Long Non-Coding RNAs Embedded in the Rb and p53 Pathways

Abstract
:1. Introduction
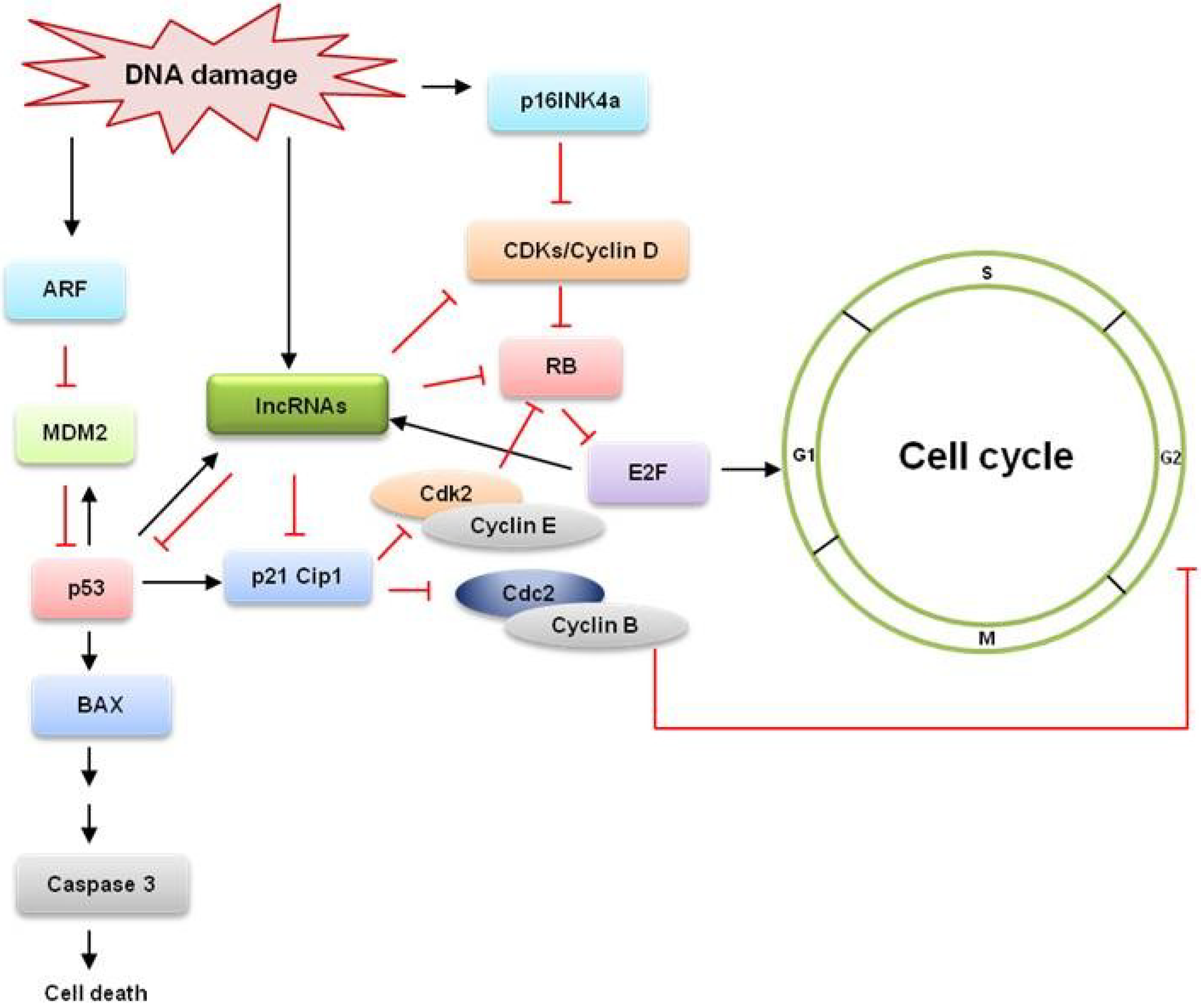

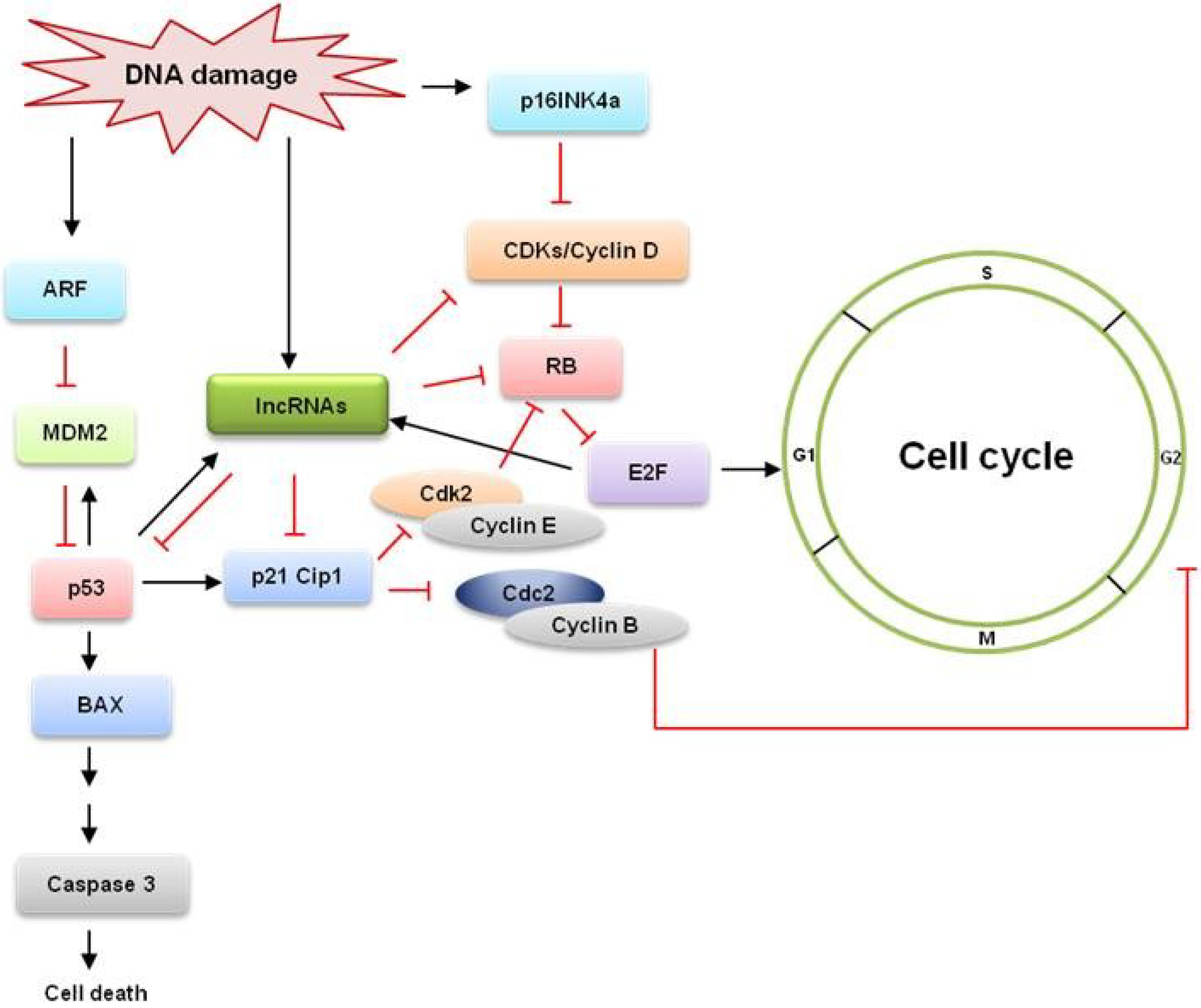{kind=link}
{kind=link}
{kind=link}
| lncRNA | Targets and mechanism of action | References |
|---|---|---|
| lncRNA-p21 | Inhibition of transcription of target genes involved in apoptosis and cell cycle through hnRNPK; also represses the translation of β-catenin and Jun B mRNA translation through HuR | [38,39,40] |
| PANDA | Interacts with the transcription factor NF-YA to limit expression of proapoptotic genes | [41] |
| MEG3 | Interacts with p53 to transactive target gene promoters | [42,43,44,45,46] |
| LincRNA-RoR | Inhibits translation of p53 mRNA | [47,48] |
| LOC285194 | Inhibits function of miR-211 | [49] |
| MALAT1 | Down-regulates p53 and target genes; also up-regulates pro-metastatic genes such as MIA2 and ROBO1 | [50,51,52,53,54] |
| p53-induced eRNAs | Promotes p53 target gene transcription | [55] |
| H19 | Down-regulates RB mRNA translation through miR-675 | [56] |
| KCNQ1OT1 | Inhibits p57 transcription | [57,58,59,60,61,62,63] |
| ANRIL | Represses the INK4b-ARF-INK4a locus with PRC1/2 | [64,65,66,67,68] |
| HULC | Sequestration of miR-372, causing derepression of PRKACB; also suppress p18 expression | [69,70] |
| ncRNACCNDI | Represses CCND1 transcription with activation of TLS | [71] |
| GADD7 | Inhibits TDP43 by sequestration of TDP43 | [72,73] |
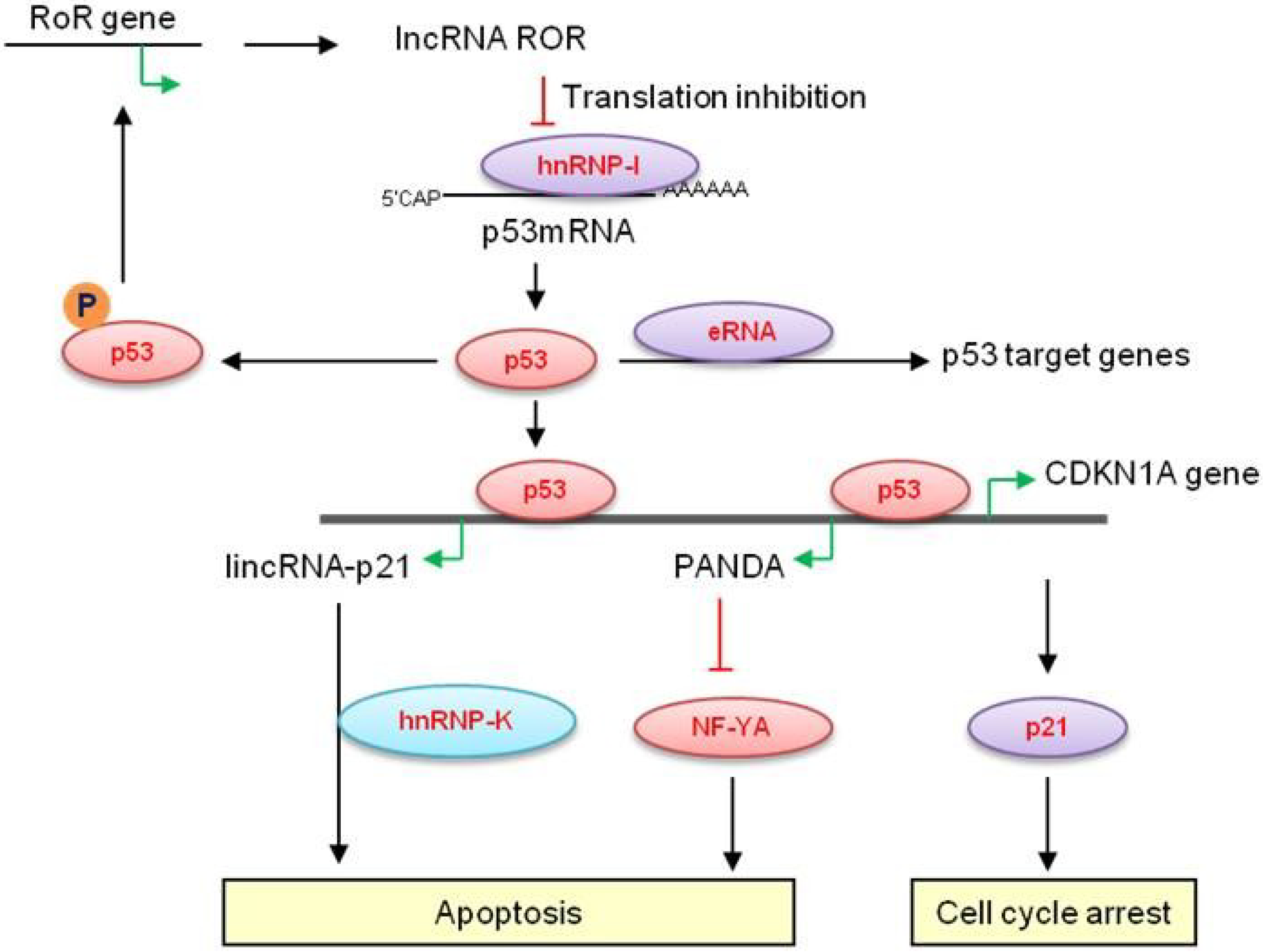
2. lncRNAs Involved in the p53 Pathway
2.1. Long Non-Coding RNAs at the CDKN1A (p21) Locus—LincRNA-p21
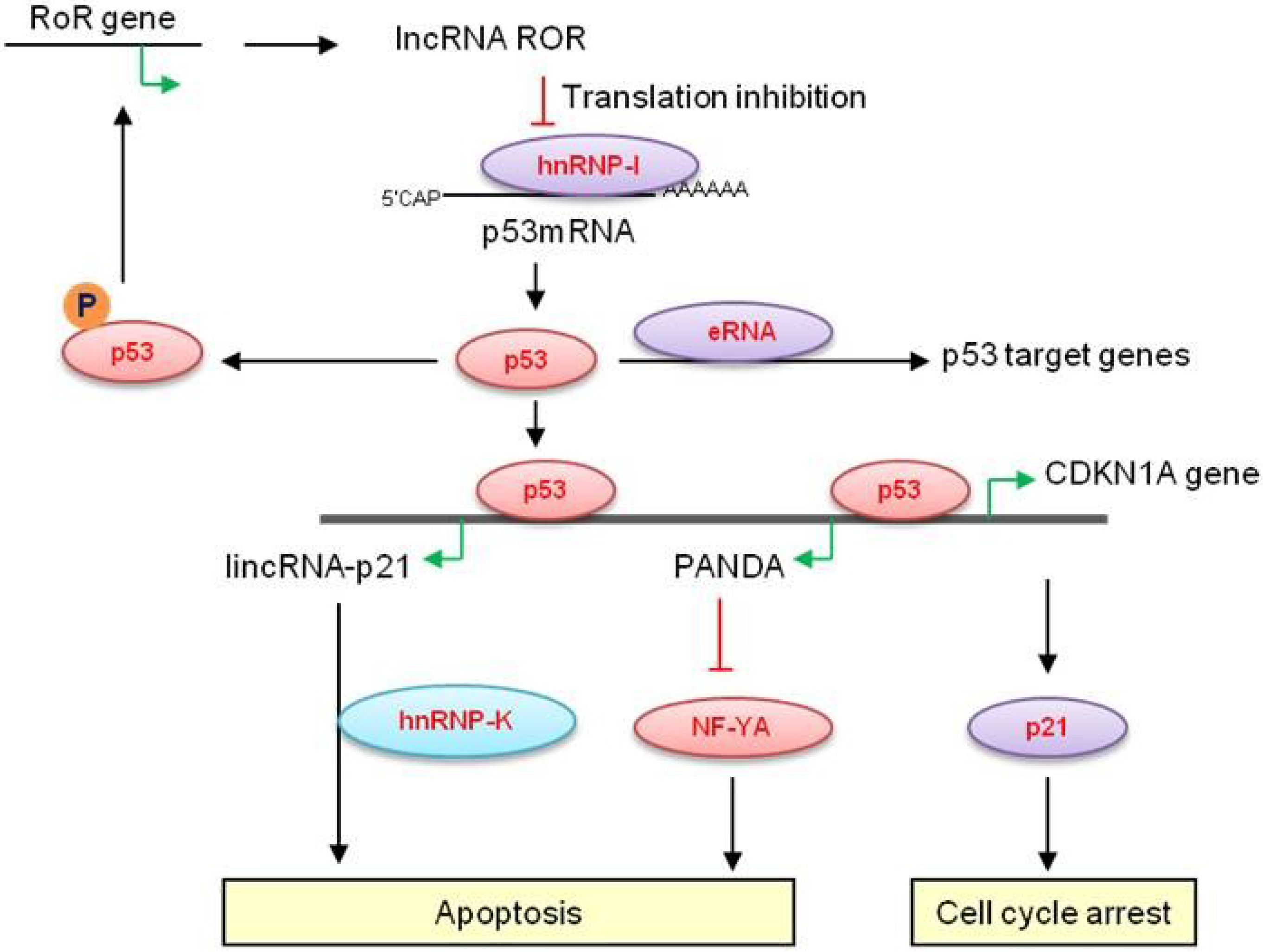
2.2. p21-Associated ncRNA DNA Damage-Activated—PANDA
2.3. MEG3
2.4. LincRNA-RoR
2.5. LOC285194
2.6. MALAT1
2.7. P53-Induced Enhancer RNAs
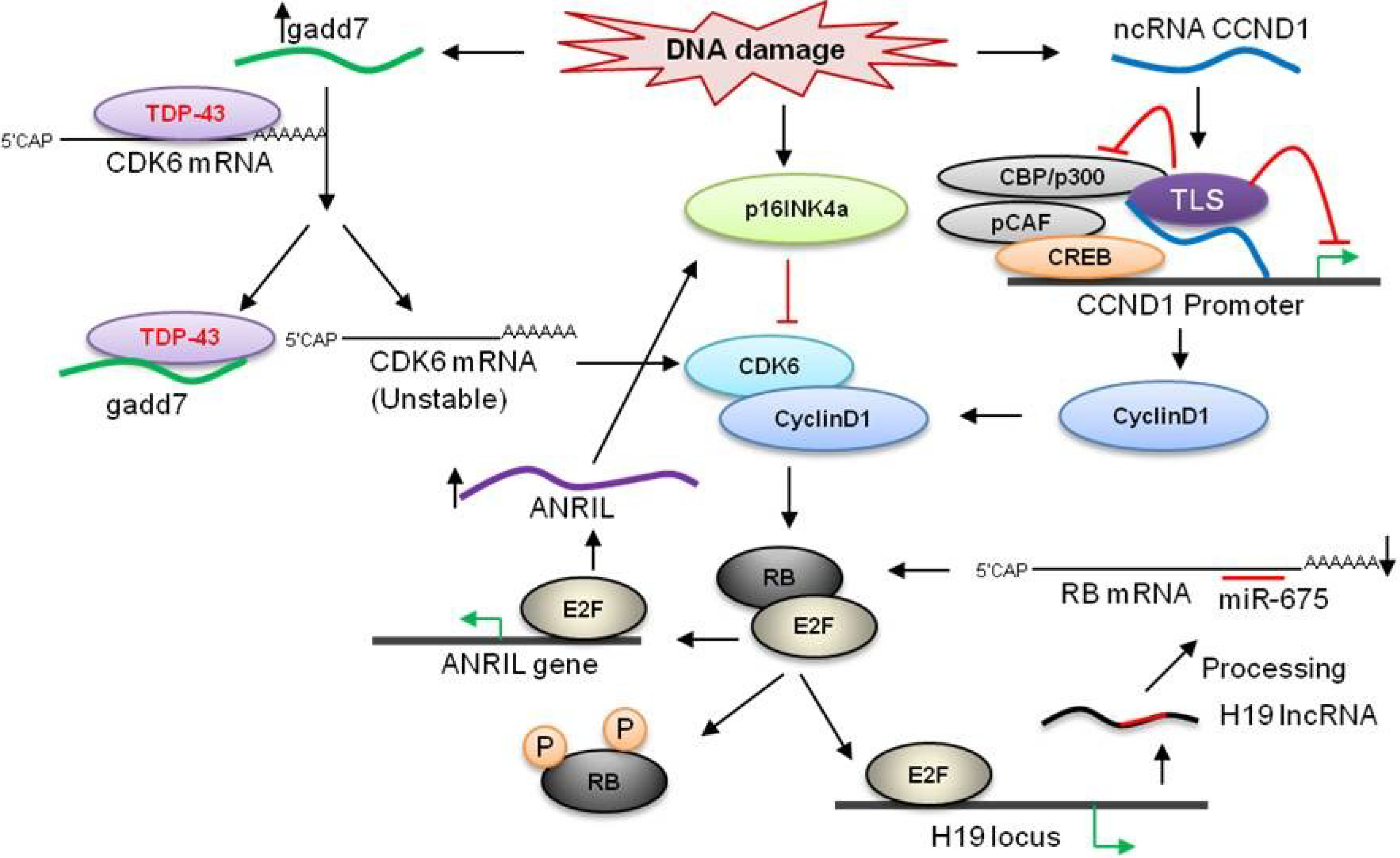
3. LncRNAs Involved in the Rb Pathway
3.1. H19
3.2. KCNQ1OT1
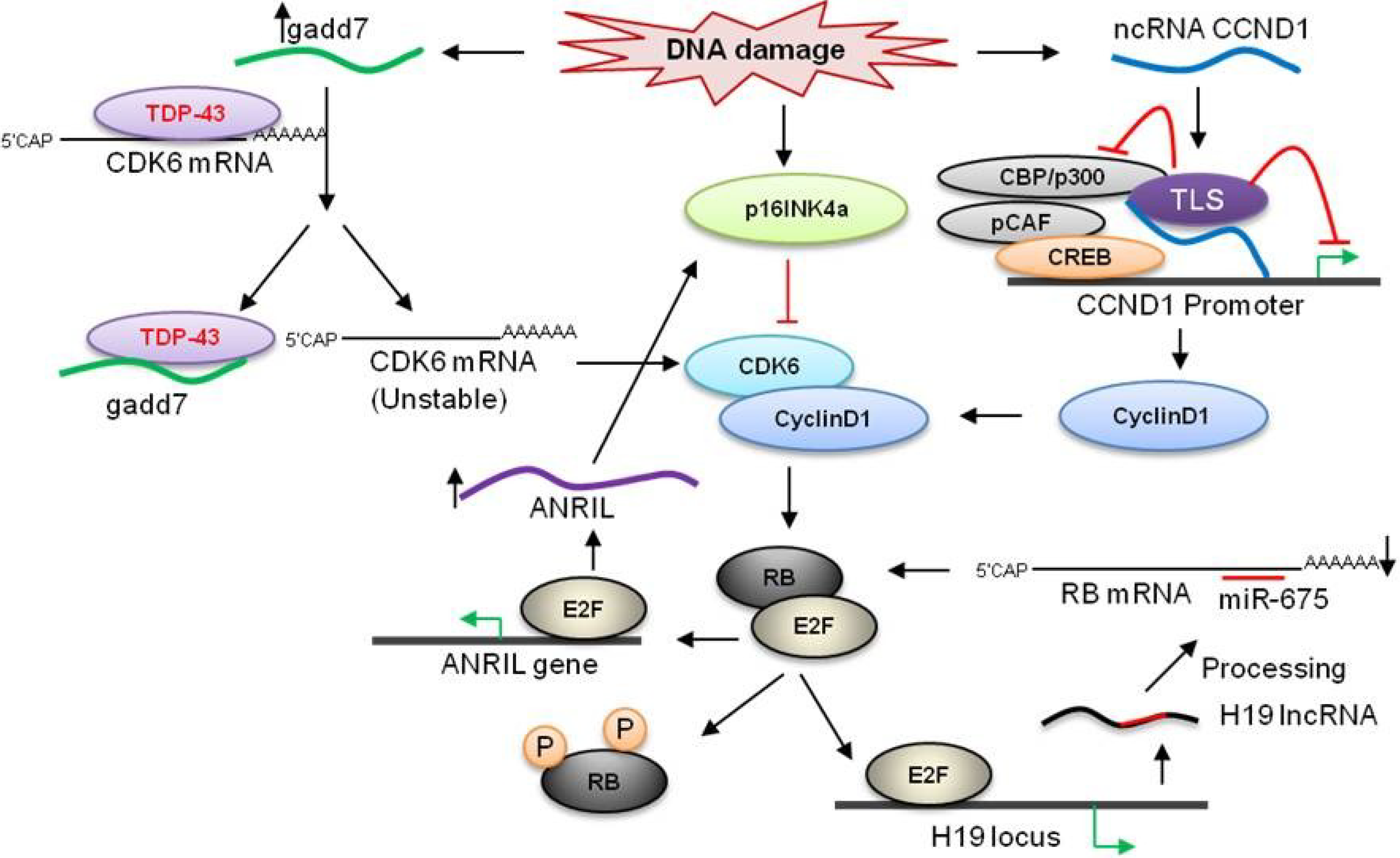
3.3. ANRIL
3.4. HULC
3.5. ncRNACCND1
3.6. Gadd7
4. Perspectives and Conclusions
Conflicts of Interest
References
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef]
- Lee, K.-H.; Li, M.; Michalowski, A.M.; Zhang, X.; Liao, H.; Chen, L.; Xu, Y.; Wu, X.; Huang, J. A genomewide study identifies the Wnt signaling pathway as a major target of p53 in murine embryonic stem cells. Proc. Natl. Acad. Sci. USA 2010, 107, 69–74. [Google Scholar]
- Li, M.; He, Y.; Dubois, W.; Wu, X.; Shi, J.; Huang, J. Distinct regulatory mechanisms and functions for p53-activated and p53-repressed DNA damage response genes in embryonic stem cells. Mol. Cell 2012, 46, 30–42. [Google Scholar] [CrossRef]
- Tokino, T.; Nakamura, Y. The role of p53-target genes in human cancer. Crit. Rev. Oncol. Hematol. 2000, 33, 1–6. [Google Scholar] [CrossRef]
- Prives, C.; Hall, P.A. The p53 pathway. J. Pathol. 1999, 187, 112–126. [Google Scholar] [CrossRef]
- Juven-Gershon, T.; Oren, M. Mdm2: The ups and downs. Mol. Med. 1999, 5, 71–83. [Google Scholar]
- Lundberg, A.S.; Weinberg, R.A. Control of the cell cycle and apoptosis. Eur. J. Cancer 1999, 35, 531–539. [Google Scholar] [CrossRef]
- Morgan, D.O. Principles of CDK regulation. Nature 1995, 374, 131–134. [Google Scholar] [CrossRef]
- Sherr, C.J.; McCormick, F. The RB and p53 pathways in cancer. Cancer Cell 2002, 2, 103–112. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995, 9, 1149–1163. [Google Scholar]
- Hsieh, J.K.; Chan, F.S.; O’Connor, D.J.; Mittnacht, S.; Zhong, S.; Lu, X. RB regulates the stability and the apoptotic function of p53 via MDM2. Mol. Cell 1999, 3, 181–193. [Google Scholar]
- Chivukula, R.R.; Mendell, J.T. Circular reasoning: microRNAs and cell-cycle control. Trends Biochem. Sci. 2008, 33, 474–481. [Google Scholar]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 2008, 102–114. [Google Scholar] [CrossRef]
- Linsley, P.S.; Schelter, J.; Burchard, J.; Kibukawa, M.; Martin, M.M.; Bartz, S.R.; Johnson, J.M.; Cummins, J.M.; Raymond, C.K.; Dai, H.; et al. Transcripts targeted by the microRNA-16 family cooperatively regulate cell cycle progression. Mol. Cell. Biol. 2007, 27, 2240–2252. [Google Scholar] [CrossRef]
- Liu, Q.; Fu, H.; Sun, F.; Zhang, H.; Tie, Y.; Zhu, J.; Xing, R.; Sun, Z.; Zheng, X. miR-16 family induces cell cycle arrest by regulating multiple cell cycle genes. Nucleic Acids Res. 2008, 36, 5391–5404. [Google Scholar] [CrossRef]
- Lal, A.; Kim, H.H.; Abdelmohsen, K.; Kuwano, Y.; Pullmann, R.; Srikantan, S.; Subrahmanyam, R.; Martindale, J.L.; Yang, X.; Ahmed, F.; et al. p16(INK4a) translation suppressed by miR-24. PLoS One 2008, 3, e1864. [Google Scholar] [CrossRef]
- Pierson, J.; Hostager, B.; Fan, R.; Vibhakar, R. Regulation of cyclin dependent kinase 6 by microRNA 124 in medulloblastoma. J. Neurooncol. 2008, 90, 1–7. [Google Scholar] [CrossRef]
- Johnson, C.D.; Esquela-Kerscher, A.; Stefani, G.; Byrom, M.; Kelnar, K.; Ovcharenko, D.; Wilson, M.; Wang, X.; Shelton, J.; Shingara, J.; et al. The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Res. 2007, 67, 7713–7722. [Google Scholar]
- Sun, F.; Fu, H.; Liu, Q.; Tie, Y.; Zhu, J.; Xing, R.; Sun, Z.; Zheng, X. Downregulation of CCND1 and CDK6 by miR-34a induces cell cycle arrest. FEBS Lett. 2008, 582, 1564–1568. [Google Scholar]
- Wu, J.; Qian, J.; Li, C.; Kwok, L.; Cheng, F.; Liu, P.; Perdomo, C.; Kotton, D.; Vaziri, C.; Anderlind, C.; et al. miR-129 regulates cell proliferation by downregulating CDK6 expression. Cell Cycle 2010, 9, 1809–1818. [Google Scholar] [CrossRef]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—microRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef]
- Bernstein, E.; Allis, C.D. RNA meets chromatin. Genes Dev. 2005, 19, 1635–1655. [Google Scholar] [CrossRef]
- Bracken, A.P.; Helin, K. Polycomb group proteins: Navigators of lineage pathways led astray in cancer. Nat. Rev. Cancer 2009, 9, 773–784. [Google Scholar] [CrossRef]
- Wilusz, J.E.; Sunwoo, H.; Spector, D.L. Long noncoding RNAs: Functional surprises from the RNA world. Genes Dev. 2009, 23, 1494–1504. [Google Scholar] [CrossRef]
- Wapinski, O.; Chang, H.Y. Long noncoding RNAs and human disease. Trends Cell Biol. 2011, 21, 354–361. [Google Scholar] [CrossRef]
- Willingham, A.T.; Orth, A.P.; Batalov, S.; Peters, E.C.; Wen, B.G.; Aza-Blanc, P.; Hogenesch, J.B.; Schultz, P.G. A strategy for probing the function of noncoding RNAs finds a repressor of NFAT. Science 2005, 309, 1570–1573. [Google Scholar] [CrossRef]
- Wong, L.H.; Brettingham-Moore, K.H.; Chan, L.; Quach, J.M.; Anderson, M.A.; Northrop, E.L.; Hannan, R.; Saffery, R.; Shaw, M.L.; Williams, E.; et al. Centromere RNA is a key component for the assembly of nucleoproteins at the nucleolus and centromere. Genome Res. 2007, 17, 1146–1160. [Google Scholar] [CrossRef]
- Lee, J.T. Lessons from X-chromosome inactivation: Long ncRNA as guides and tethers to the epigenome. Genes Dev. 2009, 23, 1831–1842. [Google Scholar] [CrossRef]
- Schoeftner, S.; Blasco, M.A. A “higher order” of telomere regulation: Telomere heterochromatin and telomeric RNAs. EMBO J. 2009, 28, 2323–2336. [Google Scholar] [CrossRef]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef]
- Li, J.; Xuan, Z.; Liu, C. Long non-coding RNAs and complex human diseases. IJMS 2013, 14, 18790–18808. [Google Scholar]
- Calin, G.A.; Liu, C.-G.; Ferracin, M.; Hyslop, T.; Spizzo, R.; Sevignani, C.; Fabbri, M.; Cimmino, A.; Lee, E.J.; Wojcik, S.E.; et al. Ultraconserved regions encoding ncRNAs are altered in human leukemias and carcinomas. Cancer Cell 2007, 12, 215–229. [Google Scholar] [CrossRef]
- Jalali, S.; Bhartiya, D.; Lalwani, M.K.; Sivasubbu, S.; Scaria, V. Systematic transcriptome wide analysis of lncRNA-miRNA interactions. PLoS One 2013, 8, e53823. [Google Scholar]
- Huarte, M.; Guttman, M.; Feldser, D.; Garber, M.; Koziol, M.J.; Kenzelmann-Broz, D.; Khalil, A.M.; Zuk, O.; Amit, I.; Rabani, M.; et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 2010, 142, 409–419. [Google Scholar] [CrossRef]
- Yoon, J.H.; Abdelmohsen, K.; Srikantan, S.; Yang, X. LincRNA-p21 suppresses target mRNA translation. Mol. Cell 2012, 47, 648–655. [Google Scholar] [CrossRef]
- Zhai, H.; Fesler, A.; Schee, K.; Fodstad, O.; Flatmark, K.; Ju, J. Clinical significance of long intergenic noncoding RNA-p21 in colorectal cancer. Clin. Colorectal Cancer 2013, 12, 261–266. [Google Scholar] [CrossRef]
- Hung, T.; Wang, Y.; Lin, M.F.; Koegel, A.K.; Kotake, Y.; Grant, G.D.; Horlings, H.M.; Shah, N.; Umbricht, C.; Wang, P.; et al. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat. Genet. 2011, 43, 621–629. [Google Scholar] [CrossRef]
- Miyoshi, N.; Wagatsuma, H.; Wakana, S.; Shiroishi, T.; Nomura, M.; Aisaka, K.; Kohda, T.; Surani, M.A.; Kaneko-Ishino, T.; Ishino, F. Identification of an imprinted gene, Meg3/Gtl2 and its human homologue MEG3, first mapped on mouse distal chromosome 12 and human chromosome 14q. Genes Cells 2000, 5, 211–220. [Google Scholar] [CrossRef]
- Hagan, J.P.; O’Neill, B.L.; Stewart, C.L.; Kozlov, S.V.; Croce, C.M. At least ten genes define the imprinted Dlk1-Dio3 cluster on mouse chromosome 12qF1. PLoS One 2009, 4, e4352. [Google Scholar]
- Zhang, X. A pituitary-derived MEG3 isoform functions as a growth suppressor in tumor cells. J. Clin. Endocrinol. Metab. 2003, 88, 5119–5126. [Google Scholar]
- Zhou, Y.; Zhong, Y.; Wang, Y.; Zhang, X.; Batista, D.L.; Gejman, R.; Ansell, P.J.; Zhao, J.; Weng, C.; Klibanski, A. Activation of p53 by MEG3 non-coding RNA. J. Biol. Chem. 2007, 282, 24731–24742. [Google Scholar]
- Zhang, X.; Rice, K.; Wang, Y.; Chen, W.; Zhong, Y.; Nakayama, Y.; Zhou, Y.; Klibanski, A. Maternally expressed gene 3 (MEG3) noncoding ribonucleic acid: Isoform structure, expression, and functions. Endocrinology 2010, 151, 939–947. [Google Scholar] [CrossRef]
- Loewer, S.; Cabili, M.N.; Guttman, M.; Loh, Y.-H.; Thomas, K.; Park, I.H.; Garber, M.; Curran, M.; Onder, T.; Agarwal, S.; et al. Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat. Genet. 2010, 42, 1113–1117. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.; Zhou, N.; Huang, J.; Liu, Q.; Fukuda, K.; Ma, D.; Lu, Z.; Bai, C.; Watabe, K.; Mo, Y.-Y. The human long non-coding RNA-RoR is a p53 repressor in response to DNA damage. Cell Res. 2013, 23, 340–350. [Google Scholar] [CrossRef]
- Liu, Q.; Huang, J.; Zhou, N.; Zhang, Z.; Zhang, A.; Lu, Z.; Wu, F.; Mo, Y.-Y. LncRNA loc285194 is a p53-regulated tumor suppressor. Nucleic Acids Res. 2013, 41, 4976–4987. [Google Scholar] [CrossRef]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef]
- Tripathi, V.; Shen, Z.; Chakraborty, A.; Giri, S.; Freier, S.M.; Wu, X.; Zhang, Y.; Gorospe, M.; Prasanth, S.G.; Lal, A.; et al. Long noncoding RNA MALAT1 controls cell cycle progression by regulating the expression of oncogenic transcription factor B-MYB. PLoS Genet. 2013, 9, e1003368. [Google Scholar] [CrossRef]
- Gutschner, T.; Hämmerle, M.; Eißmann, M.; Hsu, J.; Kim, Y.; Hung, G.; Revenko, A.; Arun, G.; Stentrup, M.; Groß, M.; et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013, 73, 1180–1189. [Google Scholar] [CrossRef]
- Nakagawa, S.; Ip, J.Y.; Shioi, G.; Tripathi, V.; Zong, X.; Hirose, T.; Prasanth, K.V. Malat1 is not an essential component of nuclear speckles in mice. RNA 2012, 18, 1487–1499. [Google Scholar] [CrossRef]
- Zhang, B.; Arun, G.; Mao, Y.S.; Lazar, Z.; Hung, G.; Bhattacharjee, G.; Xiao, X.; Booth, C.J.; Wu, J.; Zhang, C.; et al. The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep. 2012, 2, 111–123. [Google Scholar] [CrossRef]
- Melo, C.A.; Drost, J.; Wijchers, P.J.; van de Werken, H.; de Wit, E.; Oude Vrielink, J.A.F.; Elkon, R.; Melo, S.A.; Léveillé, N.; Kalluri, R.; et al. eRNAs are required for p53-dependent enhancer activity and gene transcription. Mol. Cell 2013, 49, 524–535. [Google Scholar] [CrossRef]
- Keniry, A.; Oxley, D.; Monnier, P.; Kyba, M.; Dandolo, L.; Smits, G.; Reik, W. The H19 lincRNA is a developmental reservoir of miR-675 that suppresses growth and Igf1r. Nat. Cell Biol. 2012, 14, 659–665. [Google Scholar] [CrossRef]
- Weksberg, R.; Nishikawa, J.; Caluseriu, O.; Fei, Y.L.; Shuman, C.; Wei, C.; Steele, L.; Cameron, J.; Smith, A.; Ambus, I.; et al. Tumor development in the Beckwith-Wiedemann syndrome is associated with a variety of constitutional molecular 11p15 alterations including imprinting defects of KCNQ1OT1. Hum. Mol. Genet. 2001, 10, 2989–3000. [Google Scholar]
- Arima, T. ZAC, LIT1 (KCNQ1OT1) and p57KIP2 (CDKN1C) are in an imprinted gene network that may play a role in Beckwith-Wiedemann syndrome. Nucleic Acids Res. 2005, 33, 2650–2660. [Google Scholar]
- Mancini-DiNardo, D.; Steele, S.J.S.; Levorse, J.M.; Ingram, R.S.; Tilghman, S.M. Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev. 2006, 20, 1268–1282. [Google Scholar]
- Pandey, R.R.; Mondal, T.; Mohammad, F.; Enroth, S.; Redrup, L.; Komorowski, J.; Nagano, T.; Mancini-DiNardo, D.; Kanduri, C. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell 2008, 32, 232–246. [Google Scholar] [CrossRef]
- Rodriguez, B.A.T.; Weng, Y.-I.; Liu, T.-M.; Zuo, T.; Hsu, P.-Y.; Lin, C.-H.; Cheng, A.-L.; Cui, H.; Yan, P.S.; Huang, T.H.-M. Estrogen-mediated epigenetic repression of the imprinted gene cyclin-dependent kinase inhibitor 1C in breast cancer cells. Carcinogenesis 2011, 32, 812–821. [Google Scholar] [CrossRef]
- Nakano, S.; Murakami, K.; Meguro, M.; Soejima, H.; Higashimoto, K.; Urano, T.; Kugoh, H.; Mukai, T.; Ikeguchi, M.; Oshimura, M. Expression profile of LIT1/KCNQ1OT1 and epigenetic status at the KvDMR1 in colorectal cancers. Cancer Sci. 2006, 97, 1147–1154. [Google Scholar] [CrossRef]
- Wan, J.; Huang, M.; Zhao, H.; Wang, C.; Zhao, X.; Jiang, X.; Bian, S.; He, Y.; Gao, Y. A novel tetranucleotide repeat polymorphism within KCNQ1OT1 confers risk for hepatocellular carcinoma. DNA Cell Biol. 2013, 32, 628–634. [Google Scholar] [CrossRef]
- Pasmant, E.; Laurendeau, I.; Héron, D.; Vidaud, M.; Vidaud, D.; Bièche, I. Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: Identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Res. 2007, 67, 3963–3969. [Google Scholar] [CrossRef]
- Yap, K.L.; Li, S.; Muñoz-Cabello, A.M.; Raguz, S.; Zeng, L.; Mujtaba, S.; Gil, J.; Walsh, M.J.; Zhou, M.-M. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol. Cell 2010, 38, 662–674. [Google Scholar] [CrossRef]
- Kotake, Y.; Nakagawa, T.; Kitagawa, K.; Suzuki, S.; Liu, N.; Kitagawa, M.; Xiong, Y. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene 2011, 30, 1956–1962. [Google Scholar] [CrossRef]
- Wan, G.; Mathur, R.; Hu, X.; Liu, Y.; Zhang, X.; Peng, G.; Lu, X. Long non-coding RNA ANRIL (CDKN2B-AS) is induced by the ATM-E2F1 signaling pathway. Cell. Signal. 2013, 25, 1086–1095. [Google Scholar] [CrossRef]
- Holdt, L.M.; Hoffmann, S.; Sass, K.; Langenberger, D.; Scholz, M.; Krohn, K.; Finstermeier, K.; Stahringer, A.; Wilfert, W.; Beutner, F.; et al. Alu elements in ANRIL non-coding RNA at chromosome 9p21 modulate atherogenic cell functions through trans-regulation of gene networks. PLoS Genet. 2013, 9, e1003588. [Google Scholar] [CrossRef]
- Du, Y.; Kong, G.; You, X.; Zhang, S.; Zhang, T.; Gao, Y.; Ye, L.; Zhang, X. Elevation of highly up-regulated in liver cancer (HULC) by hepatitis B virus X protein promotes hepatoma cell proliferation via down-regulating p18. J. Biol. Chem. 2012, 287, 26302–26311. [Google Scholar]
- Wang, J.; Liu, X.; Wu, H.; Ni, P.; Gu, Z.; Qiao, Y.; Chen, N.; Sun, F.; Fan, Q. CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucleic Acids Res. 2010, 38, 5366–5383. [Google Scholar] [CrossRef]
- Wang, X.; Arai, S.; Song, X.; Reichart, D.; Du, K.; Pascual, G.; Tempst, P.; Rosenfeld, M.G.; Glass, C.K.; Kurokawa, R. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 2008, 454, 126–130. [Google Scholar] [CrossRef]
- Hollander, M.C.; Alamo, I.; Fornace, A.J. A novel DNA damage-inducible transcript, gadd7, inhibits cell growth, but lacks a protein product. Nucleic Acids Res. 1996, 24, 1589–1593. [Google Scholar] [CrossRef]
- Hollander, M.C.; Zhan, Q.; Yu, K.; Fornace, A.J. Identification of several human homologs of hamster DNA damage-inducible transcripts cloning and characterization of a novel uv-inducible cDNA that codes for a putative RNA-binding protein. J. Biol. Chem. 1997, 272, 26720–26726. [Google Scholar] [CrossRef]
- Jones, M.; Lal, A. MicroRNAs, wild-type and mutant p53: More questions than answers. RNA Biol. 2012, 9, 781–791. [Google Scholar]
- Seila, A.C.; Calabrese, J.M.; Levine, S.S.; Yeo, G.W.; Rahl, P.B.; Flynn, R.A.; Young, R.A.; Sharp, P.A. Divergent transcription from active promoters. Science 2008, 322, 1849–1851. [Google Scholar]
- Olive, K.P.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004, 119, 847–860. [Google Scholar] [CrossRef]
- Özgür, E.; Mert, U.; Isin, M.; Okutan, M.; Dalay, N.; Gezer, U. Differential expression of long non-coding RNAs during genotoxic stress-induced apoptosis in HeLa and MCF-7 cells. Clin. Exp. Med. 2013, 13, 119–126. [Google Scholar]
- Gibb, E.A.; Brown, C.J.; Lam, W.L. The functional role of long non-coding RNA in human carcinomas. Mol. Cancer 2011, 10, e38. [Google Scholar]
- Pang, K.C.; Frith, M.C.; Mattick, J.S. Rapid evolution of noncoding RNAs: Lack of conservation does not mean lack of function. Trends Genet. 2006, 22, 1–5. [Google Scholar] [CrossRef]
- Pasic, I.; Shlien, A.; Durbin, A.D.; Stavropoulos, D.J.; Baskin, B.; Ray, P.N.; Novokmet, A.; Malkin, D. Recurrent focal copy-number changes and loss of heterozygosity implicate two noncoding RNAs and one tumor suppressor gene at chromosome 3q13.31 in osteosarcoma. Cancer Res. 2010, 70, 160–171. [Google Scholar] [CrossRef]
- Ji, P.; Diederichs, S.; Wang, W.; Böing, S.; Metzger, R.; Schneider, P.M.; Tidow, N.; Brandt, B.; Buerger, H.; Bulk, E.; et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003, 22, 8031–8041. [Google Scholar] [CrossRef]
- Hutchinson, J.N.; Ensminger, A.W.; Clemson, C.M.; Lynch, C.R.; Lawrence, J.B.; Chess, A. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics 2007, 8, e39. [Google Scholar] [CrossRef]
- Giannoukakis, N.; Deal, C.; Paquette, J.; Goodyer, C.G.; Polychronakos, C. Parental genomic imprinting of the human IGF2 gene. Nat. Genet. 1993, 4, 98–101. [Google Scholar] [CrossRef]
- Zhang, Y.; Tycko, B. Monoallelic expression of the human H19 gene. Nat. Genet. 1992, 1, 40–44. [Google Scholar] [CrossRef]
- Berteaux, N.; Lottin, S.; Monté, D.; Pinte, S.; Quatannens, B.; Coll, J.; Hondermarck, H.; Curgy, J.-J.; Dugimont, T.; Adriaenssens, E. H19 mRNA-like noncoding RNA promotes breast cancer cell proliferation through positive control by E2F1. J. Biol. Chem. 2005, 280, 29625–29636. [Google Scholar] [CrossRef]
- Yang, F.; Bi, J.; Xue, X.; Zheng, L.; Zhi, K.; Hua, J. Up-regulated long non-coding RNA H19 contributes to proliferation of gastric cancer cells. FEBS J. 2012, 279, 3159–3165. [Google Scholar]
- Ayesh, S.; Matouk, I.; Schneider, T.; Ohana, P.; Laster, M.; Al-Sharef, W.; de-Groot, N.; Hochberg, A. Possible physiological role of H19 RNA. Mol. Carcinog. 2002, 35, 63–74. [Google Scholar] [CrossRef]
- Matouk, I.J.; DeGroot, N.; Mezan, S.; Ayesh, S.; Abu-lail, R.; Hochberg, A.; Galun, E. The H19 non-coding RNA is essential for human tumor growth. PLoS One 2007, 2, e845. [Google Scholar] [CrossRef]
- Berteaux, N.; Aptel, N.; Cathala, G.; Genton, C.; Coll, J.; Daccache, A.; Spruyt, N.; Hondermarck, H.; Dugimont, T.; Curgy, J.-J.; et al. A novel H19 antisense RNA overexpressed in breast cancer contributes to paternal IGF2 expression. Mol. Cell. Biol. 2008, 28, 6731–6745. [Google Scholar]
- Tsang, W.P.; Ng, E.K.O.; Ng, S.S.M.; Jin, H.; Yu, J.; Sung, J.J.Y.; Kwok, T.T. Oncofetal H19-derived miR-675 regulates tumor suppressor RB in human colorectal cancer. Carcinogenesis 2010, 31, 350–358. [Google Scholar] [CrossRef]
- Hao, Y.; Crenshaw, T.; Moulton, T.; Newcomb, E.; Tycko, B. Tumour-suppressor activity of H19 RNA. Nature 1993, 365, 764–767. [Google Scholar] [CrossRef]
- Yoshimizu, T.; Miroglio, A.; Ripoche, M.-A.; Gabory, A.; Vernucci, M.; Riccio, A.; Colnot, S.; Godard, C.; Terris, B.; Jammes, H.; et al. The H19 locus acts in vivo as a tumor suppressor. Proc. Natl. Acad. Sci. USA 2008, 105, 12417–12422. [Google Scholar] [CrossRef]
- Onyango, P.; Feinberg, A.P. A nucleolar protein, H19 opposite tumor suppressor (HOTS), is a tumor growth inhibitor encoded by a human imprinted H19 antisense transcript. Proc. Natl. Acad. Sci. USA 2011, 108, 16759–16764. [Google Scholar] [CrossRef]
- Serrano, M.; Hannon, G.J.; Beach, D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993, 366, 704–707. [Google Scholar]
- Kamb, A.; Gruis, N.A.; Weaver-Feldhaus, J.; Liu, Q.; Harshman, K.; Tavtigian, S.V.; Stockert, E.; Day, R.S.; Johnson, B.E.; Skolnick, M.H. A cell cycle regulator potentially involved in genesis of many tumor types. Science 1994, 264, 436–440. [Google Scholar]
- Kamijo, T.; Zindy, F.; Roussel, M.F.; Quelle, D.E.; Downing, J.R.; Ashmun, R.A.; Grosveld, G.; Sherr, C.J. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19 Arf. Cell 1997, 91, 649–659. [Google Scholar] [CrossRef]
- Uchida, C.; Miwa, S.; Kitagawa, K.; Hattori, T.; Isobe, T.; Otani, S.; Oda, T.; Sugimura, H.; Kamijo, T.; Ookawa, K.; et al. Enhanced Mdm2 activity inhibits pRB function via ubiquitin-dependent degradation. EMBO J. 2005, 24, 160–169. [Google Scholar] [CrossRef]
- Gil, J.; Peters, G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: All for one or one for all. Nat. Rev. Mol. Cell Biol. 2006, 7, 667–677. [Google Scholar]
- Kitagawa, M.; Kitagawa, K.; Kotake, Y.; Niida, H.; Ohhata, T. Cell cycle regulation by long non-coding RNAs. Cell. Mol. Life Sci. 2013, 70, 4785–4794. [Google Scholar] [CrossRef]
- Morrison, J.; Pernet, D.; Renwick, A.; Maranian, M. Genome-wide association study identifies five new breast cancer susceptibility loci. Nat. Genet. 2010, 42, 504–507. [Google Scholar] [CrossRef]
- Stacey, S.N.; Sulem, P.; Masson, G.; Gudjonsson, S.A.; Thorleifsson, G.; Jakobsdottir, M.; Sigurdsson, A.; Gudbjartsson, D.F.; Sigurgeirsson, B.; Benediktsdottir, K.R.; et al. New common variants affecting susceptibility to basal cell carcinoma. Nat. Genet. 2009, 41, 909–914. [Google Scholar] [CrossRef]
- Bei, J.-X.; Li, Y.; Jia, W.-H.; Feng, B.-J.; Zhou, G.; Chen, L.-Z.; Feng, Q.-S.; Low, H.-Q.; Zhang, H.; He, F.; et al. A genome-wide association study of nasopharyngeal carcinoma identifies three new susceptibility loci. Nat. Genet. 2010, 42, 599–603. [Google Scholar] [CrossRef]
- Shete, S.; Hosking, F.J.; Robertson, L.B.; Dobbins, S.E.; Sanson, M.; Malmer, B.; Simon, M.; Marie, Y.; Boisselier, B.; Delattre, J.-Y.; et al. Genome-wide association study identifies five susceptibility loci for glioma. Nat. Genet. 2009, 41, 899–904. [Google Scholar] [CrossRef] [Green Version]
- Pasmant, E.; Sabbagh, A.; Vidaud, M.; Bièche, I. ANRIL, a long, noncoding RNA, is an unexpected major hotspot in GWAS. FASEB J. 2011, 25, 444–448. [Google Scholar] [CrossRef]
- Park, B.-J.; Kang, J.W.; Lee, S.W.; Choi, S.-J.; Shin, Y.K.; Ahn, Y.H.; Choi, Y.H.; Choi, D.; Lee, K.S.; Kim, S. The haploinsufficient tumor suppressor p18 upregulates p53 via interactions with ATM/ATR. Cell 2005, 120, 209–221. [Google Scholar] [CrossRef]
- Fornace, A.J.; Alamo, I.; Hollander, M.C. DNA damage-inducible transcripts in mammalian cells. Proc. Natl. Acad. Sci. USA 1988, 85, 8800–8804. [Google Scholar] [CrossRef]
- Liu, X.; Li, D.; Zhang, W.; Guo, M.; Zhan, Q. Long non-coding RNA gadd7 interacts with TDP-43 and regulates Cdk6 mRNA decay. EMBO J. 2012, 31, 4415–4427. [Google Scholar] [CrossRef]
- Penny, G.D.; Kay, G.F.; Sheardown, S.A.; Rastan, S.; Brockdorff, N. Requirement for Xist in X chromosome inactivation. Nature 1996, 379, 131–137. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The rosetta stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef]
- Yang, Z.; Zhou, L.; Wu, L.-M.; Lai, M.-C.; Xie, H.-Y.; Zhang, F.; Zheng, S.-S. Overexpression of long non-coding RNA HOTAIR predicts tumor recurrence in hepatocellular carcinoma patients following liver transplantation. Ann. Surg. Oncol. 2011, 18, 1243–1250. [Google Scholar] [CrossRef]
- Van Poppel, H.; Haese, A.; Graefen, M.; la Taille de, A.; Irani, J.; de Reijke, T.; Remzi, M.; Marberger, M. The relationship between Prostate CAncer gene 3 (PCA3) and prostate cancer significance. BJU Int. 2012, 109, 360–366. [Google Scholar] [CrossRef]
- Zhang, Z.; Hao, H.; Zhang, C.-J.; Yang, X.-Y.; He, Q.; Lin, J. Evaluation of novel gene UCA1 as a tumor biomarker for the detection of bladder cancer. Zhonghua Yi Xue Za Zhi 2012, 92, 384–387. [Google Scholar]
- Lai, M.-C.; Yang, Z.; Zhou, L.; Zhu, Q.-Q.; Xie, H.-Y.; Zhang, F.; Wu, L.-M.; Chen, L.-M.; Zheng, S.-S. Long non-coding RNA MALAT-1 overexpression predicts tumor recurrence of hepatocellular carcinoma after liver transplantation. Med. Oncol. 2012, 29, 1810–1816. [Google Scholar] [CrossRef]
- Wahlestedt, C. Targeting long non-coding RNA to therapeutically upregulate gene expression. Nat. Rev. Drug Discov. 2013, 12, 433–446. [Google Scholar] [CrossRef]
- Weinberg, M.S.; Morris, K.V. Long non-coding RNA targeting and transcriptional de-repression. Nucleic Acid Ther. 2013, 23, 9–13. [Google Scholar]
- Cheng, C.J.; Saltzman, W.M.; Slack, F.J. Canonical and non-canonical barriers facing antimiR cancer therapeutics. Curr. Med. Chem. 2013, 20, 3582–3593. [Google Scholar] [CrossRef]
- Babar, I.A.; Cheng, C.J.; Booth, C.J.; Liang, X.; Weidhaas, J.B.; Saltzman, W.M.; Slack, F.J. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc. Natl. Acad. Sci. USA 2012, 109, E1695–E1704. [Google Scholar]
- Kasinski, A.L.; Slack, F.J. MicroRNAs en route to the clinic: Progress in validating and targeting microRNAs for cancer therapy. Nat. Rev. Cancer 2011, 11, 849–864. [Google Scholar] [CrossRef]
- Broderick, J.A.; Zamore, P.D. MicroRNA therapeutics. Gene Ther. 2011, 18, 1104–1110. [Google Scholar] [CrossRef]
- Kota, J.; Chivukula, R.R.; O’Donnell, K.A.; Wentzel, E.A.; Montgomery, C.L.; Hwang, H.-W.; Chang, T.-C.; Vivekanandan, P.; Torbenson, M.; Clark, K.R.; et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell 2009, 137, 1005–1017. [Google Scholar] [CrossRef]
- Lee, M.M.; Childs-Disney, J.L.; Pushechnikov, A.; French, J.M.; Sobczak, K.; Thornton, C.A.; Disney, M.D. Controlling the specificity of modularly assembled small molecules for RNA via ligand module spacing: Targeting the RNAs that cause myotonic muscular dystrophy. J. Am. Chem. Soc. 2009, 131, 17464–17472. [Google Scholar] [CrossRef]
- Castanotto, D.; Rossi, J.J. The promises and pitfalls of RNA-interference-based therapeutics. Nature 2009, 457, 426–433. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Subramanian, M.; Jones, M.F.; Lal, A. Long Non-Coding RNAs Embedded in the Rb and p53 Pathways. Cancers 2013, 5, 1655-1675. https://doi.org/10.3390/cancers5041655
Subramanian M, Jones MF, Lal A. Long Non-Coding RNAs Embedded in the Rb and p53 Pathways. Cancers. 2013; 5(4):1655-1675. https://doi.org/10.3390/cancers5041655
Chicago/Turabian StyleSubramanian, Murugan, Matthew F. Jones, and Ashish Lal. 2013. "Long Non-Coding RNAs Embedded in the Rb and p53 Pathways" Cancers 5, no. 4: 1655-1675. https://doi.org/10.3390/cancers5041655
APA StyleSubramanian, M., Jones, M. F., & Lal, A. (2013). Long Non-Coding RNAs Embedded in the Rb and p53 Pathways. Cancers, 5(4), 1655-1675. https://doi.org/10.3390/cancers5041655