Circulating Tumor Cell Detection and Capture by Photoacoustic Flow Cytometry in Vivo and ex Vivo

Abstract
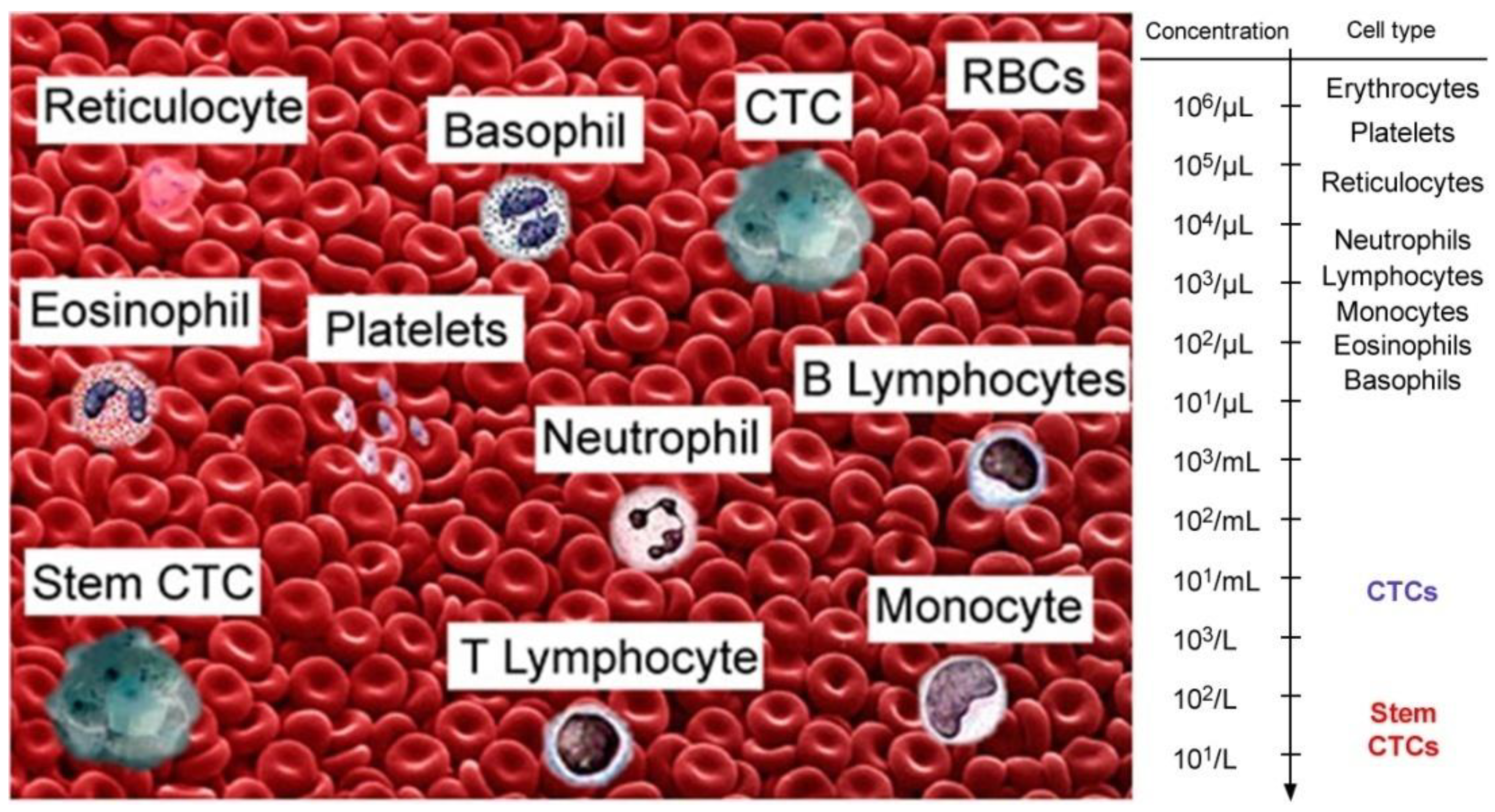
:1. Introduction
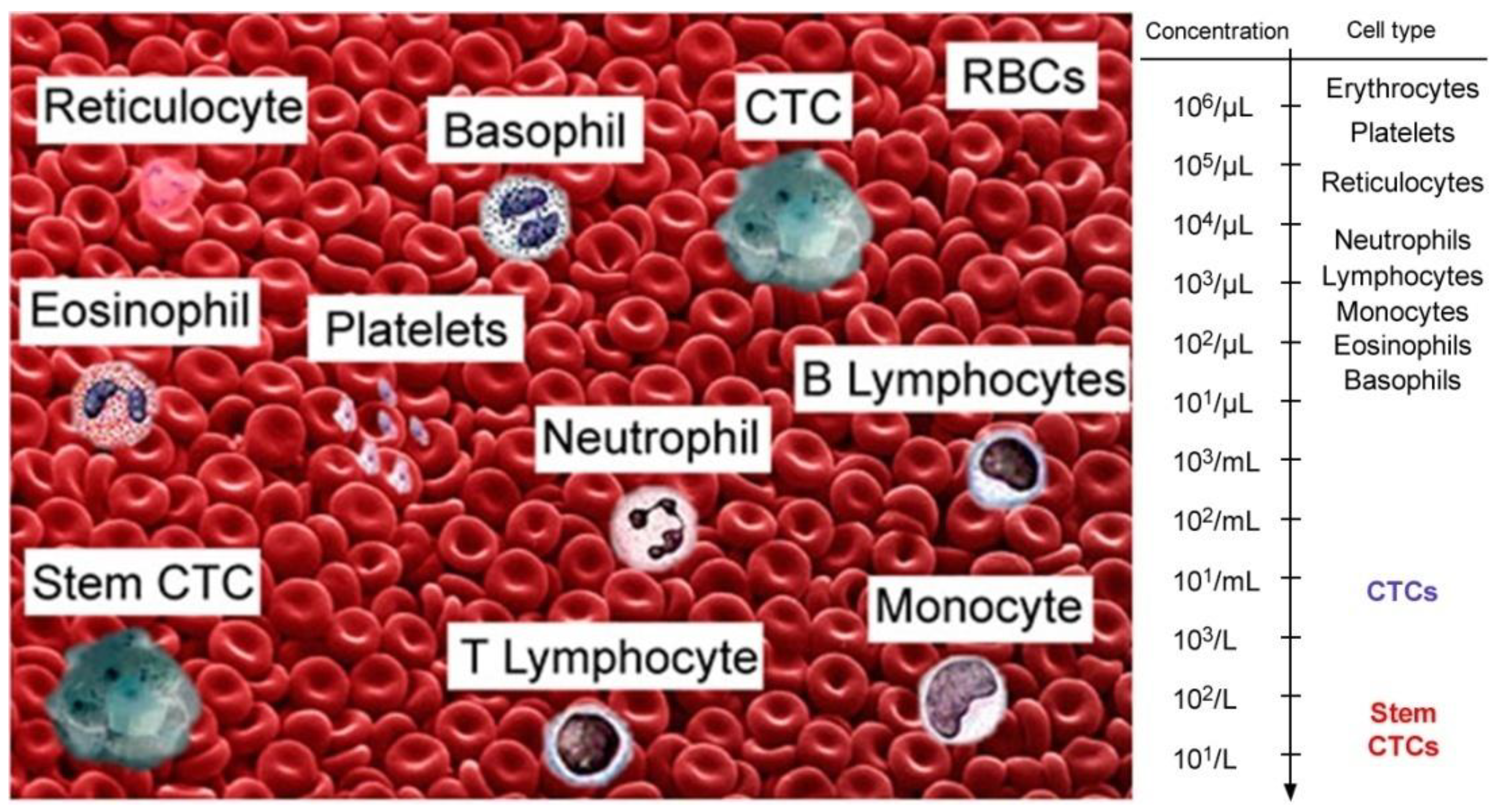
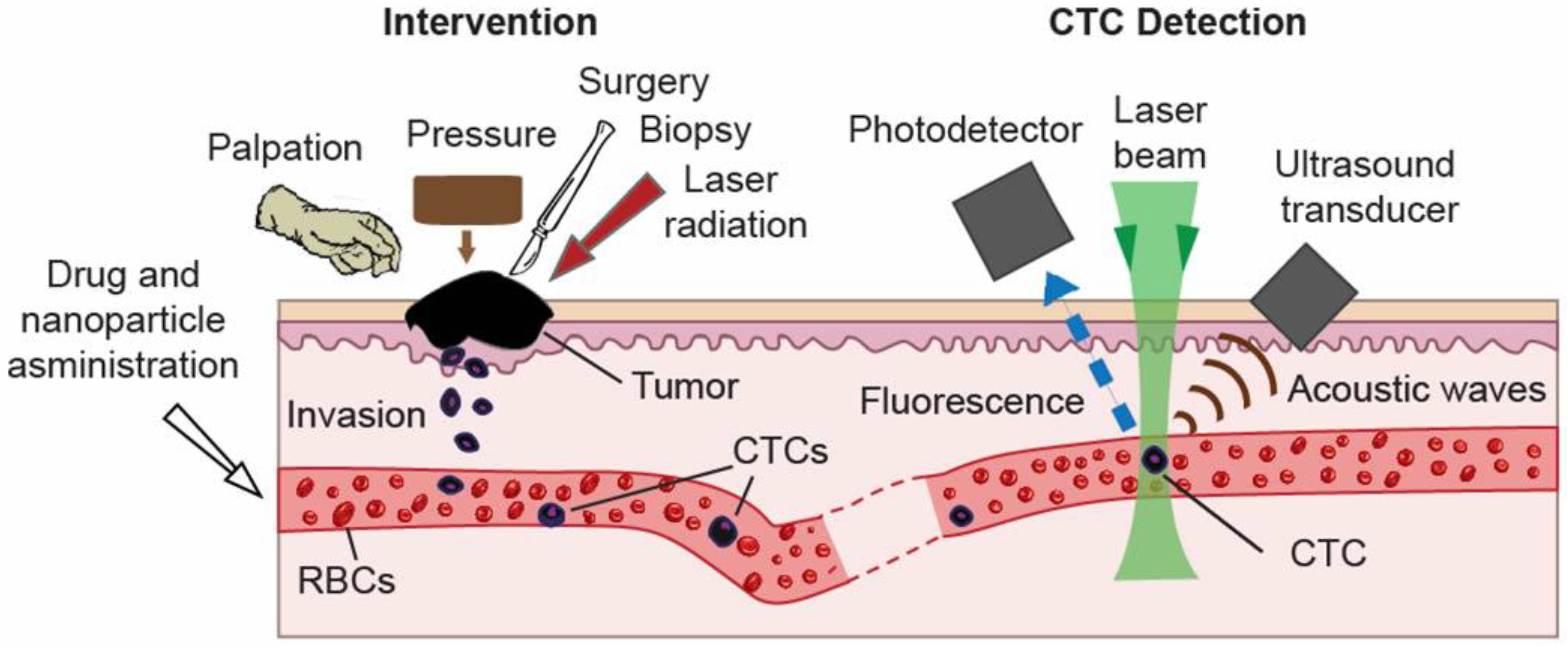
2. In Vivo Flow Cytometry
2.1. General Schematics of in Vivo Flow Cytometry
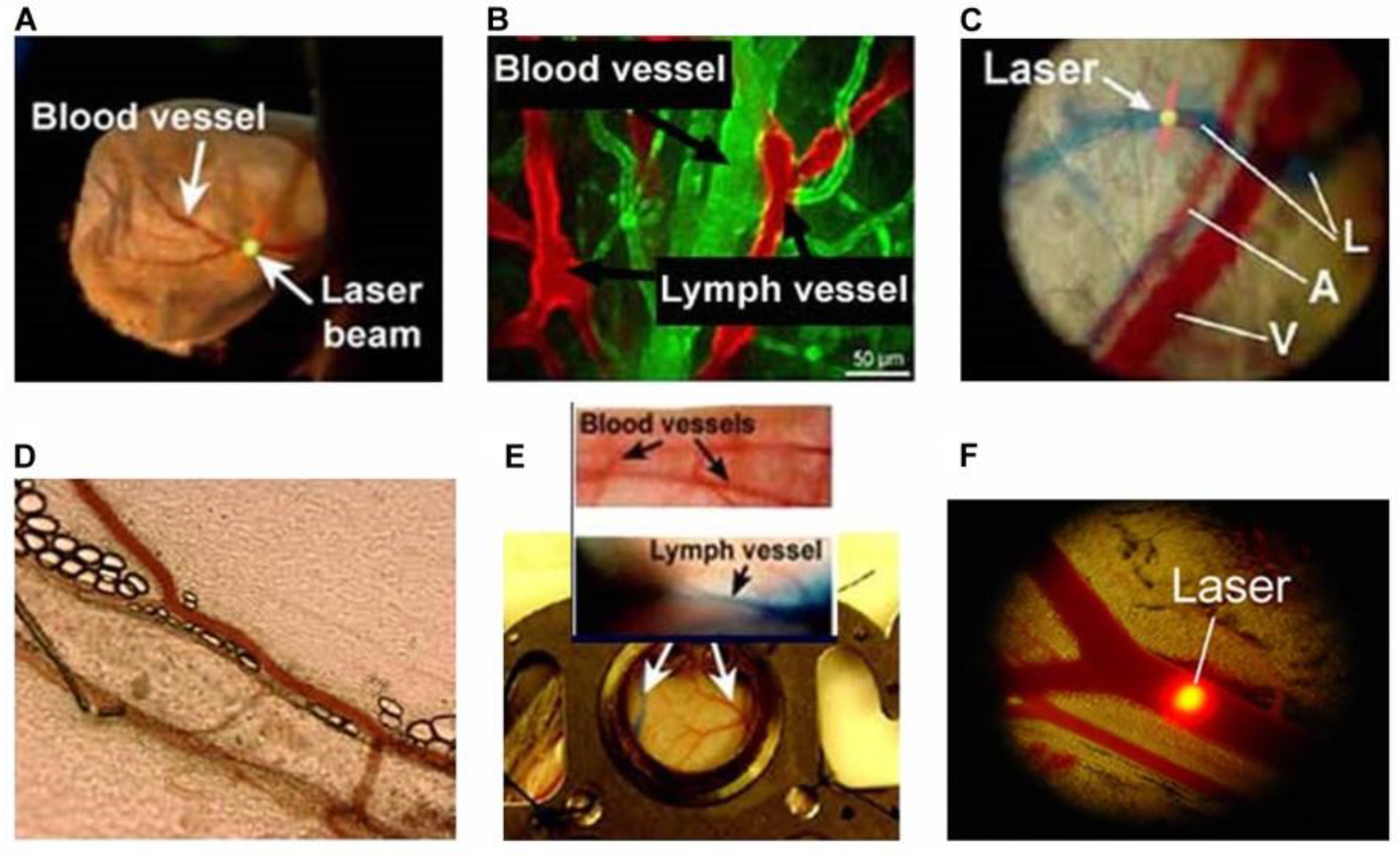
2.2. Animal Models

3. PA Flow Cytometry (PAFC)
3.1. Principle of PAFC
3.2. General Schematics of PAFC
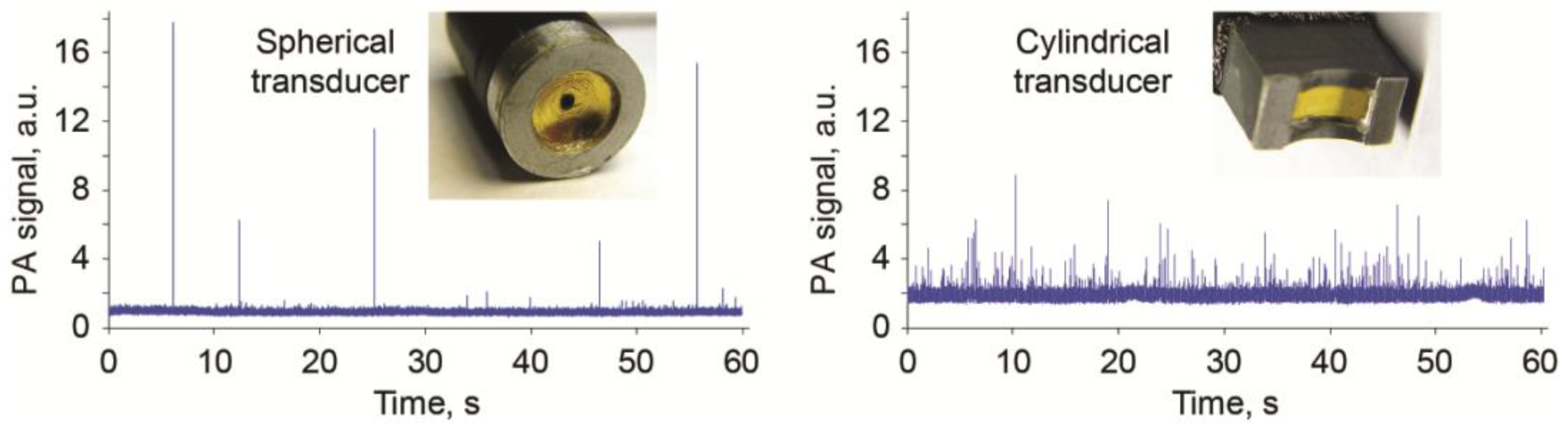
3.3. PAFC with Optical and Acoustic Resolution
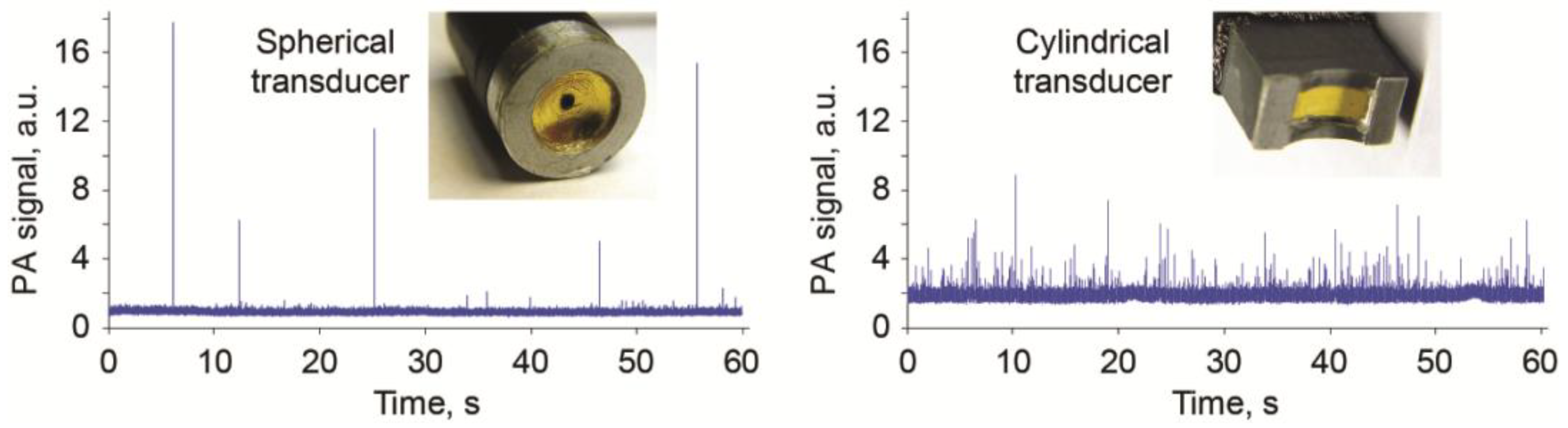
3.4. Nonlinear PAFC for CTC Contrast Enhancement

3.5. Real-Time Spectral Identification of CTCs
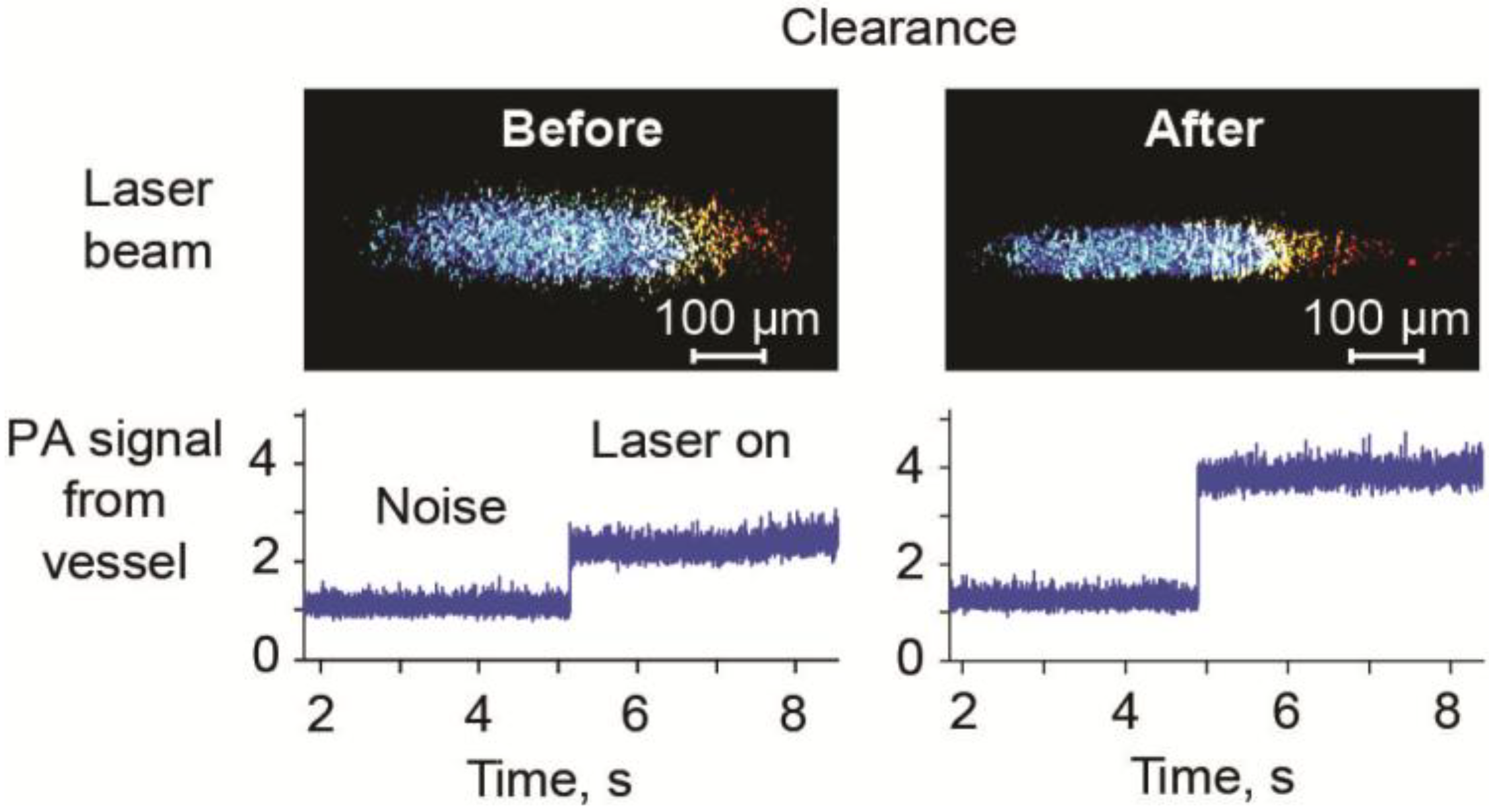
3.6. Optical Clearance in PAFC
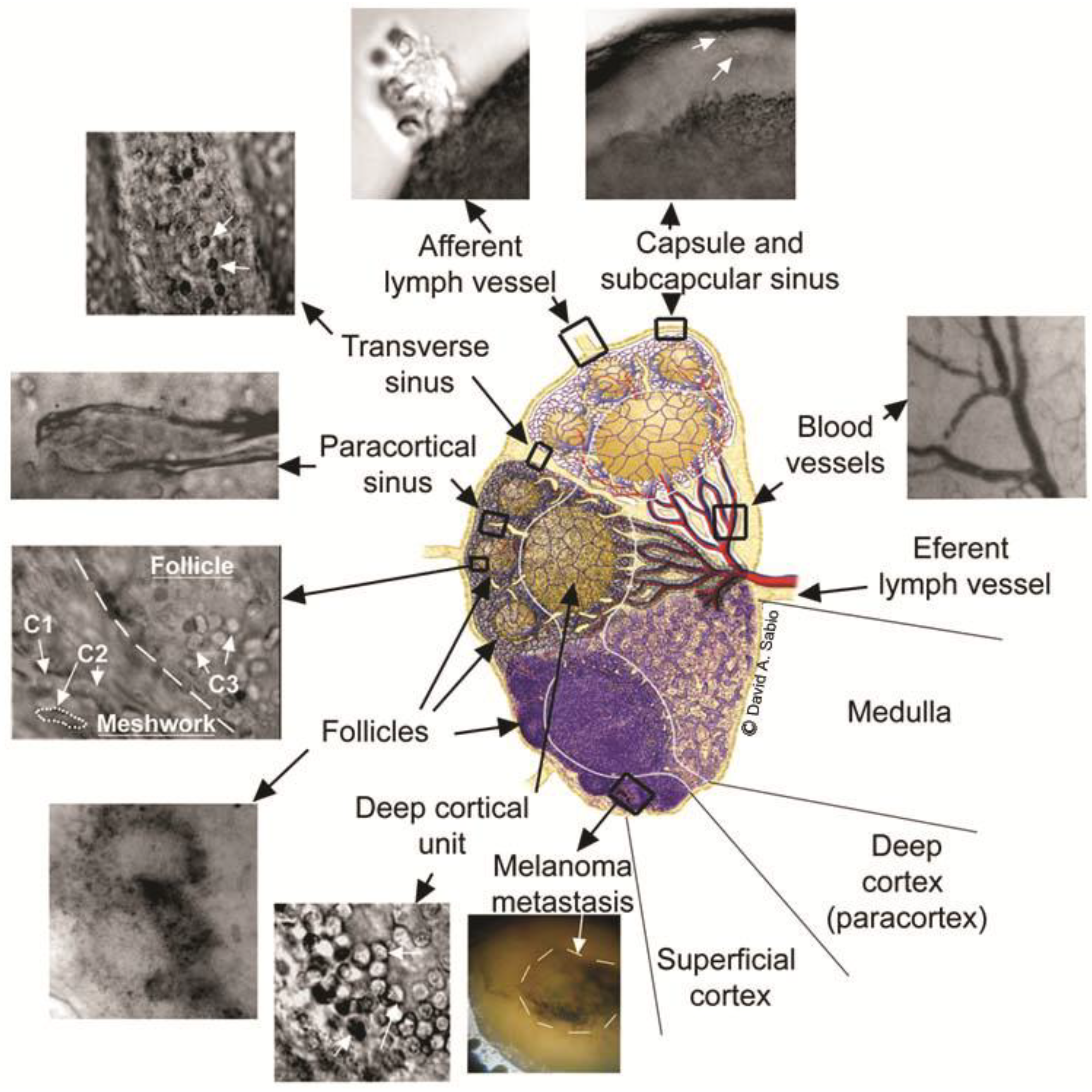
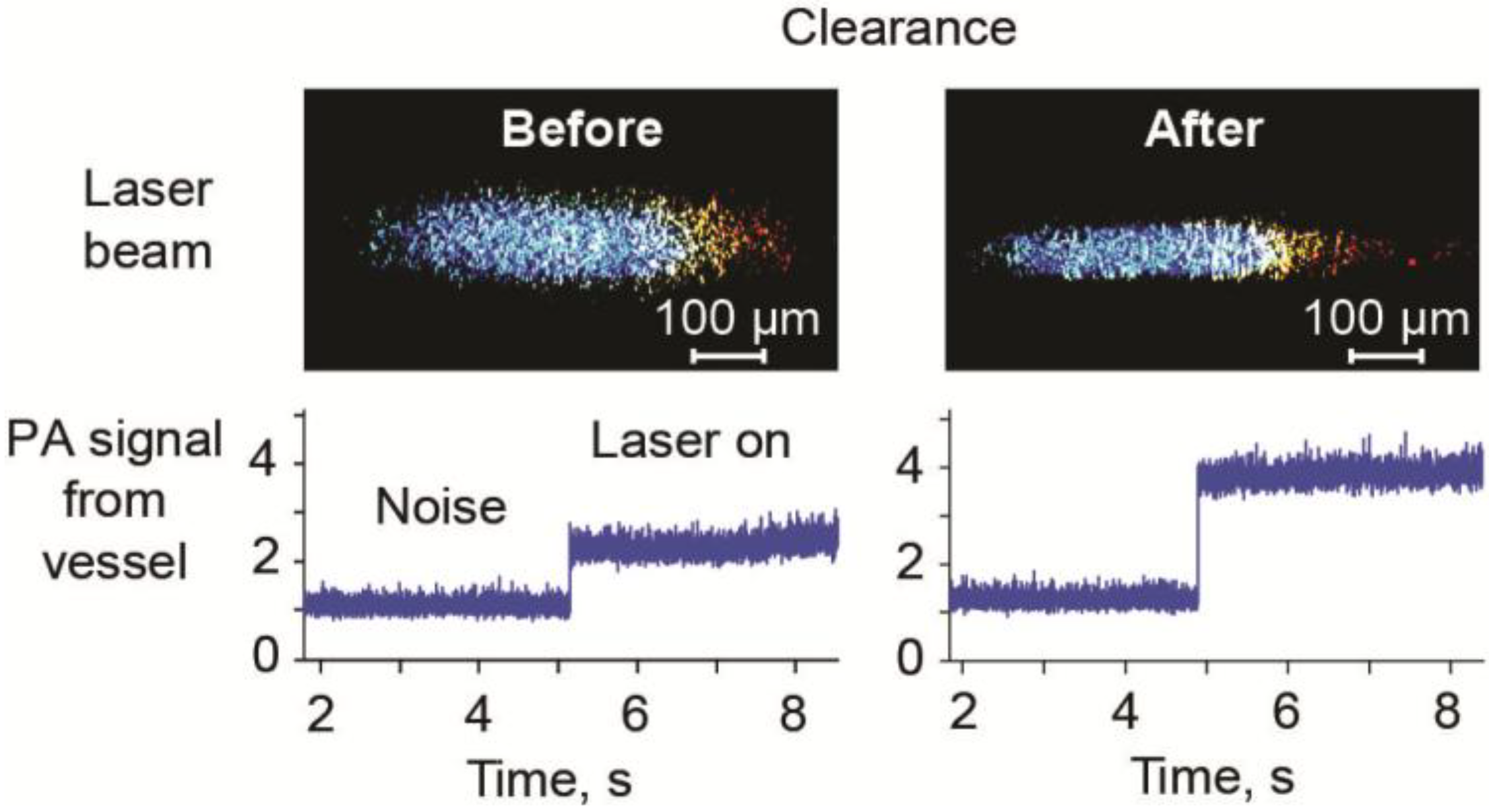
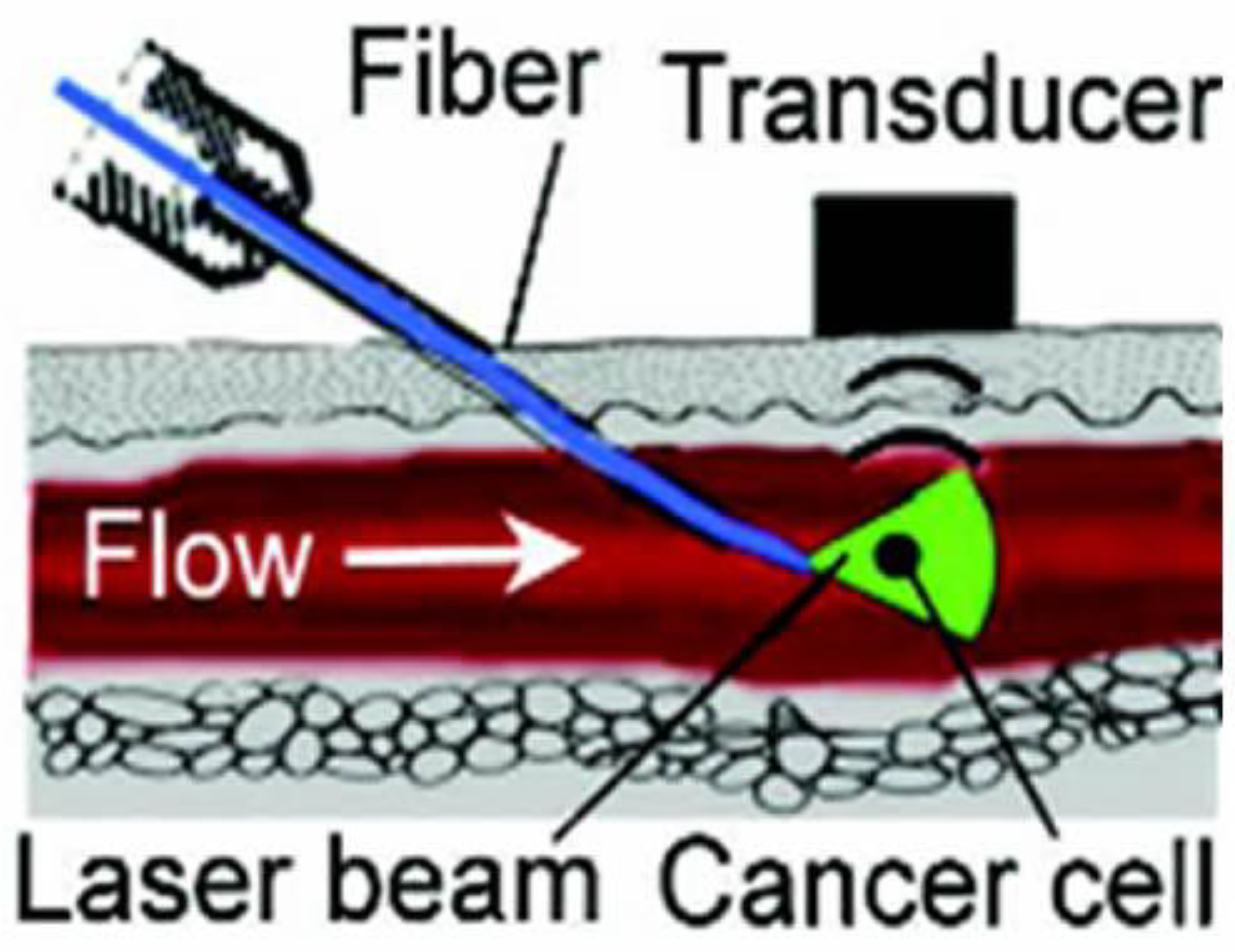
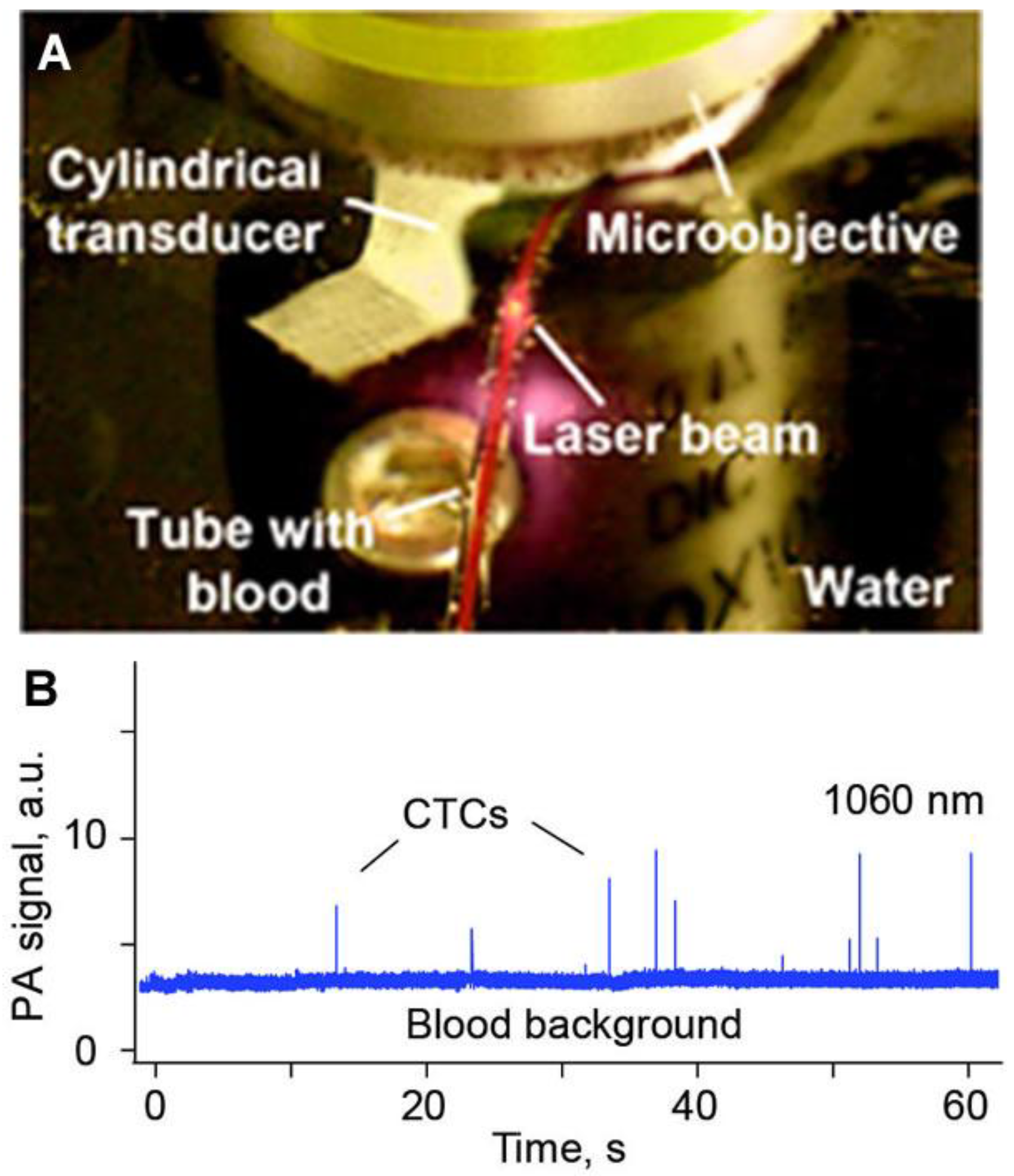
3.7. Minimally Invasive PAFC

3.8. Labeling in Vivo
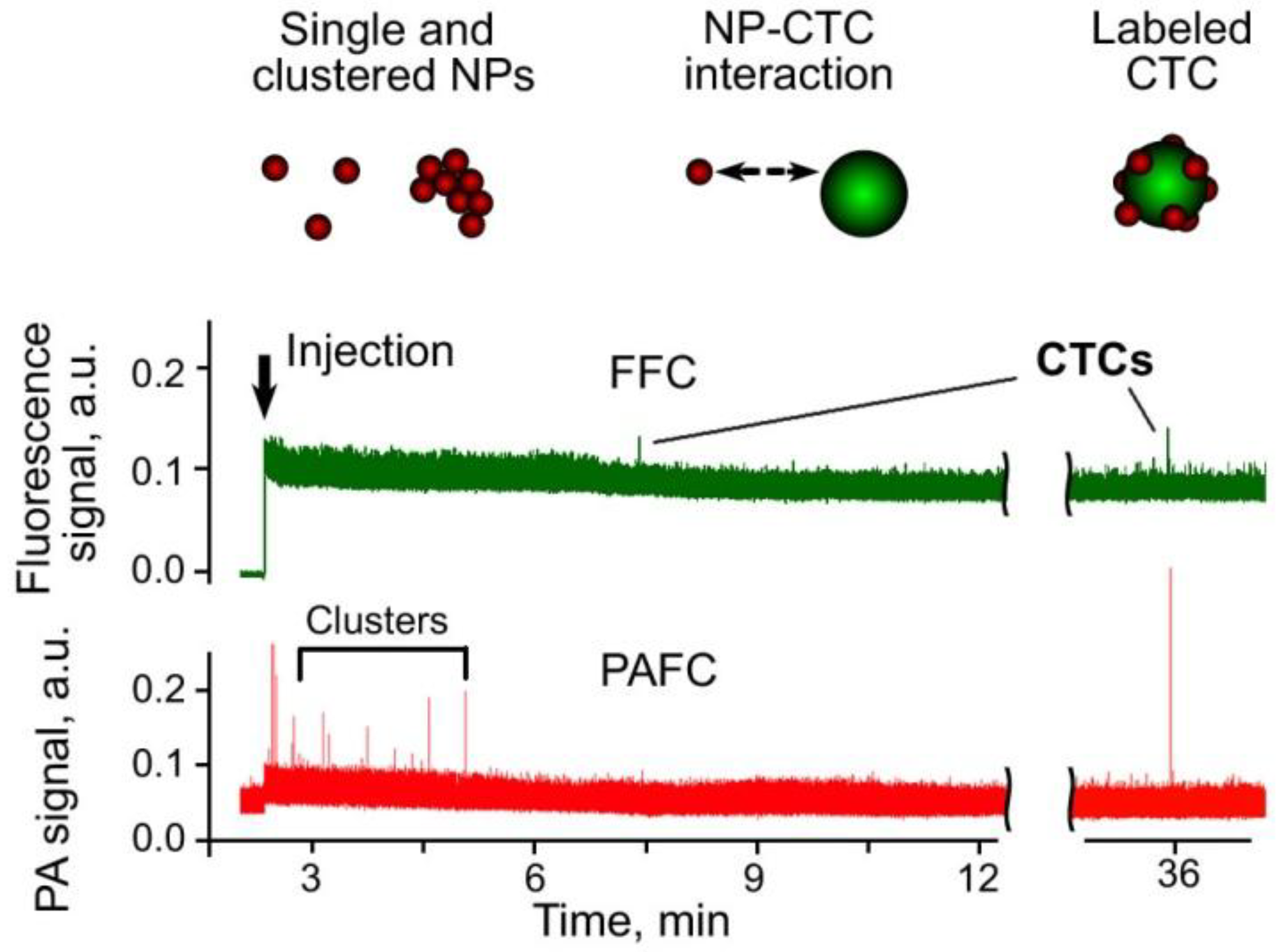
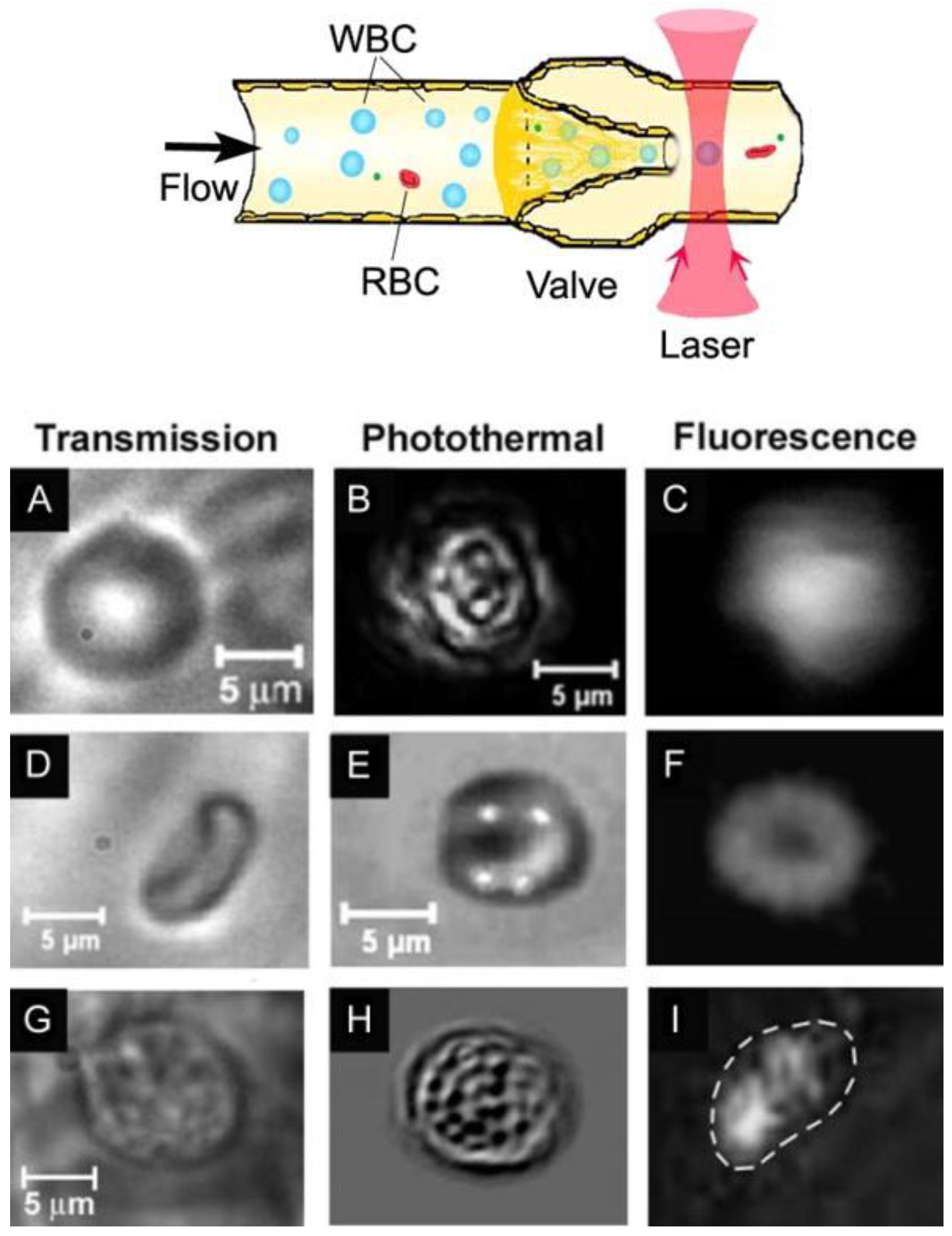
3.9. Combination of PA, PT and Fluorescence Methods
3.10. Photoswitchable PAFC
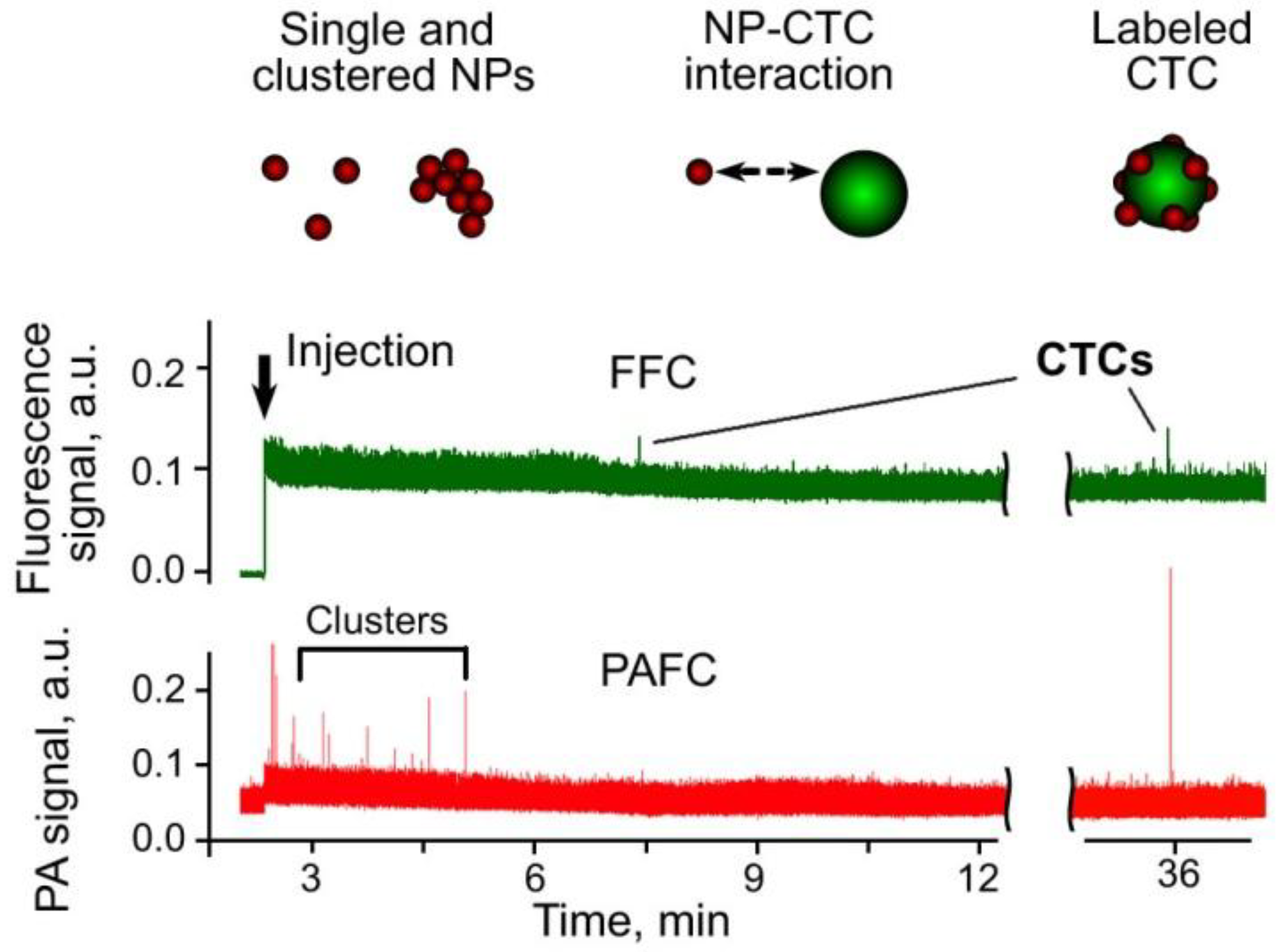
4. Application of PAFC for the Detection of CTCs
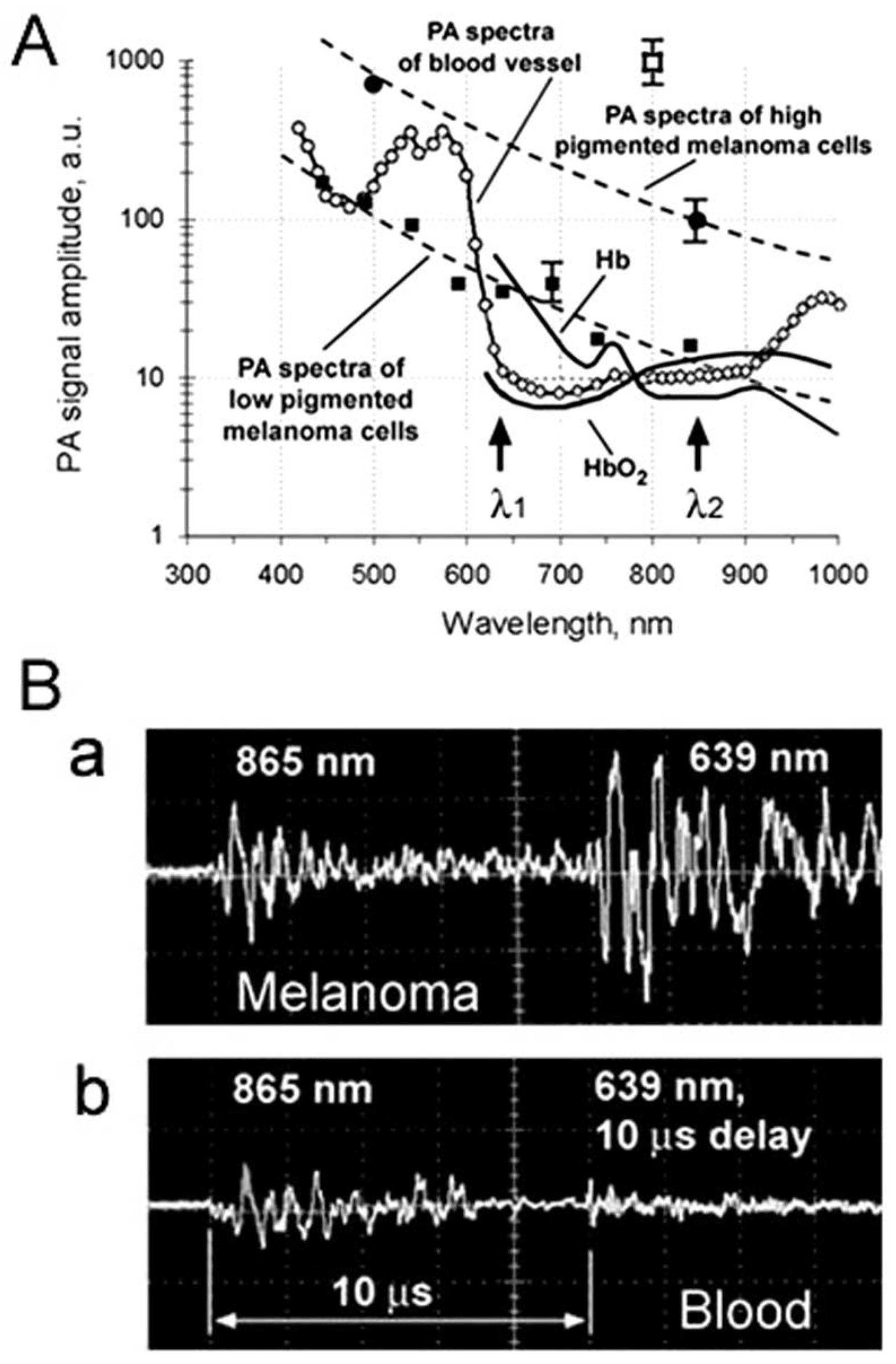
4.1. Detection of Melanoma CTCs
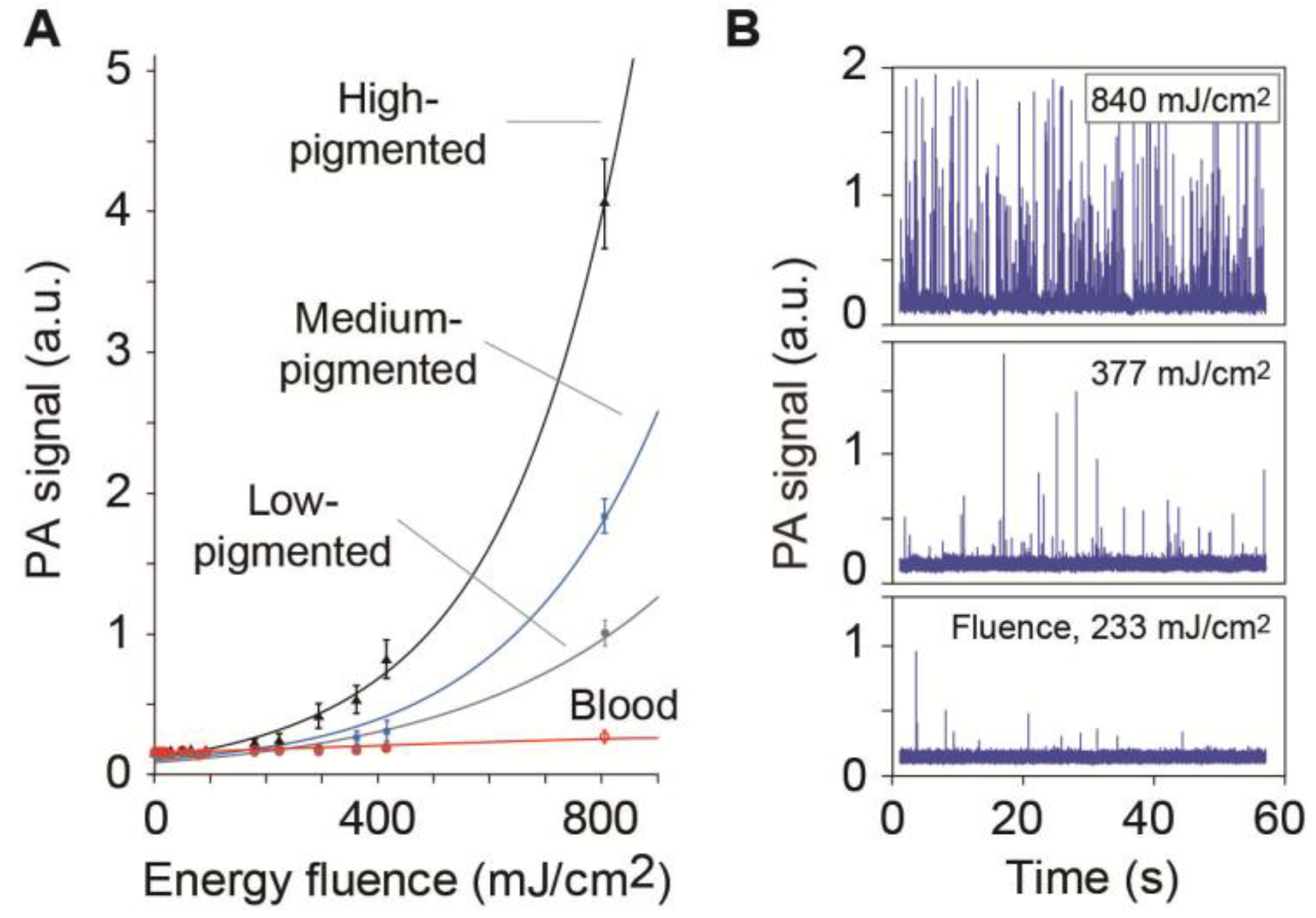
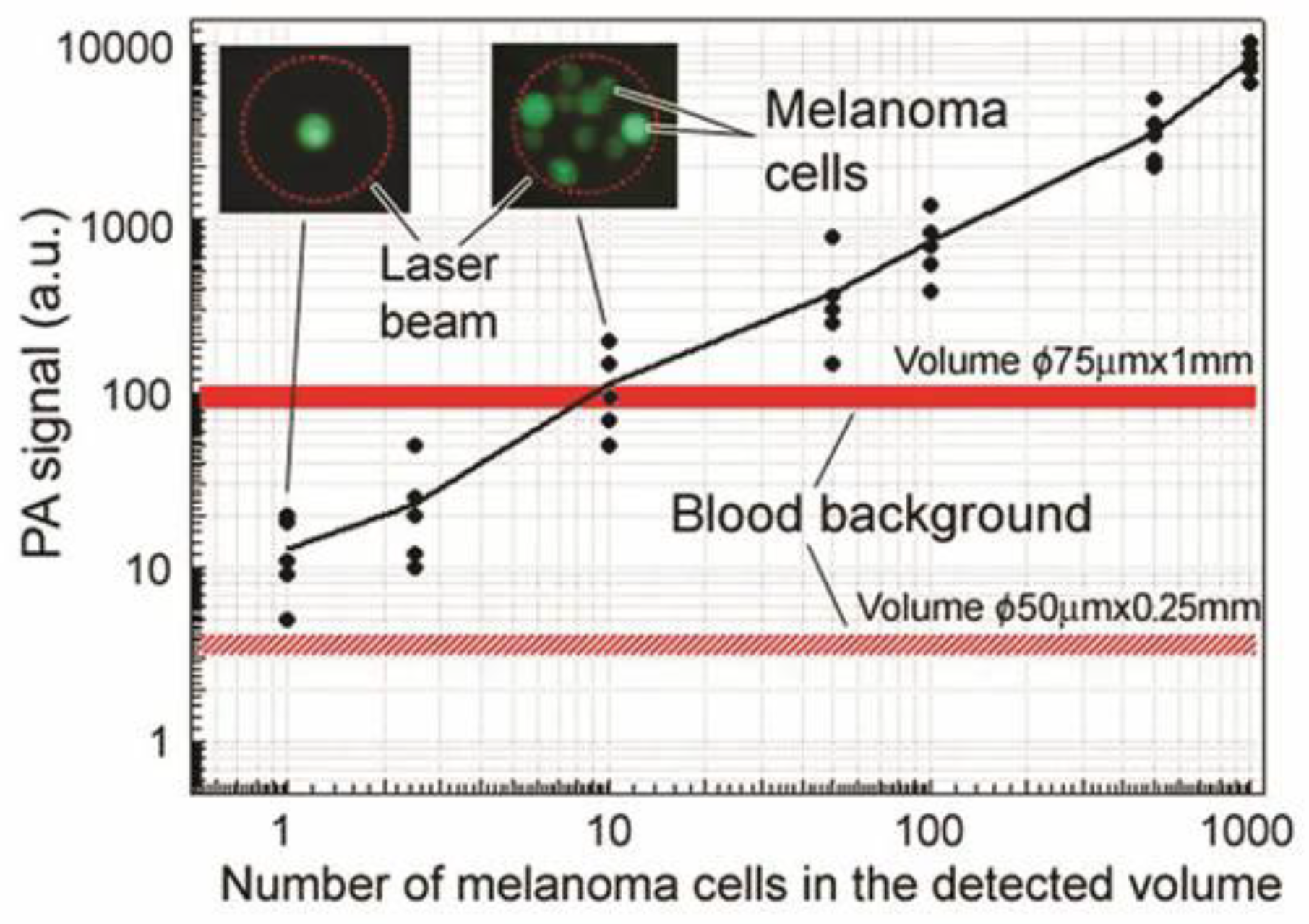
4.1.1. Label-Free Detection

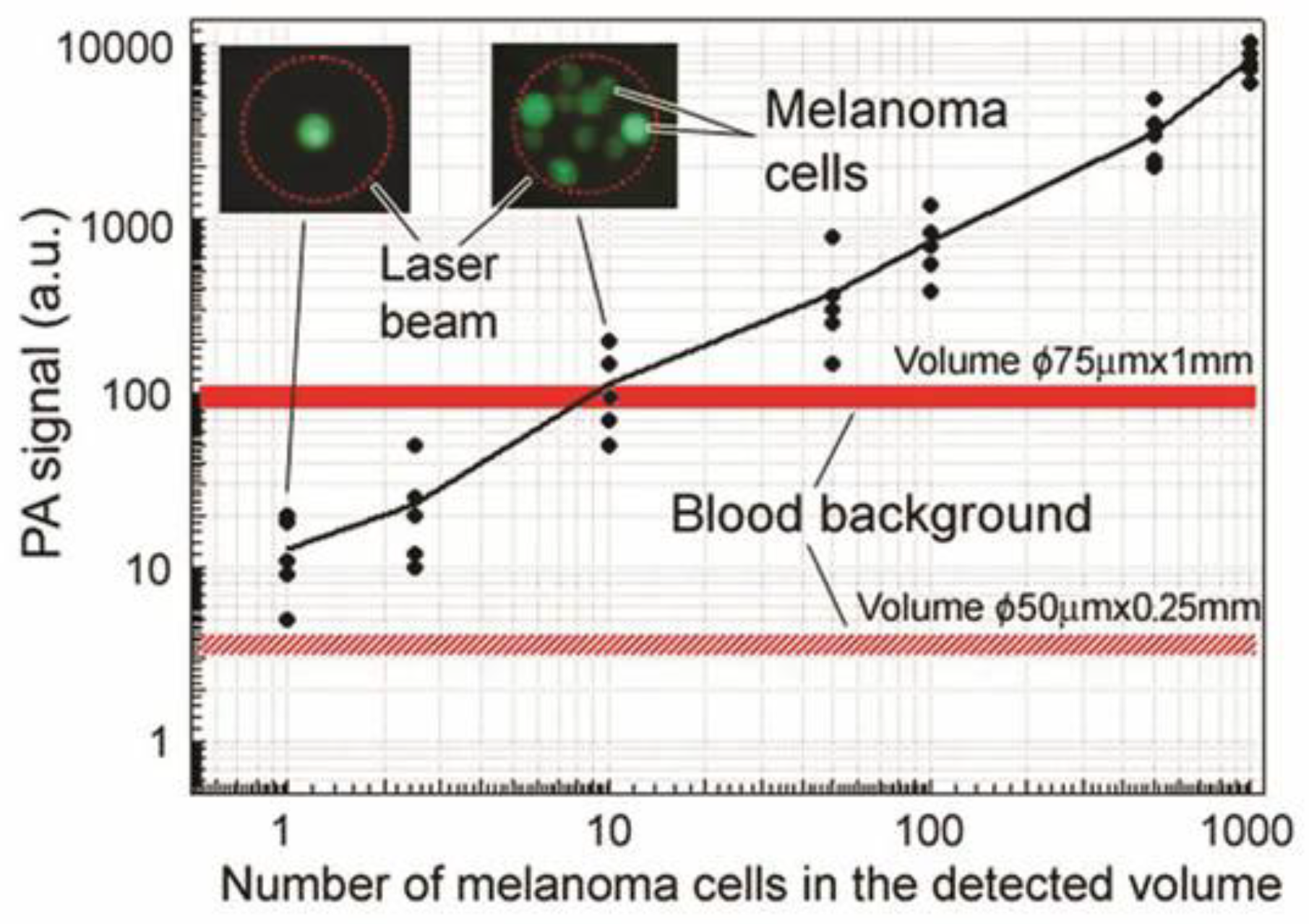
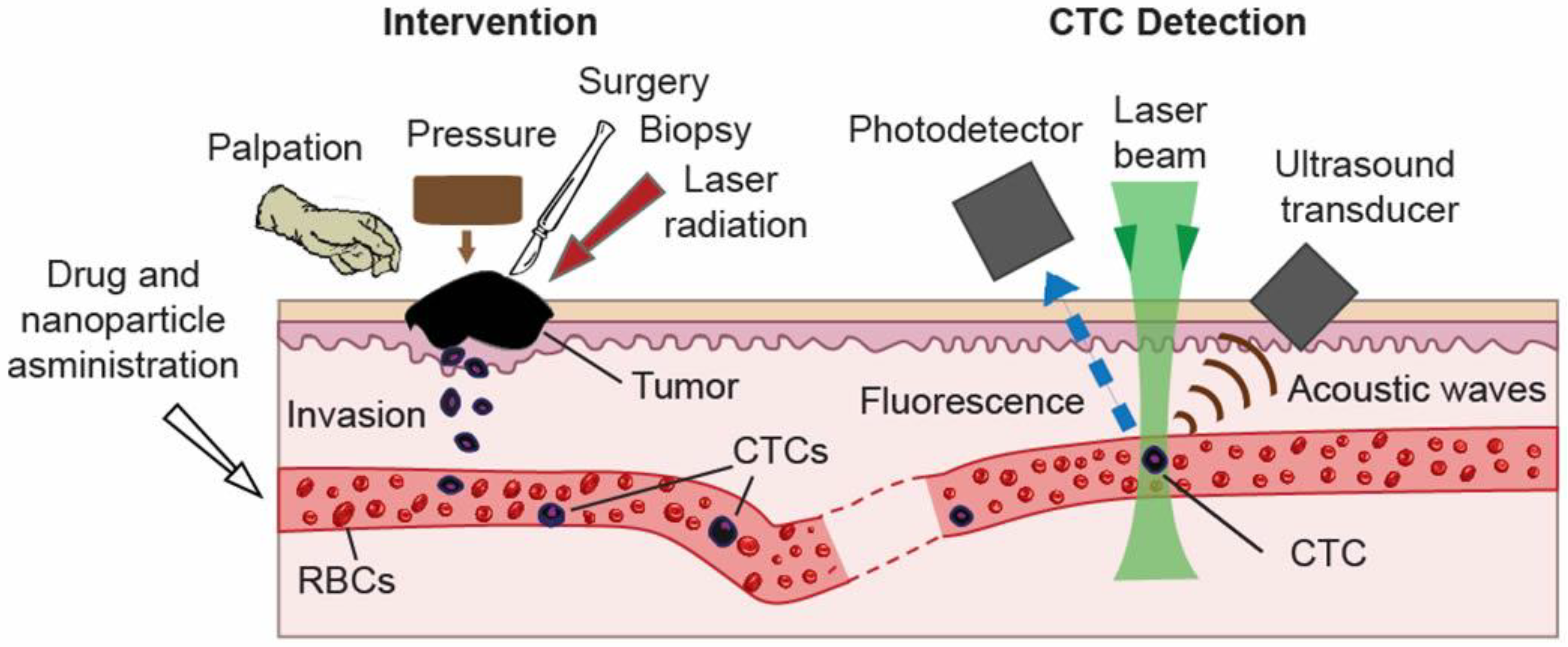
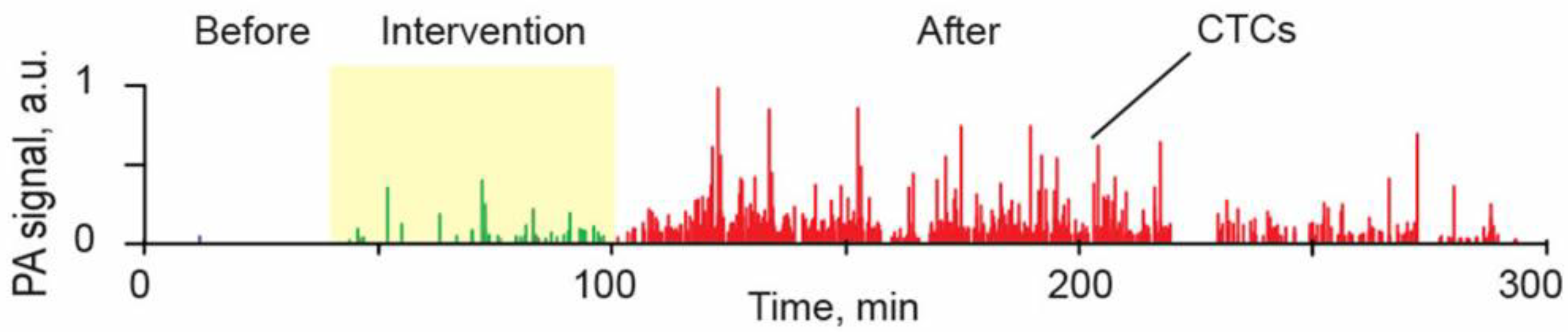
4.1.2. Study of CTC Release from the Primary Tumor during a Medical Procedure

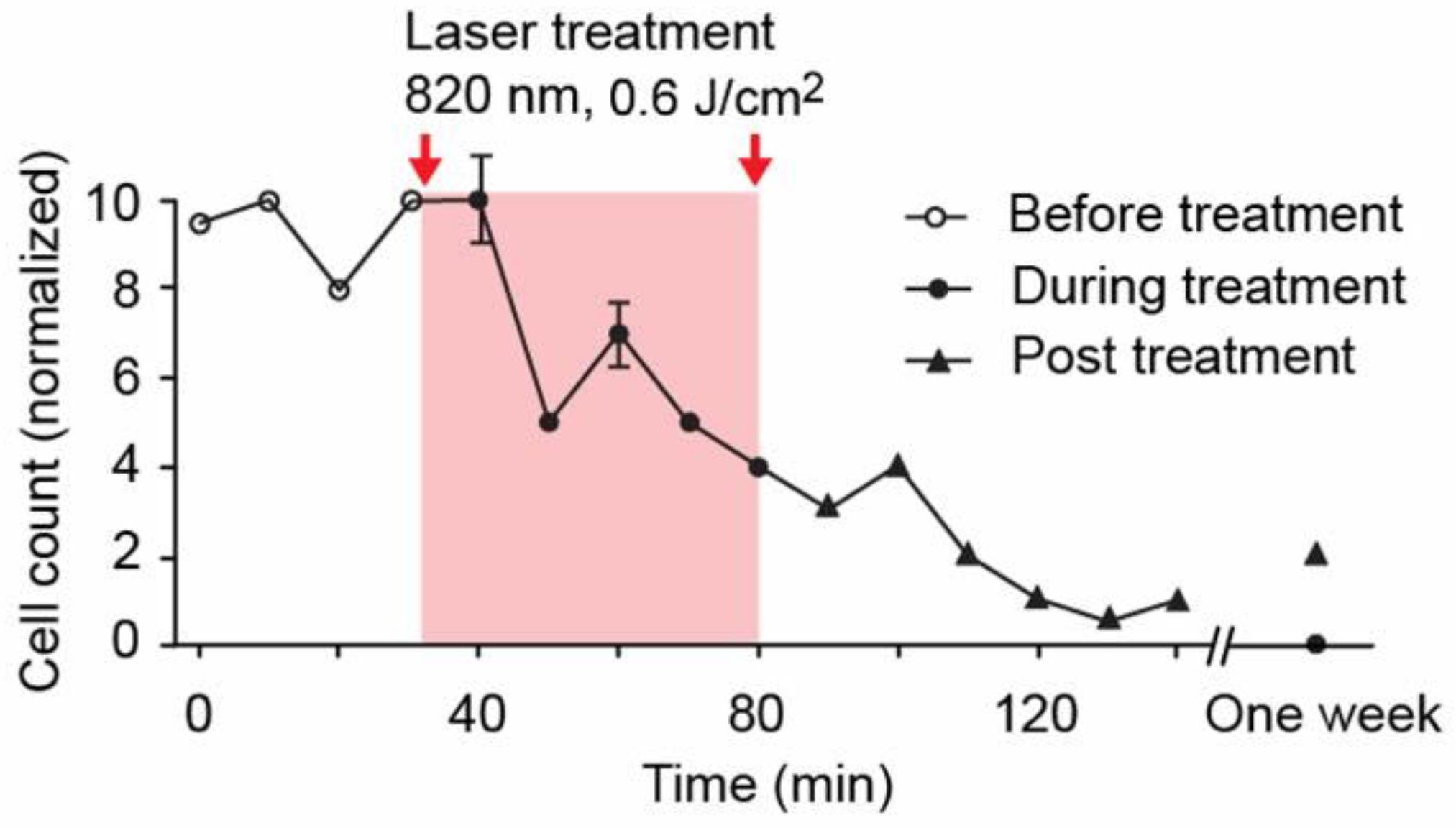
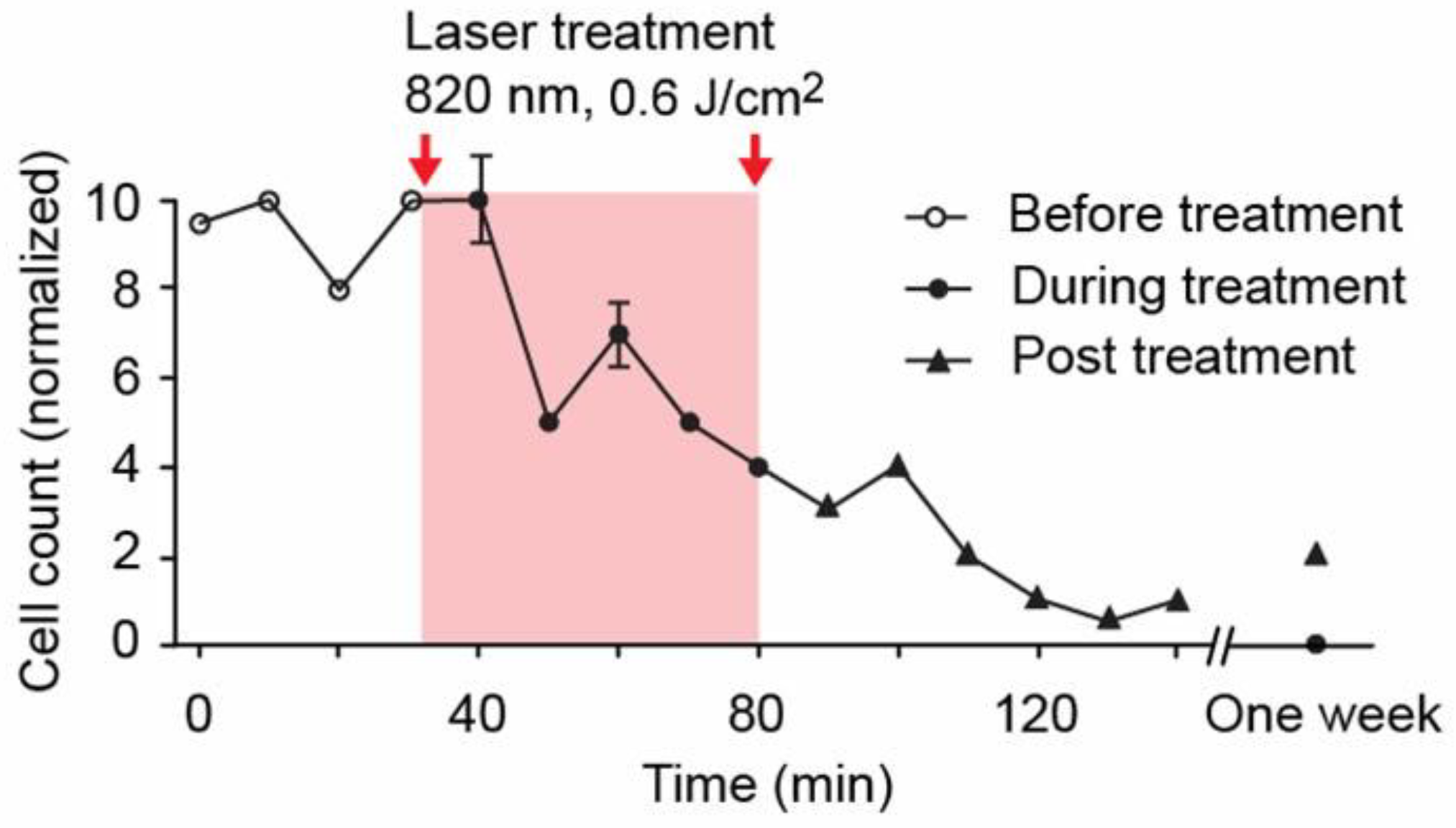
4.1.3. PA-PT Theranostics of Melanoma CTCs with Nonlinear PAFC
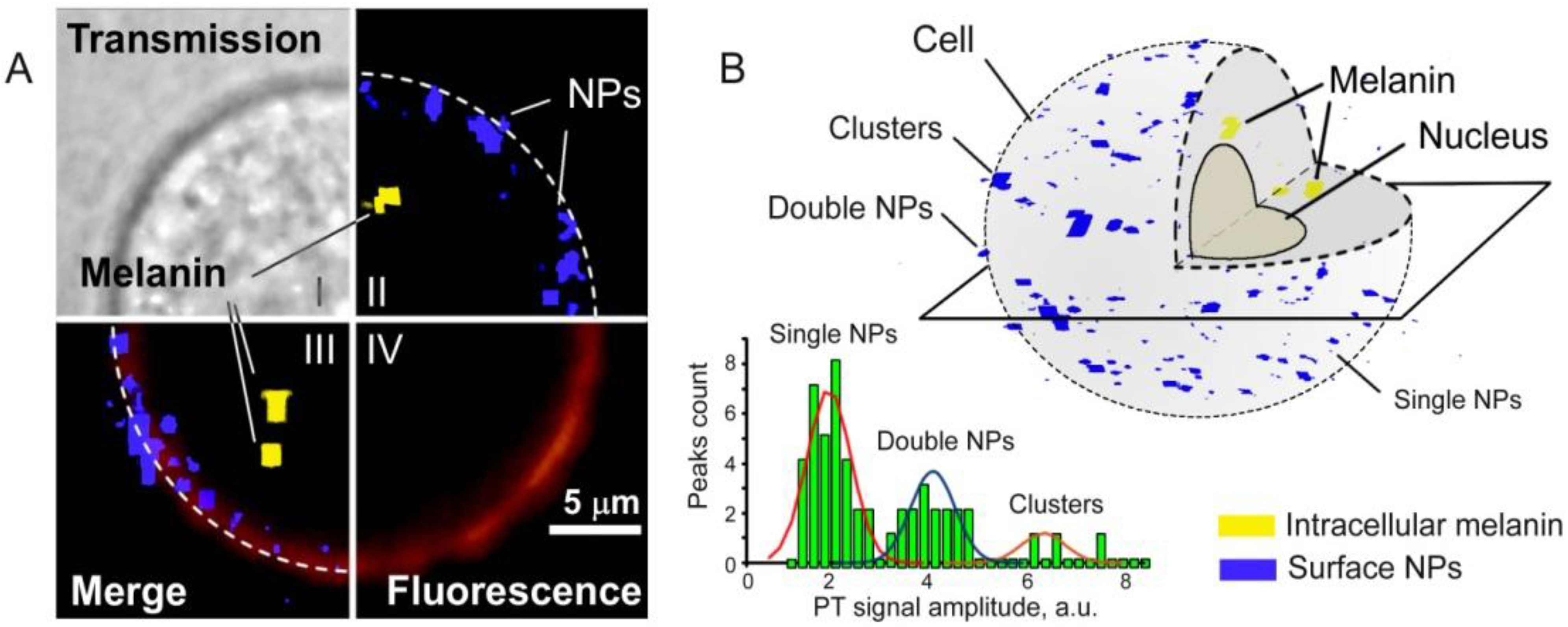
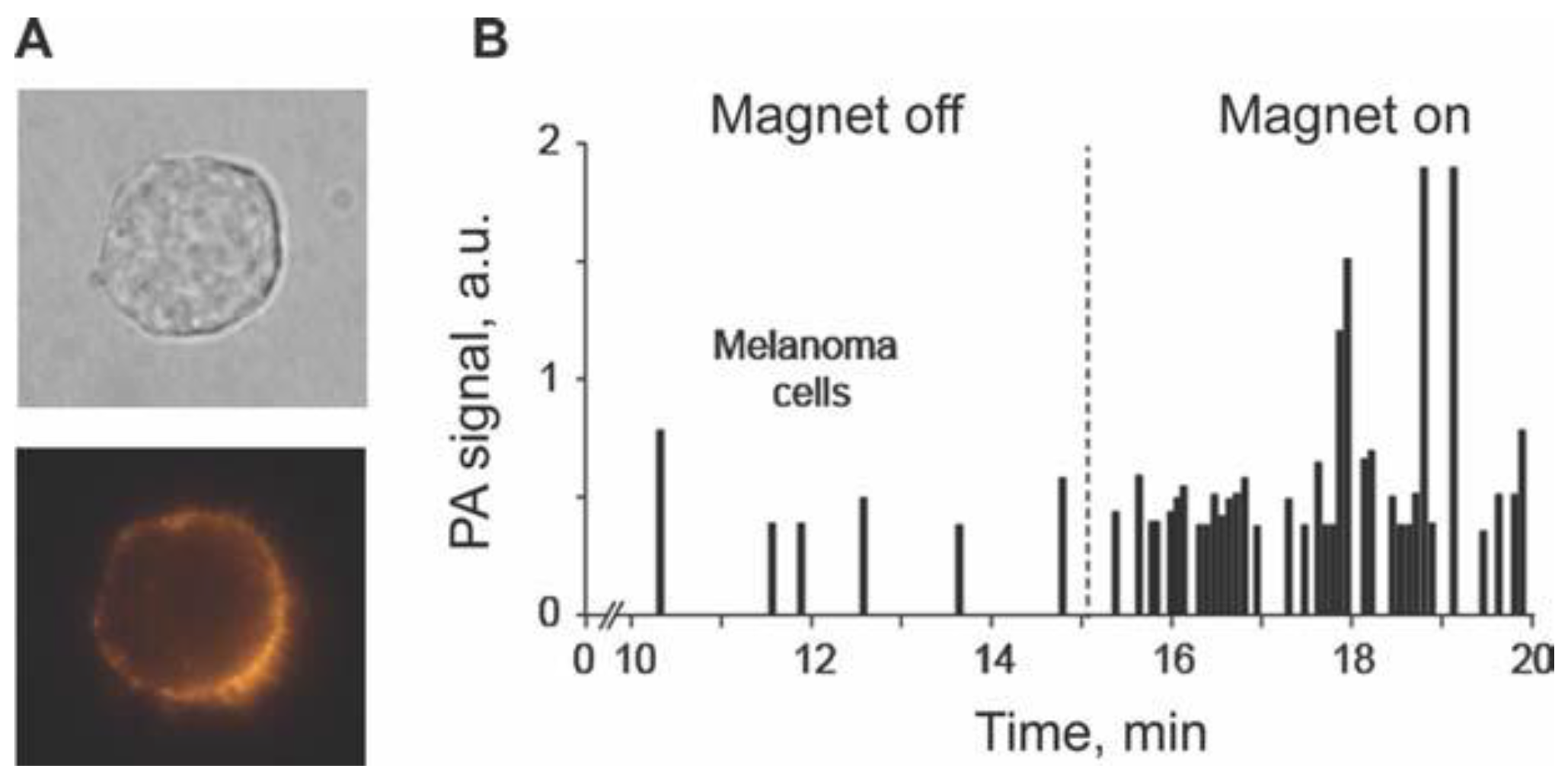
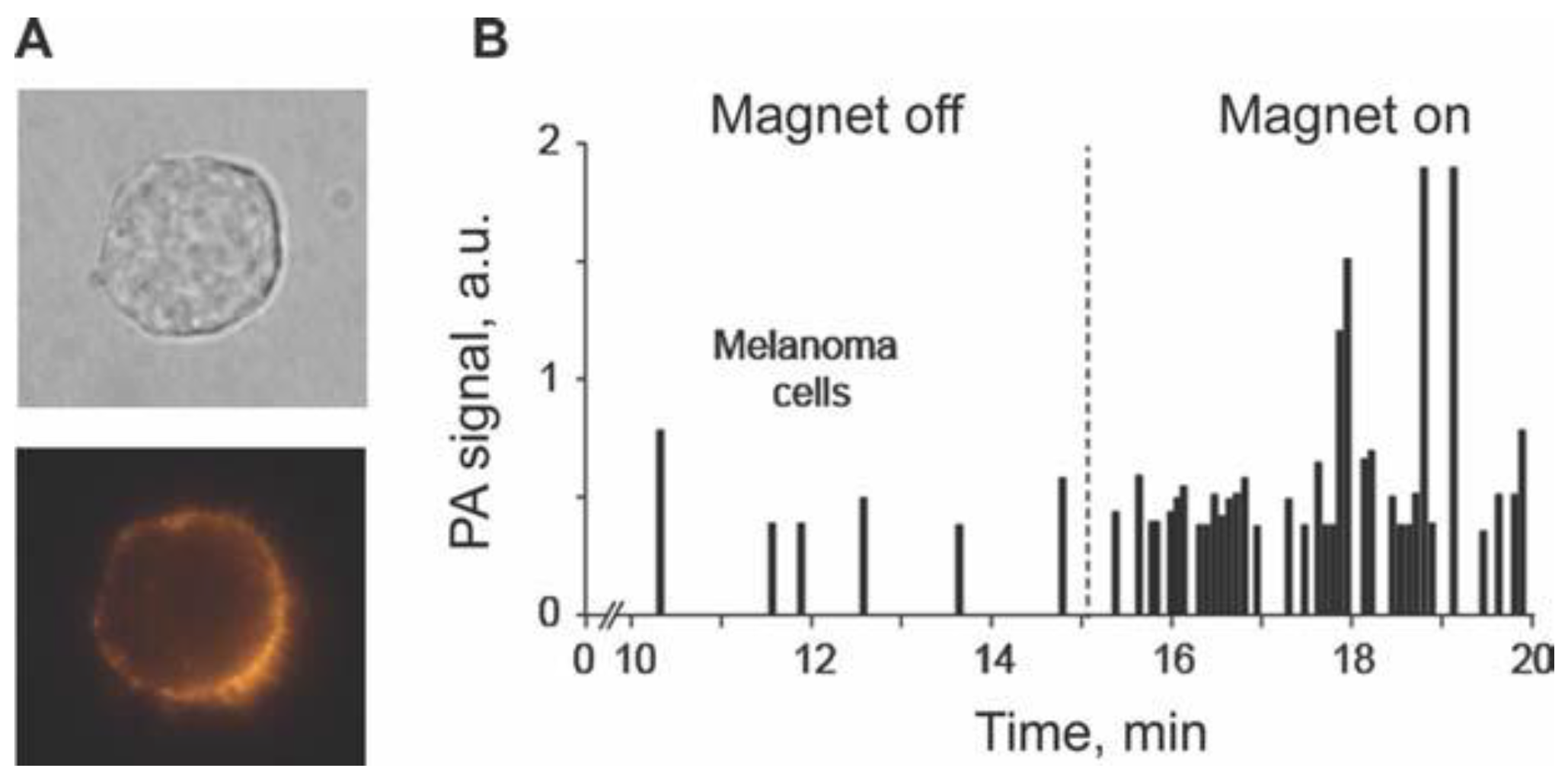
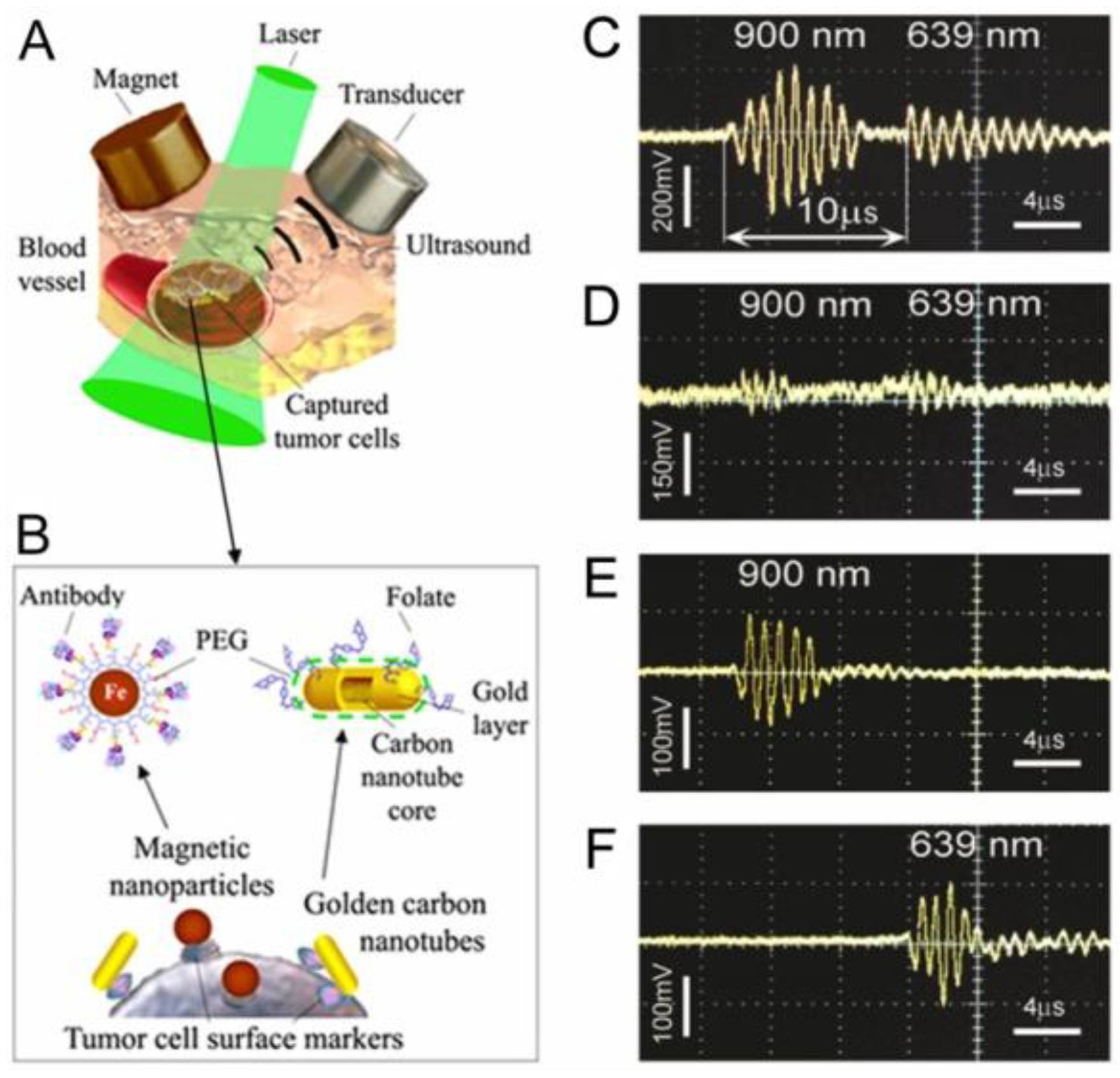
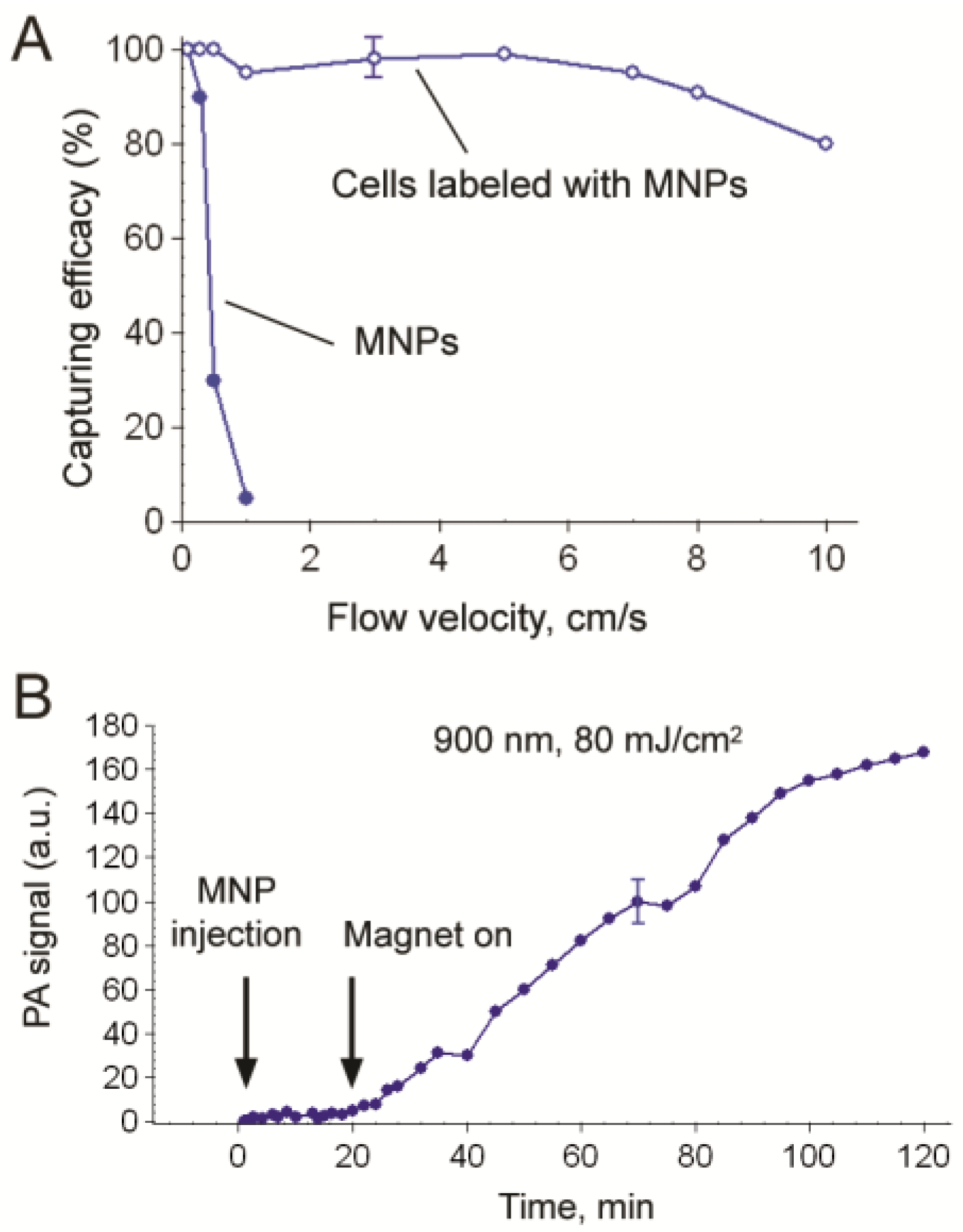
4.1.4. Magnetic Capture of Melanoma CTCs in Vivo
4.2. Detection of Breast CTCs in Blood
4.2.1. Multiplex Molecular Targeting, Magnetic Capture, and Two-Color Detection of Bulk Breast CTCs



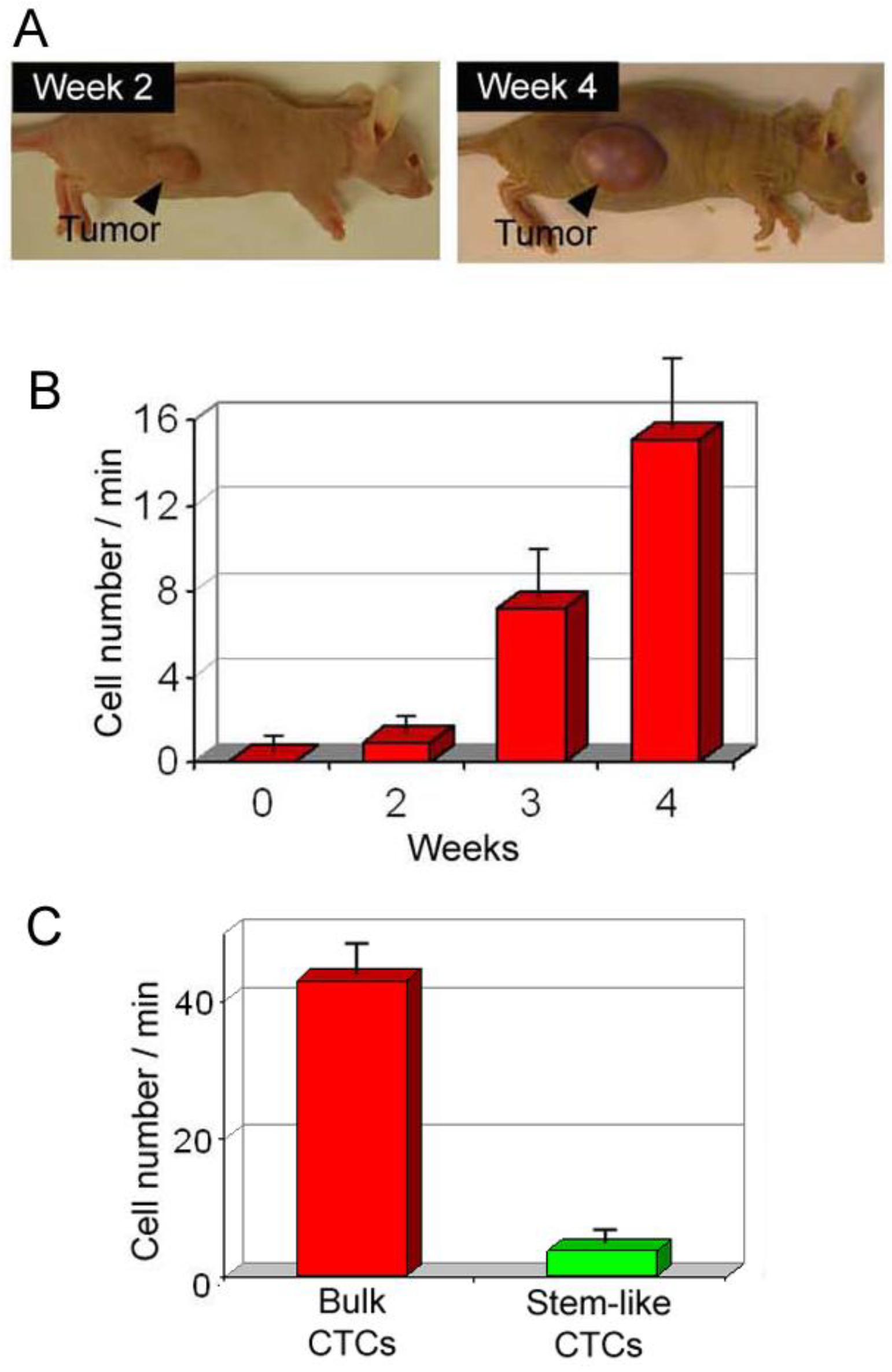
4.2.2. Flow Cytometry Platform for Detection of Circulating Cancer Stem Cells in Blood
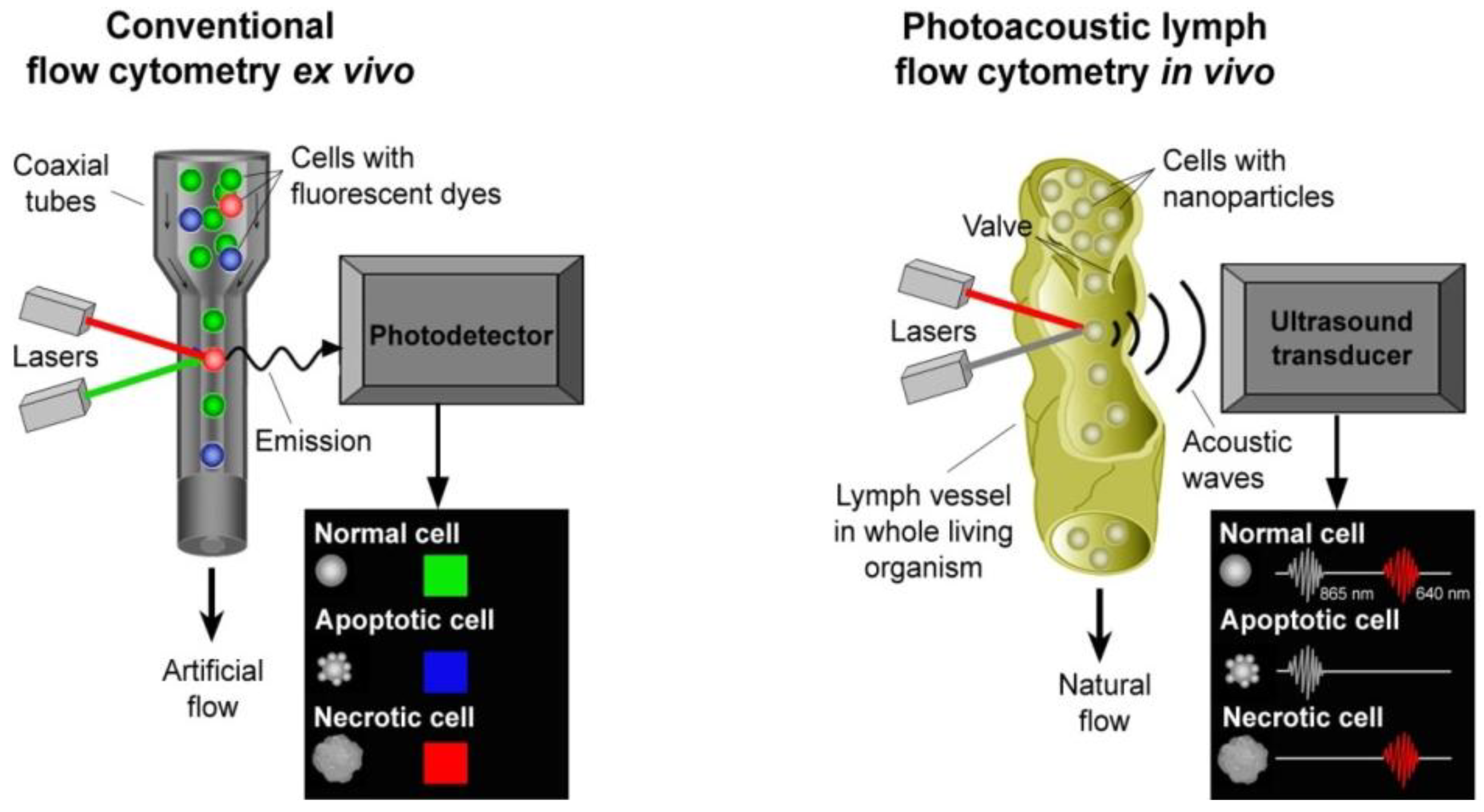
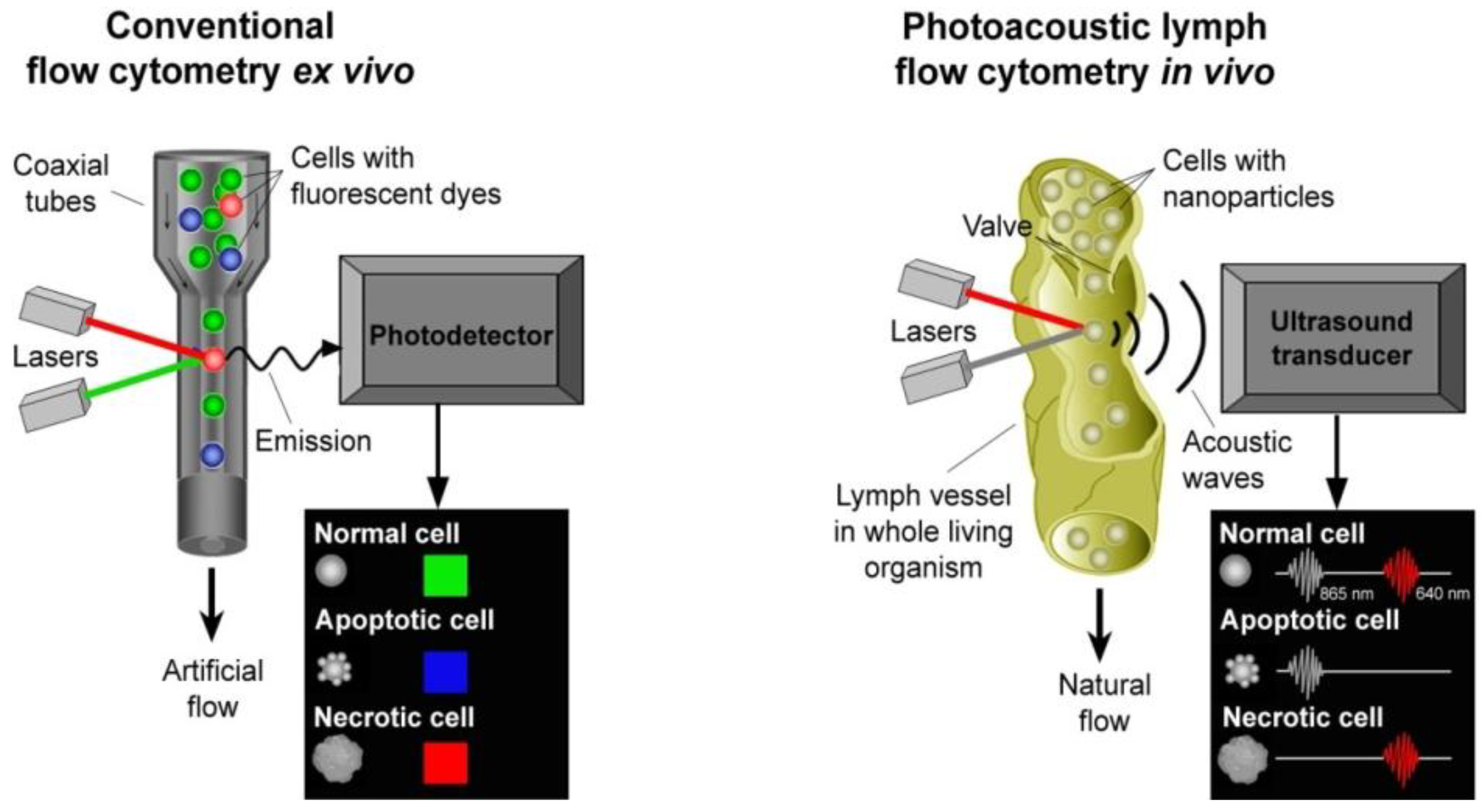
4.3. In Vivo Lymph Flow Cytometry
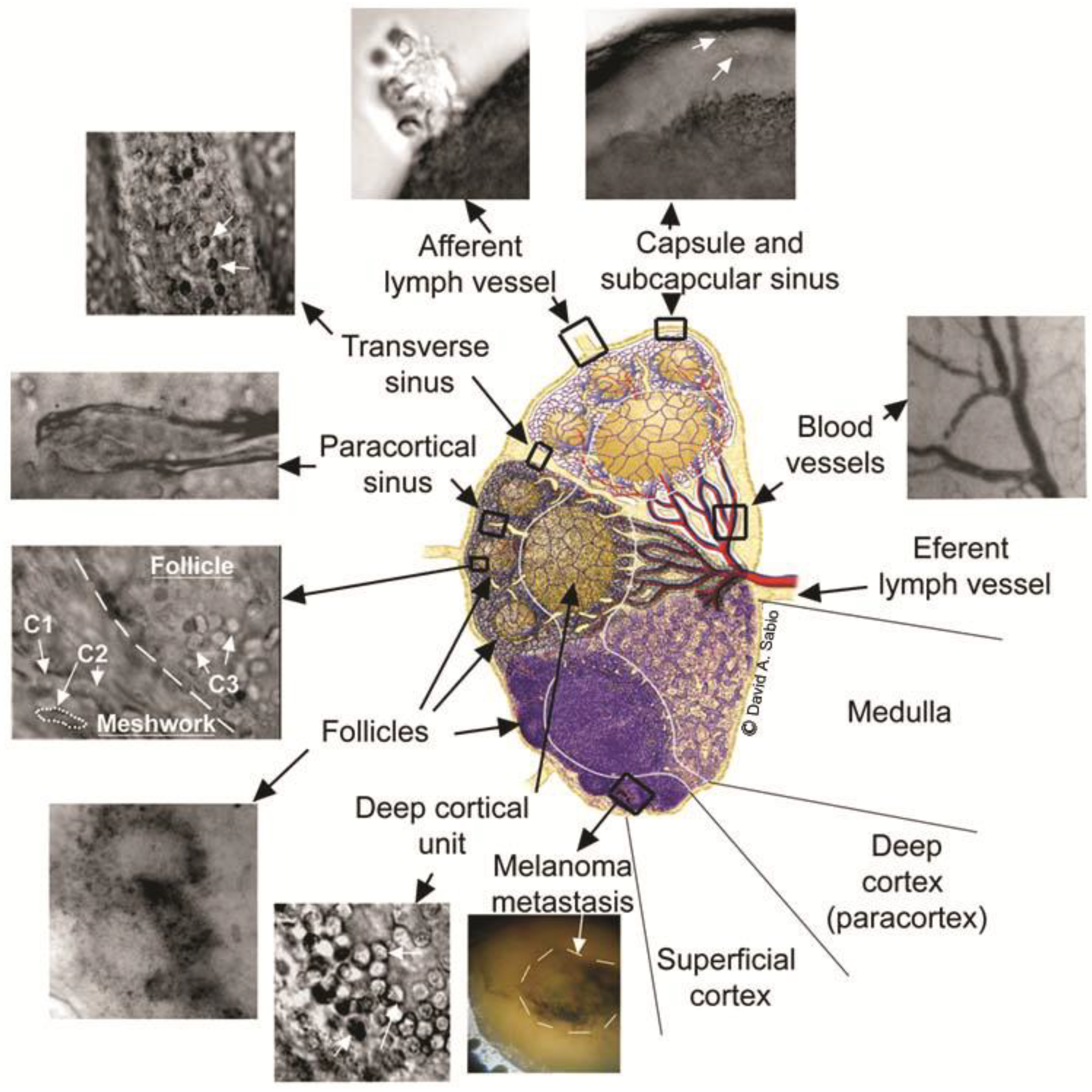
4.3.1. PA Detection of Tumor Cells in Lymph Flow and Sentinel Lymph Nodes
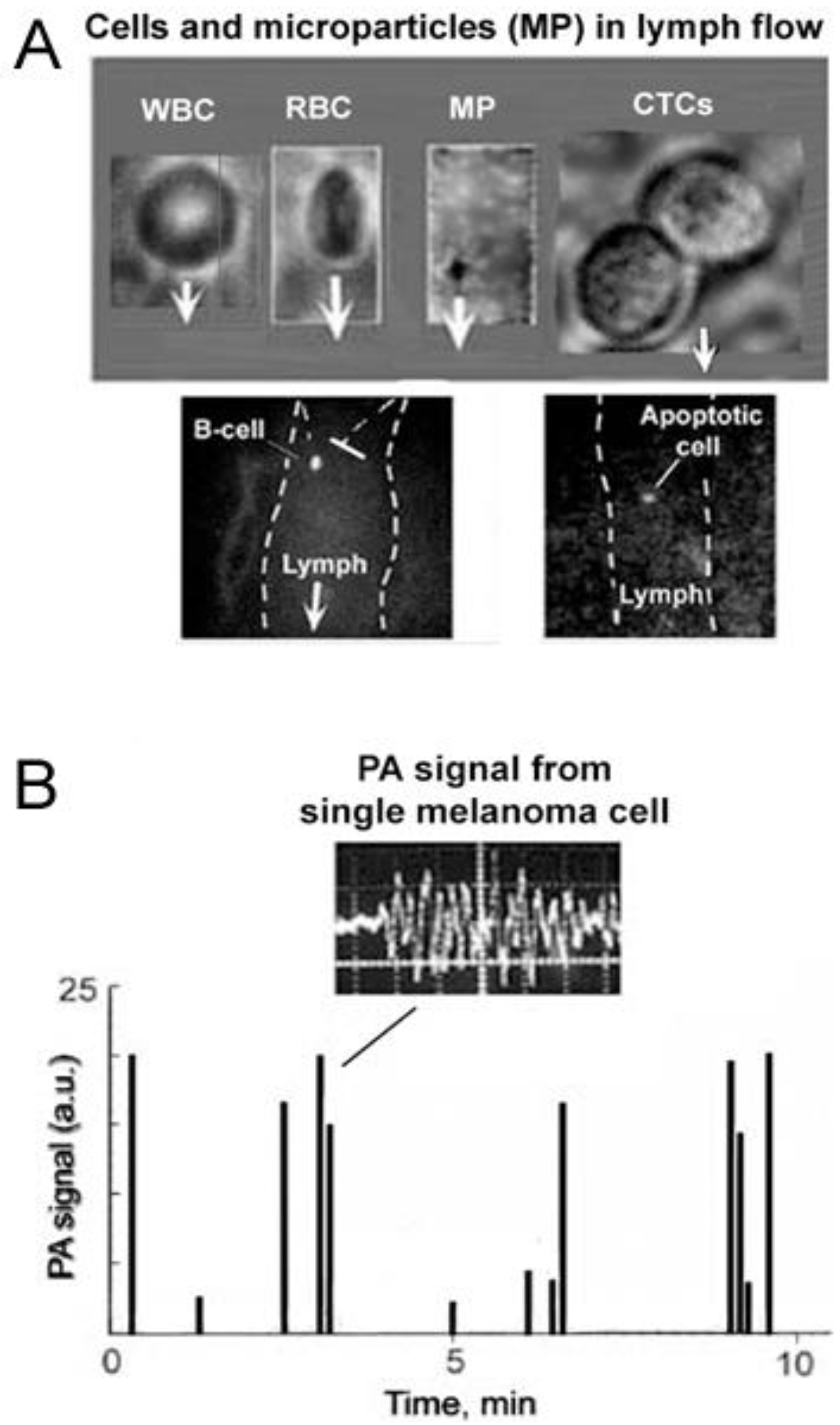
4.3.2. In Vivo PT Imaging of Cells in Lymph Flow
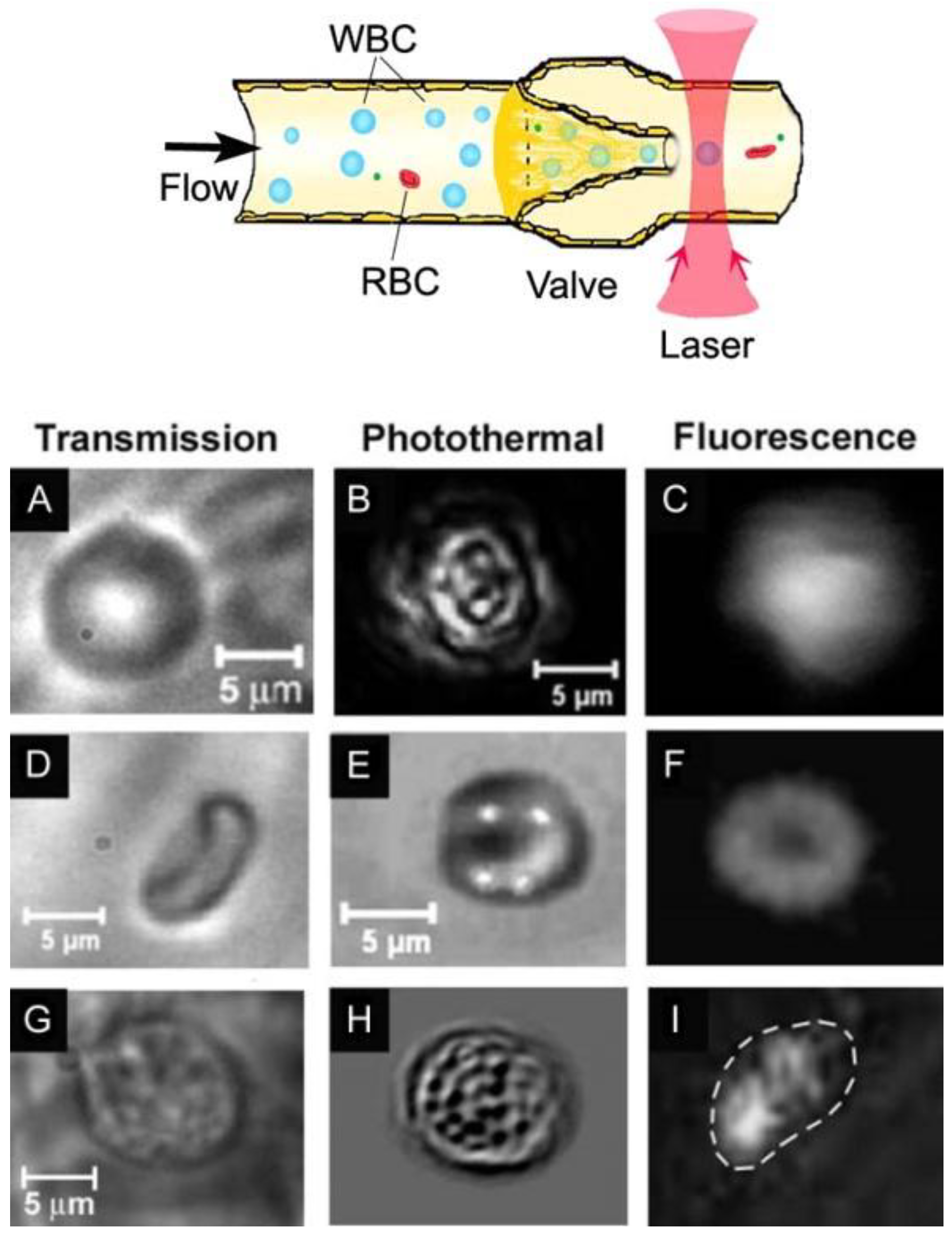
4.4. Detection of Cancer Cells in the Blood Stream, Lymph Flow and Sentinel Lymph Nodes; Theranostics of Lymphatics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
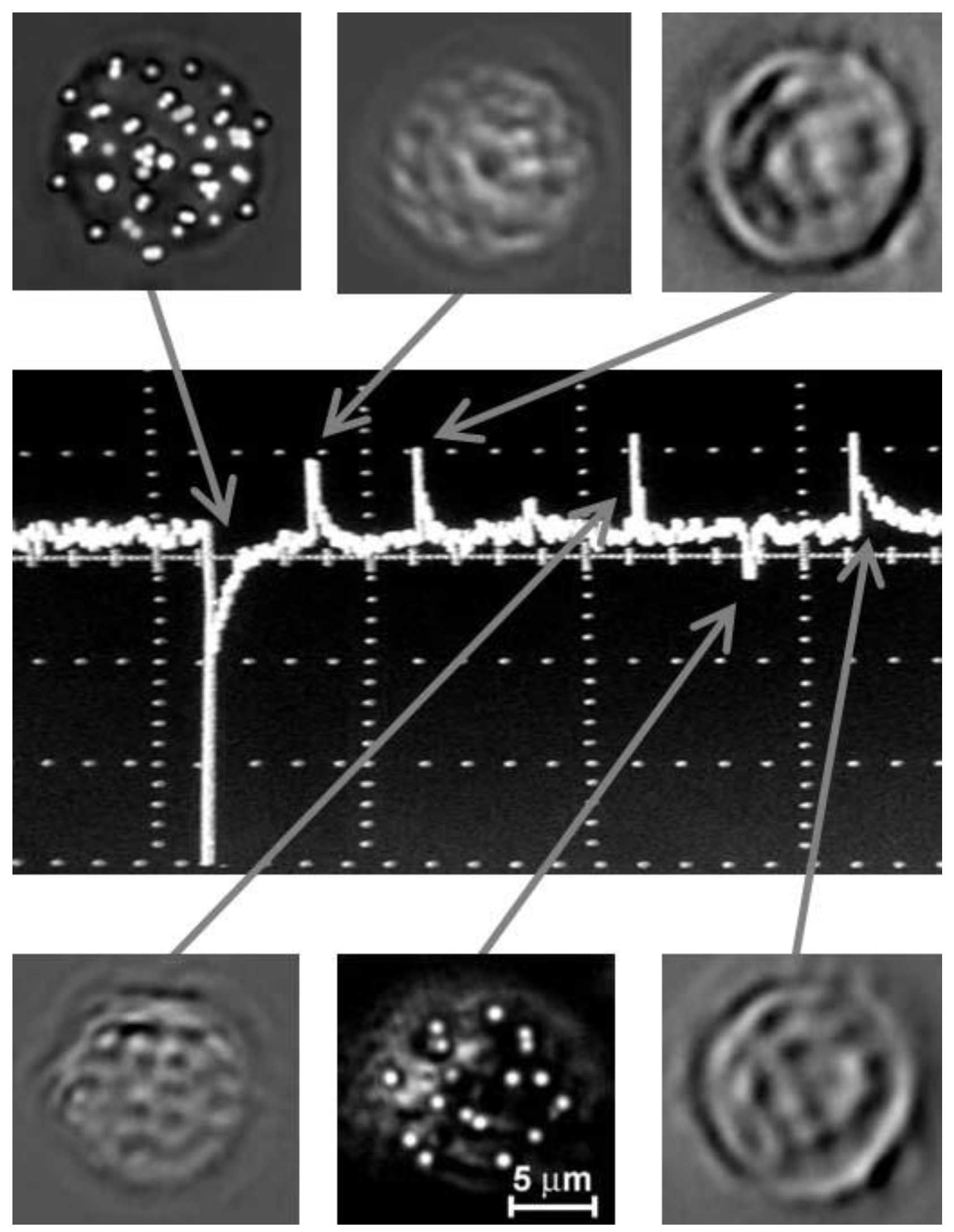
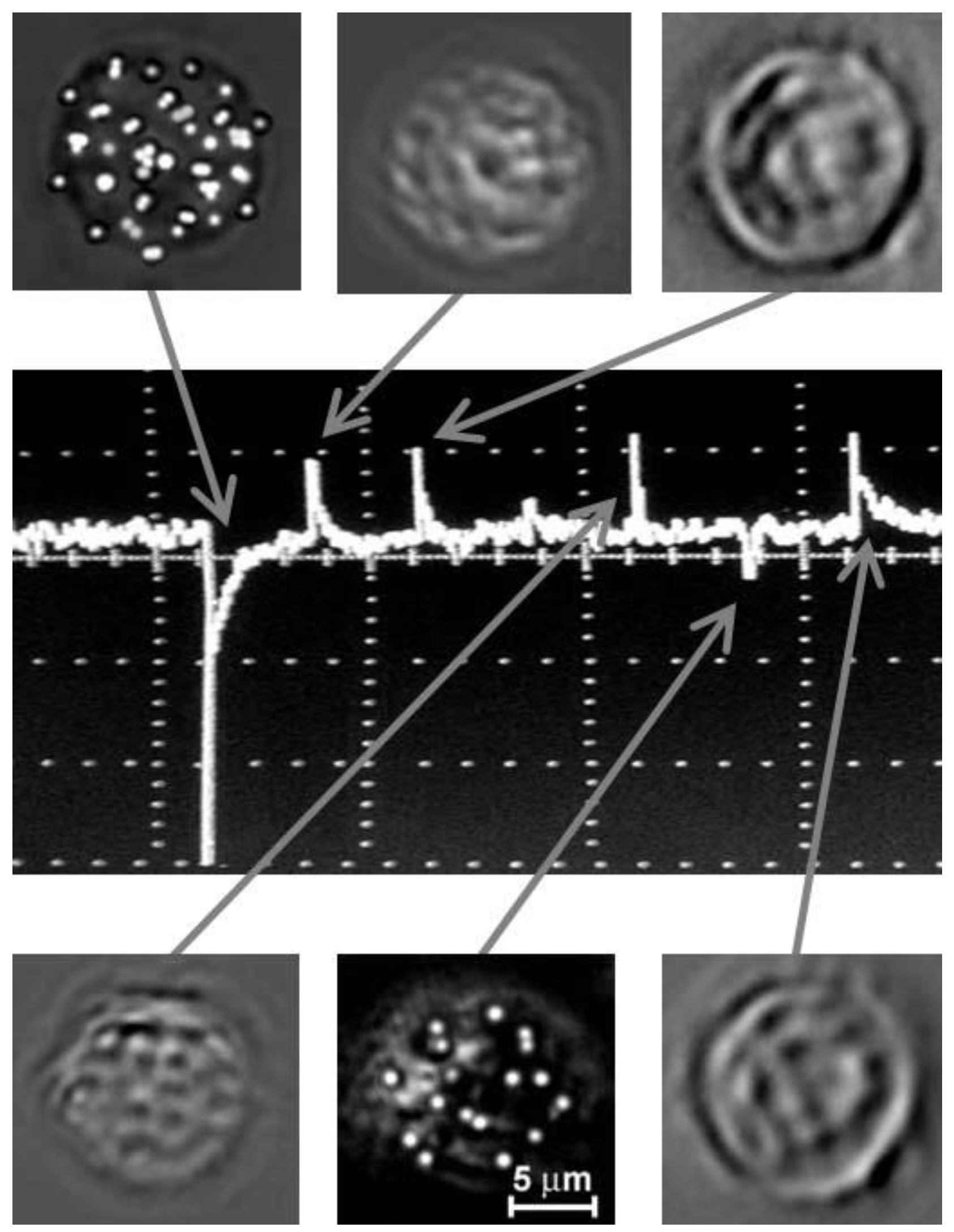{kind=link}
{kind=link}
| Time tumor growth | Tumor size, mm2 | Rate of lymph CTCs, cell/min | Rate of blood CTCs, cell/min | Number of PA signals associated with metastasis in SLNs | Histology |
|---|---|---|---|---|---|
| 1 week | 1.0 ± 0.2 | 0.26 ± 0.05 | 0.85 ± 0.03 | 493 | NO |
| 2 weeks | 3.6 ± 0.5 | 2.13 ± 0.30 | 1.07 ± 0.05 | 3,188 | YES |
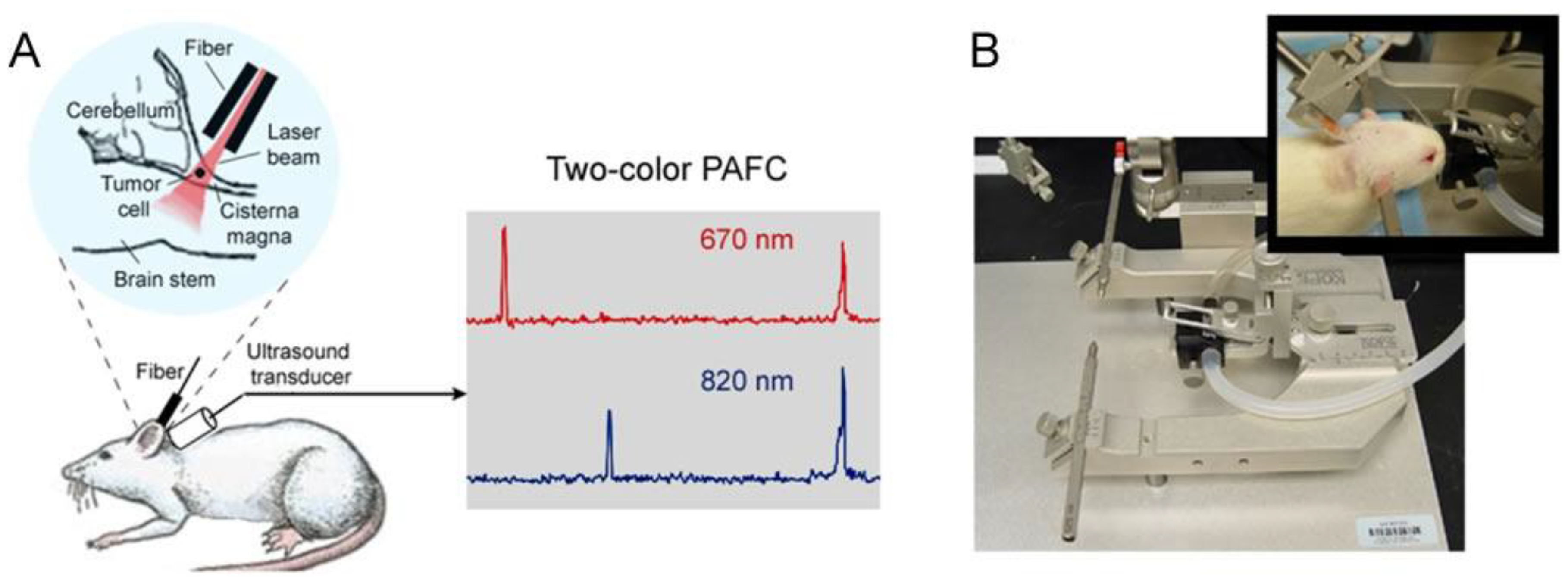
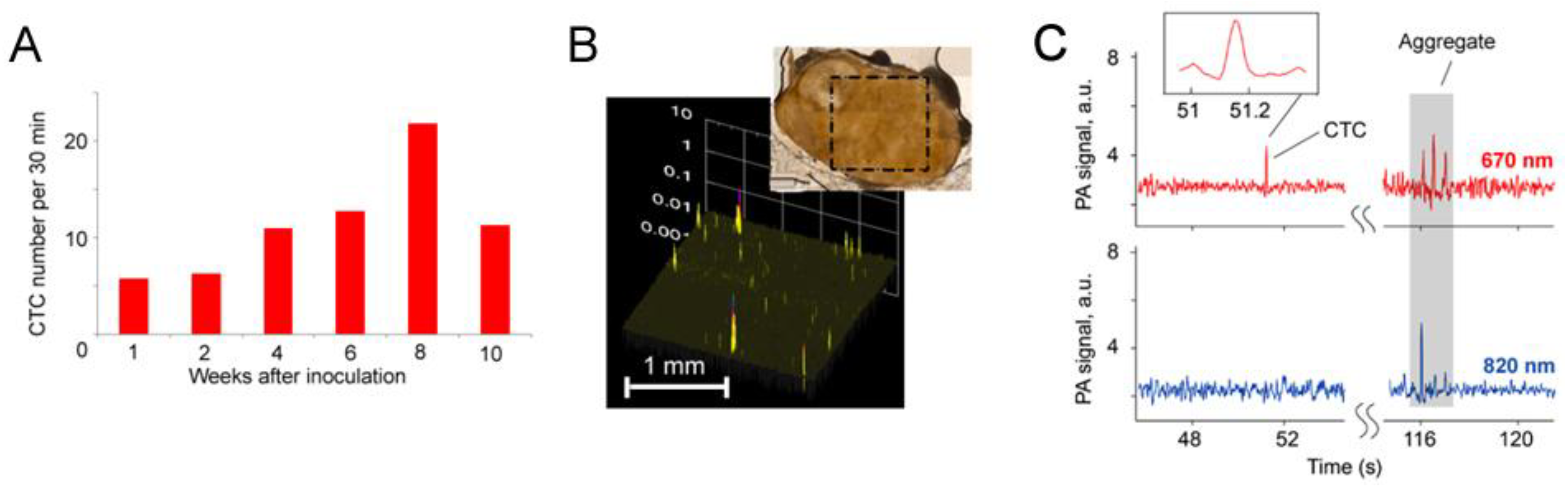
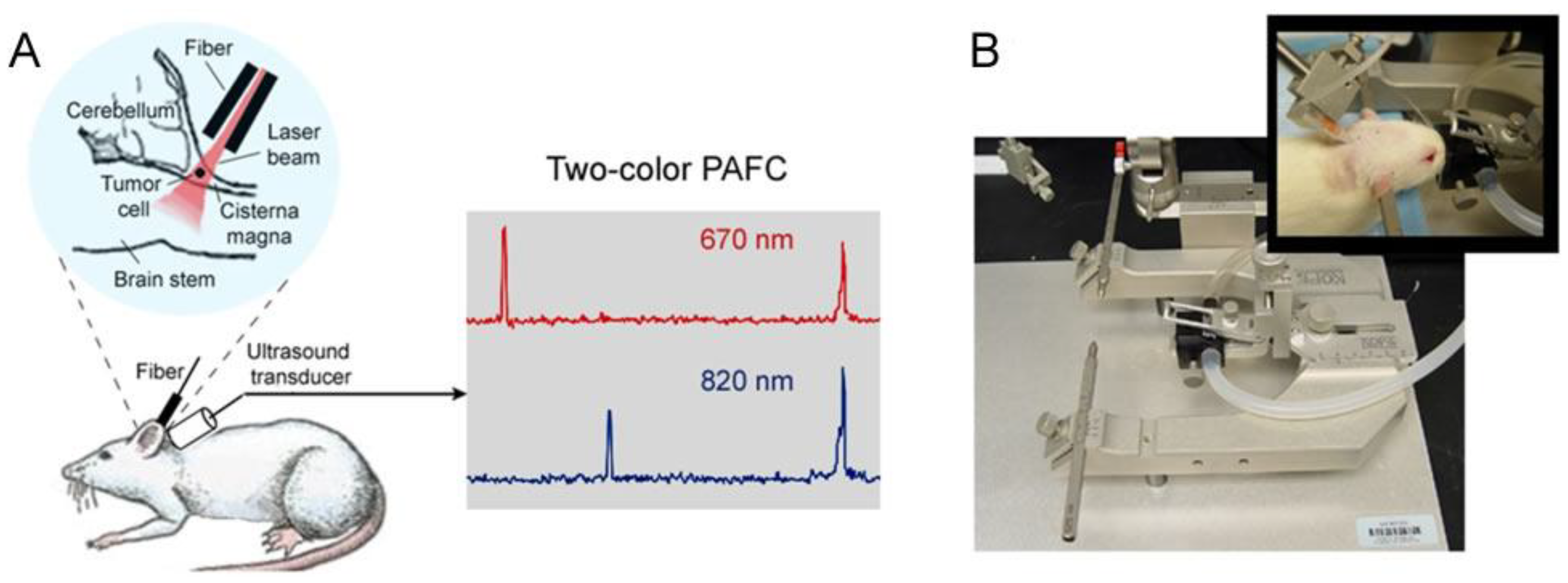
4.5. In Vivo Integrated Cerebrospinal Blood-Lymph Flow Cytometry

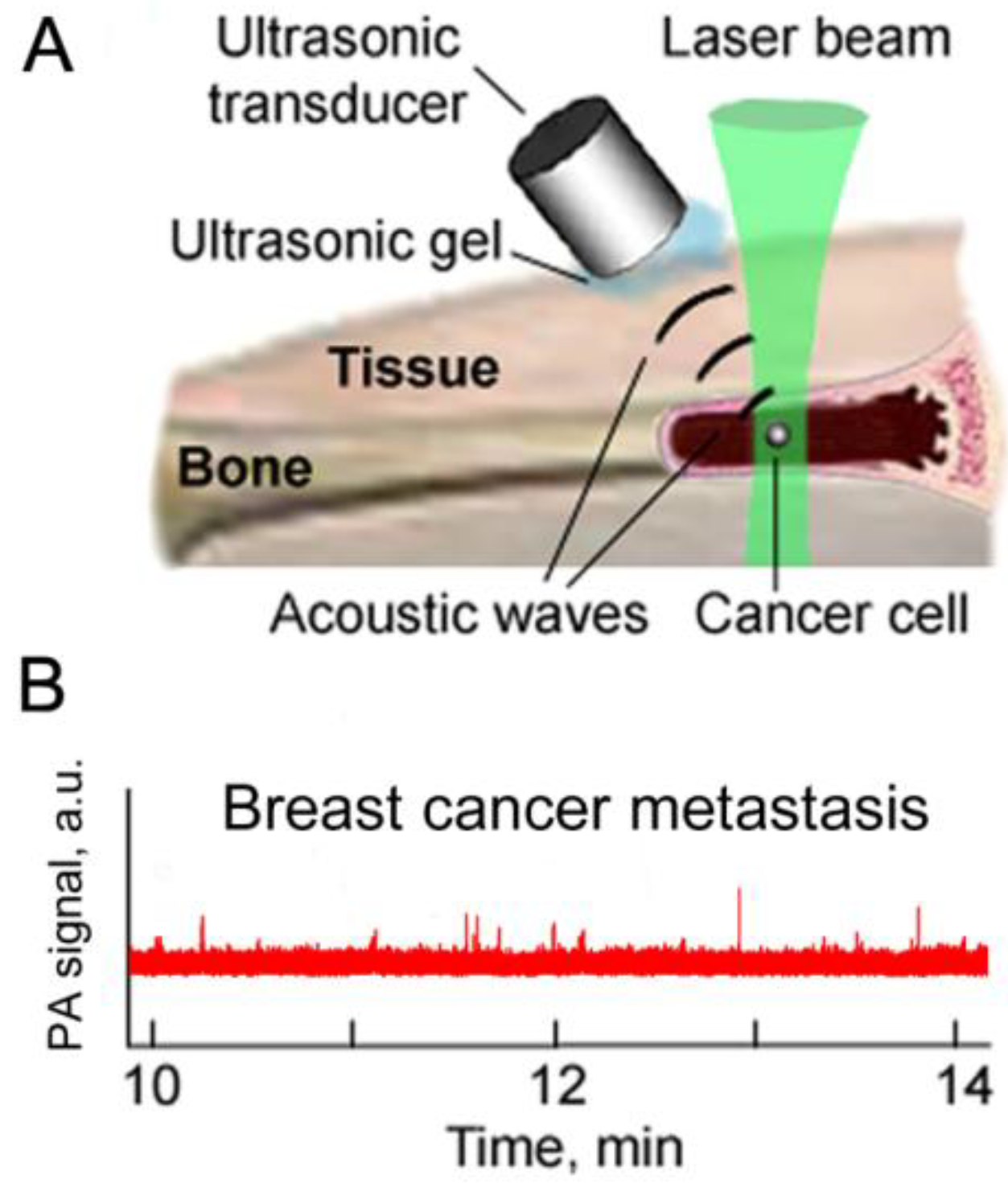
4.6. In Vivo PA Detection in Bone

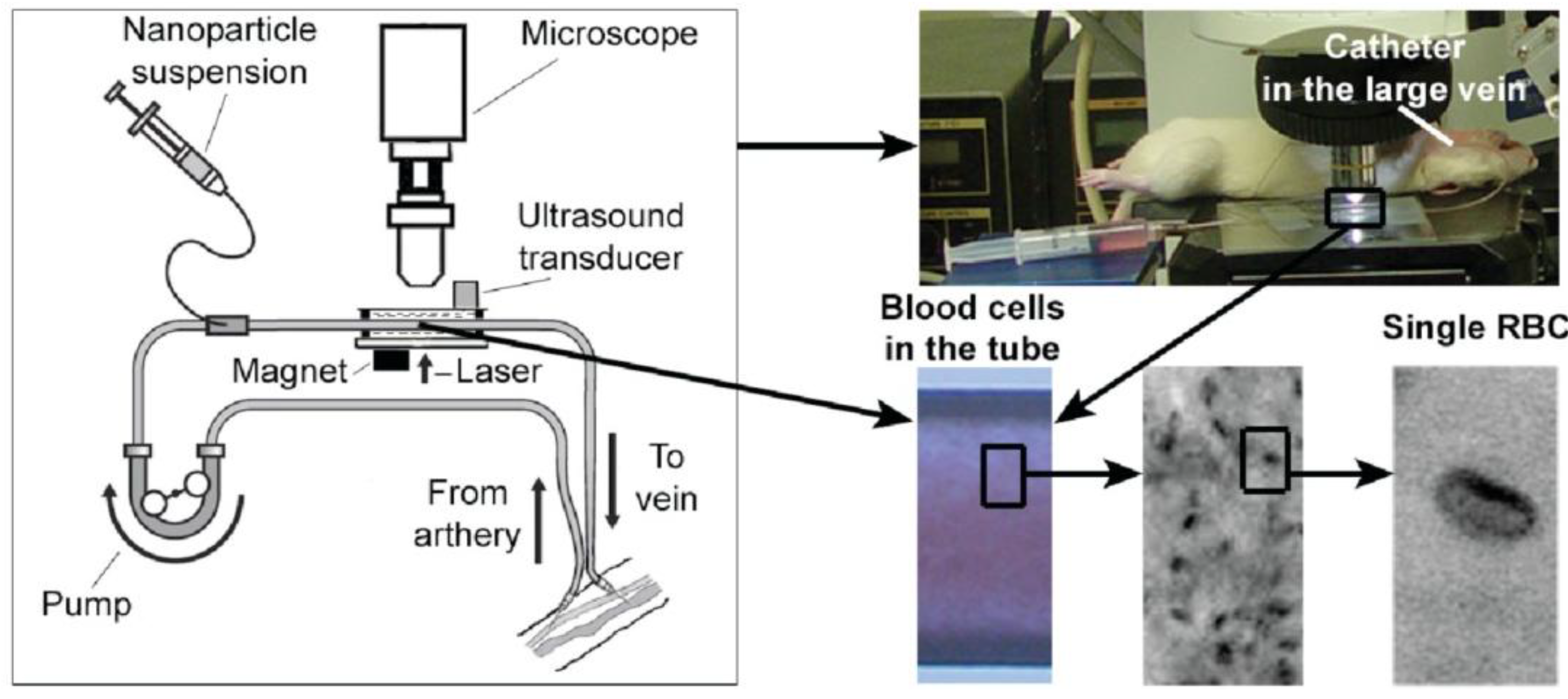
4.7. In Vivo Flow Cytometry with Bypass Schematics

4.8. In Vitro Flow Cytometry Integrating PA, PT and Fluorescence Detection Schematics
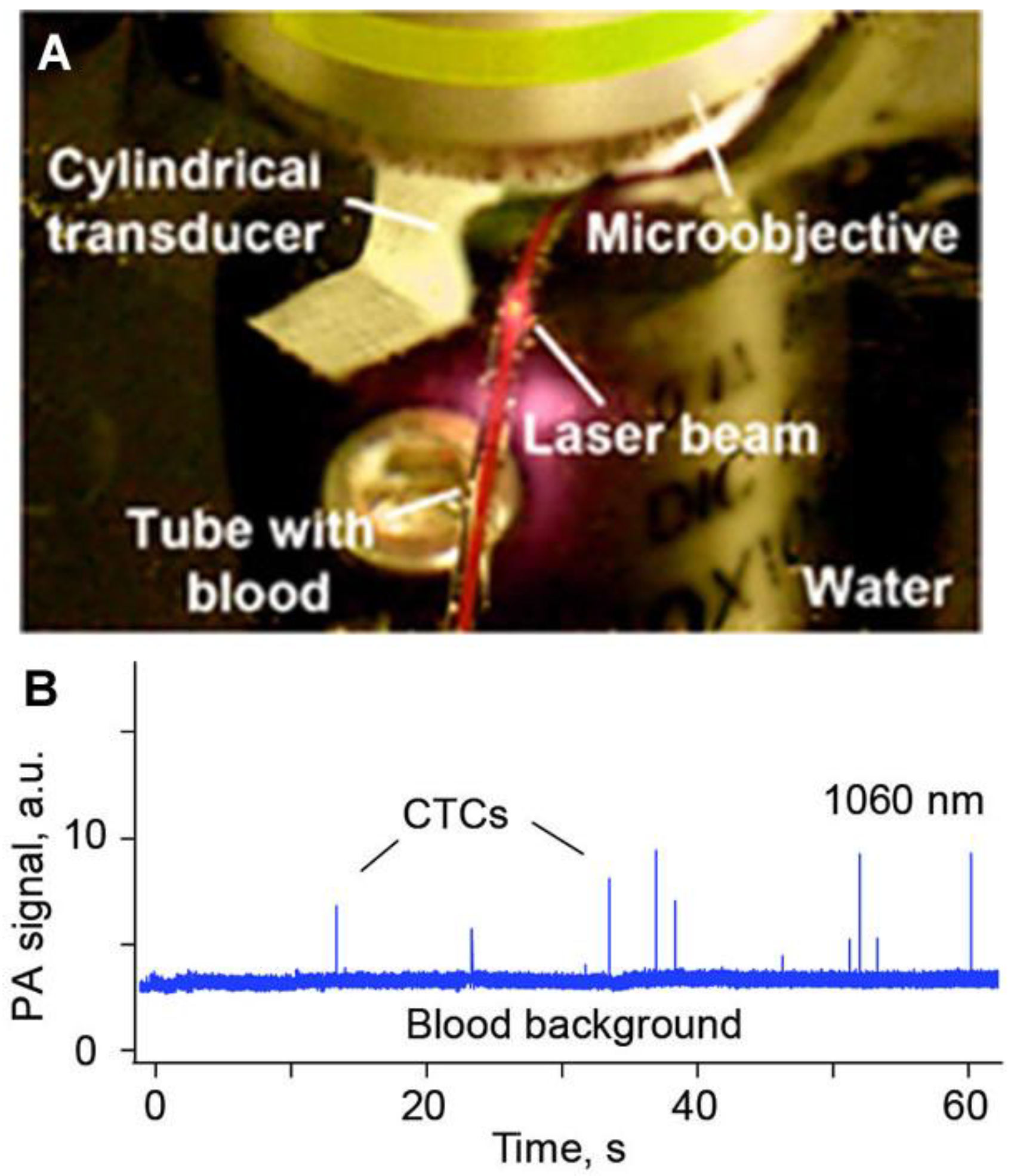
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Kuo, A.H.; Clarke, M.F. Identifying the metastatic seeds of breast cancer. Nat. Biotechnol. 2013, 31, 504–505. [Google Scholar] [CrossRef]
- Karakousis, G.; Yang, R.; Xu, X. Circulating melanoma cells as a predictive biomarker. J. Invest. Dermatol. 2013, 133, 1460–1462. [Google Scholar] [CrossRef]
- Alix-Panabieres, C.; Pantel, K. Circulating tumor cells, liquid biopsy of cancer. Clin. Chem. 2013, 59, 110–118. [Google Scholar] [CrossRef]
- Joosse, S.A.; Pantel, K. Biologic challenges in the detection of circulating tumor cells. Cancer Res. 2013, 73, 8–11. [Google Scholar] [CrossRef]
- Alix-Panabières, C.; Schwarzenbach, H.; Pantel, K. Circulating tumor cells and circulating tumor DNA. Annu. Rev. Med. 2012, 63, 199–215. [Google Scholar] [CrossRef]
- Pantel, K.; Brakenhoff, R.H.; Brandt, B. Detection, clinical relevance and specific biological properties of disseminating tumour cells. Nat. Rev. Cancer. 2008, 8, 329–340. [Google Scholar] [CrossRef]
- Kaiser, J. Cancer’s circulation problem. Science 2010, 327, 1072–1074. [Google Scholar] [CrossRef]
- Yu, M.; Stott, S.; Toner, M.; Maheswaran, S.; Haber, D.A. Circulating tumor cells: Approaches to isolation and characterization (review). J. Cell Biol. 2011, 192, 373–382. [Google Scholar] [CrossRef]
- Dietz, L.; Bruce, R. Advances in optical technologies for rare cell detection and characterization. Recent Results Cancer Res. 2012, 195, 77–85. [Google Scholar] [CrossRef]
- Nguyen, D.X.; Bos, P.D.; Massagué, J. Metastasis, from dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef]
- Krishnamurthy, S. The emerging role of circulating tumor cells in breast cancer. Cancer Cytopathol. 2012, 120, 161–166. [Google Scholar] [CrossRef]
- Riethdorf, S.; Fritsche, H.; Müller, V.; Rau, T.; Schindlbeck, C.; Rack, B.; Janni, W.; Coith, C.; Beck, K.; Jänicke, F.; et al. Detection of circulating tumor cells in peripheral blood of patients with metastatic breast cancer, a validation study of the CellSearch system. Clin. Cancer Res. 2007, 13, 920–928. [Google Scholar] [CrossRef]
- Nagrath, S.; Sequist, L.V.; Maheswaran, S.; Bell, D.W.; Irimia, D.; Ulkus, L.; Smith, M.R.; Kwak, E.L.; Digumarthy, S.; Muzikansky, A.; et al. Isolation of rare circulating tumor cells in cancer patients by microchip technology. Nature 2007, 450, 1235–1239. [Google Scholar]
- Lianidou, E.S. Circulating tumor cells-new challenges ahead. Clin. Chem. 2012, 58, 805–807. [Google Scholar] [CrossRef]
- Hou, H.W.; Warkiani, M.E.; Khoo, B.L.; Li, Z.R.; Soo, R.A.; Tan, D.S.; Lim, W.T.; Han, J.; Bhagat, A.A.; Lim, C.T. Isolation and retrieval of circulating tumor cells using centrifugal forces. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef]
- Stott, S.L.; Hsu, C.H.; Tsukrov, D.I.; Yu, M.; Miyamoto, D.T.; Waltman, B.A.; Rothenberg, S.M.; Shah, A.M.; Smas, M.E.; Korir, G.K.; et al. Isolation of circulating tumor cells using a microvertex-generating herringbone-chip. Proc. Natl. Acad. Sci. USA 2010, 107, 18392–18397. [Google Scholar] [CrossRef]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef]
- Ozkumur, E.; Shah, A.M.; Ciciliano, J.C.; Emmink, B.L.; Miyamoto, D.T.; Brachtel, E.; Yu, M.; Chen, P.; Morgan, B.; Trautwein, J.; et al. Inertial focusing for tumor antigen-dependent and -independent sorting of rare circulating tumor cells. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef]
- Liu, Z.; Fusi, A.; Klopocki, E.; Schmittel, A.; Tinhofer, I.; Nonnenmacher, A.; Keilholz, U. Negative enrichment by immunomagnetic nanobeads for unbiased characterization of circulating tumor cells from peripheral blood of cancer patients. J. Transl. Med. 2011, 9. [Google Scholar] [CrossRef]
- Klinac, D.; Gray, E.S.; Millward, M.; Ziman, M. Advances in personalized targeted treatment of metastatic melanoma and non-invasive tumor monitoring. Front. Oncol. 2013, 3. [Google Scholar] [CrossRef]
- Le Rhun, E.; Tu, Q.; de Carvalho, B.M.; Farre, I.; Mortier, L.; Cai, H.; Kohler, C.; Faure, G.C. Detection and quantification of CSF malignant cells by the CellSearch technology in patients with melanoma leptomeningeal metastasis. Med. Oncol. 2013, 30. [Google Scholar] [CrossRef]
- Adebayo, A.J.; Xu, M.C.; Wechsler, J.; Benali-Furet, N.; Cayre, Y.E.; Saranchuk, J.; Drachenberg, D.; Mai, S. Three-dimensional telomeric analysis of isolated circulating tumor cells (CTCs) defines CTC subpopulations. Transl. Oncol. 2013, 6, 51–65. [Google Scholar]
- Hofman, V.; Ilie, M.; Long-Mira, E.; Giacchero, D.; Butori, C.; Dadone, B.; Selva, E.; Tanga, V.; Passeron, T.; Poissonnet, G.; et al. Usefulness of immunocytochemistry for the detection of the BRAF(V600E) mutation in circulating tumor cells from metastatic melanoma patients. J. Invest. Dermatol. 2013, 133, 1378–1381. [Google Scholar] [CrossRef]
- Khoja, L.; Lorigan, P.; Zhou, C.; Lancashire, M.; Booth, J.; Cummings, J.; Califano, R.; Clack, G.; Hughes, A.; Dive, C. Biomarker utility of circulating tumor cells in metastatic cutaneous melanoma. J. Invest. Dermatol. 2013, 133, 1582–1590. [Google Scholar] [CrossRef]
- Hoshimoto, S.; Shingai, T.; Morton, D.L.; Kuo, C.; Faries, M.B.; Chong, K.; Elashoff, D.; Wang, H.J.; Elashoff, R.M.; Hoon, D.S. Association between circulating tumor cells and prognosis in patients with stage III melanoma with sentinel lymph node metastasis in a phase III international multicenter trial. J. Clin. Oncol. 2012, 30, 3819–3826. [Google Scholar] [CrossRef]
- Visus, C.; Andres, R.; Mayordomo, J.I.; Martinez-Lorenzo, M.J.; Murillo, L.; Saez-Gutierrez, B.; Diestre, C.; Marcos, I.; Astier, P.; Godino, J.; et al. Prognostic role of circulating melanoma cells detected by reverse transcriptase-polymerase chain reaction for tyrosinase mRNA in patients with melanoma. Melanoma Res. 2007, 17, 83–89. [Google Scholar] [CrossRef]
- Mocellin, S.; Hoon, D.; Ambrosi, A.; Nitti, D.; Rossi, C.R. The prognostic value of circulating tumor cells in patients with melanoma, a systematic review and meta-analysis. Clin. Cancer Res. 2006, 12, 4605–4613. [Google Scholar] [CrossRef]
- Galanzha, E.I.; Zharov, V.P. Photoacoustic flow cytometry. Methods 2012, 57, 28–296. [Google Scholar]
- Juratli, M.; Sarimollaoglu, M.; Siegel, E.; Nedosekin, D.A.; Galanzha, E.I.; Suen, J.Y.; Zharov, V.P. Real-time monitoring of circulating tumor cell release during tumor manipulation using in vivo photoacoustic and fluorescent flow cytometry. Head Neck 2013. [Google Scholar] [CrossRef]
- Hayashi, K.; Jiang, P.; Yamauchi, K.; Yamamoto, N.; Tsuchiya, H.; Tomita, K.; Moossa, A.R.; Bouvet, M.; Hoffman, R.M. Real-time imaging of tumor-cell shedding and trafficking in lymphatic channels. Cancer Res. 2007, 67, 8223–2248. [Google Scholar] [CrossRef]
- Shapiro, H.M. Practical Flow Cytometry, 4th ed.; Wiley Liss: New York, NY, USA, 2003. [Google Scholar]
- Cellular Diagnostics: Basic Principles, Methods and Clinical Applications of Flow Cytometry; Sack, U.; Tárnok, A.; Rothe, G. (Eds.) Karger Medical and Scientific Publishers: Basel, Switzerland, 2008.
- Tuchin, V.V.; Tárnok, A.; Zharov, V.P. Towards in vivo flow cytometry. Biophotonics 2009, 2, 457–458. [Google Scholar] [CrossRef]
- Tuchin, V.V.; Tárnok, A.; Zharov, V.P. In vivo flow cytometry, a horizon of opportunities. Cytometry 2011, 79, 737–745. [Google Scholar]
- Zharov, V.P.; Galanzha, E.I.; Tuchin, V.V. Photothermal imaging of moving cells in lymph and blood flow in vivo. Proc. SPIE 2004, 5320, 256–263. [Google Scholar]
- Zharov, V.P.; Galanzha, E.I.; Tuchin, V.V. Photothermal image flow cytometry in vivo. Opt. Lett. 2005, 30, 628–630. [Google Scholar]
- Zharov, V.; Galanzha, E.; Shashkov, E.; Khlebtsov, N.; Tuchin, V. In vivo photoacoustic flow cytometry for monitoring circulating singe cancer cells and contrast agents. Opt. Lett. 2006, 31, 3623–3625. [Google Scholar] [CrossRef]
- Novak, J.; Georgakoudi, I.; Wei, X.; Prossin, A.; Lin, C.P. An in vivo flow cytometer for real-time detection and quantification of circulating cells. Opt. Lett. 2004, 29, 77–79. [Google Scholar]
- Georgakoudi, I.; Solban, N.; Novak, J.; Rice, W.L.; Wei, X.; Hasan, T.; Lin, C.P. In vivo flow cytometry: A new method for enumerating circulating cancer cells. Cancer Res. 2004, 64, 5044–5047. [Google Scholar]
- Wang, H.; Hartmann, L.C.; Cheng, J.X.; Low, P.S. In vivo quantification of rare circulating tumor cells by multiphoton intravital flow cytometry. Proc. Natl. Acad. Sci. USA 2007, 104, 11760–11765. [Google Scholar]
- Tkaczyk, E.R.; Zhong, C.F.; Ye, J.Y.; Myc, A.; Thomas, T.; Cao, Z.; Duran-Struuck, R.; Luker, K.E.; Luker, G.D.; Norris, T.B.; et al. In vivo monitoring of multiple circulating cell populations using two-photon flow cytometry. Opt. Commun. 2008, 281, 888–894. [Google Scholar]
- Boutrus, S.; Greiner, C.; Hwu, D.; Chan, M.; Kuperwasser, C.; Lin, C.P.; Georgakoudi, I. Portable two-color in vivo flow cytometer for real-time detection of fluorescently-labeled circulating cells. J. Biomed. Opt. 2007, 12. [Google Scholar] [CrossRef]
- Tuchin, V.V.; Galanzha, E.I.; Zharov, V.P. In vivo Image Flow Cytometry. In Advanced Optical Cytometry, Methods and Disease Diagnoses; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; pp. 387–431. [Google Scholar]
- Tanev, S.; Sun, W.; Pond, J.; Tuchin, V.V.; Zharov, V.P. Optical Imaging of Cells with Gold Nanoparticle Clusters as Light Scattering Contrast Agents, A Finite-Difference Time-Domain Approach to the Modeling of Flow Cytometry Configurations. In Advance Optical Cytometry, Methods and Disease Diagnoses; Tuchin, V.V., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; pp. 35–62. [Google Scholar]
- Tuchin, V.V.; Galanzha, E.I.; Zharov, V.P. In vivo Photothermal and Photoacoustic Flow Cytometry. In Advanced Optical Flow Cytometry; Tuchin, V.V., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; pp. 501–571. [Google Scholar]
- Greiner, C.; Georgakoudi, I. Advances in fluorescence-based in vivo flow cytometry for cancer application. In Advanced Optical Cytometry, Methods and Disease Diagnoses; Tuchin, V.V., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; pp. 463–500. [Google Scholar]
- Tkaczyk, E.R.; Tkaczyk, A.H. Multiphoton flow cytometry strategies and applications. Cytometry 2011, 79, 775–788. [Google Scholar]
- Zichao, F.; Xunbin, W. In vivo flow cytometry: A powerful optical technology to detect circulating tumor cells and diagnose cancer metastasis in vivo. Photon Lasers Med. 2013, 2, 27–35. [Google Scholar]
- Nedosekin, D.A.; Sarimollaoglu, M.; Galanzha, E.I.; Sawant, R.; Torchilin, V.P.; Verkhusha, V.V.; Ma, J.; Frank, M.H.; Biris, A.S.; Zharov, V.P. Synergy of photoacoustic and fluorescence flow cytometry of circulating cells with negative and positive contrasts. J. Biophotonics 2013, 6, 425–434. [Google Scholar] [CrossRef]
- Zharov, V.P.; Galanzha, E.I.; Tuchin, V.V. Integrated photothermal flow cytometry in vivo. J. Biomed. Opt. 2005, 10. [Google Scholar] [CrossRef]
- Galanzha, E.I.; Tuchin, V.V.; Zharov, V.P. Advances in small animal mesentery models for in vivo flow cytometry, dynamic microscopy, and drug screening (review). World J. Gastroenterol. 2007, 13, 192–218. [Google Scholar]
- Galanzha, E.I.; Shashkov, E.V.; Tuchin, V.V.; Zharov, V.P. In vivo multiparameter, multispectral lymph flow cytometry with natural cell focusing, label-free detection and multicolor nanoparticle probes. Cytometry 2008, 73, 884–894. [Google Scholar]
- Zharov, V.P.; Letokhov, V.S. Laser Optoacoustic Spectroscopy; Springer: Berlin, Germany, 1986. [Google Scholar]
- Zharov, V.P. Laser optoacoustic spectroscopy in chromatography. In Laser Analytical Spectrochemistry; Adam Hilger: Bristol & Boston, MA, Boston, MA, USA, 1986; pp. 229–271. [Google Scholar]
- Oraevsky, A.A.; Karabutov, A.A. Optoacoustic Tomography. In Handbook of Biomedical Photonics; Vo-Dinh, T., Ed.; CRC Press: Boca Raton, FL, USA, 2003; pp. 34/1–34/34. [Google Scholar]
- Wang, L.V.; Hu, S. Photoacoustic tomography: In vivo imaging from organelles to organs. Science 2012, 335, 1458–1462. [Google Scholar]
- Razansky, D.; Distel, M.; Vinegoni, C.; Ma, R.; Perrimon, N.; Koster, R.W.; Ntziachristos, V. Multispectral opto-acoustic tomography of deep-seated fluorescent proteins in vivo. Nat. Photon. 2009, 3, 412–417. [Google Scholar] [CrossRef]
- Beard, P. Biomedical photoacoustic imaging. Interface Focus 2011, 1, 602–631. [Google Scholar] [CrossRef]
- Mehrmohammadi, M.; Yoon, S.J.; Yeager, D.; Emelianov, S.Y. Photoacoustic imaging for cancer detection and staging. Curr. Mol. Imaging 2013, 2, 89–105. [Google Scholar] [CrossRef]
- The thermal lens in absorption spectroscopy. In Ultrasensitive Laser Spectroscopy; Klinger, D.S. (Ed.) Acad. Press: New York, NY, USA, 1983; pp. 175–232.
- Tamaki, E.; Sato, K.; Tokeshi, M.; Aihara, M.; Kitamori, T. Single-cell analysis by a scanning thermal lens microscope with a microchip: Direct monitoring of cytochrome c distribution during apoptosis process. Ann. Chem. 2002, 74, 1560–1564. [Google Scholar] [CrossRef]
- Sato, K.; Yamanaka, M.; Hagino, T.; Tokeshi, M.; Kimura, H.; Kitamori, T. Microchip-based enzyme-linked immunosorbent assay (microELISA) system with thermal lens detection. Lab Chip. 2004, 4, 570–575. [Google Scholar] [CrossRef]
- Zharov, V.P.; Lapotko, D.O. Photothermal imaging of nanoparticles and cells. IEEE J. Sel. Topics Quant. Electron. 2005, 11, 733–751. [Google Scholar] [CrossRef]
- Van Dijk, M.A.; Tchebotareva, A.L.; Orrit, M.; Lippitz, M.; Berciaud, S.; Lasne, D.; Cognet, L.; Lounis, B. Absorption and scattering microscopy of single metal nanoparticles. Chem Phys. 2006, 8, 3486–3495. [Google Scholar]
- Blab, G.A.; Cognet, L.; Berciaud, S.; Alexandre, I.; Husar, D.; Remacle, J.; Lounis, B. Optical readout of gold nanoparticle-based DNA microarrays without silver enhancement. Biophys. J. 2006, 90, L13–L15. [Google Scholar] [CrossRef]
- Nedosekin, D.A.; Galanzha, E.I.; Ayyadevara, S.; Shmookler Reis, R.J.; Zharov, V.P. Photothermal confocal spectromicroscopy of multiple cellular chromophores and fluorophores. Biophys. J. 2012, 102, 672–681. [Google Scholar] [CrossRef]
- Nedosekin, D.A.; Galanzha, E.I.; Dervishi, E.; Biris, A.S.; Zharov, V.P. Super-resolution nonlinear photothermal microscopy. Small 2013. [Google Scholar] [CrossRef]
- Thakor, A.S.; Gambir, S.S. Nanooncology: The future of cancer diagnosis and therapy. CA Cancer J. Clin. 2013, 63, 395–418. [Google Scholar] [CrossRef]
- Gaiduk, A.; Yorulmaz, M.; Ruijgrok, P.V.; Orrit, M. Room-temperature detection of a single molecule’s absorption by photothermal contrast. Science 2010, 330, 353–356. [Google Scholar] [CrossRef]
- Berciaud, S.; Cognet, L.; Blab, G.A.; Lounis, B. Photothermal heterodyne imaging of individual nonfluorescent nanoclusters and nanocrystals. Phys. Rev. Lett. 2004, 93. [Google Scholar] [CrossRef]
- Galanzha, E.I.; Tuchin, V.V.; Zharov, V.P. In vivo integrated flow image cytometry and lymph/blood vessels dynamic microscopy. J. Biomed. Opt. 2005, 10. [Google Scholar] [CrossRef]
- Menyaev, Y.A.; Nedosekin, D.A.; Sarimollaoglu, M.; Juratli, M.A.; Galanzha, E.I.; Tuchin, V.V.; Zharov, V.P. Optical clearing in photoacoustic flow cytometry. Biomed. Opt. Express 2013, 4, 3030–3041. [Google Scholar] [CrossRef]
- Zharov, V.; Galitovsky, V.; Viegas, M. Photothermal detection of local thermal effects during selective nanophotothermolysis. Appl. Phys. Letter 2003, 83, 4897–4899. [Google Scholar] [CrossRef]
- Zharov, V.P.; Galanzha, E.I.; Menyaev, Y.A.; Tuchin, V.V. In vivo high-speed imaging of individual cells in fast blood flow. J. Biomed. Opt. 2006, 11. [Google Scholar] [CrossRef]
- Zharov, V.P.; Galanzha, E.I.; Tuchin, V.V. In vivo photothermal flow cytometry: Imaging and detection of individual cells in blood and lymph flow (review/prospect). J. Cell Biochem. 2006, 97, 916–932. [Google Scholar] [CrossRef]
- Zharov, V.P.; Galanzha, E.I.; Tuchin, V.V. Photothermal flow cytometry in vitro for detection and imaging of individual moving cells. Cytometry 2007, 71, 191–206. [Google Scholar]
- Zharov, V.P.; Galanzha, E.I.; Shashkov, E.V.; Kim, J.-W.; Khlebtsov, N.G.; Tuchin, V.V. Photoacoustic flow cytometry: Principle and application for real-time detection of circulating single nanoparticles, pathogens, and contrast dyes in vivo. J. Biomed. Opt. 2007, 12. [Google Scholar] [CrossRef]
- Galanzha, E.I.; Tuchin, V.V.; Zharov, V.P. Optical monitoring of microlympatic disturbances at experimental lymphedema, Lymphat. Res. Biol. 2007, 5, 11–27. [Google Scholar]
- Kalchenko, V.; Brill, A.; Bayewitch, M.; Fine, I.; Zharov, V.; Galanzha, E.; Tuchin, V.; Harmelin, A. In vivo dynamic light scattering imaging of blood coagulation. J. Biomed. Opt. 2007, 12. [Google Scholar] [CrossRef]
- Kim, J.-W.; Galanzha, E.I.; Shashkov, E.V.; Moon, H.-M.; Zharov, V.P. Golden carbon nanotubesas multimodal photoacoustic and photothermal high-contrast molecular agents. Nat. Nanotechnol. 2009, 4, 688–694. [Google Scholar] [CrossRef]
- Galanzha, E.I.; Shashkov, E.V.; Kelly, T.; Kim, J.-W.; Yang, L.; Zharov, V.P. In vivo magnetic enrichment and multiplex photoacoustic detection of circulating tumour cells. Nat. Nanotechnol. 2009, 4, 855–860. [Google Scholar] [CrossRef]
- Galanzha, E.I.; Shashkov, E.V.; Spring, P.; Suen, J.Y.; Zharov, V.P. In vivo, noninvasive, label-free detection and eradication of circulating metastatic melanoma cells using two-color photoacoustic flow cytometry with a diode laser. Cancer Res. 2009, 69, 7926–7934. [Google Scholar] [CrossRef]
- Galanzha, E.I.; Kokoska, M.S.; Shashkov, E.V.; Kim, J.-W.; Tuchin, V.V.; Zharov, V.P. In vivo fiber-based multicolor photoacoustic detection and photothermal purging of metastasis in sentinel lymph nodes targeted by nanoparticles. J. Biophotonics 2009, 2, 528–539. [Google Scholar] [CrossRef]
- Tanev, S.; Sun, W.; Pond, J.; Tuchin, V.V.; Zharov, V.P. Flow cytometry with gold nanoparticles and their clusters as scattering contrast agents. J. Biophotonics 2009, 2, 505–520. [Google Scholar]
- Galanzha, I.E.; Kim, J.-W.; Zharov, V.P. Nanotechnology-based molecular photoacoustic and photothermal flow cytometry platform for in vivo detection and killing of circulating cancer stem cells. J. Biophotonics 2009, 2, 725–735. [Google Scholar] [CrossRef]
- Shashkov, E.V.; Galanzha, E.I.; Zharov, V.P. Photothermal and photoacoustic Raman cytometry in vitro and in vivo. Opt. Exp. 2010, 18, 6929–6944. [Google Scholar] [CrossRef]
- Nedosekin, D.A.; Sarimollaoglu, M.; Shashkov, E.V.; Galanzha, E.I.; Zharov, V.P. Ultra-fast photoacoustic flow cytometry with a 0.5 MHz pulse repetition rate nanosecond laser. Opt. Exp. 2010, 18, 8605–8620. [Google Scholar] [CrossRef]
- Galanzha, E.I.; Zharov, V.P. In vivo photoacoustic and photothermal cytometry for monitoring multiple blood rheology parameters. Cytometry 2011, 79, 746–757. [Google Scholar]
- Nedosekin, D.A.; Khodakovskaya, M.V.; de Silva, K.; Dervishi, E.; Biris, A.S.; Galanzha, E.I.; Zharov, V.P. In vivo plant flow cytometry: A first proof-of-concept. Cytometry 2011, 79, 55–65. [Google Scholar]
- Nedosekin, D.; Sarimollaoglu, M.; Ye, J.-H.; Galanzha, E.; Zharov, V.P. In vivo ultra-fast photoacoustic flow cytometry of circulating human melanoma cells using near-infrared high-pulse rate lasers. Cytometry 2011, 79, 825–833. [Google Scholar]
- Proskurnin, M.; Galanzha, E.I.; Mock, D.M.; Zharov, V.P. In vivo multispectral photothermal and photoacoustic flow cytometry with multicolor dyes: A potential for real-time assessment of circulation, dye-cell interaction, and blood volume. Cytometry 2011, 79, 834–847. [Google Scholar]
- De la Zerda, A.; Kim, J.-W.; Galanzha, E.I.; Gambhir, S.S.; Zharov, V.P. Advanced contrast nanoagents for photoacoustic molecular imaging, cytometry, blood test, and photothermal theranostics. Contrast Media Mol. Imaging 2011, 6, 346–369. [Google Scholar] [CrossRef]
- Sarimollaoglu, M.; Nedosekin, D.A.; Simanovsky, Y.; Galanzha, E.I.; Zharov, V.P. In vivo photoacoustic time-of-flight velocity measurement of single cells and nanoparticles. Opt. Lett. 2011, 36, 4086–4088. [Google Scholar]
- Galanzha, E.I.; Shashkov, E.; Sarimollaoglu, M.; Beenken, K.E.; Basnakian, A.G.; Shirtliff, M.E.; Kim, J.W.; Smeltzer, M.S.; Zharov, V.P. In vivo magnetic enrichment, photoacoustic diagnosis, and photothermal purging of infected blood using multifunctional gold and magnetic nanoparticles. PLoS One 2012, 7, e45557. [Google Scholar]
- Kim, J.W.; Galanzha, E.I.; Zaharoff, D.A.; Griffin, R.J.; Zharov, V.P. Nanotheranostics of circulating tumor cells, infections and other pathological features in vivo. Mol. Pharm. 2013, 10, 813–830. [Google Scholar] [CrossRef]
- Shao, J.; Griffin, R.J.; Galanzha, E.I.; Kim, J.-W.; Koonce, N.; Webber, J.; Mustafa, T.; Biris, A.; Nedosekin, D.A.; Zharov, V.P. Photothermal nanodrugs: Potential of TNF-gold nanospheres for cancer theranostics. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef]
- Nedosekin, D.A.; Juratli, M.A.; Sarimollaoglu, M.; Moore, C.L.; Rusch, N.J.; Smeltzer, M.S.; Zharov, V.P.; Galanzha, E.I. Photoacoustic and photothermal detection of circulating tumor cells, bacteria and nanoparticles in cerebrospinal fluid in vivo and ex vivo. J. Biophotonics 2013, 6, 523–533. [Google Scholar] [CrossRef]
- Sarimollaoglu, M.; Nedosekin, D.A.; Galanzha, E.I.; Zharov, V.P. In vivo real-time monitoring of nanoparticle clearance rate from blood circulation using high speed flow cytometry. Proc. SPIE 2013, 8427. [Google Scholar] [CrossRef]
- Juratli, M.A.; Galanzha, E.I.; Sarimollaoglu, M.; Nedosekin, D.A.; Suen, J.Y.; Zharov, V.P. In vivo detection of circulating tumor cells during tumor manipulation. Proc. SPIE 2013, 8565. [Google Scholar] [CrossRef]
- Nedosekin, D.A.; Sarimollaoglu, M.; Foster, S.; Galanzha, E.I.; Zharov, V.P. Photoacoustic-fluorescence in vitro flow cytometry for quantification of absorption, scattering and fluorescence properties of the cells. Proc. SPIE 2013, 8581. [Google Scholar] [CrossRef]
- Juratli, M.A.; Galanzha, E.I.; Sarimollaoglu, M.; Nedosekin, D.A.; Suen, J.Y.; Zharov, V.P. Photoacoustic monitoring of circulating tumor cells released during medical procedures. Proc. SPIE 2013, 8581. [Google Scholar] [CrossRef]
- Sarimollaoglu, M.; Nedosekin, D.A.; Menyaev, Y.A.; Juratli, M.A.; Zharov, V.P. Nonlinear photoacoustic signal amplification from single targets in absorption background. Photoacoustics 2013. [Google Scholar] [CrossRef]
- Zharov, V.P. Ultrasharp nonlinear photothermal and photoacoustic resonances and holes beyond the spectral limit. Nat. Photonics 2011, 5, 110–116. [Google Scholar] [CrossRef]
- Nedosekin, D.A.; Shashkov, E.V.; Galanzha, E.I.; Hennings, L.; Zharov, V.P. Photothermal multispectral image cytometry for quantitative histology of nanoparticles and micrometastasis in intact, stained and selectively burned tissues. Cytometry 2010, 77, 1049–1058. [Google Scholar]
- Goode, B. In vivo flow cytometry enables highly sensitive diagnosis. Available online: http://www.bioopticsworld.com//articles/2009/03/in-vivo-flow-cytometry-enables-highly-sensitive-diagnosis.html (accessed on 1 December 2013).
- Wang, L.; Maslov, K.; Wang, L.V. Single-cell label-free photoacoustic flowoxigraphy in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 5759–5764. [Google Scholar] [CrossRef]
- Galanzha, E.I.; Nedosekin, D.A.; Sarimollaoglu, M.; Orza, A.I.; Biris, A.I.; Verkhusha, V.V.; Zharov, V.P. Photoacoustic and photothermal cytometry usingh photoswitchable proteins and nanoparticles with ultrasharp resonances. J. Biophotonics 2013. [Google Scholar] [CrossRef]
- Mertiri, A.; Altug, N.; Hong, M.K.; Mehta, P.; Mertz, J.; Ziegler, L.D.; Erramilli, S. Pitchfork bifurcation and Zharov splitting in nonlinear mid-infrared photothermal spectroscopy in a liquid crystal using a quantum cascade laser. Available online: http://arxiv.org/abs/1310.0123 (accessed on 1 September 2013).
- Tuchin, V.V. Optical clearing tissue and blood using immersion method. J. Phys. 2005, 38, 2497–2518. [Google Scholar]
- Zhou, Y.; Yao, J.; Wang, L.V. Optical clearing-aided photoacoustic microscopy with enhanced resolution and imaging depth. Opt Lett. 2013, 38, 2592–2595. [Google Scholar] [CrossRef]
- Chen, Y.S.; Frey, W.; Kim, S.; Kruizinga, P.; Homan, K.; Emelianov, S. Silica-coated gold nanorods as photoacoustic signal nanoamplifiers. Nano Lett. 2011, 11, 348–354. [Google Scholar] [CrossRef]
- Shashkov, E.V.; Everts, M.; Galanzha, E.I.; Zharov, V.P. Quantum dots as multimodal photoacoustic and photothermal contrast agents. Nano Lett. 2008, 8, 3953–3958. [Google Scholar] [CrossRef]
- Gutiérrez-Juárez, G.; Gupta, S.K.; Weight, R.M.; Polo-Parada, L.; Papagiorgio, C.; Bunch, J.D.; Viator, J.A. Optical photoacoustic detection of circulating melanoma cells in vitro. Int. J. Thermophys. 2010, 31, 784–792. [Google Scholar] [CrossRef]
- O’Brien, C.M.; Rood, K.D.; Bhattacharyya, K.; DeSouza, T.; Sengupta, S.; Gupta, S.K.; Mosley, J.D.; Goldschmidt, B.S.; Sharma, N.; Viator, J.A. Capture of circulating tumor cells using photoacoustic flowmetry and two phase flow. J. Biomed. Opt. 2012, 17. [Google Scholar] [CrossRef]
- Bhattacharyya, K.; Njoroge, M.; Goldschmidt, B.S.; Gaffigan, B.; Rood, K.; Viator, J.A. Detection, isolation, and capture of circulating breast cancer cells with photoacoustic flow cytometry. Proc. SPIE 2013, 8570. [Google Scholar] [CrossRef]
- Pérez-Gutiérrez, F.G.; Camacho-López, S.; Evans, R.; Guillén, G.; Goldschmidt, B.S.; Viator, J.A.; Aguilar, G. Plasma membrane integrity and survival of melanoma cells after nanosecond laser pulses. Ann. Biomed. Eng. 2010, 38, 3521–3531. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Galanzha, E.I.; Zharov, V.P. Circulating Tumor Cell Detection and Capture by Photoacoustic Flow Cytometry in Vivo and ex Vivo. Cancers 2013, 5, 1691-1738. https://doi.org/10.3390/cancers5041691
Galanzha EI, Zharov VP. Circulating Tumor Cell Detection and Capture by Photoacoustic Flow Cytometry in Vivo and ex Vivo. Cancers. 2013; 5(4):1691-1738. https://doi.org/10.3390/cancers5041691
Chicago/Turabian StyleGalanzha, Ekaterina I., and Vladimir P. Zharov. 2013. "Circulating Tumor Cell Detection and Capture by Photoacoustic Flow Cytometry in Vivo and ex Vivo" Cancers 5, no. 4: 1691-1738. https://doi.org/10.3390/cancers5041691