Tumor Metabolism of Malignant Gliomas

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
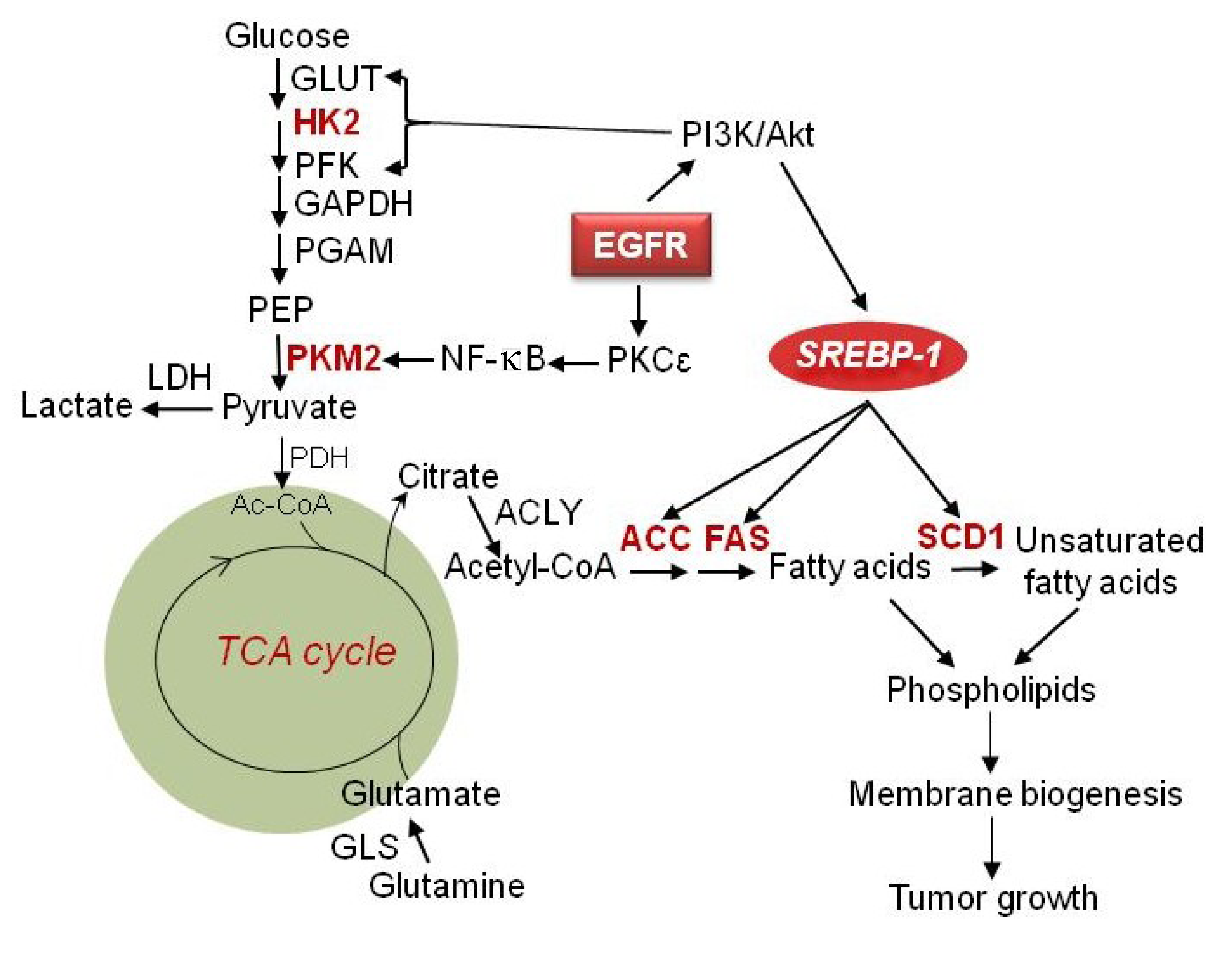
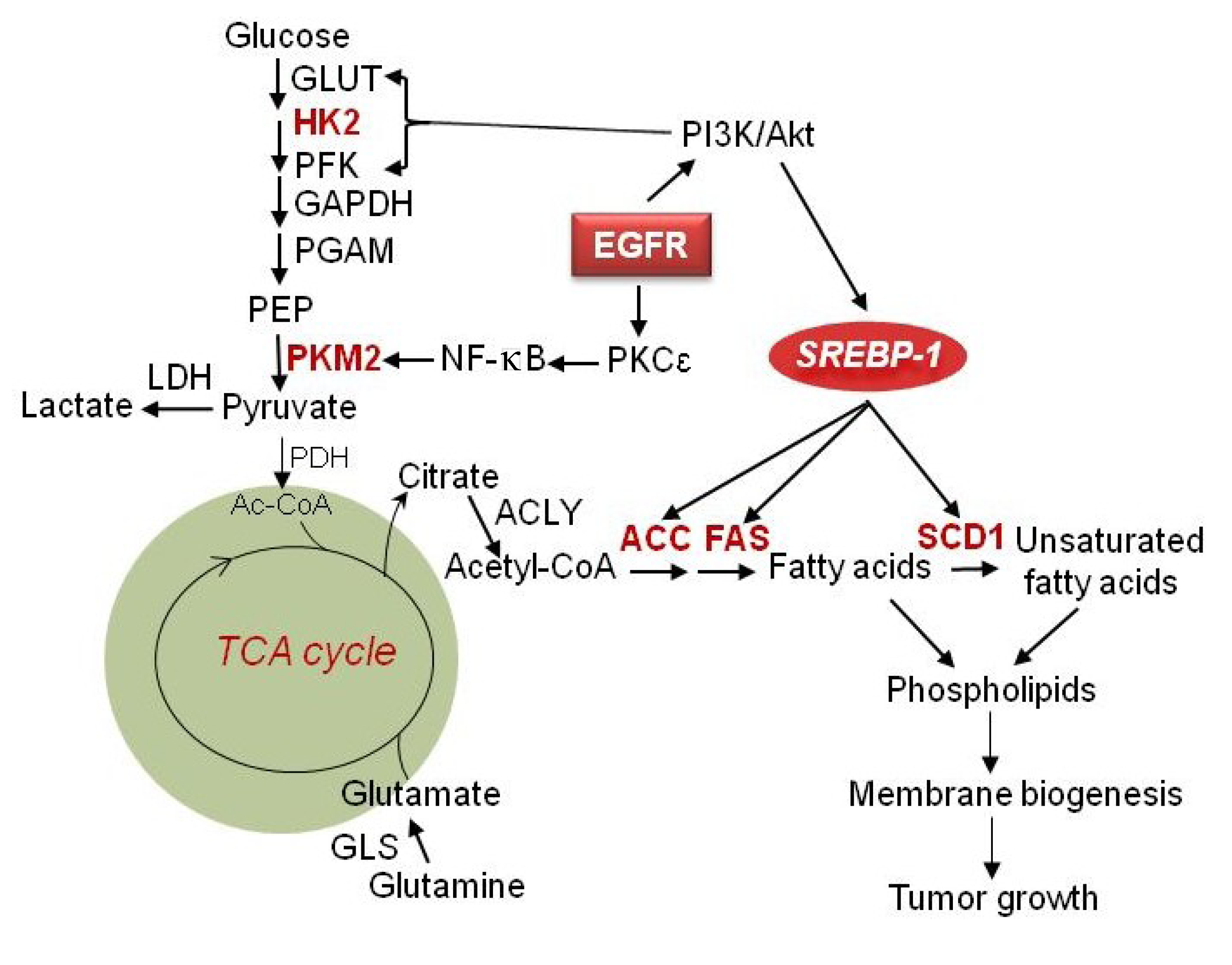
2. Glucose Metabolism in GBM
2.1. Hexokinase II
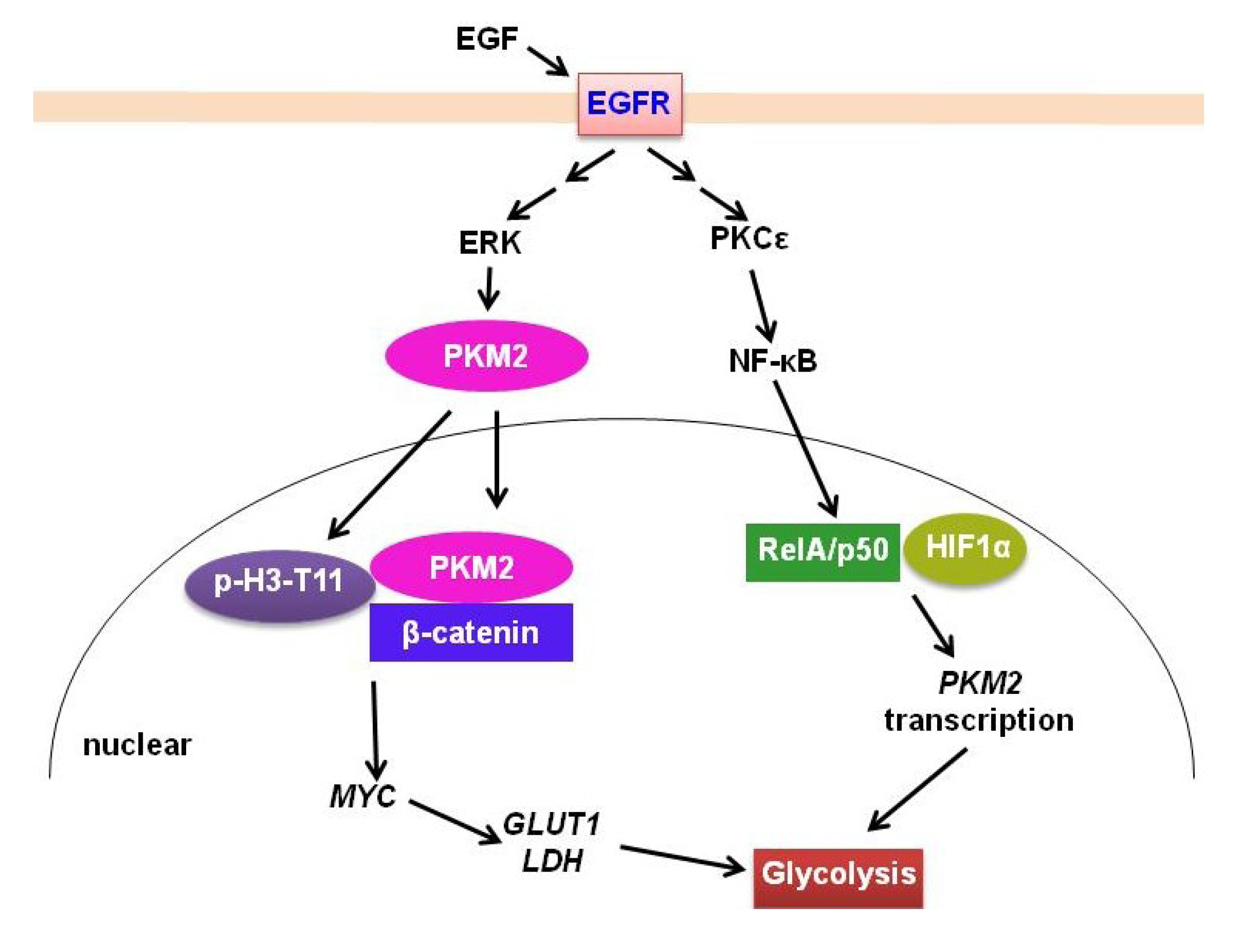
2.2. PKM2
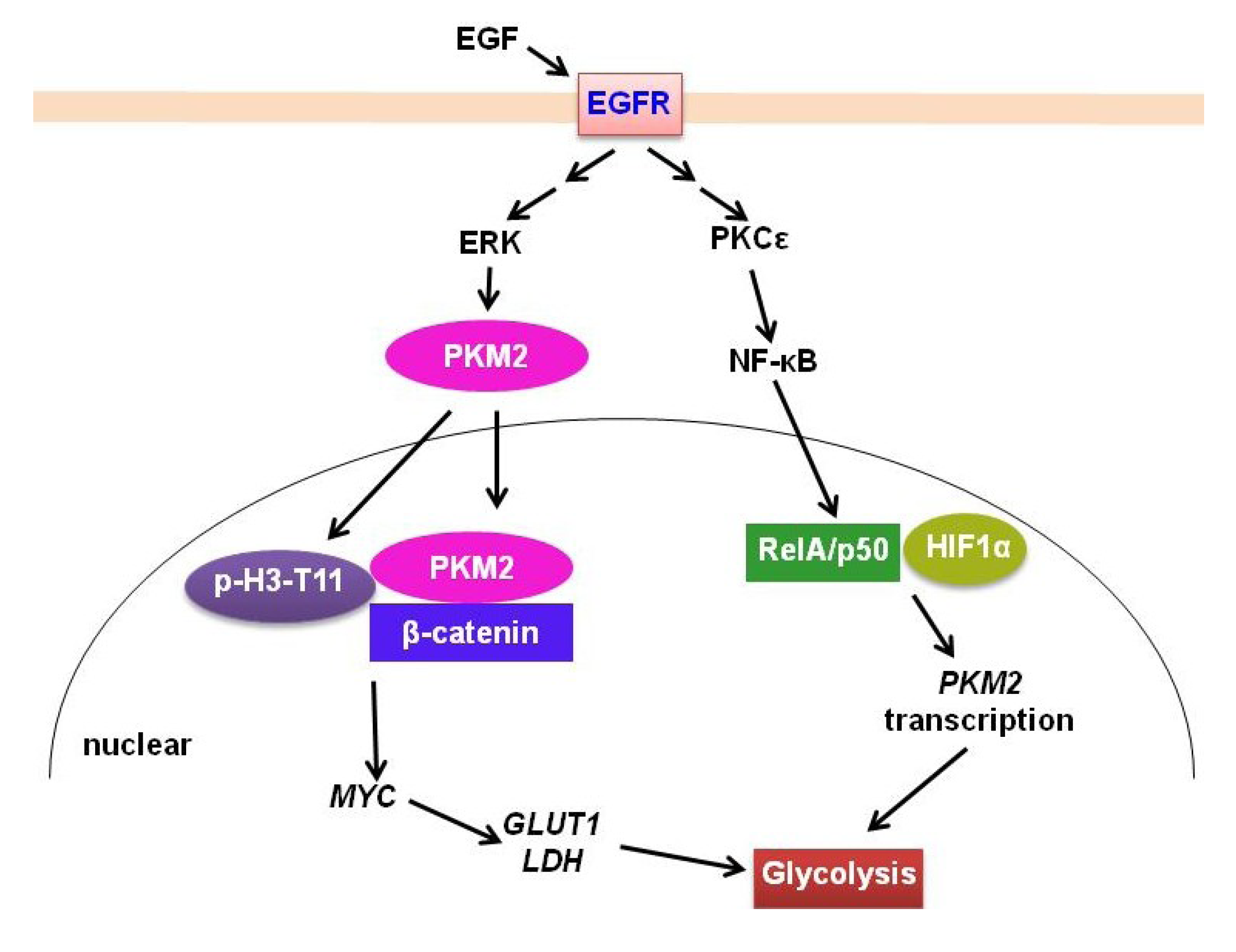
2.3. IDH
3. Glutamine Metabolism
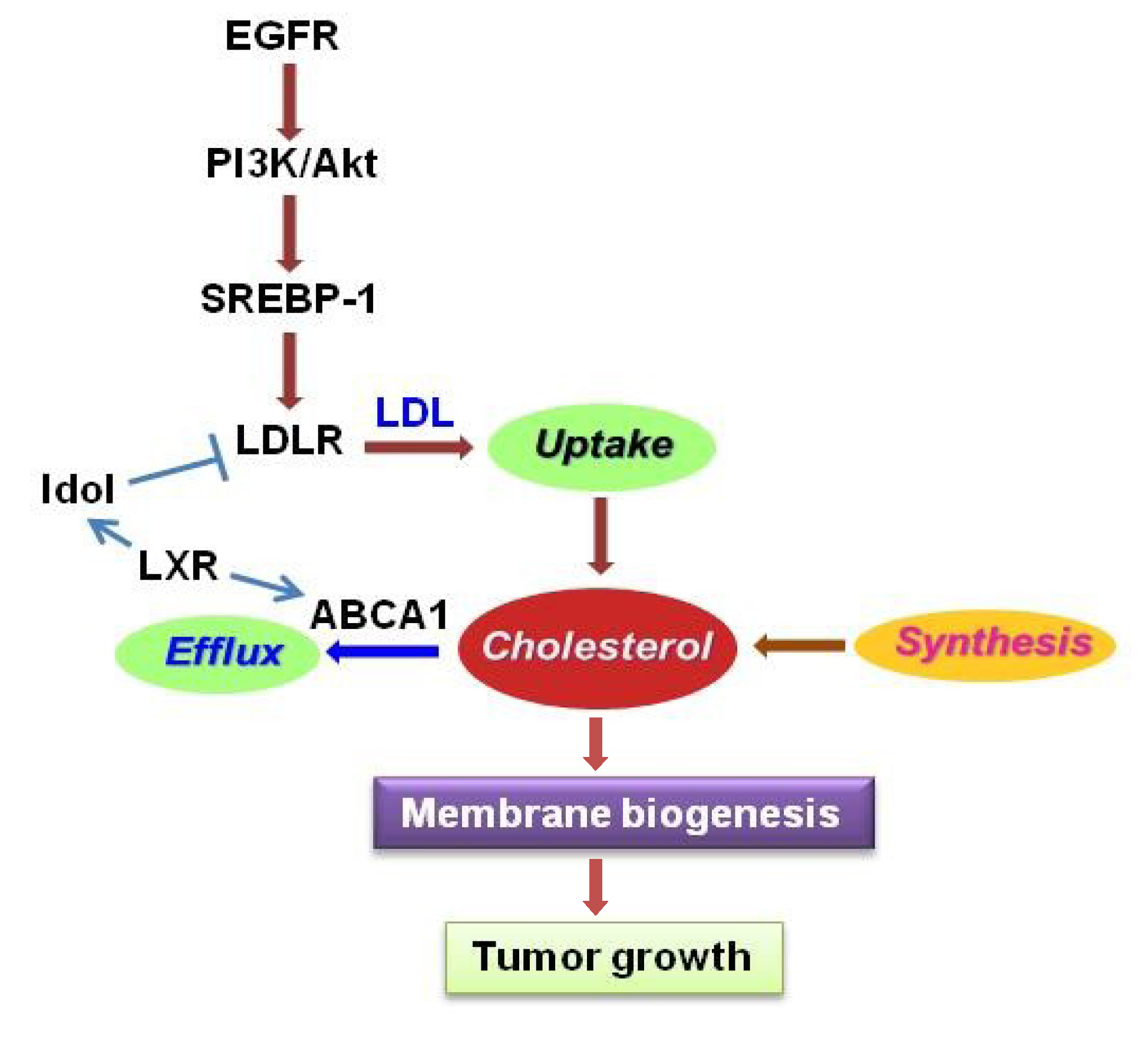
4. Lipid Metabolism in GBM
4.1. SREBP-1
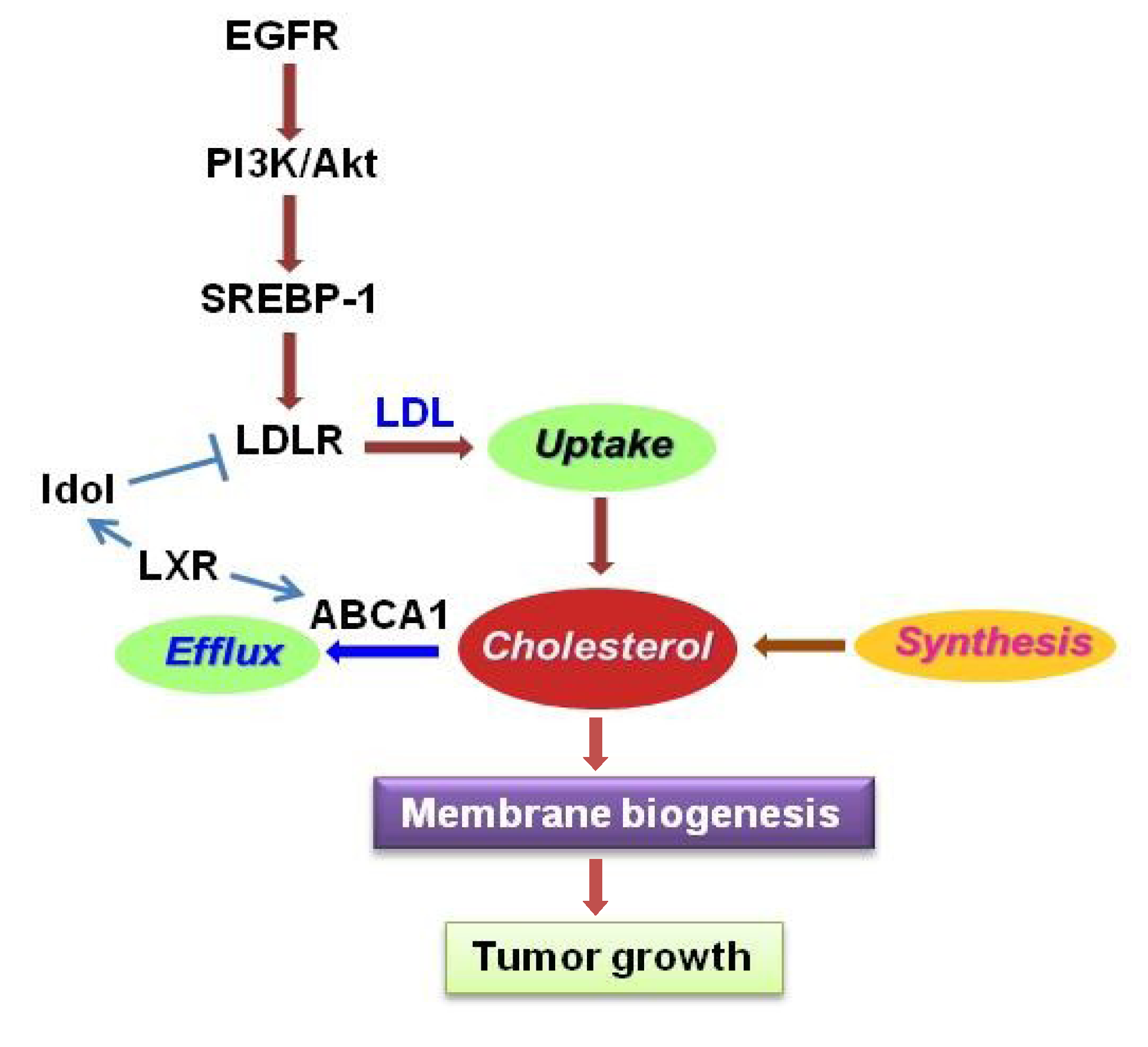
4.2. LDLR
4.3. LXR/ABCA1
5. Conclusions
Acknowledgements
Conflicts of Interest
References
- Kaplan, R.S. Supratentorial malignant gliomas: Risk patterns and therapy. J. Natl. Cancer. Inst. 1993, 85, 690–691. [Google Scholar] [CrossRef]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes. Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar]
- Dunn, G.P.; Rinne, M.L.; Wykosky, J.; Genovese, G.; Quayle, S.N.; Dunn, I.F.; Agarwalla, P.K.; Chheda, M.G.; Campos, B.; Wang, A.; et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 2012, 26, 756–784. [Google Scholar] [CrossRef]
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 15–31. [Google Scholar] [CrossRef]
- Mellinghoff, I.K.; Wang, M.Y.; Vivanco, I.; Haas-Kogan, D.A.; Zhu, S.; Dia, E.Q.; Lu, K.V.; Yoshimoto, K.; Huang, J.H.; Chute, D.J.; et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N. Engl. J. Med. 2005, 353, 2012–2024. [Google Scholar] [CrossRef]
- Wheeler, D.L.; Dunn, E.F.; Harari, P.M. Understanding resistance to EGFR inhibitors-impact on future treatment strategies. Nat. Rev. Clin. Oncol. 2010, 7, 493–507. [Google Scholar] [CrossRef]
- Chakravarti, A.; Wang, M.; Robins, H.I.; Lautenschlaeger, T.; Curran, W.J.; Brachman, D.G.; Schultz, C.J.; Choucair, A.; Dolled-Filhart, M.; Christiansen, J.; et al. RTOG 0211: A phase 1/2 study of radiation therapy with concurrent gefitinib for newly diagnosed glioblastoma patients. Int. J. Radiat. Oncol. Biol. Phys. 2013, 85, 1206–1211. [Google Scholar] [CrossRef]
- Tanaka, K.; Babic, I.; Nathanson, D.; Akhavan, D.; Guo, D.; Gini, B.; Dang, J.; Zhu, S.; Yang, H.; de Jesus, J.; et al. Oncogenic EGFR signaling activates an mTORC2-NF-kappaB pathway that promotes chemotherapy resistance. Cancer Discov. 2011, 1, 524–538. [Google Scholar] [CrossRef]
- Marin-Valencia, I.; Yang, C.; Mashimo, T.; Cho, S.; Baek, H.; Yang, X.L.; Rajagopalan, K.N.; Maddie, M.; Vemireddy, V.; Zhao, Z.; et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 2012, 15, 827–837. [Google Scholar] [CrossRef]
- Martini, M.; Ciraolo, E.; Gulluni, F.; Hirsch, E. Targeting PI3K in Cancer: Any Good News? Front. Oncol. 2013, 3, 108. [Google Scholar]
- Courtney, K.D.; Corcoran, R.B.; Engelman, J.A. The PI3K pathway as drug target in human cancer. J. Clin. Oncol. 2010, 28, 1075–1083. [Google Scholar] [CrossRef]
- Chakravarti, A.; Zhai, G.; Suzuki, Y.; Sarkesh, S.; Black, P.M.; Muzikansky, A.; Loeffler, J.S. The prognostic significance of phosphatidylinositol 3-kinase pathway activation in human gliomas. J. Clin. Oncol. 2004, 22, 1926–1933. [Google Scholar] [CrossRef]
- Ferrara, N. VEGF as a therapeutic target in cancer. Oncology 2005, 69, 11–16. [Google Scholar] [CrossRef]
- Jun, H.J.; Acquaviva, J.; Chi, D.; Lessard, J.; Zhu, H.; Woolfenden, S.; Bronson, R.T.; Pfannl, R.; White, F.; Housman, D.E.; et al. Acquired MET expression confers resistance to EGFR inhibition in a mouse model of glioblastoma multiforme. Oncogene 2012, 31, 3039–3050. [Google Scholar] [CrossRef]
- Huang, T.T.; Sarkaria, S.M.; Cloughesy, T.F.; Mischel, P.S. Targeted therapy for malignant glioma patients: Lessons learned and the road ahead. Neurotherapeutics 2009, 6, 500–512. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef]
- Deberardinis, R.J.; Thompson, C.B. Cellular metabolism and disease: What do metabolic outliers teach us? Cell 2012, 148, 1132–1144. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Schulze, A.; Harris, A.L. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 2012, 491, 364–373. [Google Scholar] [CrossRef]
- Wolf, A.; Agnihotri, S.; Guha, A. Targeting metabolic remodeling in glioblastoma multiforme. Oncotarget 2010, 1, 552–562. [Google Scholar]
- Maher, E.A.; Marin-Valencia, I.; Bachoo, R.M.; Mashimo, T.; Raisanen, J.; Hatanpaa, K.J.; Jindal, A.; Jeffrey, F.M.; Choi, C.; Madden, C.; et al. Metabolism of [U-(13) C]glucose in human brain tumors in vivo. NMR Biomed. 2012, 25, 1234–1244. [Google Scholar] [CrossRef]
- Yang, C.; Sudderth, J.; Dang, T.; Bachoo, R.M.; McDonald, J.G.; DeBerardinis, R.J. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res. 2009, 69, 7986–7993. [Google Scholar]
- Bell, E.H.; Guo, D. Biomarkers for malignant gliomas. Malig. Gliomas Radiat. Med. Rounds 2012, 3, 389–357. [Google Scholar]
- Guo, D.; Bell, E.H.; Chakravarti, A. Lipid metabolism emerges as a promising target for malignant glioma therapy. CNS Oncol. 2013, 2, 289–299. [Google Scholar] [CrossRef]
- Guo, D.; Cloughesy, T.F.; Radu, C.G.; Mischel, P.S. AMPK: A metabolic checkpoint that regulates the growth of EGFR activated glioblastomas. Cell Cycle 2010, 9, 211–212. [Google Scholar] [CrossRef]
- Guo, D.; Hildebrandt, I.J.; Prins, R.M.; Soto, H.; Mazzotta, M.M.; Dang, J.; Czernin, J.; Shyy, J.Y.; Watson, A.D.; Phelps, M.; et al. The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 12932–12937. [Google Scholar] [CrossRef]
- Guo, D.; Prins, R.M.; Dang, J.; Kuga, D.; Iwanami, A.; Soto, H.; Lin, K.Y.; Huang, T.T.; Akhavan, D.; Hock, M.B.; et al. EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci. Signal. 2009, 2, ra82. [Google Scholar] [CrossRef]
- Guo, D.; Reinitz, F.; Youssef, M.; Hong, C.; Nathanson, D.; Akhavan, D.; Kuga, D.; Amzajerdi, A.N.; Soto, H.; Zhu, S.; et al. An LXR agonist promotes GBM cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer. Discov. 2011, 1, 442–456. [Google Scholar] [CrossRef]
- Kefas, B.; Comeau, L.; Erdle, N.; Montgomery, E.; Amos, S.; Purow, B. Pyruvate kinase M2 is a target of the tumor-suppressive microRNA-326 and regulates the survival of glioma cells. Neuro. Oncol. 2010, 12, 1102–1112. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Cao, Y.; Zheng, Y.; Bu, W.; Zhang, L.; You, M.J.; Koh, M.Y.; Cote, G.; Aldape, K.; et al. EGFR-induced and PKCepsilon monoubiquitylation-dependent NF-kappaB activation upregulates PKM2 expression and promotes tumorigenesis. Mol. Cell 2012, 48, 771–784. [Google Scholar] [CrossRef]
- Dang, L.; Jin, S.; Su, S.M. IDH mutations in glioma and acute myeloid leukemia. Trends Mol. Med. 2010, 16, 387–397. [Google Scholar] [CrossRef]
- Williams, K.J.; Argus, J.P.; Zhu, Y.; Wilks, M.Q.; Marbois, B.N.; York, A.G.; Kidani, Y.; Pourzia, A.L.; Akhavan, D.; Lisiero, D.N.; et al. An Essential Requirement for the SCAP/SREBP Signaling Axis to Protect Cancer Cells from Lipotoxicity. Cancer Res. 2013, 73, 2850–2862. [Google Scholar] [CrossRef]
- Ainscow, E.K.; Brand, M.D. Top-down control analysis of ATP turnover, glycolysis and oxidative phosphorylation in rat hepatocytes. Eur. J. Biochem. 1999, 263, 671–685. [Google Scholar] [CrossRef]
- Lopez-Lazaro, M. The warburg effect: Why and how do cancer cells activate glycolysis in the presence of oxygen? Anticancer Agents Med Chem. 2008, 8, 305–312. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg's contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell. Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [Green Version]
- Babic, I.; Anderson, E.S.; Tanaka, K.; Guo, D.; Masui, K.; Li, B.; Zhu, S.; Gu, Y.; Villa, G.R.; Akhavan, D.; et al. EGFR Mutation-Induced Alternative Splicing of Max Contributes to Growth of Glycolytic Tumors in Brain Cancer. Cell Metab. 2013, 17, 1000–1008. [Google Scholar] [CrossRef]
- Kim, J.W.; Dang, C.V. Multifaceted roles of glycolytic enzymes. Trends Biochem. Sci. 2005, 30, 142–150. [Google Scholar] [CrossRef]
- Wilson, J.E. Isozymes of mammalian hexokinase: Structure, subcellular localization and metabolic function. J. Exp. Biol. 2003, 206, 2049–2057. [Google Scholar] [CrossRef]
- Postic, C.; Shiota, M.; Magnuson, M.A. Cell-specific roles of glucokinase in glucose homeostasis. Recent Prog. Horm. Res. 2001, 56, 195–217. [Google Scholar] [CrossRef]
- Printz, R.L.; Magnuson, M.A.; Granner, D.K. Mammalian glucokinase. Annu. Rev. Nutr. 1993, 13, 463–496. [Google Scholar] [CrossRef]
- Wolf, A.; Agnihotri, S.; Micallef, J.; Mukherjee, J.; Sabha, N.; Cairns, R.; Hawkins, C.; Guha, A. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J. Exp. Med. 2011, 208, 313–326. [Google Scholar] [CrossRef]
- Agnihotri, S.; Wolf, A.; Munoz, D.M.; Smith, C.J.; Gajadhar, A.; Restrepo, A.; Clarke, I.D.; Fuller, G.N.; Kesari, S.; Dirks, P.B.; et al. A GATA4-regulated tumor suppressor network represses formation of malignant human astrocytomas. J. Exp. Med. 2011, 208, 689–702. [Google Scholar] [CrossRef]
- Wolf, A.; Agnihotri, S.; Munoz, D.; Guha, A. Developmental profile and regulation of the glycolytic enzyme hexokinase 2 in normal brain and glioblastoma multiforme. Neurobiol. Dis. 2011, 44, 84–91. [Google Scholar] [CrossRef]
- Mazurek, S.; Boschek, C.B.; Hugo, F.; Eigenbrodt, E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin Cancer Biol. 2005, 15, 300–308. [Google Scholar] [CrossRef]
- Clower, C.V.; Chatterjee, D.; Wang, Z.; Cantley, L.C.; Vander Heiden, M.G.; Krainer, A.R. The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc. Natl. Acad. Sci. USA 2010, 107, 1894–1899. [Google Scholar]
- Mazurek, S. Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. Int. J. Biochem. Cell Biol. 2011, 43, 969–980. [Google Scholar] [CrossRef]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Hawke, D.; Li, X.; Liang, J.; Xing, D.; Aldape, K.; Hunter, T.; Alfred Yung, W.K.; Lu, Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 2012, 150, 685–696. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Ji, H.; Zheng, Y.; Liang, J.; Huang, W.; Gao, X.; Aldape, K.; Lu, Z. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature 2011, 480, 118–122. [Google Scholar]
- Yang, W.; Zheng, Y.; Xia, Y.; Ji, H.; Chen, X.; Guo, F.; Lyssiotis, C.A.; Aldape, K.; Cantley, L.C.; Lu, Z. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat. Cell Biol. 2012, 14, 1295–1304. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.M.; Tian, R.F.; Liu, W.P.; Fei, Z.; Long, Q.F.; Wang, X.A.; Zhang, X. The expression and significance of HIF-1alpha and GLUT-3 in glioma. Brain Res. 2009, 1304, 149–154. [Google Scholar] [CrossRef]
- Boado, R.J.; Black, K.L.; Pardridge, W.M. Gene expression of GLUT3 and GLUT1 glucose transporters in human brain tumors. Brain Res. Mol. Brain Res. 1994, 27, 51–57. [Google Scholar] [CrossRef]
- Reitman, Z.J.; Yan, H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: Alterations at a crossroads of cellular metabolism. J. Natl. Cancer Inst. 2010, 102, 932–941. [Google Scholar] [CrossRef]
- Kim, W.; Liau, L.M. IDH mutations in human glioma. Neurosurg. Clin. N. Am. 2012, 23, 471–480. [Google Scholar] [CrossRef]
- Weller, M.; Wick, W.; von Deimling, A. Isocitrate dehydrogenase mutations: A challenge to traditional views on the genesis and malignant progression of gliomas. Glia 2011, 59, 1200–1204. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- DeAngelis, L.M.; Mellinghoff, I.K. Virchow 2011 or how to ID(H) human glioblastoma. J. Clin. Oncol. 2011, 29, 4473–4474. [Google Scholar] [CrossRef]
- Watanabe, T.; Nobusawa, S.; Kleihues, P.; Ohgaki, H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am. J. Pathol. 2009, 174, 1149–1153. [Google Scholar] [CrossRef]
- Riemenschneider, M.J.; Jeuken, J.W.; Wesseling, P.; Reifenberger, G. Molecular diagnostics of gliomas: State of the art. Acta Neuropathol. 2010, 120, 567–584. [Google Scholar] [CrossRef]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef]
- Boulahbel, H.; Duran, R.V.; Gottlieb, E. Prolyl hydroxylases as regulators of cell metabolism. Biochem. Soc. Trans. 2009, 37, 291–294. [Google Scholar] [CrossRef]
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 2009, 324, 261–265. [Google Scholar] [CrossRef]
- Andronesi, O.C.; Kim, G.S.; Gerstner, E.; Batchelor, T.; Tzika, A.A.; Fantin, V.R.; Vander Heiden, M.G.; Sorensen, A.G. Detection of 2-hydroxyglutarate in IDH-mutated glioma patients by in vivo spectral-editing and 2D correlation magnetic resonance spectroscopy. Sci. Transl. Med. 2012, 4, 116ra114. [Google Scholar]
- Choi, C.; Ganji, S.K.; DeBerardinis, R.J.; Hatanpaa, K.J.; Rakheja, D.; Kovacs, Z.; Yang, X.L.; Mashimo, T.; Raisanen, J.M.; Marin-Valencia, I.; et al. 2-hydroxyglutarate detection by magnetic resonance spectroscopy in IDH-mutated patients with gliomas. Nat. Med. 2012, 18, 624–629. [Google Scholar] [CrossRef]
- Kalinina, J.; Carroll, A.; Wang, L.; Yu, Q.; Mancheno, D.E.; Wu, S.; Liu, F.; Ahn, J.; He, M.; Mao, H.; et al. Detection of “oncometabolite” 2-hydroxyglutarate by magnetic resonance analysis as a biomarker of IDH1/2 mutations in glioma. J. Mol. Med (Berl.) 2012, 90, 1161–1171. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2010, 465, 966. [Google Scholar]
- Lai, A.; Kharbanda, S.; Pope, W.B.; Tran, A.; Solis, O.E.; Peale, F.; Forrest, W.F.; Pujara, K.; Carrillo, J.A.; Pandita, A.; et al. Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J. Clin. Oncol. 2011, 29, 4482–4490. [Google Scholar] [CrossRef]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef]
- Zielke, H.R.; Zielke, C.L.; Ozand, P.T. Glutamine: A major energy source for cultured mammalian cells. Fed. Proc. 1984, 43, 121–125. [Google Scholar]
- Reitzer, L.J.; Wice, B.M.; Kennell, D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J. Biol. Chem. 1979, 254, 2669–2676. [Google Scholar]
- Cory, J.G.; Cory, A.H. Critical roles of glutamine as nitrogen donors in purine and pyrimidine nucleotide synthesis: Asparaginase treatment in childhood acute lymphoblastic leukemia. In vivo 2006, 20, 587–589. [Google Scholar]
- Butler, E.B.; Zhao, Y.; Munoz-Pinedo, C.; Lu, J.; Tan, M. Stalling the engine of resistance: Targeting cancer metabolism to overcome therapeutic resistance. Cancer Res. 2013, 73, 2709–2717. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar]
- Rajagopalan, K.N.; DeBerardinis, R.J. Role of glutamine in cancer: Therapeutic and imaging implications. J. Nucl. Med. 2011, 52, 1005–1008. [Google Scholar] [CrossRef]
- Daye, D.; Wellen, K.E. Metabolic reprogramming in cancer: Unraveling the role of glutamine in tumorigenesis. Semin. Cell Dev. Biol. 2012, 23, 362–369. [Google Scholar] [CrossRef]
- Lu, W.; Pelicano, H.; Huang, P. Cancer metabolism: Is glutamine sweeter than glucose? Cancer Cell 2010, 18, 199–200. [Google Scholar] [CrossRef]
- Dang, C.V.; Le, A.; Gao, P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin. Cancer Res. 2009, 15, 6479–6483. [Google Scholar] [CrossRef]
- Rathore, M.G.; Saumet, A.; Rossi, J.F.; de Bettignies, C.; Tempe, D.; Lecellier, C.H.; Villalba, M. The NF-kappaB member p65 controls glutamine metabolism through miR-23a. Int. J. Biochem. Cell Biol. 2012, 44, 1448–1456. [Google Scholar] [CrossRef]
- Wang, J.B.; Erickson, J.W.; Fuji, R.; Ramachandran, S.; Gao, P.; Dinavahi, R.; Wilson, K.F.; Ambrosio, A.L.; Dias, S.M.; Dang, C.V.; et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010, 18, 207–219. [Google Scholar] [CrossRef]
- Kallenberg, K.; Bock, H.C.; Helms, G.; Jung, K.; Wrede, A.; Buhk, J.H.; Giese, A.; Frahm, J.; Strik, H.; Dechent, P.; et al. Untreated glioblastoma multiforme: Increased myo-inositol and glutamine levels in the contralateral cerebral hemisphere at proton MR spectroscopy. Radiology 2009, 253, 805–812. [Google Scholar] [CrossRef]
- Rosati, A.; Marconi, S.; Pollo, B.; Tomassini, A.; Lovato, L.; Maderna, E.; Maier, K.; Schwartz, A.; Rizzuto, N.; Padovani, A.; et al. Epilepsy in glioblastoma multiforme: Correlation with glutamine synthetase levels. J. Neurooncol. 2009, 93, 319–324. [Google Scholar] [CrossRef]
- Maxfield, F.R. Plasma membrane microdomains. Curr. Opin. Cell Biol. 2002, 14, 483–487. [Google Scholar] [CrossRef]
- Mukherjee, S.; Maxfield, F.R. Membrane domains. Annu. Rev. Cell Dev. Biol. 2004, 20, 839–866. [Google Scholar] [CrossRef]
- Pomorski, T.; Hrafnsdottir, S.; Devaux, P.F.; van Meer, G. Lipid distribution and transport across cellular membranes. Semin. Cell Dev. Biol. 2001, 12, 139–148. [Google Scholar] [CrossRef]
- Van Meer, G. Membranes in motion. EMBO Rep. 2010, 11, 331–333. [Google Scholar] [CrossRef]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- Zechner, R.; Strauss, J.G.; Haemmerle, G.; Lass, A.; Zimmermann, R. Lipolysis: Pathway under construction. Curr. Opin. Lipidol. 2005, 16, 333–340. [Google Scholar] [CrossRef]
- Abramson, H.N. The lipogenesis pathway as a cancer target. J. Med. Chem. 2011, 54, 5615–5638. [Google Scholar] [CrossRef]
- Grossi-Paoletti, E.; Paoletti, P.; Fumagalli, R. Lipids in brain tumors. J. Neurosurg. 1971, 34, 454–455. [Google Scholar] [CrossRef]
- Podo, F. Tumour phospholipid metabolism. NMR Biomed. 1999, 12, 413–439. [Google Scholar] [CrossRef]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef]
- Yoon, S.; Lee, M.Y.; Park, S.W.; Moon, J.S.; Koh, Y.K.; Ahn, Y.H.; Park, B.W.; Kim, K.S. Up-regulation of acetyl-CoA carboxylase alpha and fatty acid synthase by human epidermal growth factor receptor 2 at the translational level in breast cancer cells. J. Biol. Chem. 2007, 282, 26122–26131. [Google Scholar]
- Gopal, K.; Grossi, E.; Paoletti, P.; Usardi, M. Lipid composition of human intracranial tumors: A biochemical study. Acta Neurochir. (Wien) 1963, 11, 333–347. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 2002, 109, 1125–1131. [Google Scholar]
- Shimano, H. Sterol regulatory element-binding proteins (SREBPs): Transcriptional regulators of lipid synthetic genes. Prog. Lipid Res. 2001, 40, 439–452. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997, 89, 331–340. [Google Scholar] [CrossRef]
- Nohturfft, A.; DeBose-Boyd, R.A.; Scheek, S.; Goldstein, J.L.; Brown, M.S. Sterols regulate cycling of SREBP cleavage-activating protein (SCAP) between endoplasmic reticulum and Golgi. Proc. Natl. Acad. Sci. USA 1999, 96, 11235–11240. [Google Scholar] [CrossRef]
- Sakai, J.; Duncan, E.A.; Rawson, R.B.; Hua, X.; Brown, M.S.; Goldstein, J.L. Sterol-regulated release of SREBP-2 from cell membranes requires two sequential cleavages, one within a transmembrane segment. Cell 1996, 85, 1037–1046. [Google Scholar] [CrossRef]
- Zelenski, N.G.; Rawson, R.B.; Brown, M.S.; Goldstein, J.L. Membrane topology of S2P, a protein required for intramembranous cleavage of sterol regulatory element-binding proteins. J. Biol. Chem. 1999, 274, 21973–21980. [Google Scholar]
- Espenshade, P.J.; Cheng, D.; Goldstein, J.L.; Brown, M.S. Autocatalytic processing of site-1 protease removes propeptide and permits cleavage of sterol regulatory element-binding proteins. J. Biol. Chem. 1999, 274, 22795–22804. [Google Scholar]
- Hua, X.; Yokoyama, C.; Wu, J.; Briggs, M.R.; Brown, M.S.; Goldstein, J.L.; Wang, X. SREBP-2, a second basic-helix-loop-helix-leucine zipper protein that stimulates transcription by binding to a sterol regulatory element. Proc. Natl. Acad. Sci. USA 1993, 90, 11603–11607. [Google Scholar]
- Jeon, T.I.; Osborne, T.F. SREBPs: Metabolic integrators in physiology and metabolism. Trends Endocrinol. Metab. 2012, 23, 65–72. [Google Scholar] [CrossRef]
- Shao, W.; Espenshade, P.J. Expanding Roles for SREBP in Metabolism. Cell Metab. 2012, 16, 414–419. [Google Scholar] [CrossRef]
- Ettinger, S.L.; Sobel, R.; Whitmore, T.G.; Akbari, M.; Bradley, D.R.; Gleave, M.E.; Nelson, C.C. Dysregulation of sterol response element-binding proteins and downstream effectors in prostate cancer during progression to androgen independence. Cancer Res. 2004, 64, 2212–2221. [Google Scholar] [CrossRef]
- Yang, Y.; Morin, P.J.; Han, W.F.; Chen, T.; Bornman, D.M.; Gabrielson, E.W.; Pizer, E.S. Regulation of fatty acid synthase expression in breast cancer by sterol regulatory element binding protein-1c. Exp. Cell Res. 2003, 282, 132–137. [Google Scholar] [CrossRef]
- Von Roemeling, C.A.; Marlow, L.A.; Wei, J.J.; Cooper, S.J.; Caulfield, T.R.; Wu, K.; Tan, W.W.; Tun, H.W.; Copland, J.A. Stearoyl-CoA Desaturase 1 Is a Novel Molecular Therapeutic Target for Clear Cell Renal Cell Carcinoma. Clin. Cancer Res. 2013, 19, 2368–2380. [Google Scholar] [CrossRef]
- Maxfield, F.R.; Tabas, I. Role of cholesterol and lipid organization in disease. Nature 2005, 438, 612–621. [Google Scholar] [CrossRef]
- Tosi, M.R.; Tugnoli, V. Cholesteryl esters in malignancy. Clin. Chim. Acta 2005, 359, 27–45. [Google Scholar] [CrossRef]
- Tugnoli, V.; Tosi, M.R. Cholesteryl ester detection in a human urothelial carcinoma. Clin. Chim. Acta 2005, 360, 208–210. [Google Scholar] [CrossRef]
- Tugnoli, V.; Tosi, M.R.; Tinti, A.; Trinchero, A.; Bottura, G.; Fini, G. Characterization of lipids from human brain tissues by multinuclear magnetic resonance spectroscopy. Biopolymers. 2001, 62, 297–306. [Google Scholar] [CrossRef]
- Tugnoli, V.; Bottura, G.; Fini, G.; Reggiani, A.; Tinti, A.; Trinchero, A.; Tosi, M.R. 1H-NMR and 13C-NMR lipid profiles of human renal tissues. Biopolymers 2003, 72, 86–95. [Google Scholar] [CrossRef]
- Yates, A.J.; Thompson, D.K.; Boesel, C.P.; Albrightson, C.; Hart, R.W. Lipid composition of human neural tumors. J. Lipid Res. 1979, 20, 428–436. [Google Scholar]
- Rudling, M.J.; Angelin, B.; Peterson, C.O.; Collins, V.P. Low density lipoprotein receptor activity in human intracranial tumors and its relation to the cholesterol requirement. Cancer Res. 1990, 50, 483–487. [Google Scholar]
- Goldstein, J.L.; Brown, M.S. The LDL receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef]
- Zelcer, N.; Tontonoz, P. Liver X receptors as integrators of metabolic and inflammatory signaling. J. Clin. Invest. 2006, 116, 607–614. [Google Scholar] [CrossRef]
- Calkin, A.C.; Tontonoz, P. Liver x receptor signaling pathways and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1513–1518. [Google Scholar] [CrossRef]
- Vedin, L.L.; Lewandowski, S.A.; Parini, P.; Gustafsson, J.A.; Steffensen, K.R. The oxysterol receptor LXR inhibits proliferation of human breast cancer cells. Carcinogenesis 2009, 30, 575–579. [Google Scholar] [CrossRef]
- Zelcer, N.; Hong, C.; Boyadjian, R.; Tontonoz, P. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science 2009, 325, 100–104. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ru, P.; Williams, T.M.; Chakravarti, A.; Guo, D. Tumor Metabolism of Malignant Gliomas. Cancers 2013, 5, 1469-1484. https://doi.org/10.3390/cancers5041469
Ru P, Williams TM, Chakravarti A, Guo D. Tumor Metabolism of Malignant Gliomas. Cancers. 2013; 5(4):1469-1484. https://doi.org/10.3390/cancers5041469
Chicago/Turabian StyleRu, Peng, Terence M. Williams, Arnab Chakravarti, and Deliang Guo. 2013. "Tumor Metabolism of Malignant Gliomas" Cancers 5, no. 4: 1469-1484. https://doi.org/10.3390/cancers5041469