3.2. Synthesis
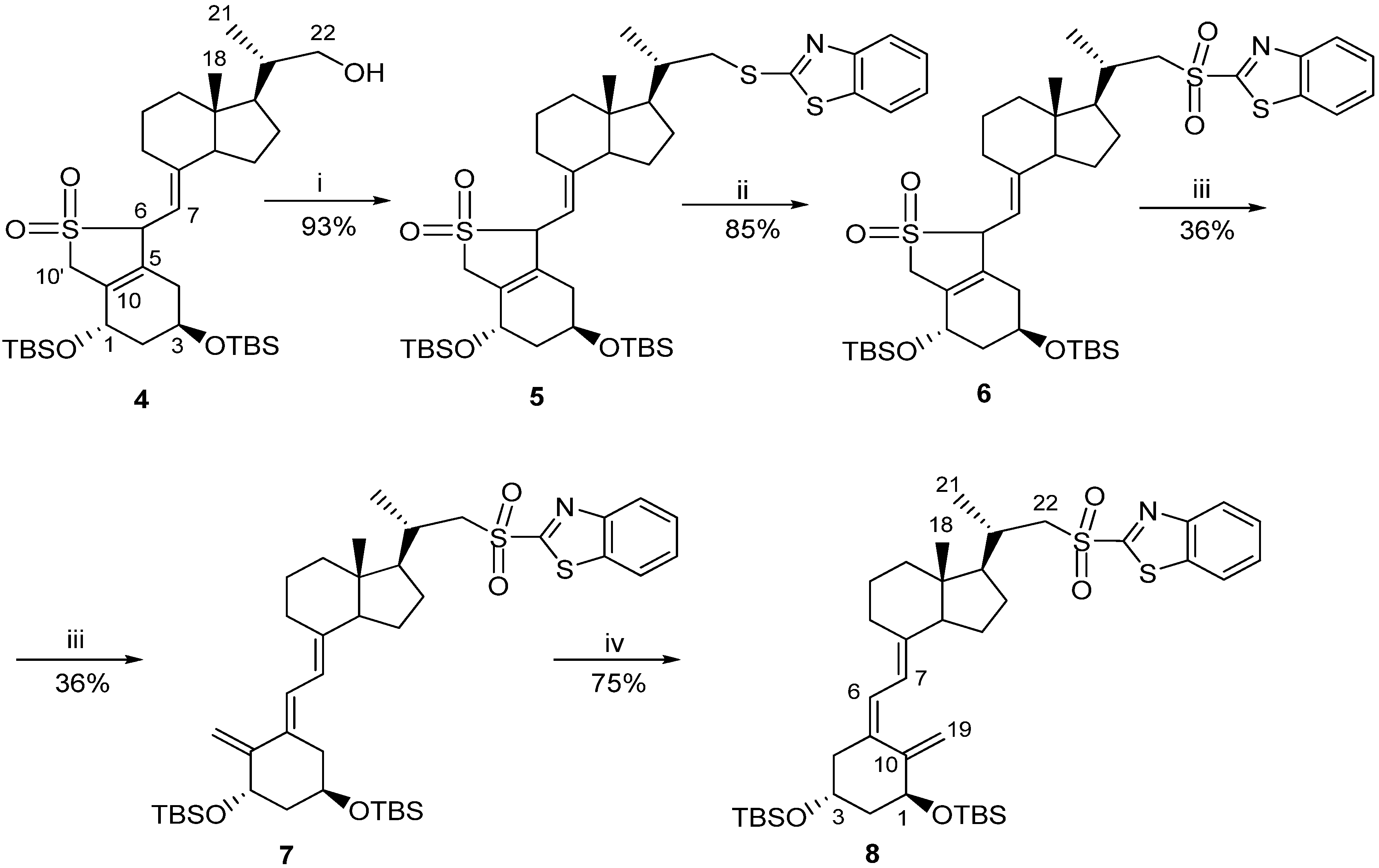
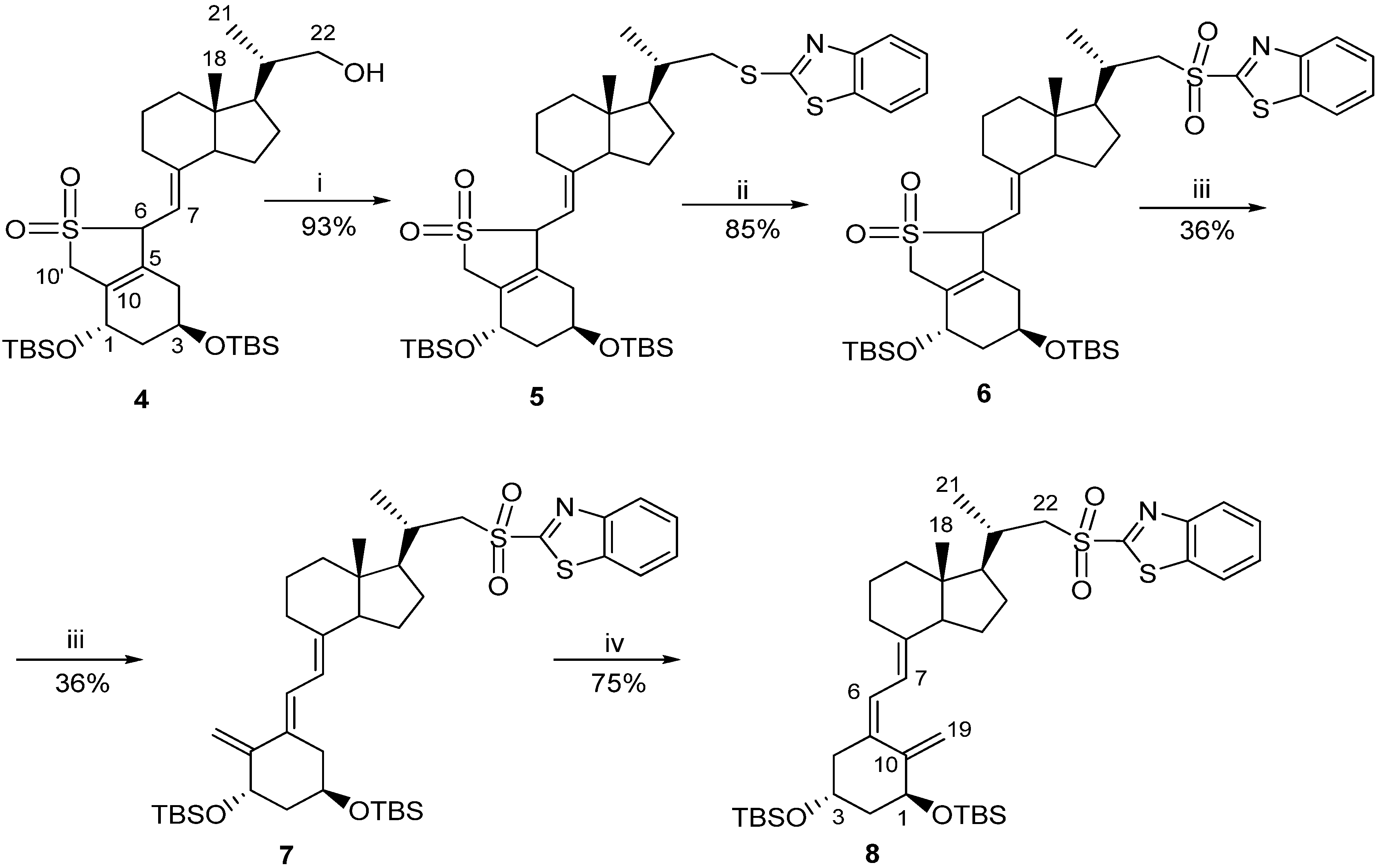
3.2.1. (6R/S)-SO2-(5E,7E)-(1S,3R)-1,3-bis[t-butyl(dimethylsilyl)oxy]-22-thiobenzothiazolyl-23,24-dinor-9,10-secochola-5(10),7-diene adduct (5)
A solution of the crude alcohol 4 (87 g, 0.14 M) in CH2Cl2 (500 mL) was prepared and placed in a dropping funnel. A suspension of 2-thiobenzothiazole (45 g, 0.27 M) in CH2Cl2 (500 mL) was prepared and placed in a cooling bath (0 °C) with a magnetic stirring. Triphenylphosphine (71 g, 0.27 M) was added with stirring in a single portion to this suspension and the solution of the alcohol 4 in CH2Cl2 was slowly added dropwise. Then diisopropylazadicarboxylate (45 mL, 0.23 M) was added dropwise. The mixture was vigorously stirred for 90 min. at 0 °C. The cooling bath was removed and brine (4 L) and water (200 mL) were added. The organic phase was separated and the residue was extracted with CH2Cl2 (2 × 200 mL). The combined organic phases were dried over anhydrous Na2SO4 (80 g). The solution was filtered and concentrated under reduced pressure. The residue was chromatographed on the column with silica gel (230-00 mesh, 800 g, 2%–8% hexane/ethyl acetate). The fractions containing the pure sulphide 5 were concentrated and dried in vacuo. The residue (284 g) was dissolved in 2:1 hexane–toluene mixture (600 mL). The suspension was filtered under reduced pressure. The filtrate was concentrated under reduced pressure and thoroughly dried on a vacuum pump. The crude sulphide 5 (100 g, 0.13 M, 93%) was obtained and used for the next step without further purification. 1H-NMR (δ, ppm) 0.06 (12H, m, 2Si(CH3)2), 0.69 (3H, s, 18-CH3), 0.88 (18H, m, 2Si-C(CH3)3), 1.14 (3H, d, J = 6.5 Hz, 21-CH3), 2.62 and 3.01 (3H, m, 6-H and 22-CH2), 3.94 (1H, m, 7-H), 4.19 and 4.37 (2H, m, 1-H and 3-H), 4.69 (2H, m, 10'-CH2), 7.34 and 7.77 (4H, m, Ar-H).
3.2.2. (6RS)-SO2-(5E,7E)-(1S,3R)-1,3-bis[Tert-butyl(dimethylsilyl)oxy]-22-sulfonylbenzothiazolyl-23,24-dinor-9,10-secochola-5(10),7-diene adduct (6)
A three neck flask fitted with a mechanical stirrer and a dropping funnel was placed in a water bath. The solution of the crude sulphide 5 (100 g, 0.13 M) in CH2Cl2 (400 mL) and C2H5OH (1200 mL) was added. A solution of ammonium heptamolybdenate hydrate (AHT, 22 g, 18.8 mM) in H2O2 (35%, 150 mL) was dropped in over 5 min. with stirring. The water bath was heated up to 65 °C and the stirring was continued for 2.5 h until the complete disappearance of the substrate (monitored by TLC). The mixture was cooled down to 0 °C and the solution of Na2SO3 (10%, 900 mL) was dropped in until the disappearance of peroxides (paper indicator). The solvents were removed under reduced pressure, and ethyl acetate (600 mL) was added to the residue. The organic phase was separated and the water phase was extracted twice with ethyl acetate (2 × 300 mL). The combined organic phases were dried over anhydrous Na2SO4 (40 g) and filtered. The solvents were removed under reduced pressure and the residue was dried on a vacuum pump. The crude sulfone 6 (90 g, 0.11 mol, 85%) was obtained as yellow oil; IR, ν, 2952, 2928, 2883, 2856, 1624, 1472, 1381, 1360, 1323, 1253, 1148, 1121, 1083, 834, 760 cm−1; 1H-NMR (δ, ppm) 0.06 (12H, m, 2Si(CH3)2), 0.65 (3H, s, 18-CH3), 0.87 (18H, m, 2Si-C(CH3)3), 1.28 (3H, d, J = 6.5 Hz, 21-CH3), 3.28 and 3.65 (3H, m, 6-H and 22-CH2), 3.94 (1H, m, 7-H), 4.17 and 4.36 (2H, m, 1-H and 3-H), 4.65 (2H, m, 10'-CH2), 7.61, 8.02 and 8.22 (4H, m, Ar-H).
3.2.3. (5E,7E)-(1S,3R)-1,3-bis[Tert-butyl (dimethylosilyl)oxy]-22-sulfonylbenzothiazolyl-23,24-dinor-9,10-secochola-5,7,10(19)-triene (7)
NaHCO3 (70 g) was added to the solution of the sulfone 6 (90 g, 0.11 M) in ethanol (1,200 mL) and the mixture was stirred under reflux for 3 h. The mixture was cooled down and the solvents were removed under reduced pressure. Then water (600 mL) and ethyl acetate (600 mL) were added. The organic phase was separated and the residue was extracted with ethyl acetate (2 × 200 mL). The combined organic phases were dried over anhydrous Na2SO4 (80 g) and filtered. The solvents were removed under reduced pressure. The residue was dissolved in toluene and placed on the chromatographic column with aluminium oxide (1:10) in hexane. Hexane was used to wash out toluene from the column. The product was washed with a 10% mixture of hexane-ethyl acetate. The solvents were removed under reduced pressure to give 42 g of light yellow oily mixture of the crude product 7. Then ethyl acetate (30 mL) was added and the mixture was heated up near the boiling point and methanol (800 mL) was added in one portion. The mixture was left at −20 °C for 24 h. The precipitate was filtered and washed with cold methanol (90 mL). The precipitate was dried to the constant weight on a vacuum pump. The crude triene 7 (30 g, 0.04 M, 36%) was obtained and used in the next reaction without further purification; UV λmax (EtOH) 271.0, 240.6, 207.0 nm, λmin 245.6, 231.0 nm; IR, ν, 2951, 2928, 2883, 2856, 1636, 1554, 1472, 1324, 1252, 1147, 1083, 834, 760 cm−1; 1H-NMR (δ, ppm) 0.06 (12H, m, 2Si(CH3)2), 0.56 (3H, s, 18-CH3), 0.85 (18H, m, 2Si-C(CH3)3), 1.26 (3H, d, J = 6.7 Hz, 21-CH3), 3.31 and 3.62 (2H, m, 22-CH2), 4.22 (1H, m, 3-H), 4.53 (1H, m, 1-H), 4.97 (2H, m, 19E-H and 19Z-H), 5.79 (1H, d, J = 11.5 Hz, 7-H), 6.42 (1H, d, J = 11.5 Hz, 6-H), 7.62, 8.04, 8.23 (4H, m, Ar-H).
3.2.4. (5Z,7E)-(1S,3R)-1,3-bis[Tert-butyl(dimethylsilyl)oxy]-22-sulfonylbenzotiazolyl-23,24-dinor-9,10-secochola-5,7,10(19)-triene (8)
The triene 7 (24 g, 31.8 M) was dissolved in a 5:1 mixture of toluene-methanol (3 L) saturated with argon. Then anthracene (24 g, 134.8 M) was added. The solution was placed in a UV irradiation apparatus and the circulating pump and power supply for the UV lamps were turned on. The irradiation was carried out for 3.5 h at 18–20 °C. The solvents were removed under reduced pressure. Then hexane (400 mL) was added and the residue was left for 6 h at −20 °C. The mixture was filtered in vacuo. The residue was dissolved in toluene (450 mL) and the filtered solution of maleic anhydride (2.4 g) in toluene (50 mL) was added. The mixture was saturated with argon and stirred for 12 h on a magnetic stirrer at room temperature. The solvents were removed under reduced pressure. The residue was dissolved in the mixture of toluene (15 mL) and hexane (15 mL) and put on the chromatographic column filled with 350 g of silica gel 230–400 mesh. Mixtures of hexane-ethyl acetate (1%–4%) were used as eluents. The crude product (21 g) was obtained as yellow precipitate. The precipitate was dissolved in ethyl acetate (30 mL) heated up near the boiling point and methanol (700 mL) was added. The solution was left at −20 °C for 24 h. The precipitate was separated using a Buchner funnel and dried to the constant weight on a vacuum pump. The triene 8 (18 g, 23.8 mM, 75%) was obtained as colorless fluffy powder; UV λmax (EtOH) 268.2, 240.0, 214.4 nm, λmin 245.6, 231.0 nm; IR, ν, 2951, 2928, 2883, 2856, 1636, 1554, 1472, 1324, 1252, 1147, 1083, 834, 760 cm−1; 1H-NMR (δ, ppm) 0.06 (12H, m, 2Si(CH3)2, 0.55 (3H, s, 18-CH3), 0.87 (18H, m, 2Si-C(CH3)3), 1.26 (3H, d, J = 6.6 Hz, 21-CH3), 3.28 and 3.65 (2H, m, 22-CH2), 4.18 (1H, m, 3-H), 4.36 (1H, m, 1-H), 4.83 (1H, m, 19Z-H), 5.16 (1H, m, 19E-H), 5.99 (1H, d, J = 11.4 Hz, 7-H), 6.21 (1H, d, J = 11.4 Hz, 6-H), 7.61, 8.02, 8.22 (4H, m, Ar-H).
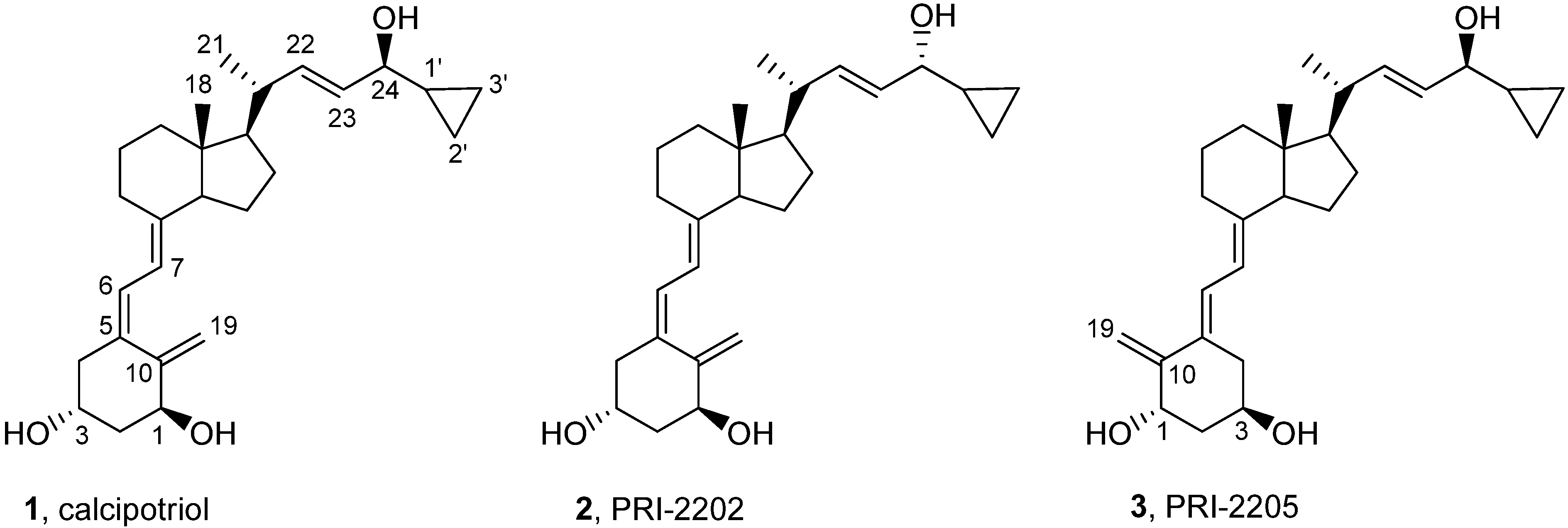
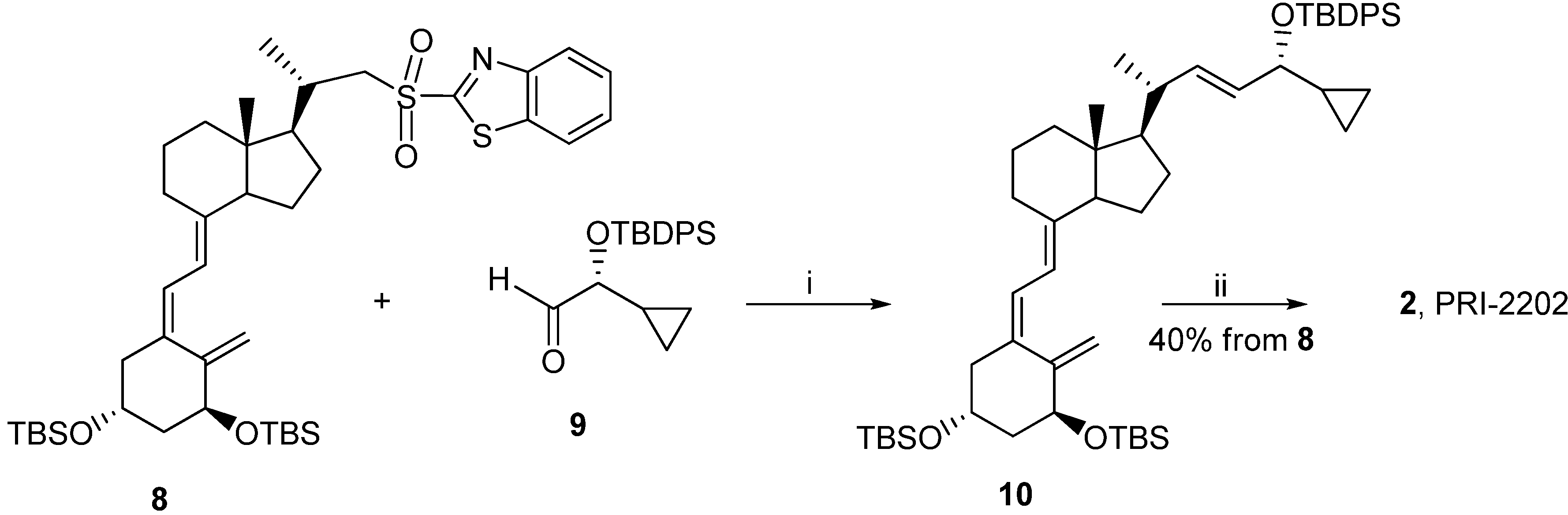
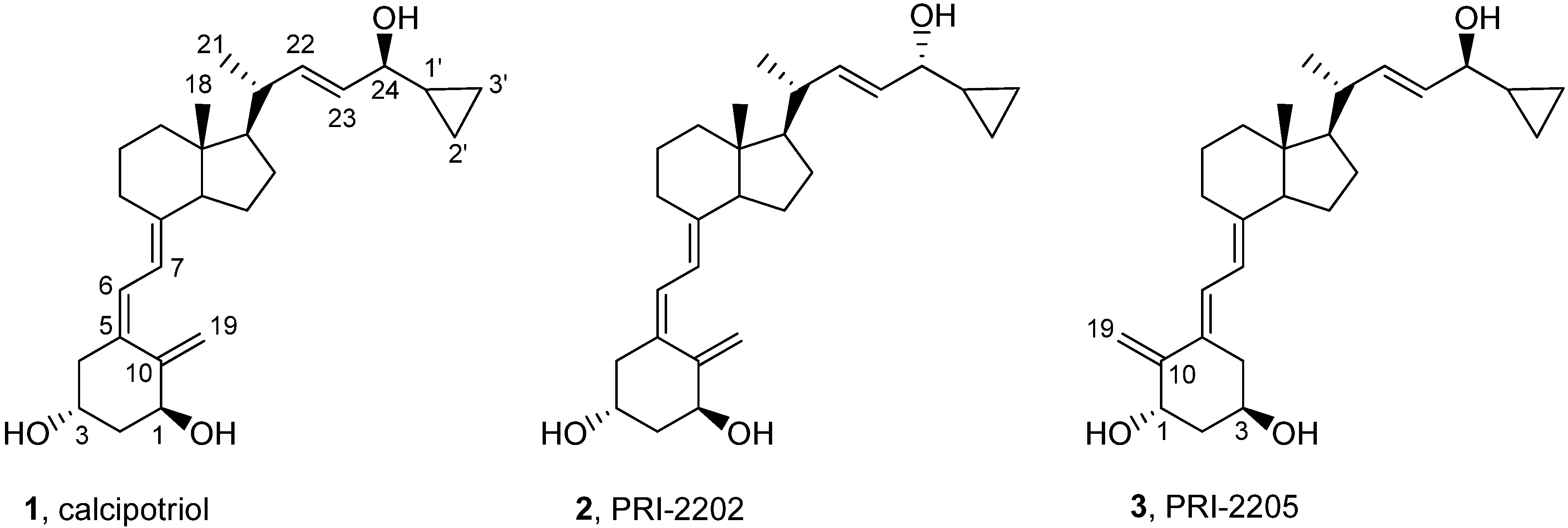
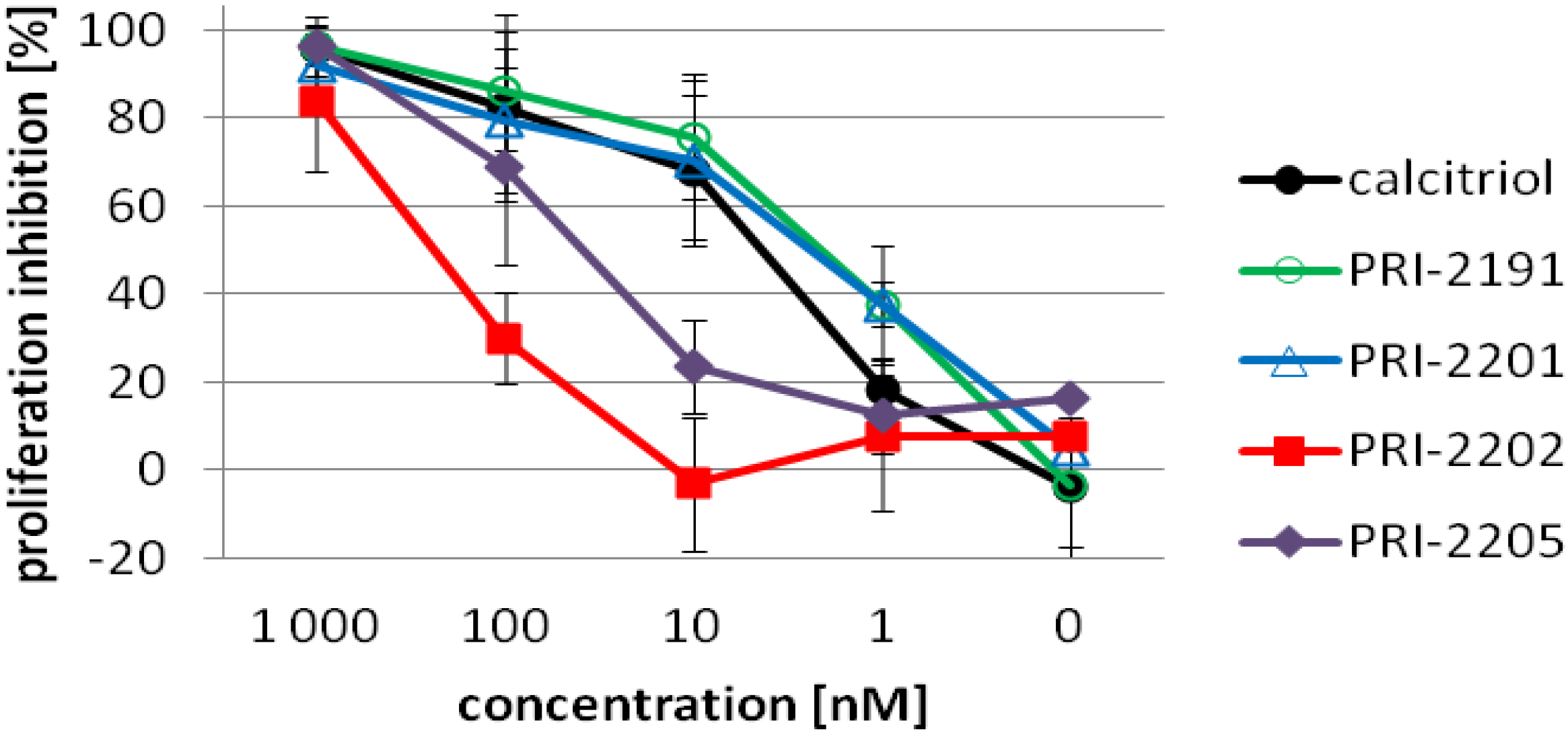
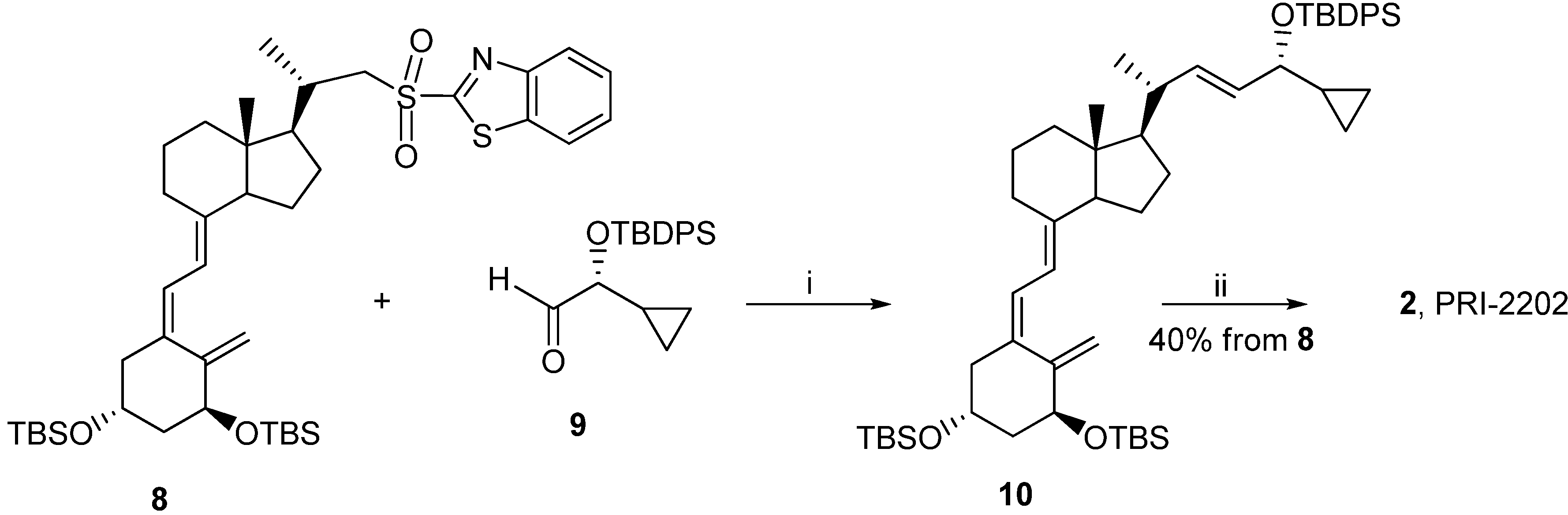
3.2.5. (5Z,7E,22E)-(1S,3R,24R)-24-Cyclopropyl-9,10-secochola-5,7,10(19),22-tetraen-1,3,24-triol (PRI-2202, 2)
The triene 8 (4.0 g, 5.3 M) was dissolved in 1,2-dimethoxyethane (DME, 40 mL). The flask was placed in a cooling bath (−68 °C) on a magnetic stirrer under argon. A solution of lithium bis(trimethylsilyl)amide (1.0 M in THF, 5.6 mL, 5.6 M)) was added drop-wise with a syringe with stirring. The stirring was continued for 30 min. at −68 °C. Then the (R)-2-(tert-butyldiphenylsilyloxy)-2-cyclopropylacetaldehyde 9 (2.0 g, 5.9 M) was slowly added dropwise. The cooling bath was removed after 30 min. and the reaction mixture was stirred for additional 24 h at ambient temp. Brine (20 mL) was added, the organic phase was separated and dried over Na2SO4 (10 g). The solvents were removed under reduced pressure. The crude intermediate 10 (4.1 g, 5.6 mM) was used for the next step without further purification. The intermediate 10 (4.1 g, 5.6 mM) was dissolved in THF (40 mL) under argon. The solution was warmed up to 60 °C with stirring. Tetrabutylammonium fluoride solution (1 M in THF, 15.7 mL, 15.7 M) was added dropwise and the stirring was continued for 2.0 h. The solution was cooled down to 20 °C and 20 mL of brine was added. The organic phase was separated, dried over Na2SO4 (10 g), filtered and condensed under vacuum. The residue was filtered through silica gel (50 g). The solvents were removed under vacuum and the resulting solid was crystallized from ethyl acetate (30 mL). The triol 2 was obtained (850 mg, 2.1 mM, 40% from 8), as colorless crystals of 99.5% purity (HPLC); UV λmax (EtOH) 264.6, 213.4 nm, λmin 228.6 nm; IR ν 3401, 2949, 2927, 2869, 1631, 1432, 1376, 1325, 1246, 1156, 1117, 1064, 981, 911, 797 cm−1; 1H-NMR (500 MHz, CDC13) δ ppm: 0.23, 0.32, 0.51 (4H, m, 2'-H and 3'-H ), 0.57 (3H, s, 18-CH3), 0.98 (1H, m, 1'-H), 1,04 (3H, d, J = 6.9 Hz), 3.47 (1H, dd: 6.9, 6.8, 24-H), 4.23 (1H, m, 3-H), 4.43(1H, m, 1-OH), 5.00 (1H, bs, 19Z-H), 5.33 (1H, bs, 19E-H), 5.46 (1H, dd, J = 6.0, 14.5 Hz, 23-H), 5.54 (1H, dd, J = 8.0, 15.5 Hz, 22-H), 6.01 (1H, d, J = 11.3 Hz, 7-H), 6.37 (1H, d, J = 11.3 Hz, 6-H); 13C-NMR (500 MHz, CDCl3) δ: 1.82, 2.98, 12.27, 14.19, 17.53, 20.53, 22.23, 23.55, 27.63, 29.06, 39.78, 40.36, 42.89, 45.29, 45.91, 56.19, 56.33, 66.86, 70.83, 111.76, 117.12, 124.94, 128.92, 132.98, 137.77, 142.95, 147.66; HRMS (EI): calcd for C27H4003 [M+]: calc. 412.2977, found 412.2979.
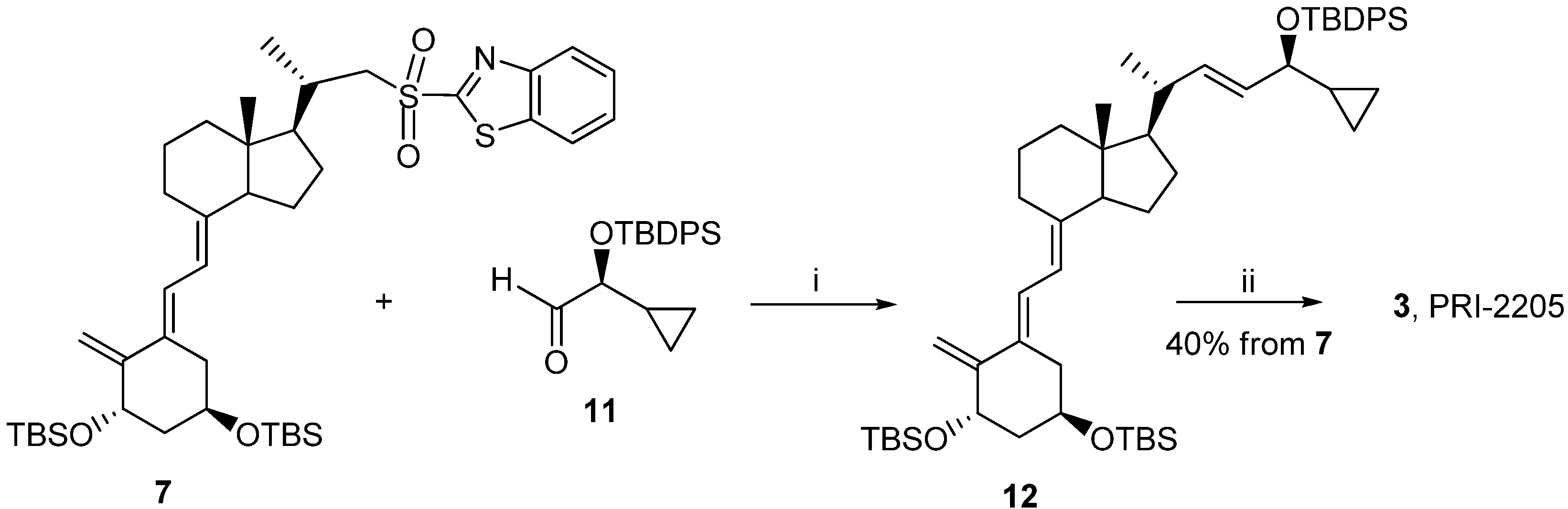
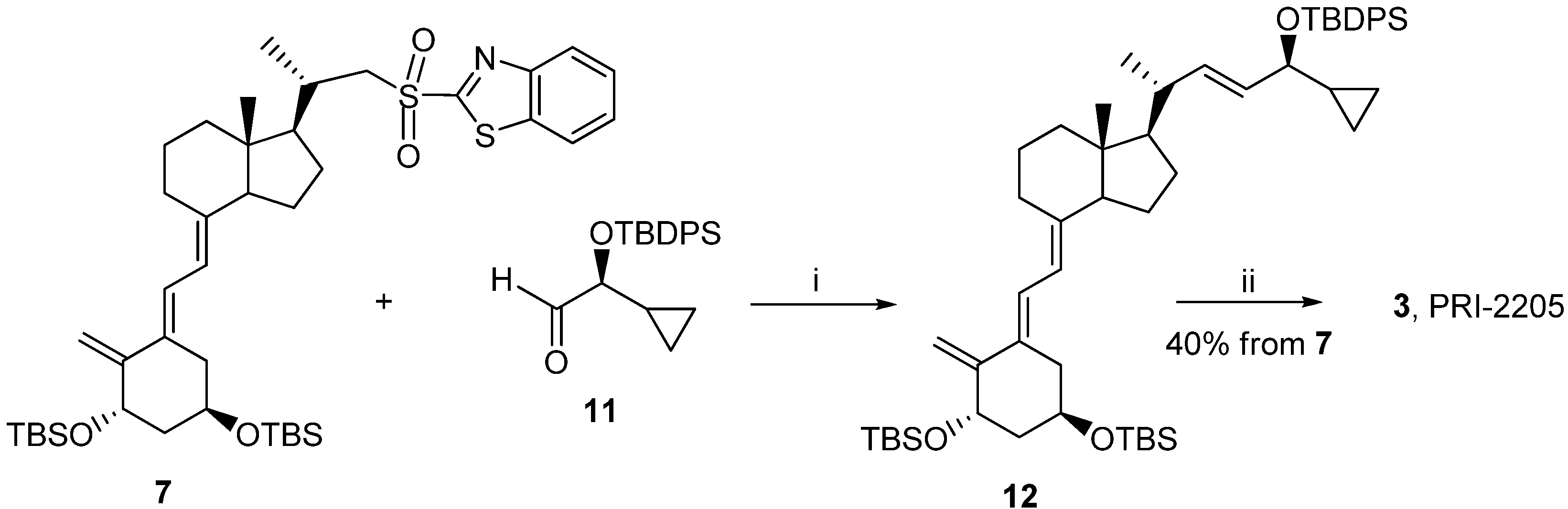
3.2.6. (5E,7E,22E)-(1S,3R,24S)-24-Cyclopropyl-9,10-secochola-5,7,10(19),22-tetraen-1,3,24-triol (PRI-2205, 3)
A solution of LiHMDS (1.0 M in THF, 2.5 mL, 2.5 mM)) was added dropwise using a syringe with stirring to the solution of the triene 7 (1.8 g, 2.4 mM) in THF (10 mL) at −68 °C under argon. The stirring was continued for 30 min. (S)-2-(tert-Butyldiphenylsilyloxy)-2-cyclopropylacetaldehyde 11 (0.9 g, 2.9 mM) was slowly added dropwise. The cooling bath was removed after 30 min. and the reaction mixture was stirred for additional 24 h at ambient temperature. Brine (5 mL) was added, the organic phase was separated and dried over Na2SO4 (8 g). The solvents were removed under reduced pressure. The crude intermediate 12 (2.0 g, 2.3 mM) was used for the next step without further purification. The intermediate 12 (2.0 g, 2.3 mM) was dissolved in THF (10 mL) under argon. The solution was warmed up to 60 °C with stiring. A solution of tetrabutylammonium fluoride (1 M in THF, 7.0 mL, 7.0 mM) was added dropwise and the stirring was continued for 2.0 h. The solution was cooled down to 20 °C and 5 mL of brine was added. The organic phase was separated, dried over Na2SO4 (5 g), filtered and condensed under vacuum. The residue was filtered through silica gel (15 g). The solvents were removed and the resulting solid was crystallized from ethyl acetate (20 mL). The triol 3 was obtained (430 mg, 1.04 mM, 43%) as colorless crystals of 99.2% purity (HPLC); UV λmax (EtOH) 273,4, 209,2 nm, λmin 231,6 nm; IR ν 3570, 3447, 2945, 2871, 1623, 1433, 1373, 1292, 1078, 1227, 1052, 1024, 983, 951, 901, 890, 871 cm−1; 1H-NMR (600 MHz, CDC13) δ ppm: 0.13, 0.26, 0.40, 0.48 (4H, m, 2'-H and 3'-H ), 0.51 (3H, s, 18-CH3), 0.89 (1H, m, 1'-H), 0.98 (3H, d, J = 6.6 Hz), 3.34 (1H, dd: 7.8, 7.2, 24-H), 4.09 (1H, m, 3-H), 4.39(1H, m, 1-OH), 4.89 (1H, s, 19Z-H), 5.04 (1H, s, 19E-H), 5.20, 5.39 (2H, m, 23-H and 22-H), 5.82 (1H, d, J = 11.4 Hz, 7-H), 6.49 (1H, d, J = 11.3 Hz, 6-H); 13C-NMR (600 MHz, CDCl3) δ: 1.73, 3.05, 12.22, 15.34, 17.34, 20.39, 22.12, 23.40, 27.58, 28.90, 36.34, 39.87, 40.24, 41.66, 45.78, 55.95, 56.39, 65.31, 70.55, 109.38, 116.02, 122.72, 128.87, 133.30, 137.79, 144.51, 151.79; HRMS calcd for C27H4003Na [M + Na]+: 435.2872, found 435.2875.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}