Control of Oxidative Stress and Generation of Induced Pluripotent Stem Cell-like Cells by Jun Dimerization Protein 2



 ,
, 
Abstract
:1. Introduction
2. Experimental Section
2.1. Reagents, Cell Culture, and Plasmid Preparation
2.2. Measurement of H2O2 Concentrations in Culture Medium
2.3. Preparation of Hydrogen Peroxide
2.4. Analysis of 7,8-Dihydro-8-oxo-8-2′-deoxyguanosine (8-oxo-dGuo), Glutathione, and Cellular ROS
2.5. Transient Transfection and Luciferase Reporter Assay
2.6. Western Blot Analysis
2.7. Chromatin Immunoprecipitation (ChIP) Assay
2.8. Immunofluorescence

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer’s name | Sequences of primers | Product size |
|---|---|---|
| Mouse E1 NQO1 | Forward: 5'-GCACGAATTCATTTCACACGAGG-3' | 128 bp |
| Reverse: 5'-GGAAGTCACCTTTGCACGCTAG-3' | ||
| Mouse E1 HO-1 | Forward: 5'-AGCGGCTGGAATGCTGAGT-3' | 184 bp |
| Reverse: 5'-CCTTCTGCCTGCCGTTCCGG-3' | ||
| Mouse E2 HO-1 | Forward: 5'-GGGCTAGCATGCGAAGTGAG-3' | 201 bp |
| Reverse: 5'-GACTCCGCCCCTAAGGGTTC-3' |
2.9. Reprograming and Generation of DAOY Medulloblastoma iPSC
2.10. RT-PCR and Quantitative PCR Analysis of iPSCs
2.11. Teratoma and Tumor Formation in SCID Mice
2.12. Statistical Analyses
| Gene | Sequence (5'→3') |
|---|---|
| OCT4-F | CCCCCTGTCTCCGTCACCAC |
| OCT4-R | CCACATAGCGTAAAAGGAGCA |
| SOX2-F | ACACTGCCCCTCTCACACATG |
| SOX2-R | CCACATAGCGTAAAAGGAGCA |
| KLF4-F | GACCACCTCGCCTTACACATG |
| KLF4-R | CCACATAGCGTAAAAGGAGCA |
| c-MYC-F | CAGCTACGGAACTCTTGTGC |
| c-MYC-R | CCACATAGCGTAAAAGGAGCA |
| NANOG-F | CAGTCTGGACACTGGCTGAA |
| NANOG-R | CTCGCTGATTAGGCTCCAAC |
| NODAL-F | GGGCAAGAGGCACCGTCGACATCA |
| NODAL-R | GGGACTCGGTGGGGCTGGTAACGTTTC |
| DPPA2-F | CCGTCCCCGCAATCTCCTTCCATC |
| DPPA2-R | ATGATGCCAACATGGCTCCCGGTG |
| DPPA4-F | GGAGCCGCCTGCCCTGGAAAATTC |
| DPPA4-R | TTTTTCCTGATATTCTATTCCCAT |
| hTERT-F | CCTGCTCAAGCTGACTCGACACCGTG |
| hTERT-R | GGAAAAGCTGGCCCTGGGGTGGAGC |
| EBAF-F | GCTGGAGCTGCACACCCTGGACCTCAG |
| EBAF-R | GGGCAGCGAGGCAGTCTCCGAGGC |
| ESG1-F | ATATCCCGCCGTGGGTGAAAGTTC |
| ESG1-R | ACTCAGCCATGGACTGGAGCATCC |
| DNMT3 β-F | TGCTGCTCACAGGGCCCGATACTTC |
| DNMT3 β-R | TCCTTTCGAGCTCAGTGCACCACAAAAC |
| UTF-1-F | CCGTCGCTGAACACCGCCCTGCTG |
| UTF-1-R | CGCGCTGCCCAGAATGAAGCCCAC |
| GAPDH-F | GACTCCGACCTTCACCTTC |
| GAPDH-R | GAAATCCCATCACCATCTTC |
| Gene | Sequence (5'→3') |
|---|---|
| OCT4-F | GGGTTTTTGGGATTAAGTTCTTCA |
| OCT4-R | GCCCCCACCCTTTGTGTT |
| SOX2-F | CAAAAATGGCCATGCAGGTT |
| SOX2-R | AGTTGGGATCGAACAAAAGCTATT |
| KLF4-F | AGCCTAAATGATGGTGCTTGGT |
| KLF4-R | TTGAAAACTTTGGCTTCCTTGTT |
| c-MYC-F | CGGGCGGGCATTTG |
| c-MYC-R | GGAGAGTCGCGTCCTTGCT |
| NANOG-F | CAGTCTGGACACTGGCTAA |
| NANOG-R | CTCGCTGATTAGGCTCCAAC |
| GAPDH-F | GAAGGTGAAGGTCGGAGTC |
| GAPDH-R | GAAGATGGTGATGGGATTTC |
3. Results
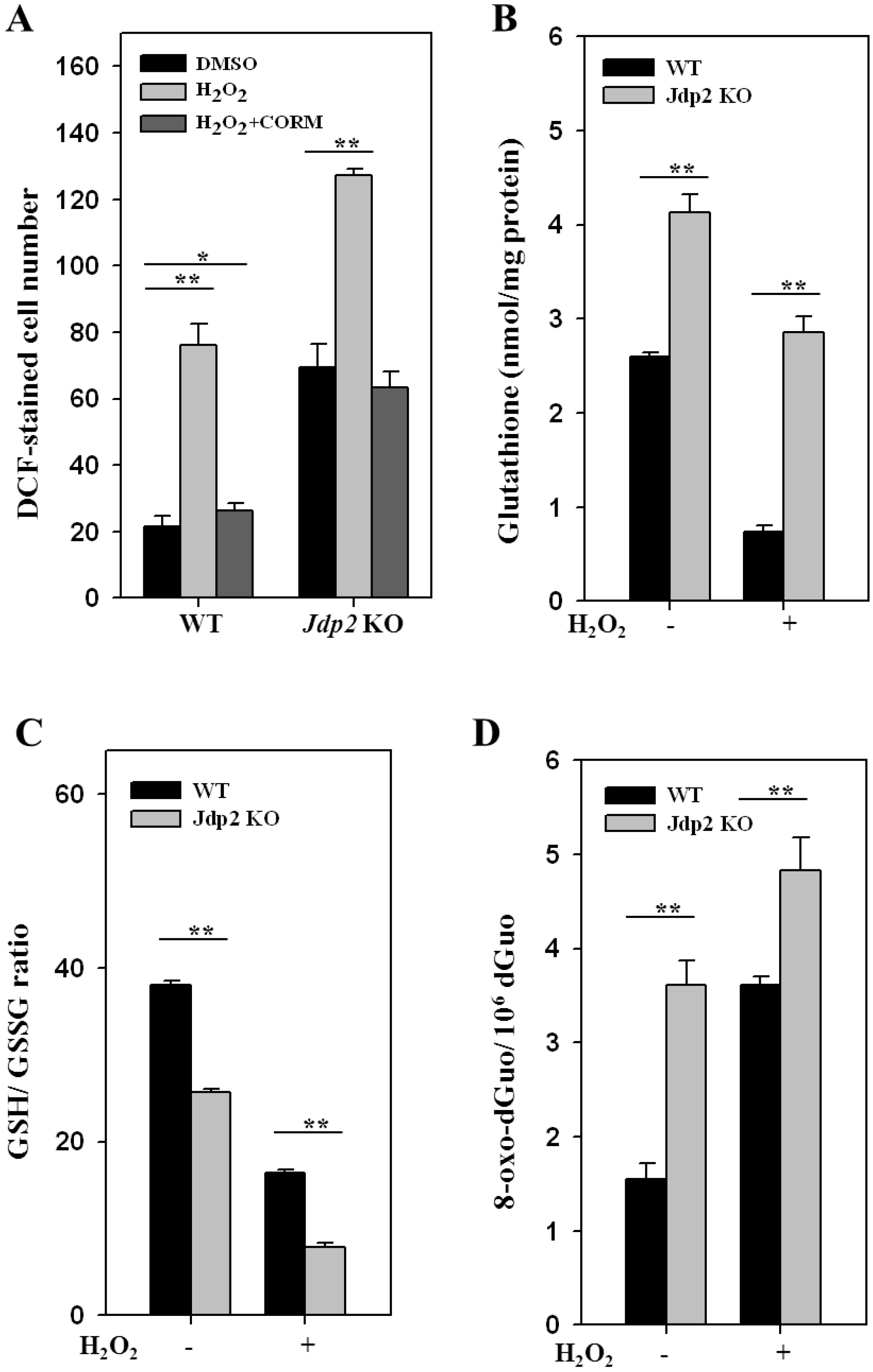
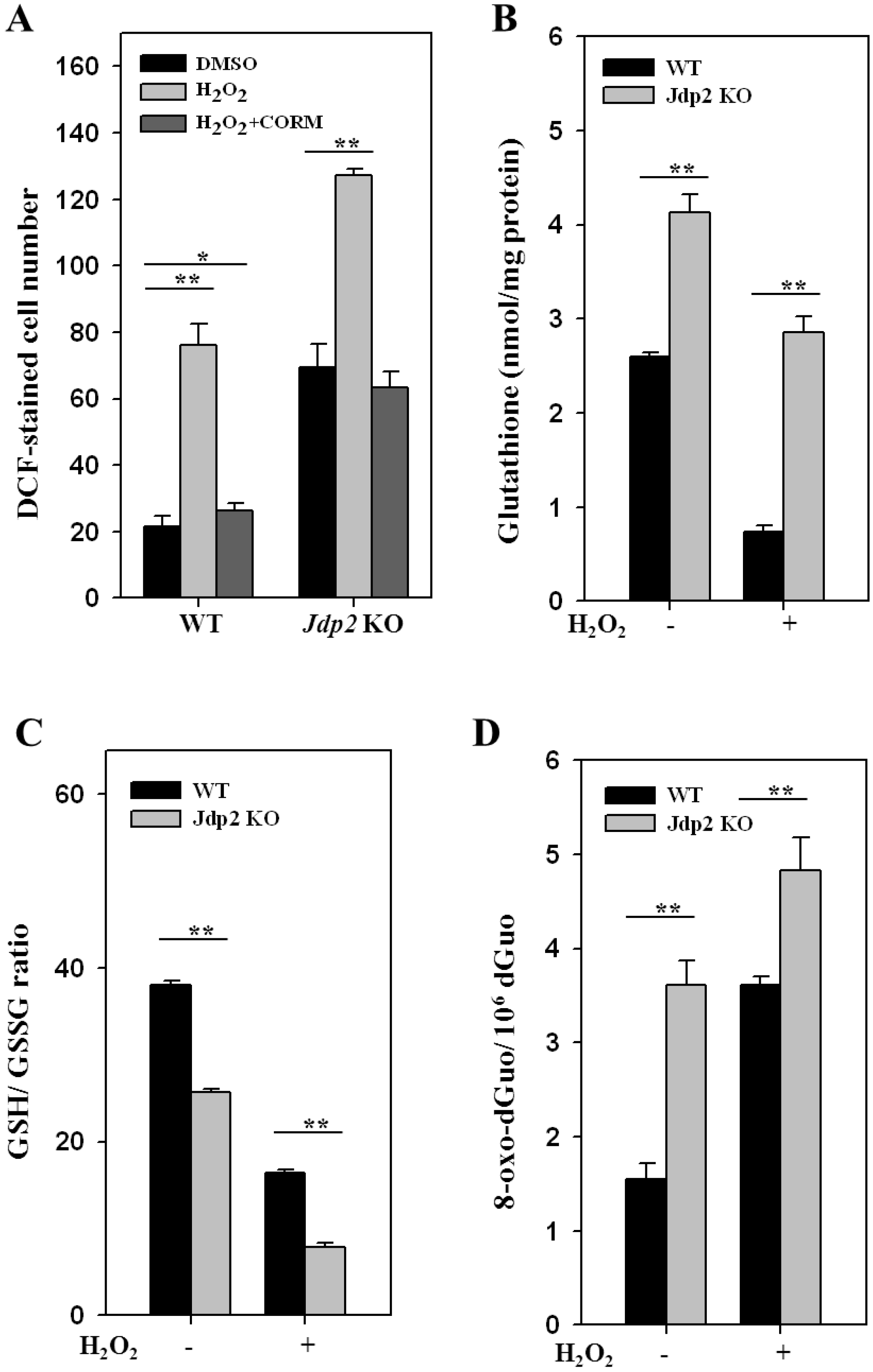
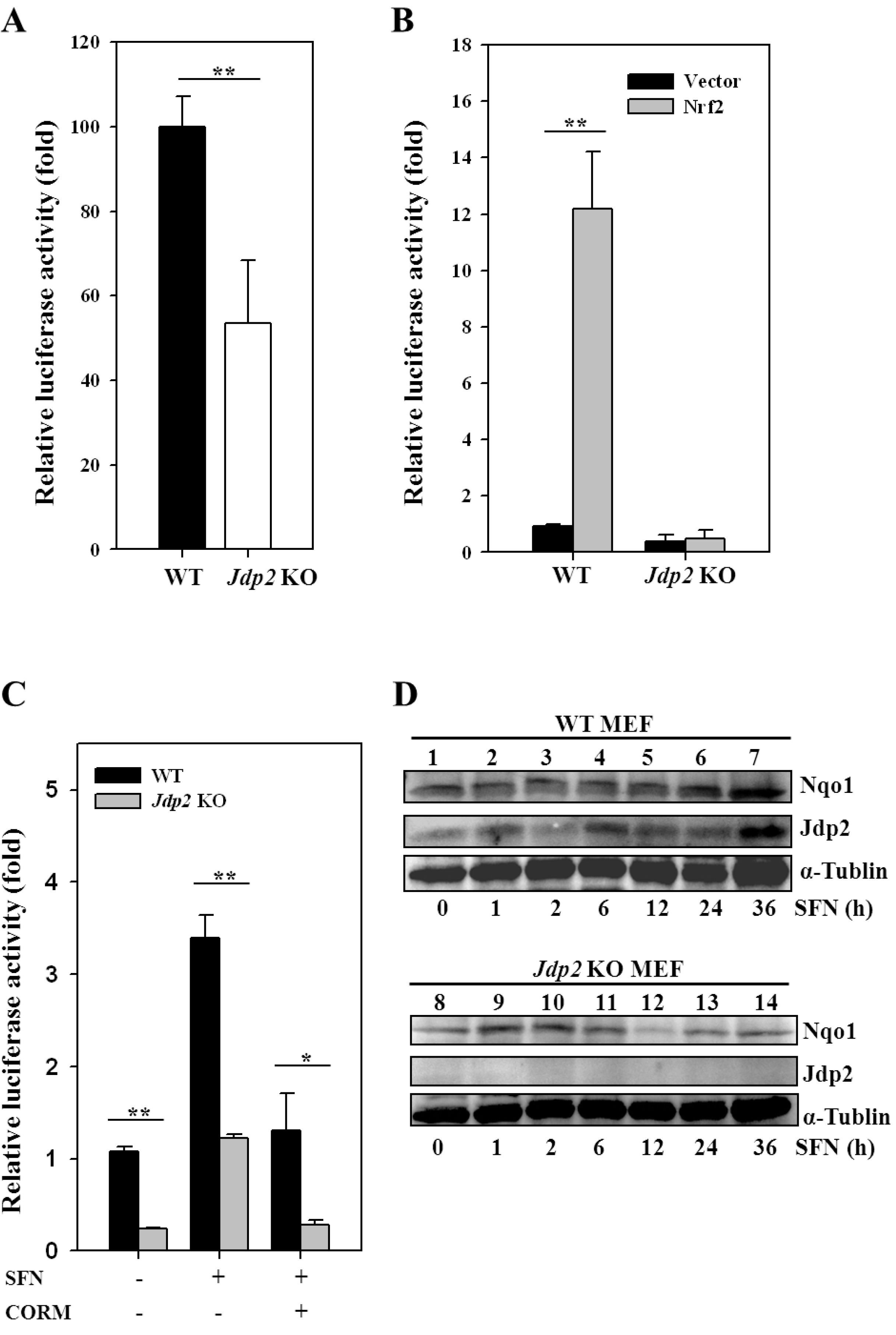
3.1. JDP2 Reduces H2O2-Mediated Production of ROS and Increases Antioxidant Activity
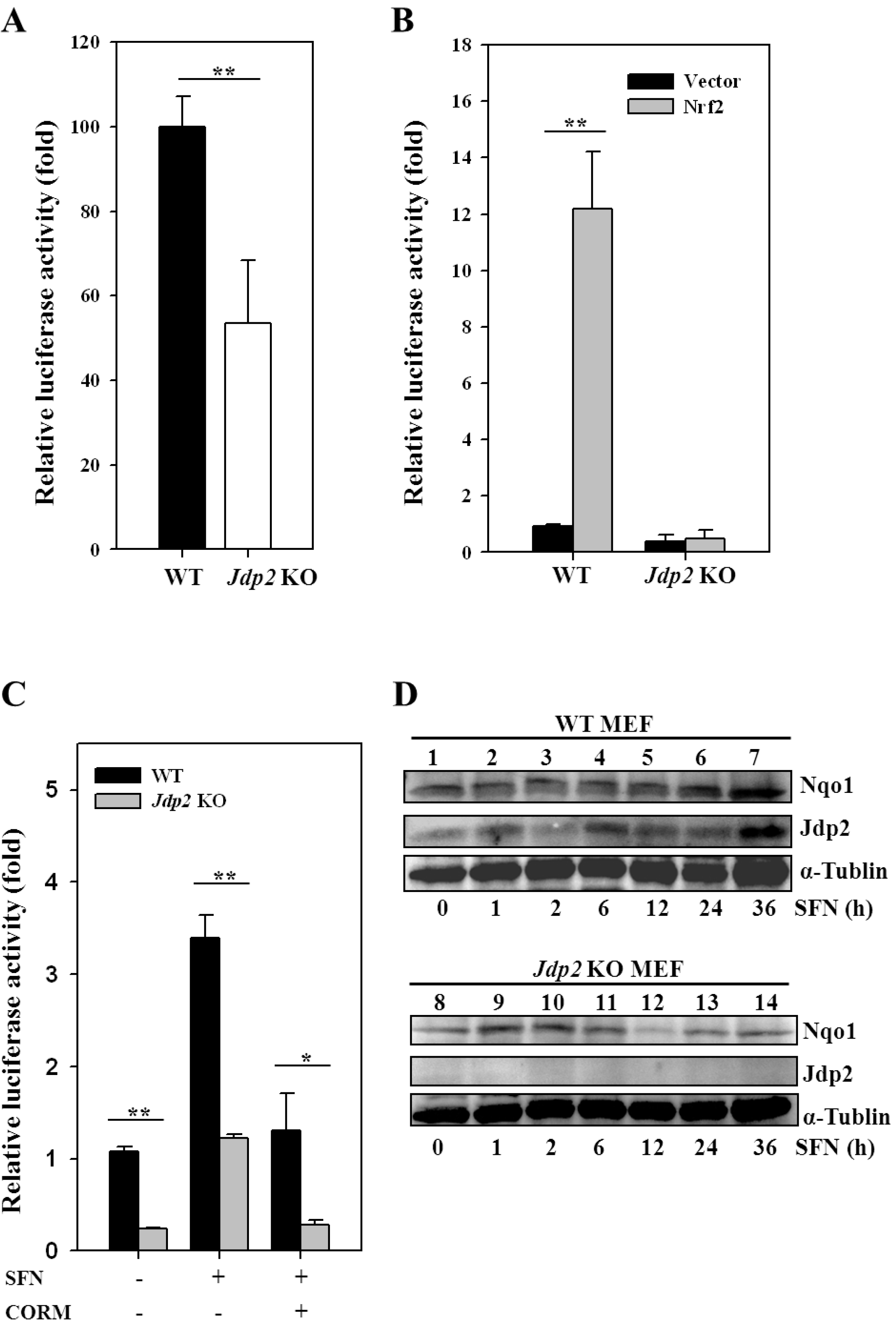
3.2. JDP2 Is Involved in ARE Transactivation
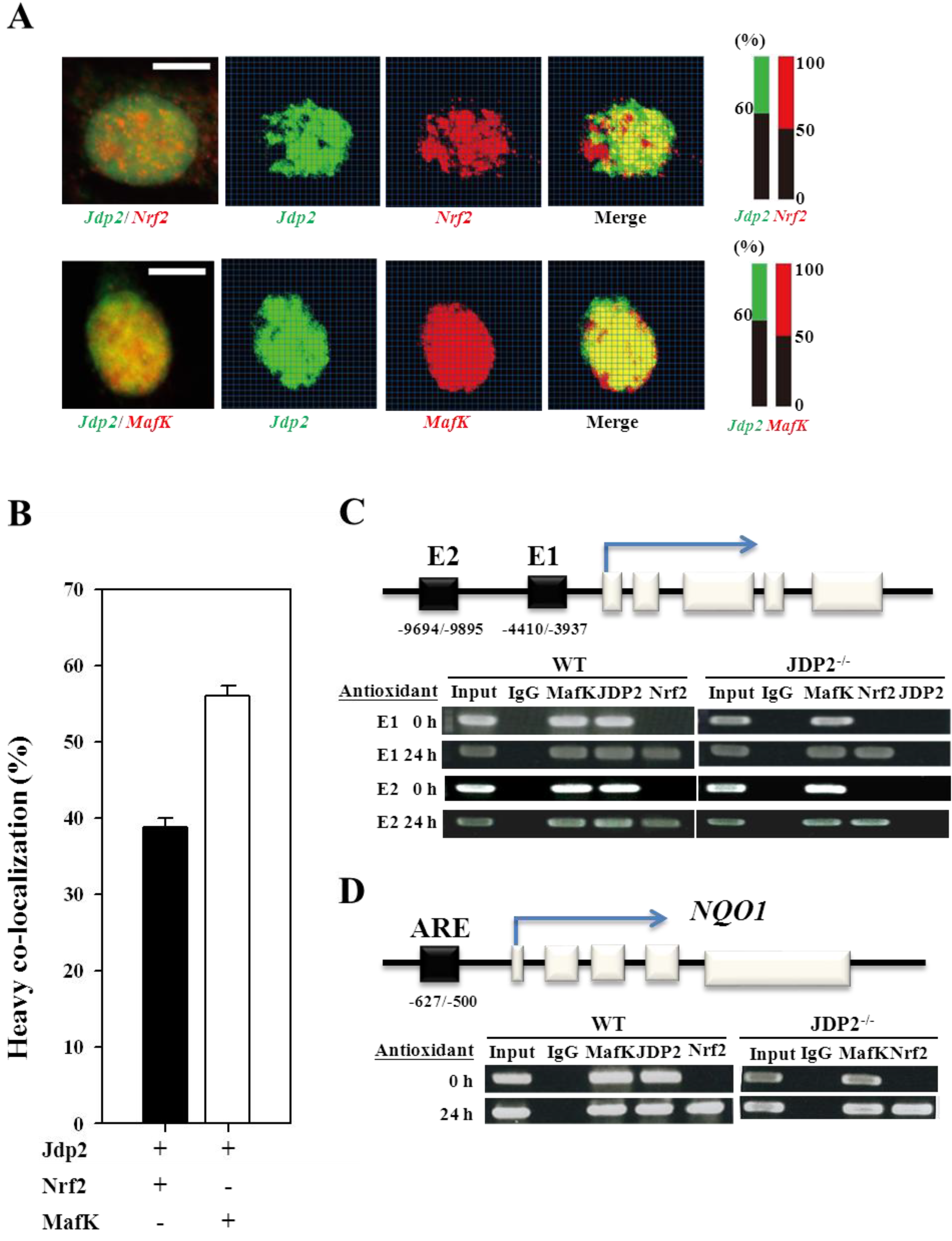
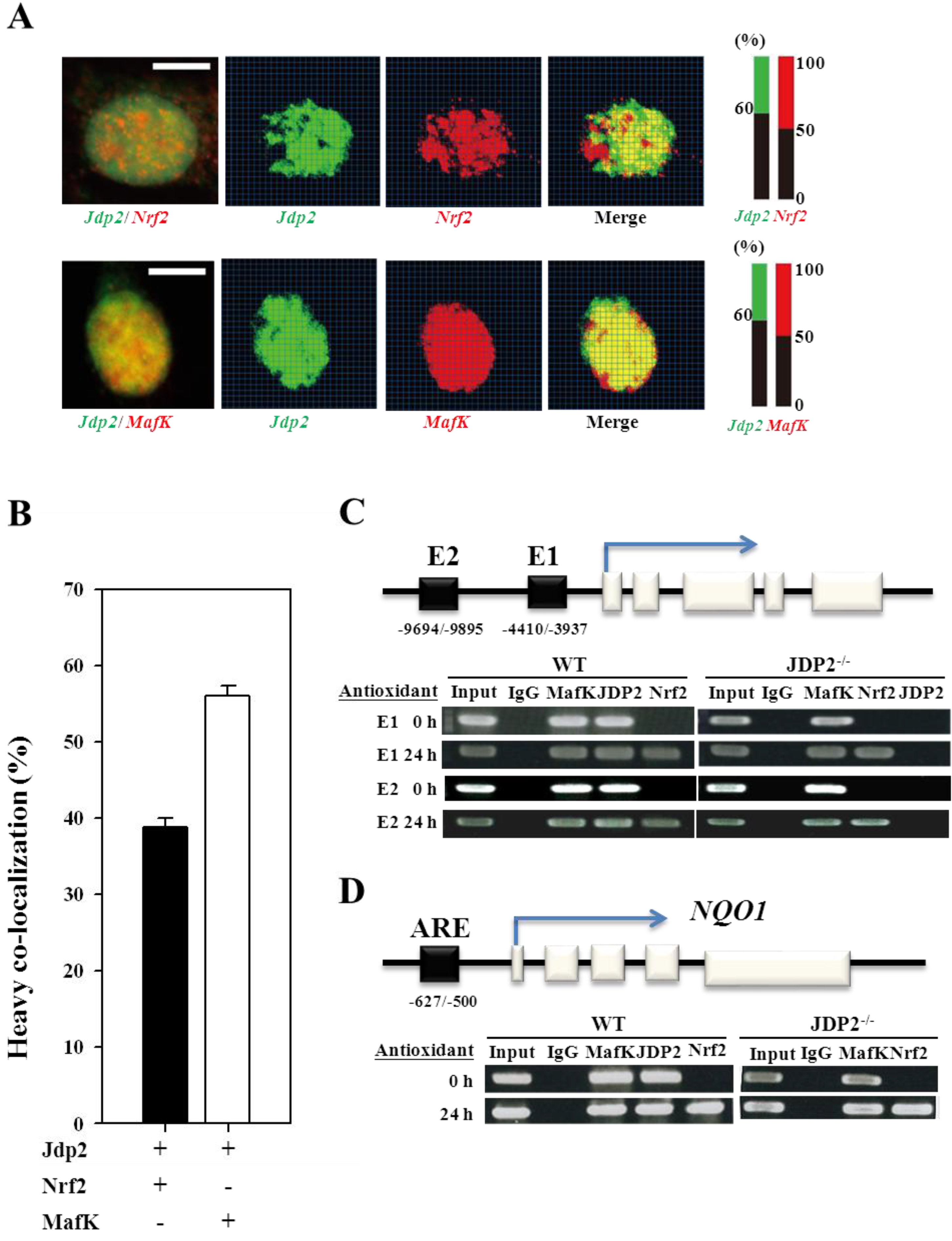
3.3. Colocalization and Recruitment of Nrf2, MafK, and JDP2
3.4. Nrf2, MafK, and JDP2 Proteins Are Recruited to the ARE
3.5. Expression of Jdp2 Is Restricted in the Cerebellum
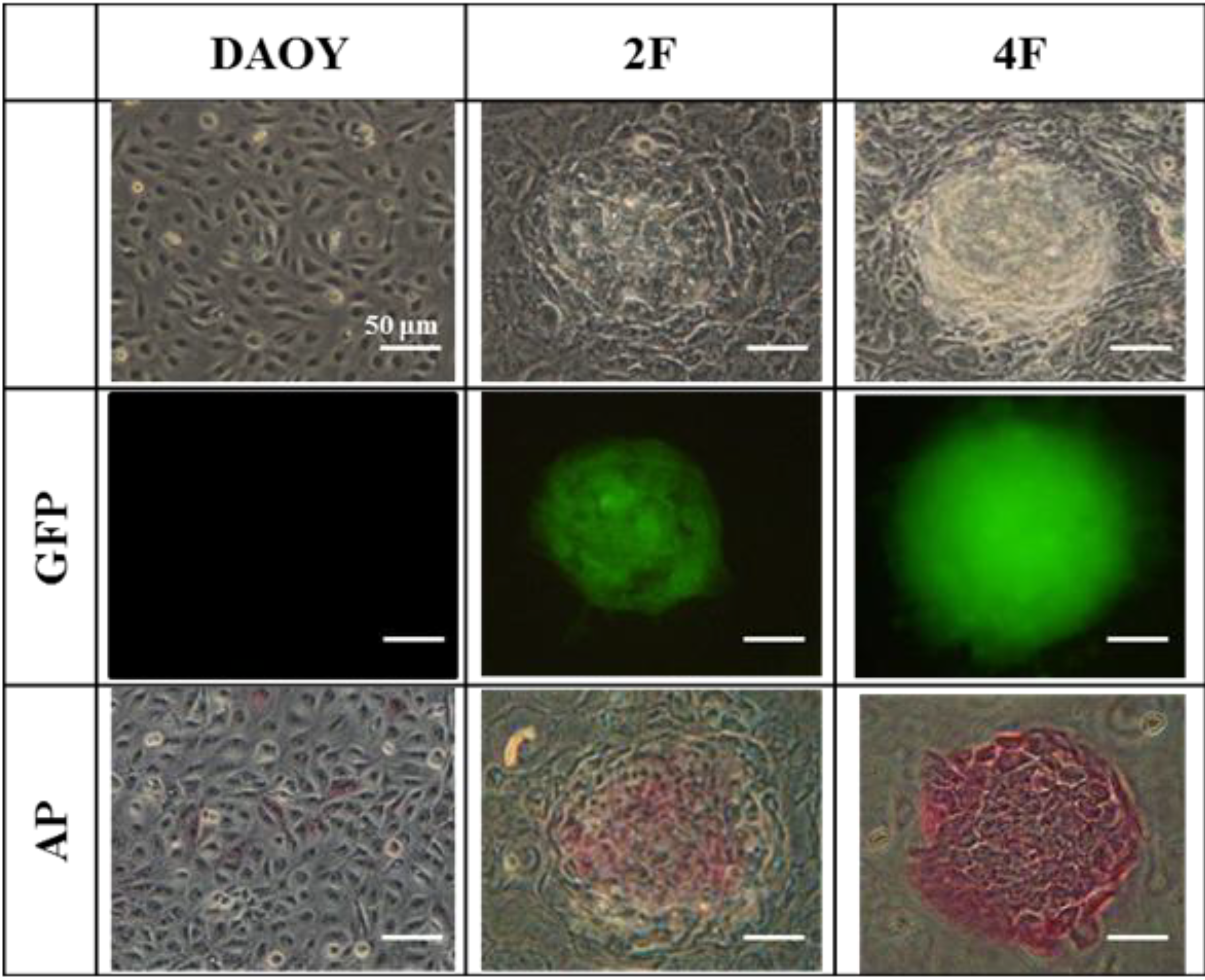
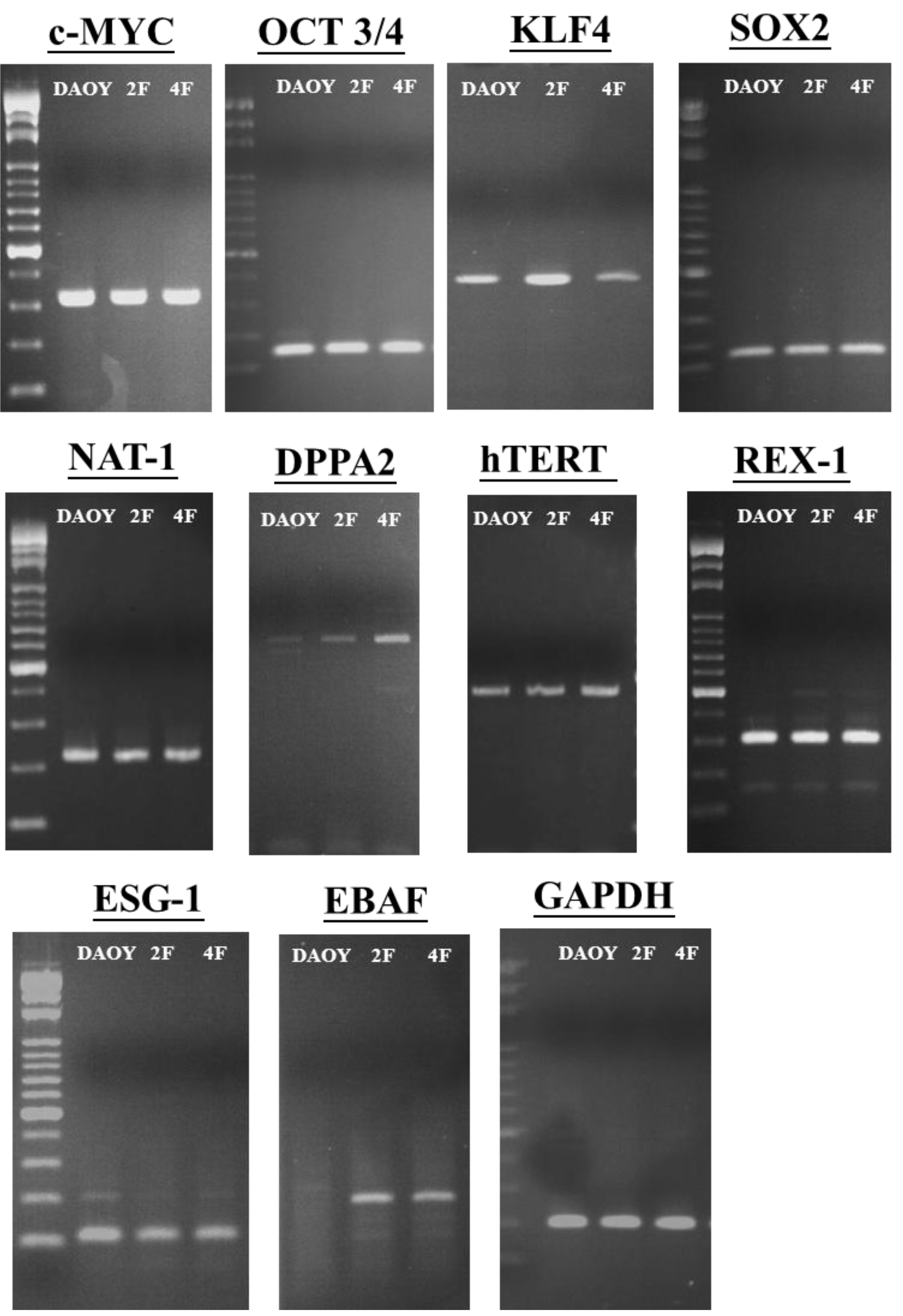
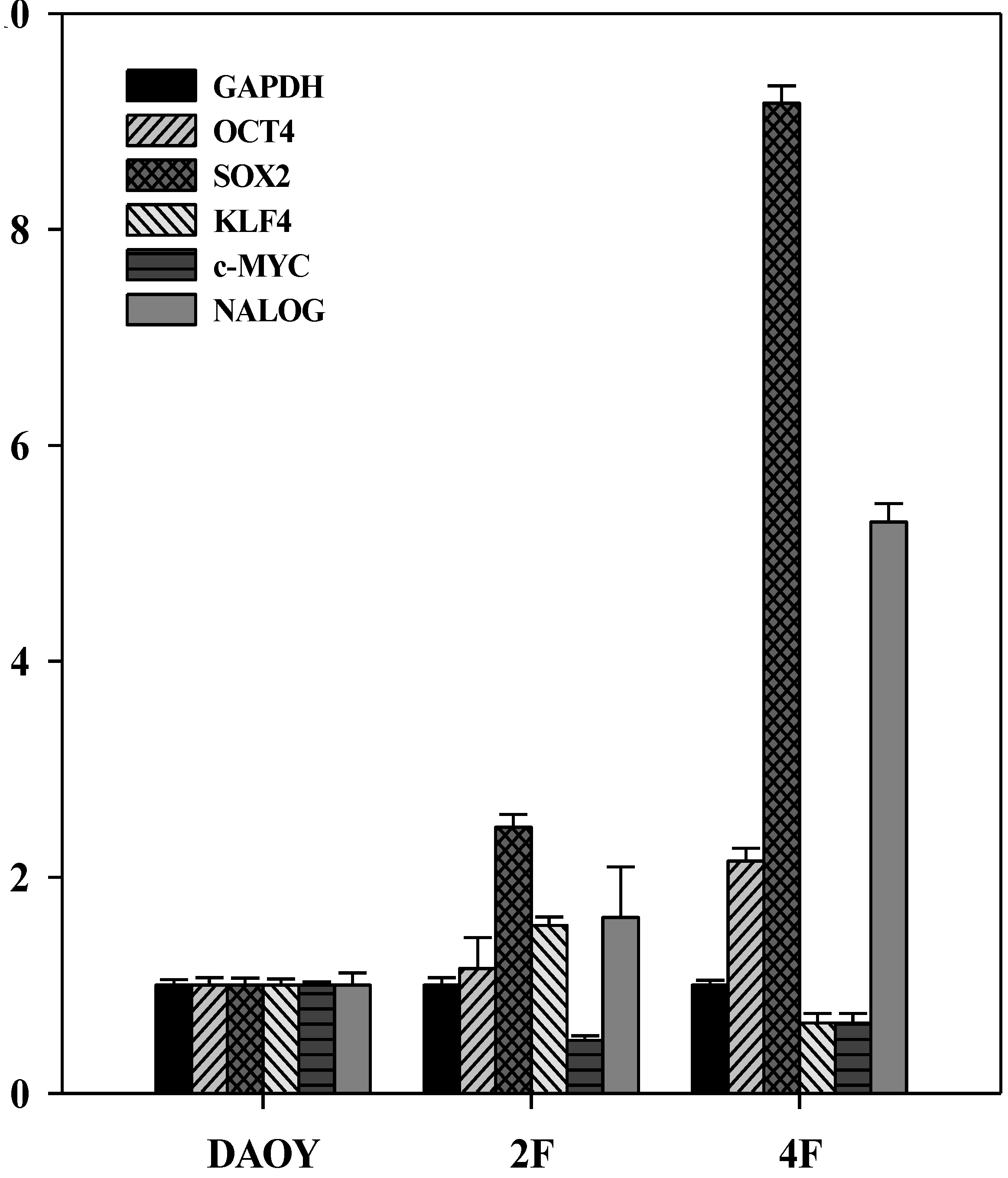
3.6. JDP2 and OCT4 Cause Nuclear Reprogramming of Medduloblastoma Cells
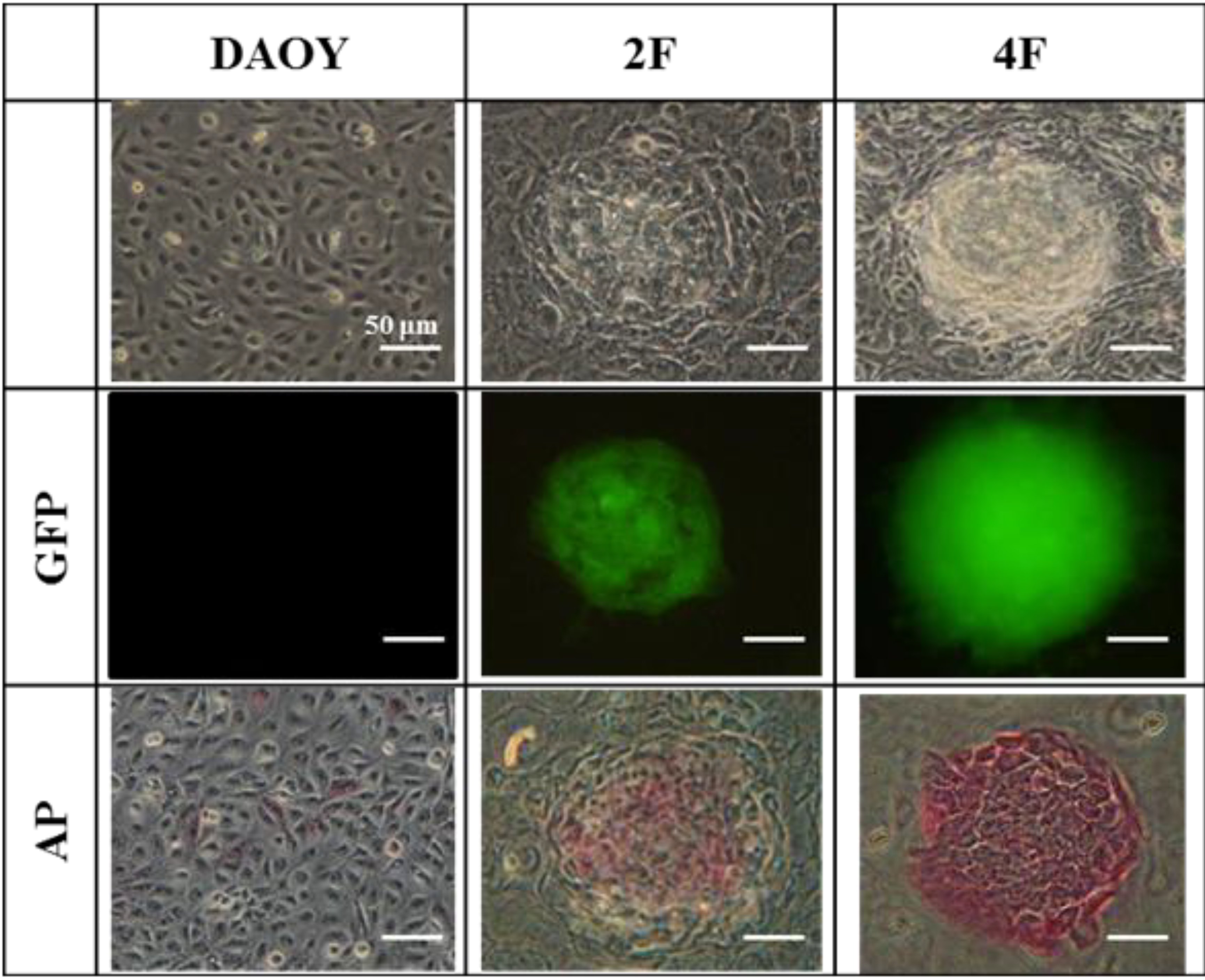
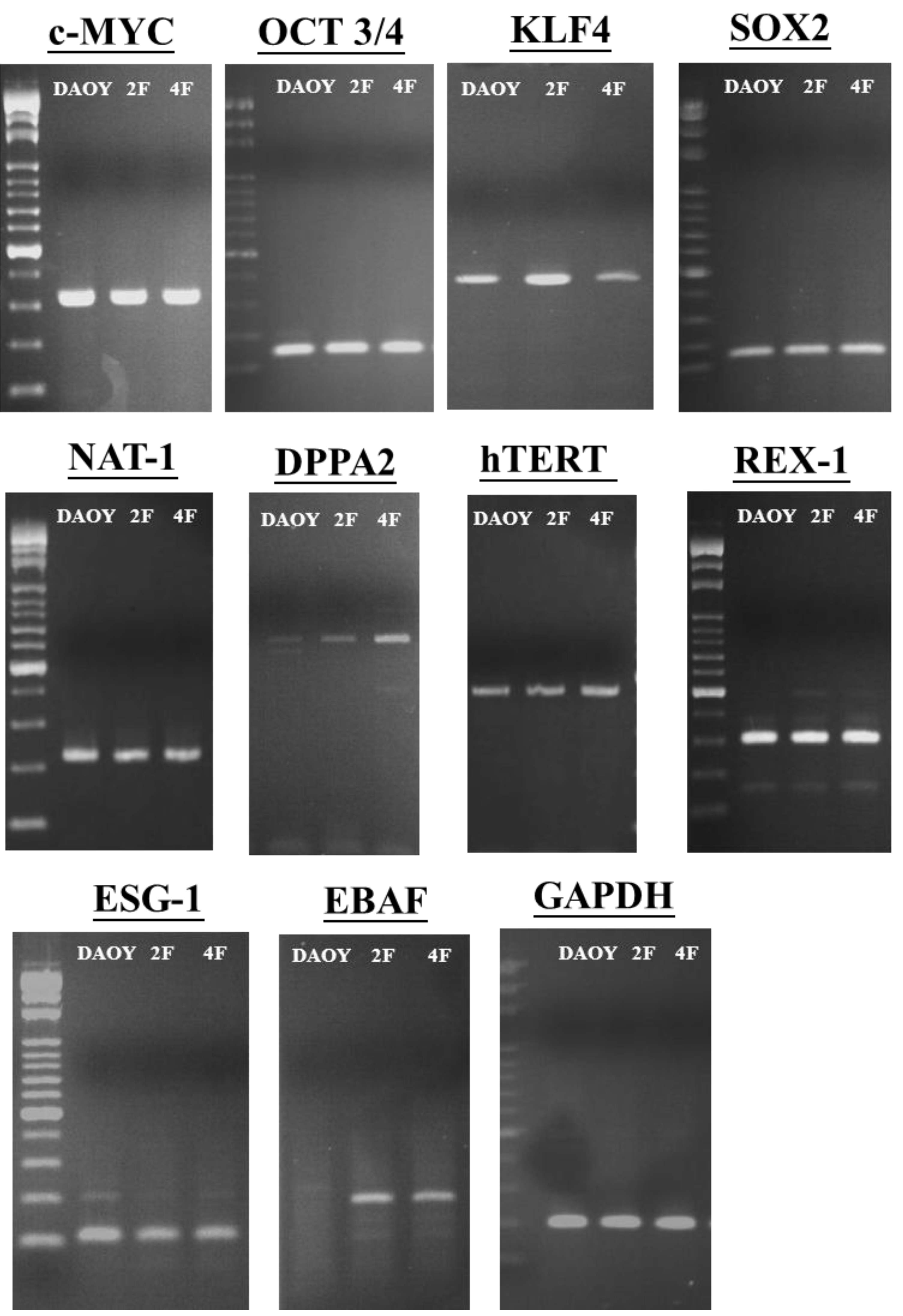
3.7. iPSCs Exhibit Pluripotency
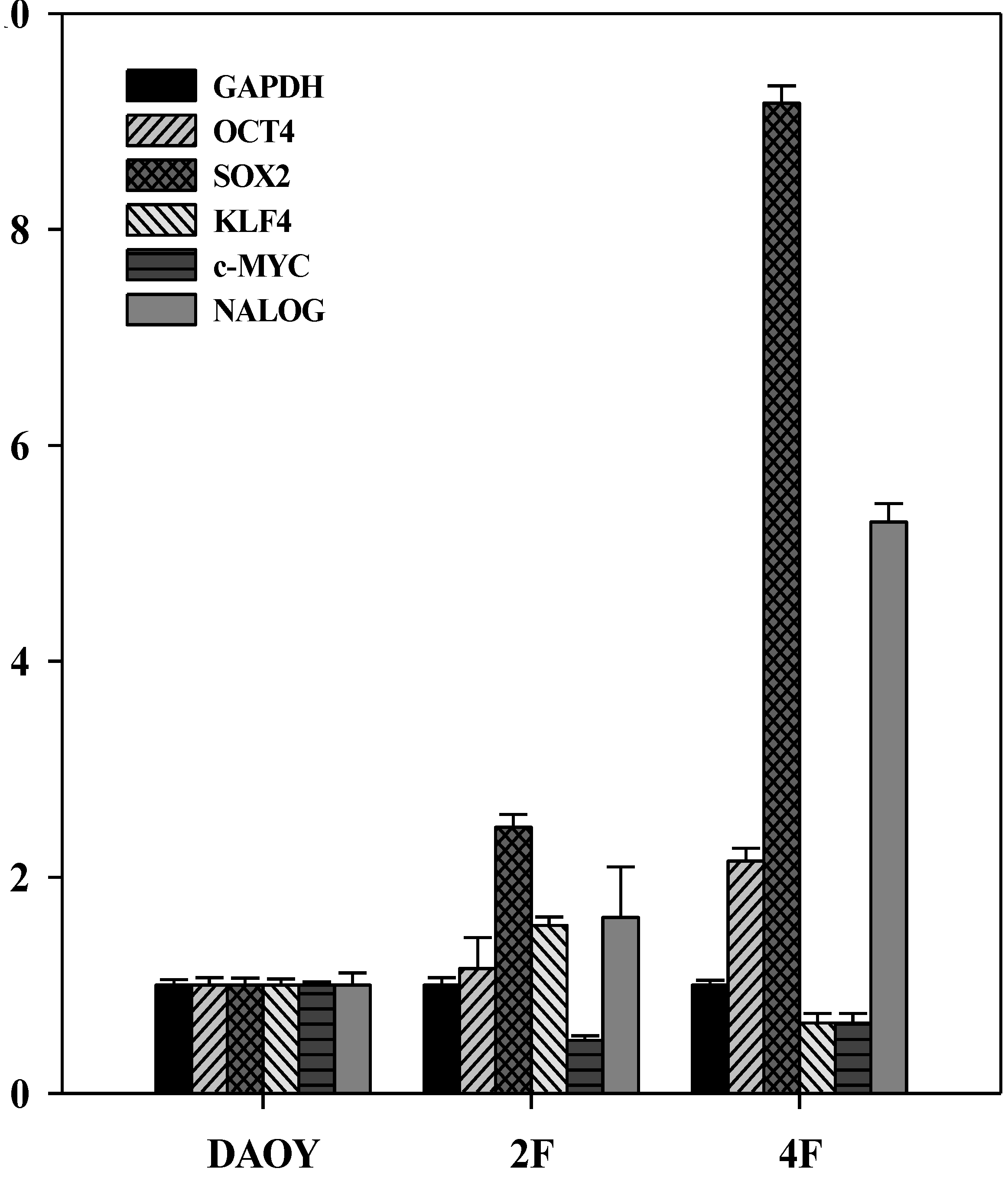
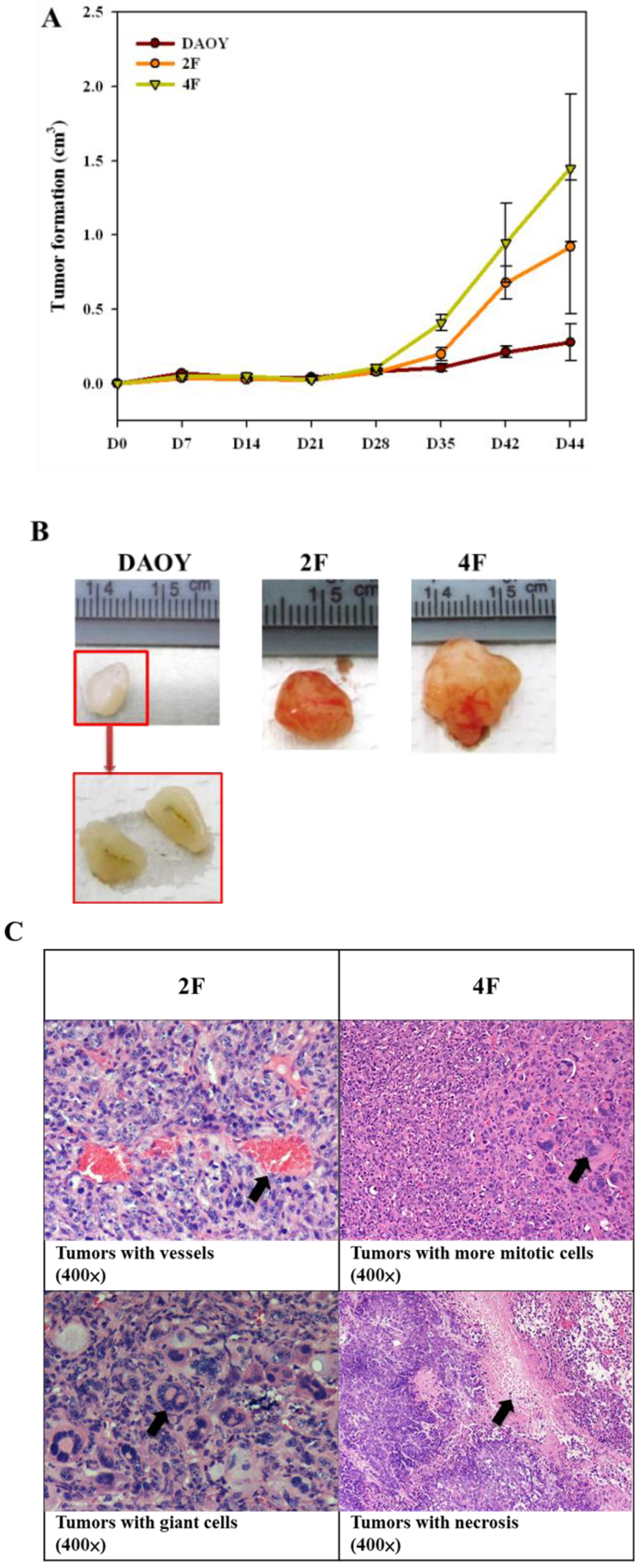
3.8. Tumor Formation
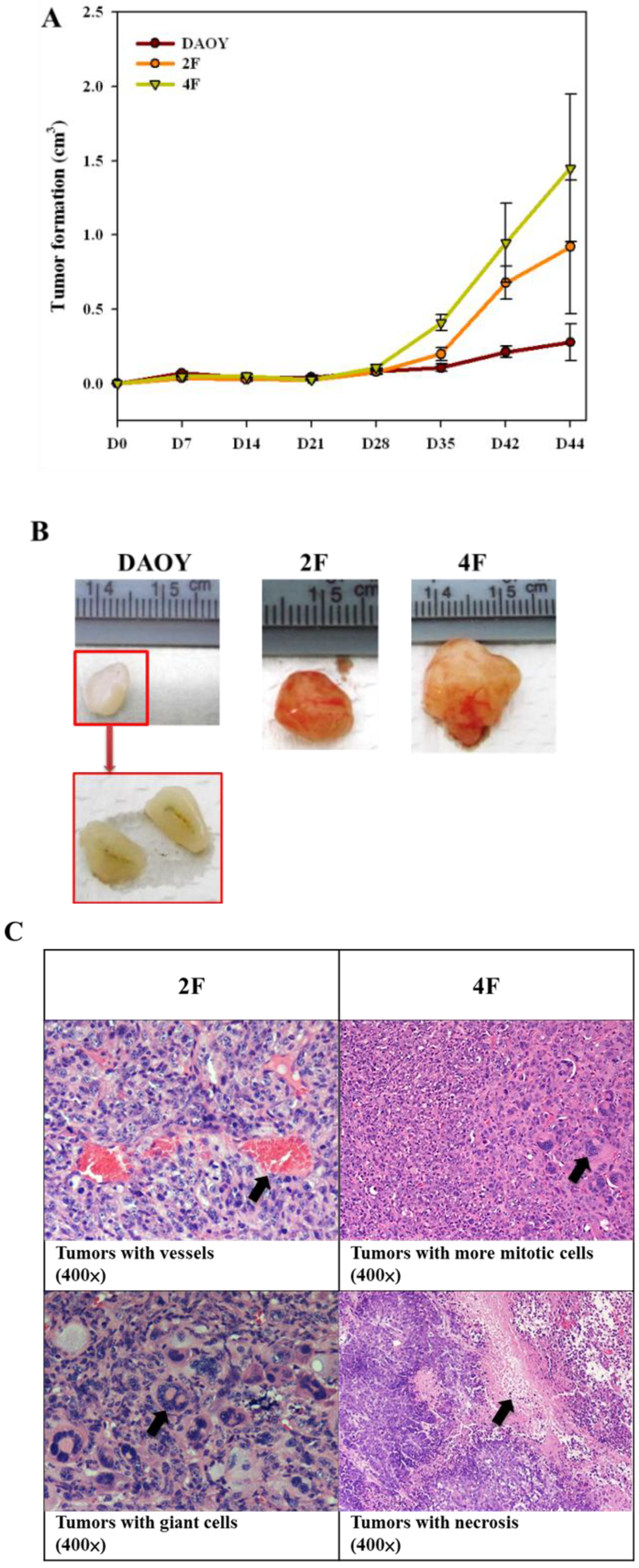
4. Discussion
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Giudice, A.; Montella, M. Activation of the Nrf2-ARE signaling pathway: A promising strategy in cancer prevention. Bioessays 2006, 28, 169–181. [Google Scholar] [CrossRef]
- Khor, T.O.; Huang, M.T.; Kwon, K.H.; Chan, J.Y.; Reddy, B.S.; Kong, A.N. Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium-induced colitis. Cancer Res. 2006, 66, 11580–11584. [Google Scholar]
- Hou, D.X.; Fukuda, M.; Johnson, J.A.; Miyamori, K.; Ushikai, M.; Fujii, M. Fisetin induces transcription of NADPH: Quinone oxidoreductase gene through an antioxidant responsive element-involved activation. Int. J. Oncol. 2001, 18, 1175–1179. [Google Scholar]
- Kaspar, J.W.; Niture, S.K.; Jaiswal, A.K. Nrf2: INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 2009, 47, 1304–1309. [Google Scholar] [CrossRef]
- Rushmore, T.H.; King, R.G.; Paulson, K.E.; Pickett, C.B. Regulation of glutathione S-transferase Ya subunit gene expression: Identification of a unique xenobiotic-responsive element controlling inducible expression by planar aromatic compounds. Proc. Natl. Acad. Sci. USA 1990, 87, 3826–3830. [Google Scholar] [CrossRef]
- Inamdar, N.M.; Ahn, Y.I.; Alam, J. The heme-responsive element of the mouse heme oxygenase-1gene is an extended AP-1 binding site that resembles the recognition sequences for MAF and NF-E2 transcription factors. Biochem. Biophys. Res. Commun. 1996, 221, 570–576. [Google Scholar] [CrossRef]
- Cheung, H.-H.; Liu, X.; Rennert, O.M. Apoptosis: Reprogramming and the fate of mature cells. ISRN Cell Biol. 2012. ID 685852. [Google Scholar]
- Katsuoka, F.; Motohashi, H.; Ishii, T.; Aburatani, H.; Engel, J.D.; Yamamoto, M. Genetic evidence that small maf proteins are essential for the activation of antioxidant response element-dependent genes. Mol. Cell. Biol. 2005, 25, 8044–8051. [Google Scholar] [CrossRef]
- Aronheim, A.; Zandi, E.; Hennemann, H.; Elledge, S.; Karin, M. Isolation of an AP-1 repressor by a novel method for detecting protein-protein interactions. Mol. Cell. Biol. 1997, 17, 3094–3102. [Google Scholar]
- Jin, C.; Ugai, H.; Song, J.; Murata, T.; Nili, F.; Sun, K.; Horikoshi, M.; Yokoyama, K.K. Identification of mouse Jun dimerization protein 2 as a novel repressor of ATF-2. FEBS Lett. 2001, 489, 34–41. [Google Scholar] [CrossRef]
- Pan, J.; Nakade, K.; Huang, Y.C.; Zhu, Z.W.; Masuzaki, S.; Hasegawa, H.; Murata, T; Yoshiki, A.; Yamaguchi, N.; Lee, C.H.; et al. Suppression of cell-cycle progression by Jun dimerization proten-2 (JDP2) involves downregulation of cycling-A2. Oncogene 2010, 29, 6245–6256. [Google Scholar] [CrossRef]
- Weidenfeld-Baranboim, K.; Bitton-Worms, K.; Aronheim, A. TRE-dependent transcription activation by JDP2-CHOP10 association. Nucleic Acids Res. 2008, 36, 3608–3619. [Google Scholar] [CrossRef]
- Jin, C.; Li, H.; Murata, T.; Sun, K.; Horikoshi, M.; Chiu, R.; Yokoyama, K.K. JDP2, a repressor of AP-1, recruits a histone deacetylase 3 complex to inhibit the retinoic acid-induced differentiation of F9 cells. Mol. Cell. Biol. 2002, 22, 4815–4826. [Google Scholar] [CrossRef]
- Jin, C.; Kato, K.; Chimura, T.; Yamasaki, T.; Nakade, K.; Murata, T.; Li, H.; Pan, J.; Zhao, M.; Sun, K.; et al. Regulation of histone acetylation and nucleosome assembly by transcription factor JDP2. Nat. Struct. Mol. Biol. 2006, 13, 331–338. [Google Scholar] [CrossRef]
- Katz, S.; Heinrich, R.; Aronheim, A. The AP-1 repressor, JDP2, is a bona fide substrate for the c-Jun N-terminal kinase. FEBS Lett. 2001, 506, 196–200. [Google Scholar] [CrossRef]
- Nakade, K.; Pan, J.; Yoshiki, A.; Ugai, H.; Kimura, M.; Liu, B.; Li, H.; Obata, Y.; Iwama, M.; Itohara, S.; et al. JDP2 suppresses adipocyte differentiation by regulating histone acetylation. Cell Death Differ. 2007, 14, 1398–1405. [Google Scholar] [CrossRef]
- Nakade, K.; Pan, J.; Yamasaki, T.; Murata, T.; Wasylyk, B.; Yokoyama, K.K. JDP2 (Jun dimerization protein 2)-deficient mouse embryonic fibroblasts are resistant to replicative senescence. J. Biol. Chem. 2009, 284, 10808–10817. [Google Scholar]
- Rasmussen, M.H.; Wang, B.; Wable, M.; Nielsen, A.L.; Pedersen, F.S. Activation of alternative Jdp2 promoters and functional protein isoforms in T-cell lymphomas by retroviral insertion mutagenesis. Nucleic Acids Res. 2009, 37, 4657–4671. [Google Scholar] [CrossRef]
- Heinrich, R.; Livne, E.; Ben-Izhak, O.; Aronheim, A. The c-Jun dimerization protein 2 inhibits cell transformation and acts as a tumor suppressor gene. J. Biol. Chem. 2004, 279, 5708–5715. [Google Scholar]
- Yuanhong, X.; Feng, X.; Qingchang, L.; Jianpeng, F.; Zhe, L.; Kejian, G. Downregulation of AP-1 repressor JDP2 is associated with tumor metastasis and poor prognosis in patients with pancreatic carcinoma. Int. J. Biol. Markers 2010, 25, 136–140. [Google Scholar]
- Van der Weyden, L.; Rust, A.G.; McIntyre, R.E.; Robles-Espinoza, C.D.; del Castillo Velasco-Herrera, M.; Strogantsev, R.; Ferguson-Smith, A.C.; McCarthy, S.; Keane, T.M.; et al. Jdp2 downregulates Trp53 transcription to promote leukaemogenesis in the context of Trp53 heterozygosity. Oncogene 2013, 32, 397–402. [Google Scholar] [CrossRef]
- Bitton-Worms, K.; Pikarsky, E.; Aronheim, A. The AP-1 repressor protein, JDP2, potentiates hepatocellular carcinoma in mice. Mol. Cancer 2010, 9, 54. [Google Scholar] [CrossRef]
- Motohashi, H.; O’Connor, T.; Katsuoka, F.; Engel, J.D.; Yamamoto, M. Integration and diversity of the regulatory network composed of Maf and CNC families of transcription factors. Gene 2002, 294, 1–12. [Google Scholar] [CrossRef]
- Newman, J.R.; Keating, A.E. Comparative identification of human bZIP interactions with coiled-coil arrays. Science 2003, 300, 2097–2101. [Google Scholar] [CrossRef]
- Blank, V. Small Maf Proteins in Mammalian Gene Control: Mere Dimerization Partners or Dynamic Transcriptional Regulators? J. Mol. Biol. 2008, 376, 913–925. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 26, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Stadtfeld, M.; Hochedlinger, K. Induced pluripotency: History, mechanisms, and applications. Genes Dev. 2010, 24, 2239–2263. [Google Scholar] [CrossRef]
- Galach, M.; Utikal, J. From skin to the treatment of diseases—The possibilities of iPS cell research in dermatology. Exp. Dermatol. 2011, 20, 523–528. [Google Scholar] [CrossRef]
- Utikal, J.; Maherali, N.; Kulalert, W.; Hochedlinger, K. Sox2 is dispensable for the reprogramming of melanocytes and melanoma cells into induced pluripotent stem cells. J. Cell Sci. 2009, 122, 3502–3510. [Google Scholar] [CrossRef]
- Hu, K.; Yu, J.; Suknuntha, K.; Tian, S.; Montgomery, K.; Choi, K.D.; Stewart, R.; Thomson, J.A.; Sulkvin, I.I. Efficient generation of transgene-free induced pluripotent stem cells from normal and neoplastic bone marrow and cord blood mononuclear cells. Blood 2011, 117, e109–e119. [Google Scholar] [CrossRef]
- Carette, J.E.; Pruszak, J.; Varadarajan, M.; Blomen, V.A.; Gokhale, S.; Camargo, F.D.; Wernig, M.; Jaenisch, R.; Brummelkamp, T.R. Generation of iPSCs from cultured human malignant cells. Blood 2010, 115, 4039–4042. [Google Scholar] [CrossRef]
- Miyoshi, N.; Ishii, H.; Nagai, K.-I.; Hoshino, H.; Mimori, K.; Tanaka, F.; Nagano, H.; Sekimoto, M.; Doki, Y.; Mori, M. Defined factors induce reprogramming of gastrointestinal cancer cells. Proc. Natl. Acad. Sci. USA 2010, 107, 40–45. [Google Scholar] [CrossRef]
- Bernhardt, M.; Galach, M.; Novak, D.; Utikal, J. Mediators of induced pluripotency and their role in cancer cells—Current scientific knowledge and future perspectives. Biotechnol. J. 2012, 7, 810–821. [Google Scholar] [CrossRef]
- Li, H.; Collado, M.; Villasante, A.; Strati, K.; Ortega, S.; Cañamero, M.; Blasco, M.A.; Serrano, M. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature 2009, 460, 1136–1139. [Google Scholar] [CrossRef]
- Yoshida, Y.; Takahashi, K.; Okita, K.; Ichisaka, T.; Yamanaka, S. Hypoxia enhances the generation of induced pluripotent stem cells. Cell Stem Cell 2009, 5, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Mah, N.; Wang, Y.; Liao, M.C.; Prigione, A.; Jozefczuk, J.; Lichtner, B.; Wolfrum, K.; Haltmeier, M.; Flöttmann, M.; Schaefer, M.; et al. Molecular insights into reprogramming-initiation events mediated by the OSKM gene regulatory network. PLoS One 2011, 6, e24351. [Google Scholar] [CrossRef]
- Miyata, H.; Ikawa, E.; Ohama, E. Medulloblastoma in an adult suggestive of external granule cells as its origin: A histological and immunohistochemical study. Brain Tumor Pathol. 1998, 15, 31–35. [Google Scholar] [CrossRef]
- Eriksson, J.W. Metabolic stress in insulin’s target cells leads to ROS accumulation-a hypothetical common pathway causing insulin resistance. FEBS Lett. 2007, 581, 3734–3742. [Google Scholar] [CrossRef]
- Winterbourn, C.C.; Garcia, R.C.; Segal, A.W. Production of the superoxide adduct of myeloperoxidase (compound III) by stimulated human neutrophils and its reactivity with hydrogen peroxide and chloride. Biochem. J. 1985, 228, 583–592. [Google Scholar]
- Zhu, P.; Oe, T.; Blair, I.-A. Determination of cellular redox status by stable isotope dilution liquid chromatography/mass spectrometry analysis of glutathione and glutathione disulfide. Rapid Commun. Mass Spectrom 2008, 22, 432–440. [Google Scholar] [CrossRef]
- Kotake, K.; Cao, R.; Viatour, P.; Sage, J.; Zhang, Y.; Xiong, Y. pRB family proteins are required for H3K27 trimethylation and Polycomb repression complexes binding to and silencing p16INK4alpha tumor suppressor gene. Genes Dev. 2007, 21, 49–54. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.; Sherratt, P.J.; Nioi, P.; Yang, C.S.; Pickett, C.B. Nrf2 controls constitutive and inducible expression of ARE-driven genes through a dynamic pathway involving nucleocytoplasmic shuttling by Keap1. J. Biol. Chem. 2005, 280, 32485–32492. [Google Scholar] [CrossRef]
- Juge, N.; Mithen, R.F.; Traka, M. Molecular basis for chemoprevention by sulforaphane: A comprehensive review. Cell. Mol. Life Sci. 2007, 64, 1105–1127. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Sun, J.; Brand, M.; Zenke, Y.; Tashiro, S.; Goudline, M.; Igarashi, K. Heme regulates the dynamic exchange of bach1 and NF-E2-related factors in the Maf transcription factor network. Proc. Natl. Acad. Sci. USA. 2004, 101, 1261–1466. [Google Scholar]
- Kataoka, K.; Nod, M.; Nishizawa, M. Maf nuclear oncoprotein recognizes sequences related to an AP-1 site and forms heterodimers with both Fos and Jun. Mol. Cell. Biol. 1994, 14, 700–712. [Google Scholar]
- Soriano, P. Generalized LacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 1999, 21, 70–71. [Google Scholar] [CrossRef]
- Novak, A.; Guo, C.; Yang, W.; Nagy, A.; Lobe, C.G. Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genetics 2000, 28, 147–155. [Google Scholar]
- Plath, K.; Lowry, W.E. Progress in understanding reprogramming to the induced pluripotent state. Nat. Rev. Genet. 2009, 5, 237–241. [Google Scholar]
- Kim, J.B.; Sebastiano, V.; Wu, G.; Arauzo-Gravo, M.J.; Sasse, P.; Gentile, L.; Ko, K.; Ruau, D.; Ehrlich, M.; van den Zaehres, H.; et al. Oct4-induced pluripotency in adult neural stem cells. Cell 2009, 136, 411–419. [Google Scholar]
- Lensch, M.W.; Ince, T.S. The terminology of teratocarcinomas and teratomas. Nat. Biotechnol. 2007, 25, 1212. [Google Scholar]
- Banito, A.; Rashid, S.T.; Acosta, J.C.; Li, S.; Pereira, C.F.; Geti, I.; Pinho, S.; Silva, J.C.; Azuara, V.; Walsh, M.; et al. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 2009, 23, 2134–2139. [Google Scholar] [CrossRef]
- Collado, M.; Blasco, M.A.; Serrano, M. Cellular senescence in cancer and aging. Cell 2009, 130, 223–233. [Google Scholar] [CrossRef]
- Mazumdar, J.; O’Brien, W.T.; Johnson, R.S.; LaManna, J.C.; Chavez, J.C.; Klein, P.S.; Simon, M.C. O2 regulates stem cells through Wnt/β-catenin signaling. Nat. Cell Biol. 2010, 12, 1007–1013. [Google Scholar] [CrossRef]
- Martello, G.; Sugimoto, T.; Diamanti, E.; Joshi, A.; Hannah, R.; Ohtsuka, S.; Göttgens, B.; Niwa, H.; Smith, A. Esrrb is a pivotal target of the Gsk3/Tcf3 axis regulating embryonic stem cell self-renewal. Cell Stem Cell 2012, 11, 491–504. [Google Scholar] [CrossRef]
- Pardo, M.; Lang, B.; Yu, L.; Prosser, H.; Bradley, A.; Babu, M.M.; Choudhary, J. An expanded Oct4 interaction network: implications for stem cell biology, development, and disease. Cell Stem Cell 2010, 6, 382–395. [Google Scholar] [CrossRef]
- Nagamatsu, G.; Saito, S.; Kosaka, T.; Takubo, K.; Kinoshita, T.; Oya, M.; Horimoto, K.; Suda, T. Optimal ratio of transcription factors for somatic cell reprogramming. J. Biol. Chem. 2012, 287, 36273–36282. [Google Scholar]
- Papp, B.; Plath, K. Epigenetics of reprogramming to induced pluripotency. Cell 2013, 152, 1324–1343. [Google Scholar] [CrossRef]
- Silva, J.; Nichols, J.; Theunissen, T.W.; Guo, G.; van Oosten, A.L.; Barrandon, O.; Wray, J.; Yamanaka, S.; Chambers, I.; Smith, A. Nanog is the gateway to the pluripotent ground state. Cell 2009, 138, 722–737. [Google Scholar] [CrossRef]
- Choi, S.-C.; Choi, J.-H.; Park, C.-Y.; Ahn, C.-M.; Hong, S.-J.; Lim, D.-S. Nanog regulates molecules involved in stemness and cell cycle-signaling pathway for maintenance of pluripotency of P19 embryonal carcinoma stem cells. J. Cell Physiol. 2012, 227, 3678–3693. [Google Scholar] [CrossRef]
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef]
Supplementary Files
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chiou, S.-S.; Wang, S.S.-W.; Wu, D.-C.; Lin, Y.-C.; Kao, L.-P.; Kuo, K.-K.; Wu, C.-C.; Chai, C.-Y.; Lin, C.-L.S.; Lee, C.-Y.; et al. Control of Oxidative Stress and Generation of Induced Pluripotent Stem Cell-like Cells by Jun Dimerization Protein 2. Cancers 2013, 5, 959-984. https://doi.org/10.3390/cancers5030959
Chiou S-S, Wang SS-W, Wu D-C, Lin Y-C, Kao L-P, Kuo K-K, Wu C-C, Chai C-Y, Lin C-LS, Lee C-Y, et al. Control of Oxidative Stress and Generation of Induced Pluripotent Stem Cell-like Cells by Jun Dimerization Protein 2. Cancers. 2013; 5(3):959-984. https://doi.org/10.3390/cancers5030959
Chicago/Turabian StyleChiou, Shyh-Shin, Sophie Sheng-Wen Wang, Deng-Chyang Wu, Ying-Chu Lin, Li-Pin Kao, Kung-Kai Kuo, Chun-Chieh Wu, Chee-Yin Chai, Cheng-Lung Steve Lin, Cheng-Yi Lee, and et al. 2013. "Control of Oxidative Stress and Generation of Induced Pluripotent Stem Cell-like Cells by Jun Dimerization Protein 2" Cancers 5, no. 3: 959-984. https://doi.org/10.3390/cancers5030959