking cellular responses to DNA damage. Curr. Opin. Genet. Dev. 2001, 11, 71–77. [Google Scholar] [CrossRef]
- Bartek, J.; Bartkova, J.; Lukas, J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene 2007, 26, 7773–7779. [Google Scholar]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef]
- Lavin, M.F.; Kozlov, S. ATM activation and DNA damage response. Cell Cycle 2007, 6, 931–942. [Google Scholar] [CrossRef]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef]
- Harper, J.W.; Elledge, S.J. The DNA damage response: Ten years after. Mol. Cell 2007, 28, 739–745. [Google Scholar] [CrossRef]
- Andreassen, P.R.; Ho, G.P.; D’Andrea, A.D. DNA damage responses and their many interactions with the replication fork. Carcinogenesis 2006, 27, 883–892. [Google Scholar] [CrossRef]
- Zhou, B.B.; Bartek, J. Targeting the checkpoint kinases: Chemosensitization versus chemoprotection. Nat. Rev. Cancer 2004, 4, 216–225. [Google Scholar] [CrossRef]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [CrossRef]
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.; Lukas, J.; Jackson, S.P. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 37–45. [Google Scholar] [CrossRef]
- Branzei, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef]
- Karlsson-Rosenthal, C.; Millar, J.B. CDC25: Mechanisms of checkpoint inhibition and recovery. Trends Cell Biol. 2006, 16, 285–292. [Google Scholar] [CrossRef]
- Antoni, L.; Sodha, N.; Collins, I.; Garrett, M.D. CHK2 kinase: Cancer susceptibility and cancer therapy—Two sides of the same coin? Nat. Rev. Cancer 2007, 7, 925–936. [Google Scholar] [CrossRef]
- Zaugg, K.; Su, Y.W.; Reilly, P.T.; Moolani, Y.; Cheung, C.C.; Hakem, R.; Hirao, A.; Liu, Q.; Elledge, S.J.; Mak, T.W. Cross-talk between Chk1 and Chk2 in double-mutant thymocytes. Proc. Natl. Acad. Sci. USA 2007, 104, 3805–3810. [Google Scholar] [CrossRef]
- Stiff, T.; Walker, S.A.; Cerosaletti, K.; Goodarzi, A.A.; Petermann, E.; Concannon, P.; O’Driscoll, M.; Jeggo, P.A. ATR-dependent phosphorylation and activation of ATM in response to UV treatment or replication fork stalling. EMBO J. 2006, 25, 5775–5782. [Google Scholar] [CrossRef]
- Ashwell, S.; Zabludoff, S. DNA damage detection and repair pathways—Recent advances with inhibitors of checkpoint kinases in cancer therapy. Clin. Cancer Res. 2008, 14, 4032–4037. [Google Scholar] [CrossRef]
- Ashwell, S.; Janetka, J.W.; Zabludoff, S. Keeping checkpoint kinases in line: New selective inhibitors in clinical trials. Expert Opin. Investig. Drugs 2008, 17, 1331–1340. [Google Scholar] [CrossRef]
- Damia, G.; D’Incalci, M. Targeting DNA repair as a promising approach in cancer therapy. Eur. J. Cancer 2007, 43, 1791–1801. [Google Scholar] [CrossRef]
- Pollard, J.M.; Gatti, R.A. Clinical radiation sensitivity with DNA repair disorders: An overview. Int. J. Radiat. Oncol. Biol. Phys. 2009, 74, 1323–1331. [Google Scholar] [CrossRef]
- Schmidt-Ullrich, R.K.; Mikkelsen, R.B.; Dent, P.; Todd, D.G.; Valerie, K.; Kavanagh, B.D.; Contessa, J.N.; Rorrer, W.K.; Chen, P.B. Radiation-induced proliferation of the human A431 squamous carcinoma cells is dependent on EGFR tyrosine phosphorylation. Oncogene 1997, 15, 1191–1197. [Google Scholar]
- Schmidt-Ullrich, R.K.; Valerie, K.; Fogleman, P.B.; Walters, J. Radiation-induced autophosphorylation of epidermal growth factor receptor in human malignant mammary and squamous epithelial cells. Radiat. Res. 1996, 145, 81–85. [Google Scholar] [CrossRef]
- Contessa, J.N.; Reardon, D.B.; Todd, D.; Dent, P.; Mikkelsen, R.B.; Valerie, K.; Bowers, G.D.; Schmidt-Ullrich, R.K. The inducible expression of dominant-negative epidermal growth factor receptor-CD533 results in radiosensitization of human mammary carcinoma cells. Clin. Cancer Res. 1999, 5, 405–411. [Google Scholar]
- Dent, P.; Reardon, D.B.; Park, J.S.; Bowers, G.; Logsdon, C.; Valerie, K.; Schmidt-Ullrich, R. Radiation-induced release of transforming growth factor alpha activates the epidermal growth factor receptor and mitogen-activated protein kinase pathway in carcinoma cells, leading to increased proliferation and protection from radiation-induced cell death. Mol. Biol. Cell 1999, 10, 2493–2506. [Google Scholar] [CrossRef]
- Lammering, G.; Hewit, T.H.; Hawkins, W.T.; Contessa, J.N.; Reardon, D.B.; Lin, P.S.; Valerie, K.; Dent, P.; Mikkelsen, R.B.; Schmidt-Ullrich, R.K. Epidermal growth factor receptor as a genetic therapy target for carcinoma cell radiosensitization. J. Natl. Cancer Inst. 2001, 93, 921–929. [Google Scholar] [CrossRef]
- Bandyopadhyay, D.; Mandal, M.; Adam, L.; Mendelsohn, J.; Kumar, R. Physical interaction between epidermal growth factor receptor and DNA-dependent protein kinase in mammalian cells. J. Biol. Chem. 1998, 273, 1568–1573. [Google Scholar] [CrossRef]
- Huang, S.M.; Harari, P.M. Modulation of radiation response after epidermal growth factor receptor blockade in squamous cell carcinomas: Inhibition of damage repair, cell cycle kinetics, and tumor angiogenesis. Clin. Cancer Res. 2000, 6, 2166–2174. [Google Scholar]
- Yacoub, A.; McKinstry, R.; Hinman, D.; Chung, T.; Dent, P.; Hagan, M.P. Epidermal growth factor and ionizing radiation up-regulate the DNA repair genes XRCC1 and ERCC1 in DU145 and LNCaP prostate carcinoma through MAPK signaling. Radiat. Res. 2003, 159, 439–452. [Google Scholar] [CrossRef]
- Dittmann, K.; Mayer, C.; Rodemann, H.P. Inhibition of radiation-induced EGFR nuclear import by C225 (Cetuximab) suppresses DNA-PK activity. Radiother. Oncol. 2005, 76, 157–161. [Google Scholar] [CrossRef]
- Dittmann, K.; Mayer, C.; Fehrenbacher, B.; Schaller, M.; Raju, U.; Milas, L.; Chen, D.J.; Kehlbach, R.; Rodemann, H.P. Radiation-induced epidermal growth factor receptor nuclear import is linked to activation of DNA-dependent protein kinase. J. Biol. Chem. 2005, 280, 31182–31189. [Google Scholar] [CrossRef]
- Li, L.; Wang, H.; Yang, E.S.; Arteaga, C.L.; Xia, F. Erlotinib attenuates homologous recombinational repair of chromosomal breaks in human breast cancer cells. Cancer Res. 2008, 68, 9141–9146. [Google Scholar] [CrossRef]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Azarnia, N.; Shin, D.M.; Cohen, R.B.; Jones, C.U.; Sur, R.; Raben, D.; Jassem, J.; et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2006, 354, 567–578. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Bell, D.W.; Lynch, T.J.; Haserlat, S.M.; Harris, P.L.; Okimoto, R.A.; Brannigan, B.W.; Sgroi, D.C.; Muir, B.; Riemenschneider, M.J.; Iacona, R.B.; et al. Epidermal growth factor receptor mutations and gene amplification in non-small-cell lung cancer: Molecular analysis of the IDEAL/INTACT gefitinib trials. J. Clin. Oncol. 2005, 23, 8081–8092. [Google Scholar] [CrossRef]
- Shigematsu, H.; Gazdar, A.F. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int. J. Cancer 2006, 118, 257–262. [Google Scholar] [CrossRef]
- Amann, J.; Kalyankrishna, S.; Massion, P.P.; Ohm, J.E.; Girard, L.; Shigematsu, H.; Peyton, M.; Juroske, D.; Huang, Y.; Stuart Salmon, J.; et al. Aberrant epidermal growth factor receptor signaling and enhanced sensitivity to EGFR inhibitors in lung cancer. Cancer Res. 2005, 65, 226–235. [Google Scholar]
- Arao, T.; Fukumoto, H.; Takeda, M.; Tamura, T.; Saijo, N.; Nishio, K. Small in-frame deletion in the epidermal growth factor receptor as a target for ZD6474. Cancer Res. 2004, 64, 9101–9104. [Google Scholar] [CrossRef]
- Engelman, J.A.; Janne, P.A.; Mermel, C.; Pearlberg, J.; Mukohara, T.; Fleet, C.; Cichowski, K.; Johnson, B.E.; Cantley, L.C. ErbB-3 mediates phosphoinositide 3-kinase activity in gefitinib-sensitive non-small cell lung cancer cell lines. Proc. Natl. Acad. Sci. USA 2005, 102, 3788–3793. [Google Scholar] [CrossRef]
- Sordella, R.; Bell, D.W.; Haber, D.A.; Settleman, J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science 2004, 305, 1163–1167. [Google Scholar] [CrossRef]
- Das, A.K.; Sato, M.; Story, M.D.; Peyton, M.; Graves, R.; Redpath, S.; Girard, L.; Gazdar, A.F.; Shay, J.W.; Minna, J.D.; et al. Non-small-cell lung cancers with kinase domain mutations in the epidermal growth factor receptor are sensitive to ionizing radiation. Cancer Res. 2006, 66, 9601–9608. [Google Scholar] [CrossRef]
- Das, A.K.; Chen, B.P.; Story, M.D.; Sato, M.; Minna, J.D.; Chen, D.J.; Nirodi, C.S. Somatic mutations in the tyrosine kinase domain of epidermal growth factor receptor (EGFR) abrogate EGFR-mediated radioprotection in non-small cell lung carcinoma. Cancer Res. 2007, 67, 5267–5274. [Google Scholar] [CrossRef]
- Miller, B.S.; Yee, D. Type I insulin-like growth factor receptor as a therapeutic target in cancer. Cancer Res. 2005, 65, 10123–10127. [Google Scholar] [CrossRef]
- Larsson, O.; Girnita, A.; Girnita, L. Role of insulin-like growth factor 1 receptor signalling in cancer. Br. J. Cancer 2005, 92, 2097–2101. [Google Scholar] [CrossRef]
- Pollak, M.N.; Schernhammer, E.S.; Hankinson, S.E. Insulin-like growth factors and neoplasia. Nat. Rev. Cancer 2004, 4, 505–518. [Google Scholar] [CrossRef]
- Tao, Y.; Pinzi, V.; Bourhis, J.; Deutsch, E. Mechanisms of disease: Signaling of the insulin-like growth factor 1 receptor pathway—Therapeutic perspectives in cancer. Nat. Clin. Pract. Oncol. 2007, 4, 591–602. [Google Scholar]
- Werner, H.; Le Roith, D. The insulin-like growth factor-I receptor signaling pathways are important for tumorigenesis and inhibition of apoptosis. Crit. Rev. Oncog. 1997, 8, 71–92. [Google Scholar] [CrossRef]
- Turner, B.C.; Haffty, B.G.; Narayanan, L.; Yuan, J.; Havre, P.A.; Gumbs, A.A.; Kaplan, L.; Burgaud, J.L.; Carter, D.; Baserga, R.; et al. Insulin-like growth factor-I receptor overexpression mediates cellular radioresistance and local breast cancer recurrence after lumpectomy and radiation. Cancer Res. 1997, 57, 3079–3083. [Google Scholar]
- Macaulay, V.M.; Salisbury, A.J.; Bohula, E.A.; Playford, M.P.; Smorodinsky, N.I.; Shiloh, Y. Downregulation of the type 1 insulin-like growth factor receptor in mouse melanoma cells is associated with enhanced radiosensitivity and impaired activation of Atm kinase. Oncogene 2001, 20, 4029–4040. [Google Scholar] [CrossRef]
- Peretz, S.; Jensen, R.; Baserga, R.; Glazer, P.M. ATM-dependent expression of the insulin-like growth factor-I receptor in a pathway regulating radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 1676–1681. [Google Scholar] [CrossRef]
- Rotman, G.; Shiloh, Y. ATM: A mediator of multiple responses to genotoxic stress. Oncogene 1999, 18, 6135–6144. [Google Scholar] [CrossRef]
- Trojanek, J.; Ho, T.; del Valle, L.; Nowicki, M.; Wang, J.Y.; Lassak, A.; Peruzzi, F.; Khalili, K.; Skorski, T.; Reiss, K. Role of the insulin-like growth factor I/insulin receptor substrate 1 axis in Rad51 trafficking and DNA repair by homologous recombination. Mol. Cell Biol. 2003, 23, 7510–7524. [Google Scholar] [CrossRef]
- Allen, G.W.; Saba, C.; Armstrong, E.A.; Huang, S.M.; Benavente, S.; Ludwig, D.L.; Hicklin, D.J.; Harari, P.M. Insulin-like growth factor-I receptor signaling blockade combined with radiation. Cancer Res. 2007, 67, 1155–1162. [Google Scholar] [CrossRef]
- Riesterer, O.; Yang, Q.; Raju, U.; Torres, M.; Molkentine, D.; Patel, N.; Valdecanas, D.; Milas, L.; Ang, K.K. Combination of anti-IGF-1R antibody A12 and ionizing radiation in upper respiratory tract cancers. Int. J. Radiat. Oncol. Biol. Phys. 2011, 79, 1179–1187. [Google Scholar] [CrossRef]
- Laird, A.D.; Vajkoczy, P.; Shawver, L.K.; Thurnher, A.; Liang, C.; Mohammadi, M.; Schlessinger, J.; Ullrich, A.; Hubbard, S.R.; Blake, R.A.; et al. Cherrington SU6668 is a potent antiangiogenic and antitumor agent that induces regression of established tumors. Cancer Res. 2000, 60, 4152–4160. [Google Scholar]
- Griffin, R.J.; Williams, B.W.; Wild, R.; Cherrington, J.M.; Park, H.; Song, C.W. Simultaneous inhibition of the receptor kinase activity of vascular endothelial, fibroblast, and platelet-derived growth factors suppresses tumor growth and enhances tumor radiation response. Cancer Res. 2002, 62, 1702–1706. [Google Scholar]
- Ning, S.; Laird, D.; Cherrington, J.M.; Knox, S.J. The antiangiogenic agents SU5416 and SU6668 increase the antitumor effects of fractionated irradiation. Radiat. Res. 2002, 157, 45–51. [Google Scholar] [CrossRef]
- Abdollahi, A.; Lipson, K.E.; Han, X.; Krempien, R.; Trinh, T.; Weber, K.J.; Hahnfeldt, P.; Hlatky, L.; Debus, J.; Howlett, A.R.; et al. SU5416 and SU6668 attenuate the angiogenic effects of radiation-induced tumor cell growth factor production and amplify the direct anti-endothelial action of radiation in vitro. Cancer Res. 2003, 63, 3755–3763. [Google Scholar]
- Fong, T.A.; Shawver, L.K.; Sun, L.; Tang, C.; App, H.; Powell, T.J.; Kim, Y.H.; Schreck, R.; Wang, X.; Risau, W.; et al. SU5416 is a potent and selective inhibitor of the vascular endothelial growth factor receptor (Flk-1/KDR) that inhibits tyrosine kinase catalysis, tumor vascularization, and growth of multiple tumor types. Cancer Res. 1999, 59, 99–106. [Google Scholar]
- Mendel, D.B.; Laird, A.D.; Smolich, B.D.; Blake, R.A.; Liang, C.; Hannah, A.L.; Shaheen, R.M.; Ellis, L.M.; Weitman, S.; Shawver, L.K.; et al. Development of SU5416, a selective small molecule inhibitor of VEGF receptor tyrosine kinase activity, as an anti-angiogenesis agent. Anticancer Drug Des. 2000, 15, 29–41. [Google Scholar]
- Timke, C.; Zieher, H.; Roth, A.; Hauser, K.; Lipson, K.E.; Weber, K.J.; Debus, J.; Abdollahi, A.; Huber, P.E. Combination of vascular endothelial growth factor receptor/platelet-derived growth factor receptor inhibition markedly improves radiation tumor therapy. Clin. Cancer Res. 2008, 14, 2210–2219. [Google Scholar] [CrossRef]
- De Bacco, F.; Luraghi, P.; Medico, E.; Reato, G.; Girolami, F.; Perera, T.; Gabriele, P.; Comoglio, P.M.; Boccaccio, C. Induction of MET by ionizing radiation and its role in radioresistance and invasive growth of cancer. J. Natl. Cancer Inst. 2011, 103, 645–661. [Google Scholar] [CrossRef]
- Medova, M.; Aebersold, D.M.; Blank-Liss, W.; Streit, B.; Medo, M.; Aebi, S.; Zimmer, Y. MET inhibition results in DNA breaks and synergistically sensitizes tumor cells to DNA-damaging agents potentially by breaching a damage-induced checkpoint arrest. Genes Cancer 2010, 1, 1053–1062. [Google Scholar] [CrossRef]
- Ganapathipillai, S.S.; Medova, M.; Aebersold, D.M.; Manley, P.W.; Berthou, S.; Streit, B.; Blank-Liss, W.; Greiner, R.H.; Rothen-Rutishauser, B.; Zimmer, Y. Coupling of mutated Met variants to DNA repair via Abl and Rad51. Cancer Res. 2008, 68, 5769–5777. [Google Scholar] [CrossRef]
- Medova, M.; Aebersold, D.M.; Zimmer, Y. MET inhibition in tumor cells by PHA665752 impairs homologous recombination repair of DNA double strand breaks. Int. J. Cancer 2012, 130, 728–734. [Google Scholar] [CrossRef]
- Welsh, J.W.; Mahadevan, D.; Ellsworth, R.; Cooke, L.; Bearss, D.; Stea, B. The c-Met receptor tyrosine kinase inhibitor MP470 radiosensitizes glioblastoma cells. Radiat. Oncol. 2009, 4, 69. [Google Scholar] [CrossRef]
- Fan, S.; Wang, J.A.; Yuan, R.Q.; Rockwell, S.; Andres, J.; Zlatapolskiy, A.; Goldberg, I.D.; Rosen, E.M. Scatter factor protects epithelial and carcinoma cells against apoptosis induced by DNA-damaging agents. Oncogene 1998, 17, 131–141. [Google Scholar]
- Fan, S.; Ma, Y.X.; Wang, J.A.; Yuan, R.Q.; Meng, Q.; Cao, Y.; Laterra, J.J.; Goldberg, I.D.; Rosen, E.M. The cytokine hepatocyte growth factor/scatter factor inhibits apoptosis and enhances DNA repair by a common mechanism involving signaling through phosphatidyl inositol 3' kinase. Oncogene 2000, 19, 2212–2223. [Google Scholar] [CrossRef]
- Fan, S.; Ma, Y.X.; Gao, M.; Yuan, R.Q.; Meng, Q.; Goldberg, I.D.; Rosen, E.M. The multisubstrate adapter Gab1 regulates hepatocyte growth factor (scatter factor)-c-Met signaling for cell survival and DNA repair. Mol. Cell Biol. 2001, 21, 4968–4984. [Google Scholar] [CrossRef]
- Maroun, C.R.; Moscatello, D.K.; Naujokas, M.A.; Holgado-Madruga, M.; Wong, A.J.; Park, M. A conserved inositol phospholipid binding site within the pleckstrin homology domain of the Gab1 docking protein is required for epithelial morphogenesis. J. Biol. Chem. 1999, 274, 31719–31726. [Google Scholar] [CrossRef]
- Yuan, R.; Fan, S.; Achary, M.; Stewart, D.M.; Goldberg, I.D.; Rosen, E.M. Altered gene expression pattern in cultured human breast cancer cells treated with hepatocyte growth factor/scatter factor in the setting of DNA damage. Cancer Res. 2001, 61, 8022–8031. [Google Scholar]
- Sheng-Hua, C.; Yan-Bin, M.; Zhi-An, Z.; Hong, Z.; Dong-Fu, F.; Zhi-Qiang, L.; Xian-Hou, Y. Radiation-enhanced hepatocyte growth factor secretion in malignant glioma cell lines. Surg. Neurol. 2007, 68, 610–613. [Google Scholar] [CrossRef]
- Qian, L.W.; Mizumoto, K.; Inadome, N.; Nagai, E.; Sato, N.; Matsumoto, K.; Nakamura, T.; Tanaka, M. Radiation stimulates HGF receptor/c-Met expression that leads to amplifying cellular response to HGF stimulation via upregulated receptor tyrosine phosphorylation and MAP kinase activity in pancreatic cancer cells. Int. J. Cancer 2003, 104, 542–549. [Google Scholar] [CrossRef]
- Goetsch, L.; Caussanel, V.; Corvaia, N. Biological significance and targeting of c-Met tyrosine kinase receptor in cancer. Front. Biosci. (Landmark Ed.) 2013, 18, 454–473. [Google Scholar] [CrossRef]
- Peters, S.; Adjei, A.A. MET: A promising anticancer therapeutic target. Nat. Rev. Clin. Oncol. 2012, 9, 314–326. [Google Scholar] [CrossRef]
- Aebersold, D.M.; Kollar, A.; Beer, K.T.; Laissue, J.; Greiner, R.H.; Djonov, V. Involvement of the hepatocyte growth factor/scatter factor receptor c-met and of Bcl-xL in the resistance of oropharyngeal cancer to ionizing radiation. Int. J. Cancer 2001, 96, 41–54. [Google Scholar] [CrossRef]
- Kim, Y.J.; Go, H.; Wu, H.G.; Jeon, Y.K.; Park, S.W.; Lee, S.H. Immunohistochemical study identifying prognostic biomolecular markers in nasopharyngeal carcinoma treated by radiotherapy. Head Neck 2011, 33, 1458–1466. [Google Scholar] [CrossRef]
- Gao, J.; Inagaki, Y.; Song, P.; Qu, X.; Kokudo, N.; Tang, W. Targeting c-Met as a promising strategy for the treatment of hepatocellular carcinoma. Pharmacol. Res. 2012, 65, 23–30. [Google Scholar] [CrossRef]
- Raghav, K.P.; Wang, W.; Liu, S.; Chavez-MacGregor, M.; Meng, X.; Hortobagyi, G.N.; Mills, G.B.; Meric-Bernstam, F.; Blumenschein, G.R., Jr.; Gonzalez-Angulo, A.M. cMET and phospho-cMET protein levels in breast cancers and survival outcomes. Clin. Cancer Res. 2012, 18, 2269–2277. [Google Scholar] [CrossRef]
- Kim, C.H.; Koh, Y.W.; Han, J.H.; Kim, J.W.; Lee, J.S.; Baek, S.J.; Hwang, H.S.; Choi, E.C. c-Met expression as an indicator of survival outcome in patients with oral tongue carcinoma. Head Neck 2010, 32, 1655–1664. [Google Scholar] [CrossRef]
- Di Renzo, M.F.; Olivero, M.; Martone, T.; Maffe, A.; Maggiora, P.; Stefani, A.D.; Valente, G.; Giordano, S.; Cortesina, G.; Comoglio, P.M. Somatic mutations of the MET oncogene are selected during metastatic spread of human HNSC carcinomas. Oncogene 2000, 19, 1547–1555. [Google Scholar] [CrossRef]
- Aebersold, D.M.; Landt, O.; Berthou, S.; Gruber, G.; Beer, K.T.; Greiner, R.H.; Zimmer, Y. Prevalence and clinical impact of Met Y1253D-activating point mutation in radiotherapy-treated squamous cell cancer of the oropharynx. Oncogene 2003, 22, 8519–8523. [Google Scholar] [CrossRef]
- Sattler, M.; Reddy, M.M.; Hasina, R.; Gangadhar, T.; Salgia, R. The role of the c-Met pathway in lung cancer and the potential for targeted therapy. Ther. Adv. Med. Oncol. 2011, 3, 171–184. [Google Scholar] [CrossRef]
- Eder, J.P.; vande Woude, G.F.; Boerner, S.A.; LoRusso, P.M. Novel therapeutic inhibitors of the c-Met signaling pathway in cancer. Clin. Cancer Res. 2009, 15, 2207–2214. [Google Scholar] [CrossRef]
- Lal, B.; Xia, S.; Abounader, R.; Laterra, J. Targeting the c-Met pathway potentiates glioblastoma responses to gamma-radiation. Clin. Cancer Res. 2005, 11, 4479–4486. [Google Scholar] [CrossRef]
- Chu, S.H.; Zhu, Z.A.; Yuan, X.H.; Li, Z.Q.; Jiang, P.C. In vitro and in vivo potentiating the cytotoxic effect of radiation on human U251 gliomas by the c-Met antisense oligodeoxynucleotides. J. Neurooncol. 2006, 80, 143–149. [Google Scholar] [CrossRef]
- Yu, H.; Li, X.; Sun, S.; Gao, X.; Zhou, D. c-Met inhibitor SU11274 enhances the response of the prostate cancer cell line DU145 to ionizing radiation. Biochem. Biophys. Res. Commun. 2012, 427, 659–665. [Google Scholar] [CrossRef]
- Bhardwaj, V.; Zhan, Y.; Cortez, M.A.; Ang, K.K.; Molkentine, D.; Munshi, A.; Raju, U.; Komaki, R.; Heymach, J.V.; Welsh, J. C-Met inhibitor MK-8003 radiosensitizes c-Met-expressing non-small-cell lung cancer cells with radiation-induced c-Met-expression. J. Thorac. Oncol. 2012, 7, 1211–1217. [Google Scholar] [CrossRef]
- Lin, C.I.; Whang, E.E.; Donner, D.B.; Du, J.; Lorch, J.; He, F.; Jiang, X.; Price, B.D.; Moore, F.D., Jr.; Ruan, D.T. Autophagy induction with RAD001 enhances chemosensitivity and radiosensitivity through Met inhibition in papillary thyroid cancer. Mol. Cancer Res. 2010, 8, 1217–1226. [Google Scholar] [CrossRef]
- Buchanan, I.M.; Scott, T.; Tandle, A.T.; Burgan, W.E.; Burgess, T.L.; Tofilon, P.J.; Camphausen, K. Radiosensitization of glioma cells by modulation of Met signalling with the hepatocyte growth factor neutralizing antibody, AMG102. J. Cell. Mol. Med. 2011, 15, 1999–2006. [Google Scholar] [CrossRef]
- Khanna, K.K.; Jackson, S.P. DNA double-strand breaks: Signaling, repair and the cancer connection. Nat. Genet. 2001, 27, 247–254. [Google Scholar] [CrossRef]
- Chen, G.; Yuan, S.S.; Liu, W.; Xu, Y.; Trujillo, K.; Song, B.; Cong, F.; Goff, S.P.; Wu, Y.; Arlinghaus, R.; et al. Radiation-induced assembly of Rad51 and Rad52 recombination complex requires ATM and c-Abl. J. Biol. Chem. 1999, 274, 12748–12752. [Google Scholar] [CrossRef]
- Takizawa, Y.; Kinebuchi, T.; Kagawa, W.; Yokoyama, S.; Shibata, T.; Kurumizaka, H. Mutational analyses of the human Rad51-Tyr315 residue, a site for phosphorylation in leukaemia cells. Genes Cells 2004, 9, 781–790. [Google Scholar] [CrossRef]
- Cook, P.J.; Ju, B.G.; Telese, F.; Wang, X.; Glass, C.K.; Rosenfeld, M.G. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature 2009, 458, 591–596. [Google Scholar] [CrossRef]
- Xiao, A.; Li, H.; Shechter, D.; Ahn, S.H.; Fabrizio, L.A.; Erdjument-Bromage, H.; Ishibe-Murakami, S.; Wang, B.; Tempst, P.; Hofmann, K.; et al. WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature 2009, 457, 57–62. [Google Scholar] [CrossRef]
- Brahimi-Horn, M.C.; Chiche, J.; Pouyssegur, J. Hypoxia and cancer. J. Mol. Med. (Berl.) 2007, 85, 1301–1307. [Google Scholar] [CrossRef]
- Brahimi-Horn, M.C.; Chiche, J.; Pouyssegur, J. Hypoxia signalling controls metabolic demand. Curr. Opin. Cell Biol. 2007, 19, 223–229. [Google Scholar] [CrossRef]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef]
- Brizel, D.M.; Sibley, G.S.; Prosnitz, L.R.; Scher, R.L.; Dewhirst, M.W. Tumor hypoxia adversely affects the prognosis of carcinoma of the head and neck. Int. J. Radiat. Oncol. Biol. Phys. 1997, 38, 285–289. [Google Scholar] [CrossRef]
- Hockel, M.; Schlenger, K.; Aral, B.; Mitze, M.; Schaffer, U.; Vaupel, P. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res. 1996, 56, 4509–4515. [Google Scholar]
- Kaanders, J.H.; Wijffels, K.I.; Marres, H.A.; Ljungkvist, A.S.; Pop, L.A.; van den Hoogen, F.J.; de Wilde, P.C.; Bussink, J.; Raleigh, J.A.; van der Kogel, A.J. Pimonidazole binding and tumor vascularity predict for treatment outcome in head and neck cancer. Cancer Res. 2002, 62, 7066–7074. [Google Scholar]
- Nordsmark, M.; Bentzen, S.M.; Rudat, V.; Brizel, D.; Lartigau, E.; Stadler, P.; Becker, A.; Adam, M.; Molls, M.; Dunst, J.; et al. Prognostic value of tumor oxygenation in 397 head and neck tumors after primary radiation therapy. An international multi-center study. Radiother. Oncol. 2005, 77, 18–24. [Google Scholar] [CrossRef]
- Vaupel, P. Tumor microenvironmental physiology and its implications for radiation oncology. Semin. Radiat. Oncol. 2004, 14, 198–206. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef]
- Bindra, R.S.; Schaffer, P.J.; Meng, A.; Woo, J.; Maseide, K.; Roth, M.E.; Lizardi, P.; Hedley, D.W.; Bristow, R.G.; Glazer, P.M. Alterations in DNA repair gene expression under hypoxia: Elucidating the mechanisms of hypoxia-induced genetic instability. Ann. N. Y. Acad. Sci. 2005, 1059, 184–195. [Google Scholar]
- Tang, N.; Wang, L.; Esko, J.; Giordano, F.J.; Huang, Y.; Gerber, H.P.; Ferrara, N.; Johnson, R.S. Loss of HIF-1alpha in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell 2004, 6, 485–495. [Google Scholar] [CrossRef]
- Gustafsson, M.V.; Zheng, X.; Pereira, T.; Gradin, K.; Jin, S.; Lundkvist, J.; Ruas, J.L.; Poellinger, L.; Lendahl, U.; Bondesson, M. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev. Cell 2005, 9, 617–628. [Google Scholar] [CrossRef]
- Tredan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug resistance and the solid tumor microenvironment. J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar] [CrossRef]
- Hockel, M.; Vaupel, P. Biological consequences of tumor hypoxia. Semin. Oncol. 2001, 28, 36–41. [Google Scholar] [CrossRef]
- Hockel, M.; Vaupel, P. Tumor hypoxia: Definitions and current clinical, biologic, and molecular aspects. J. Natl. Cancer Inst. 2001, 93, 266–276. [Google Scholar] [CrossRef]
- Nordsmark, M.; Overgaard, M.; Overgaard, J. Pretreatment oxygenation predicts radiation response in advanced squamous cell carcinoma of the head and neck. Radiother. Oncol. 1996, 41, 31–39. [Google Scholar]
- Gatenby, R.A.; Kessler, H.B.; Rosenblum, J.S.; Coia, L.R.; Moldofsky, P.J.; Hartz, W.H.; Broder, G.J. Oxygen distribution in squamous cell carcinoma metastases and its relationship to outcome of radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 1988, 14, 831–838. [Google Scholar] [CrossRef]
- Brizel, D.M.; Dodge, R.K.; Clough, R.W.; Dewhirst, M.W. Oxygenation of head and neck cancer: Changes during radiotherapy and impact on treatment outcome. Radiother. Oncol. 1999, 53, 113–117. [Google Scholar] [CrossRef]
- Aebersold, D.M.; Burri, P.; Beer, K.T.; Laissue, J.; Djonov, V.; Greiner, R.H.; Semenza, G.L. Expression of hypoxia-inducible factor-1alpha: A novel predictive and prognostic parameter in the radiotherapy of oropharyngeal cancer. Cancer Res. 2001, 61, 2911–2916. [Google Scholar]
- Bachtiary, B.; Schindl, M.; Potter, R.; Dreier, B.; Knocke, T.H.; Hainfellner, J.A.; Horvat, R.; Birner, P. Overexpression of hypoxia-inducible factor 1alpha indicates diminished response to radiotherapy and unfavorable prognosis in patients receiving radical radiotherapy for cervical cancer. Clin. Cancer Res. 2003, 9, 2234–2240. [Google Scholar]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef]
- Hara, S.; Nakashiro, K.; Klosek, S.K.; Ishikawa, T.; Shintani, S.; Hamakawa, H. Hypoxia enhances c-Met/HGF receptor expression and signaling by activating HIF-1alpha in human salivary gland cancer cells. Oral Oncol. 2006, 42, 593–598. [Google Scholar] [CrossRef]
- Chen, H.H.; Su, W.C.; Lin, P.W.; Guo, H.R.; Lee, W.Y. Hypoxia-inducible factor-1alpha correlates with MET and metastasis in node-negative breast cancer. Breast Cancer Res. Treat. 2007, 103, 167–175. [Google Scholar]
- Scarpino, S.; Cancellario d'Alena, F.; Di Napoli, A.; Pasquini, A.; Marzullo, A.; Ruco, L.P. Increased expression of Met protein is associated with up-regulation of hypoxia inducible factor-1 (HIF-1) in tumour cells in papillary carcinoma of the thyroid. J. Pathol. 2004, 202, 352–358. [Google Scholar] [CrossRef]
- Ide, T.; Kitajima, Y.; Miyoshi, A.; Ohtsuka, T.; Mitsuno, M.; Ohtaka, K.; Koga, Y.; Miyazaki, K. Tumor-stromal cell interaction under hypoxia increases the invasiveness of pancreatic cancer cells through the hepatocyte growth factor/c-Met pathway. Int. J. Cancer 2006, 119, 2750–2759. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Medová, M.; Aebersold, D.M.; Zimmer, Y. The Molecular Crosstalk between the MET Receptor Tyrosine Kinase and the DNA Damage Response—Biological and Clinical Aspects. Cancers 2014, 6, 1-27. https://doi.org/10.3390/cancers6010001
Medová M, Aebersold DM, Zimmer Y. The Molecular Crosstalk between the MET Receptor Tyrosine Kinase and the DNA Damage Response—Biological and Clinical Aspects. Cancers. 2014; 6(1):1-27. https://doi.org/10.3390/cancers6010001
Chicago/Turabian StyleMedová, Michaela, Daniel M. Aebersold, and Yitzhak Zimmer. 2014. "The Molecular Crosstalk between the MET Receptor Tyrosine Kinase and the DNA Damage Response—Biological and Clinical Aspects" Cancers 6, no. 1: 1-27. https://doi.org/10.3390/cancers6010001
APA StyleMedová, M., Aebersold, D. M., & Zimmer, Y. (2014). The Molecular Crosstalk between the MET Receptor Tyrosine Kinase and the DNA Damage Response—Biological and Clinical Aspects. Cancers, 6(1), 1-27. https://doi.org/10.3390/cancers6010001