HEXIM1, a New Player in the p53 Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
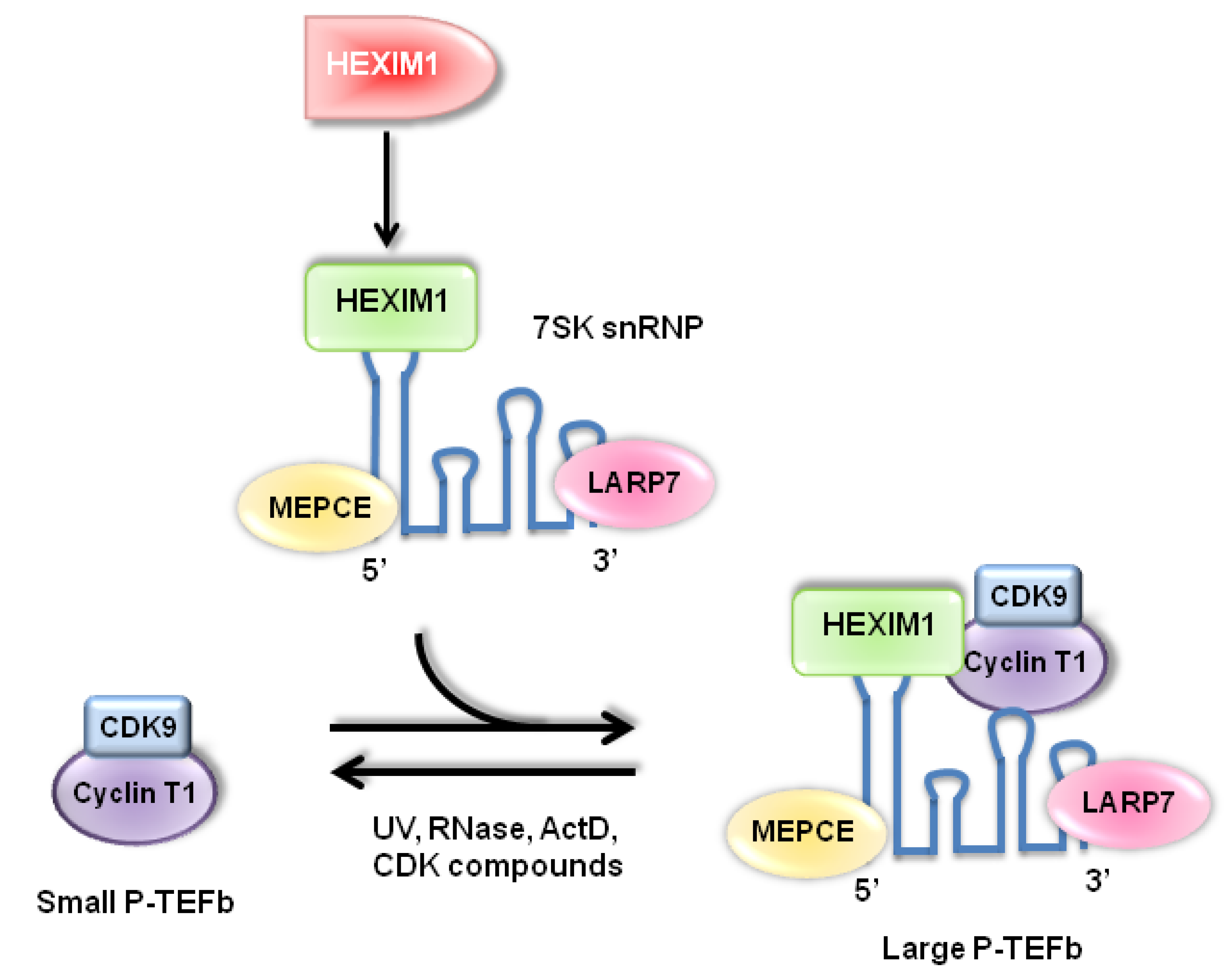
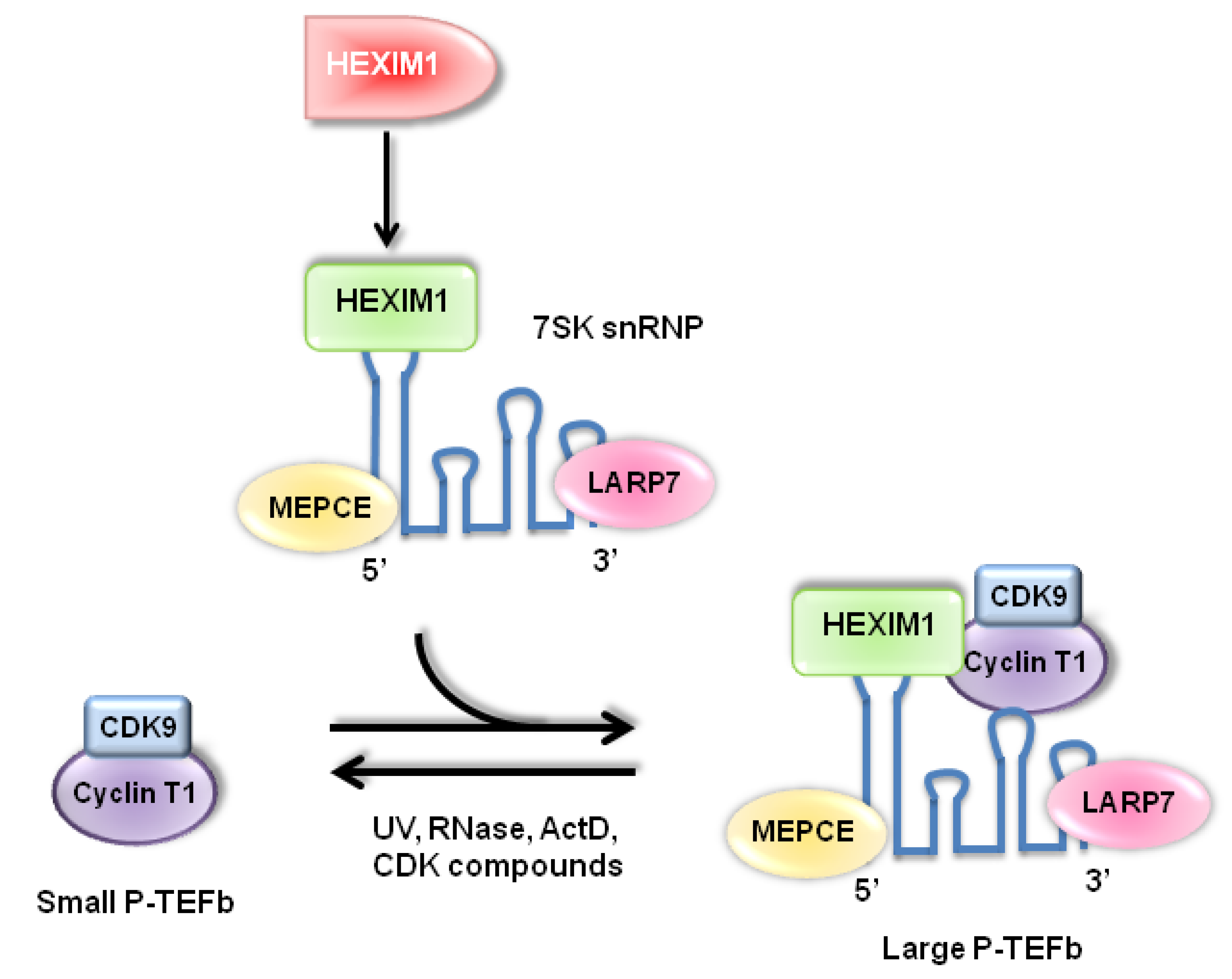
:1. Introduction
2. p53 and Its Regulators, HDM2 and NPM
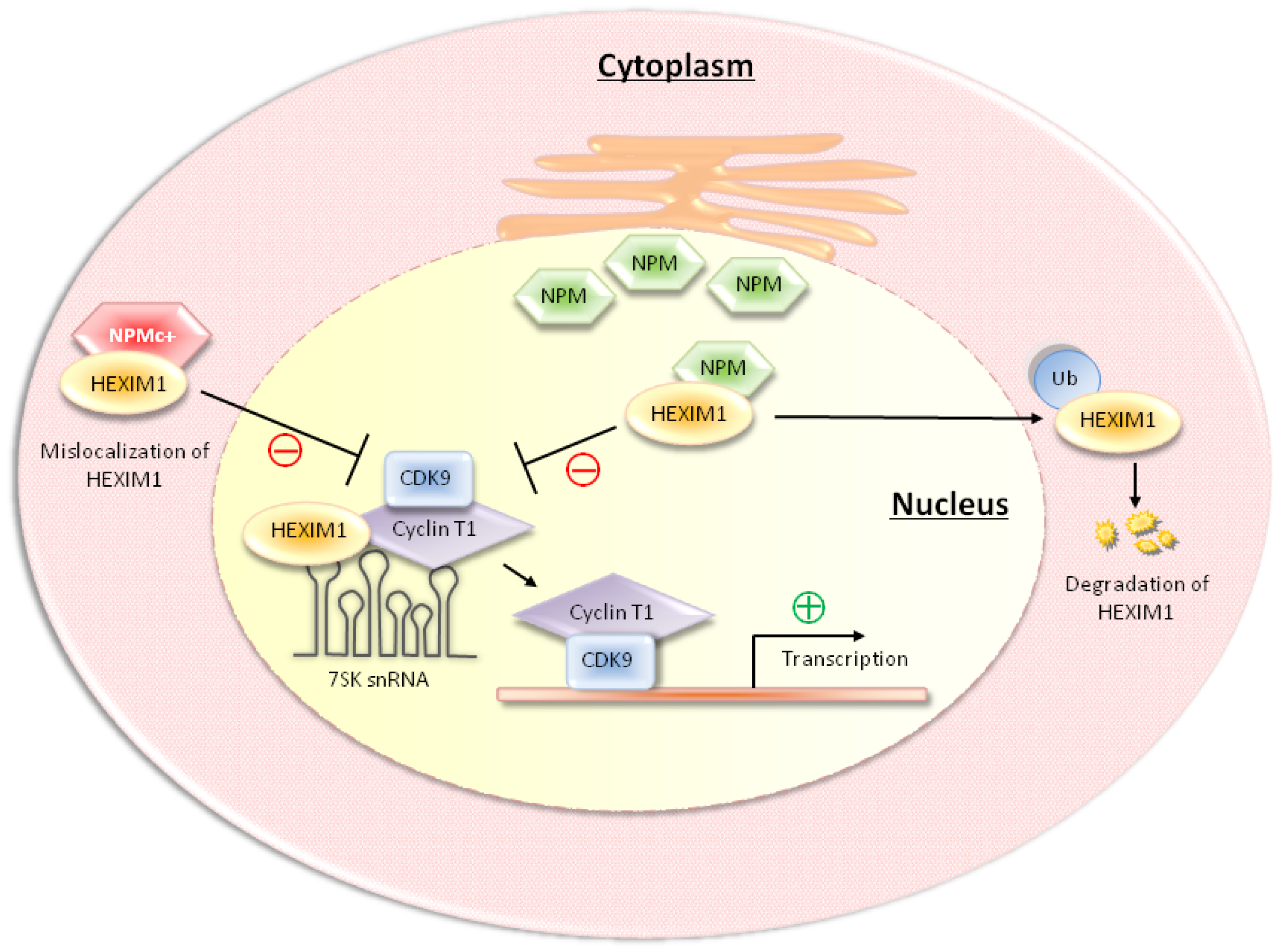
3. NPM and NPMc+ Regulate P-TEFb Activity through the Interaction with HEXIM1
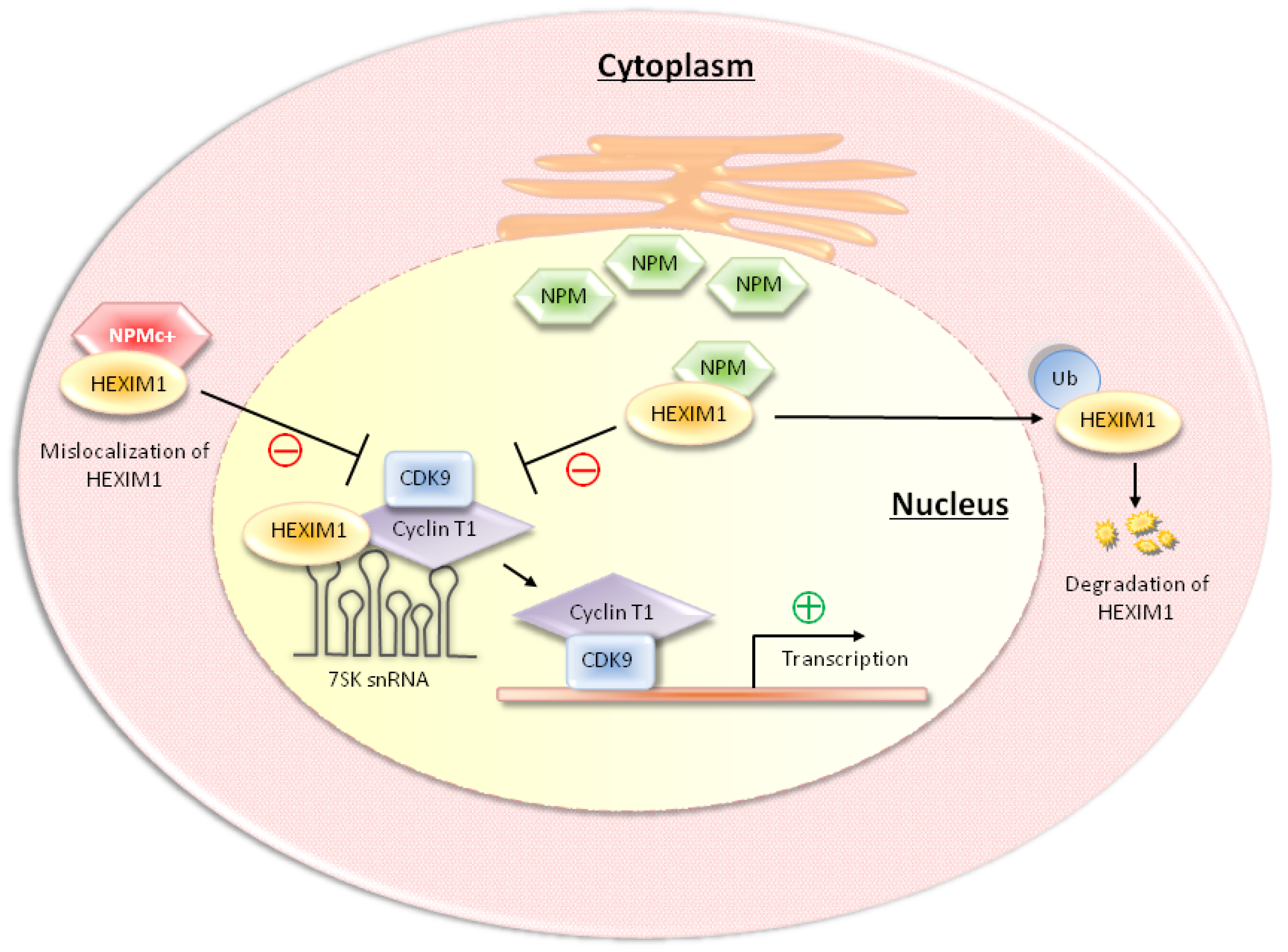
4. HDM2 Regulates P-TEFb Activity through the Ubiquitination of HEXIM1
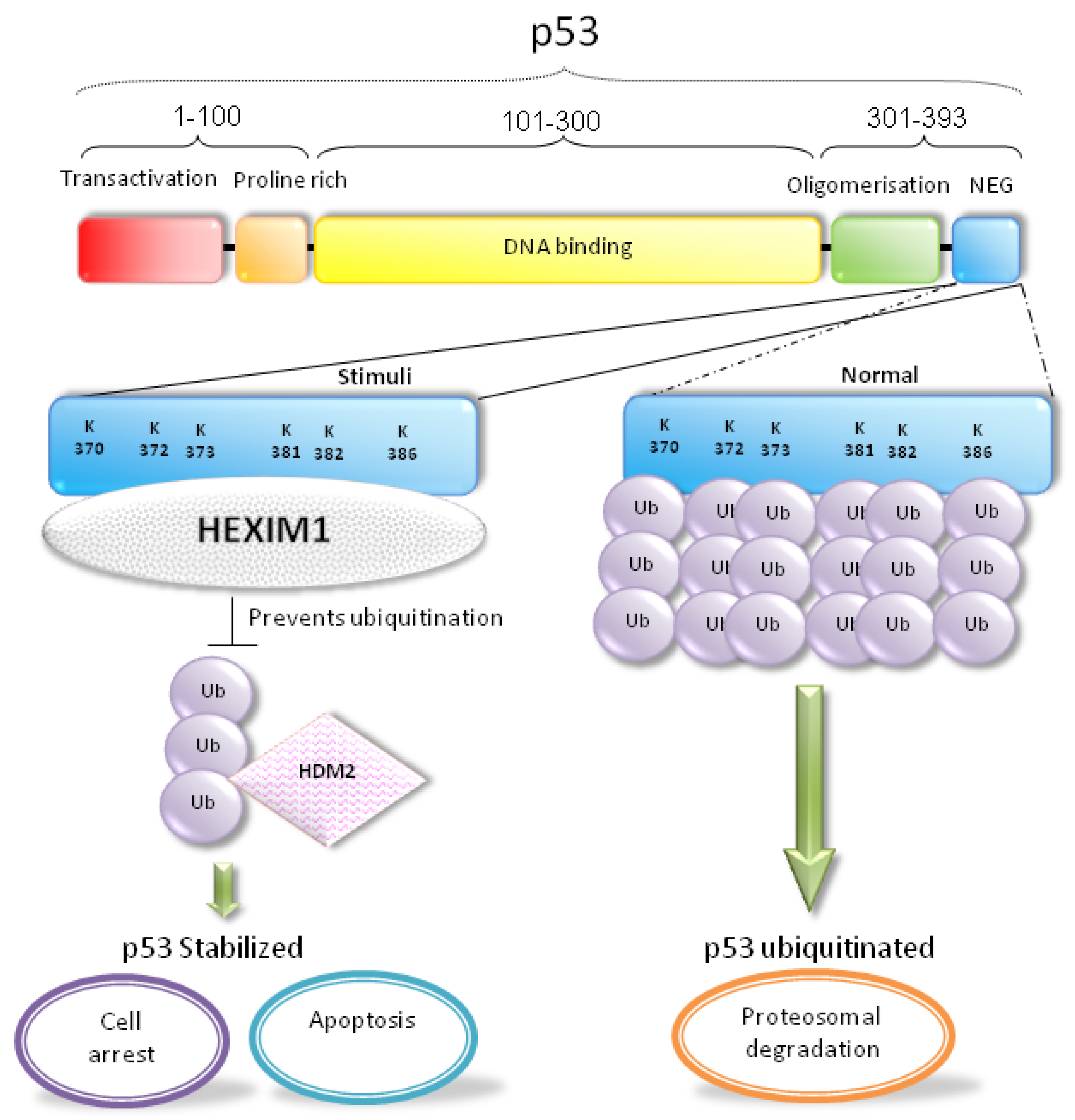
5. HEXIM1 Stabilizes p53 through the Protein-Protein Interaction with p53
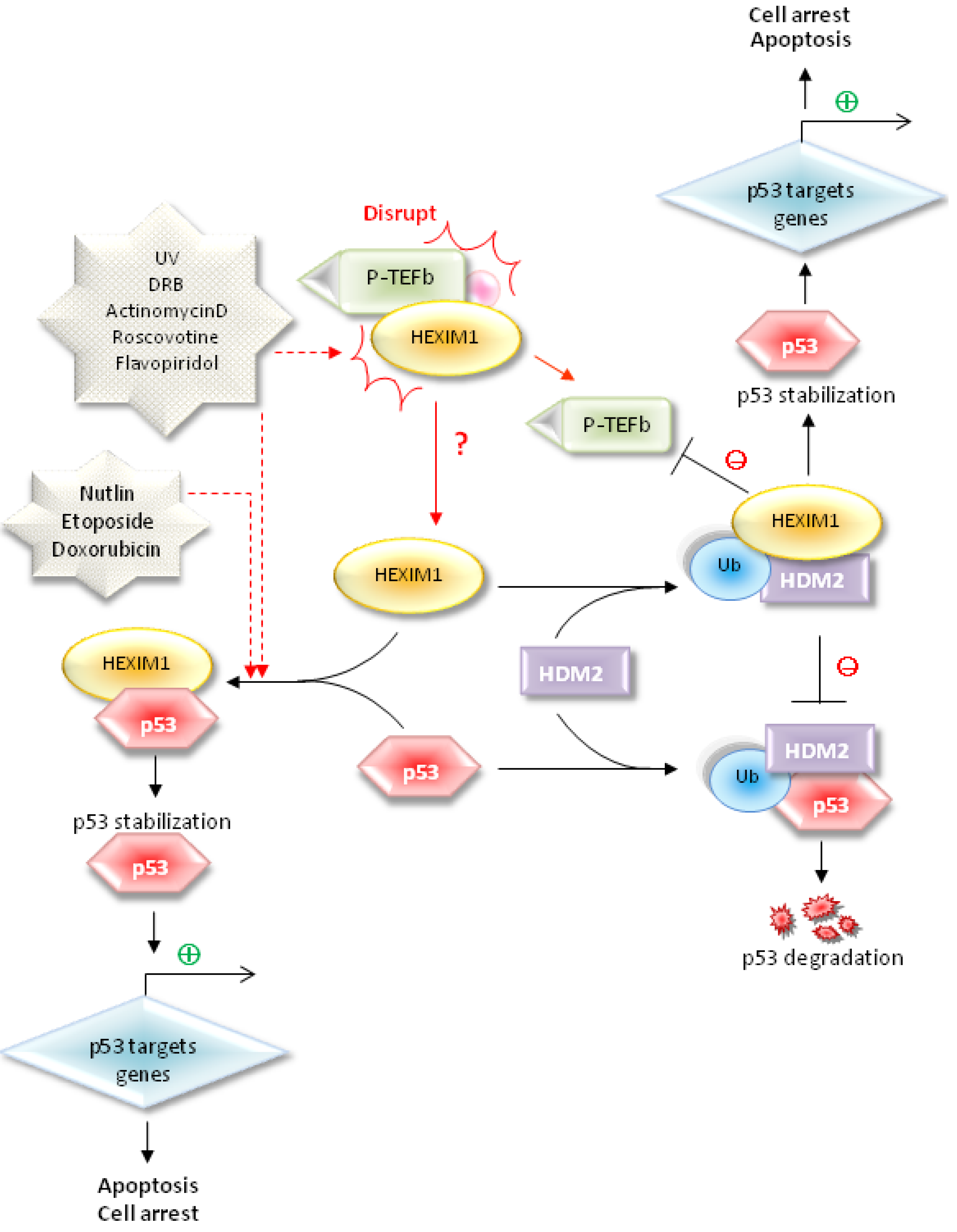
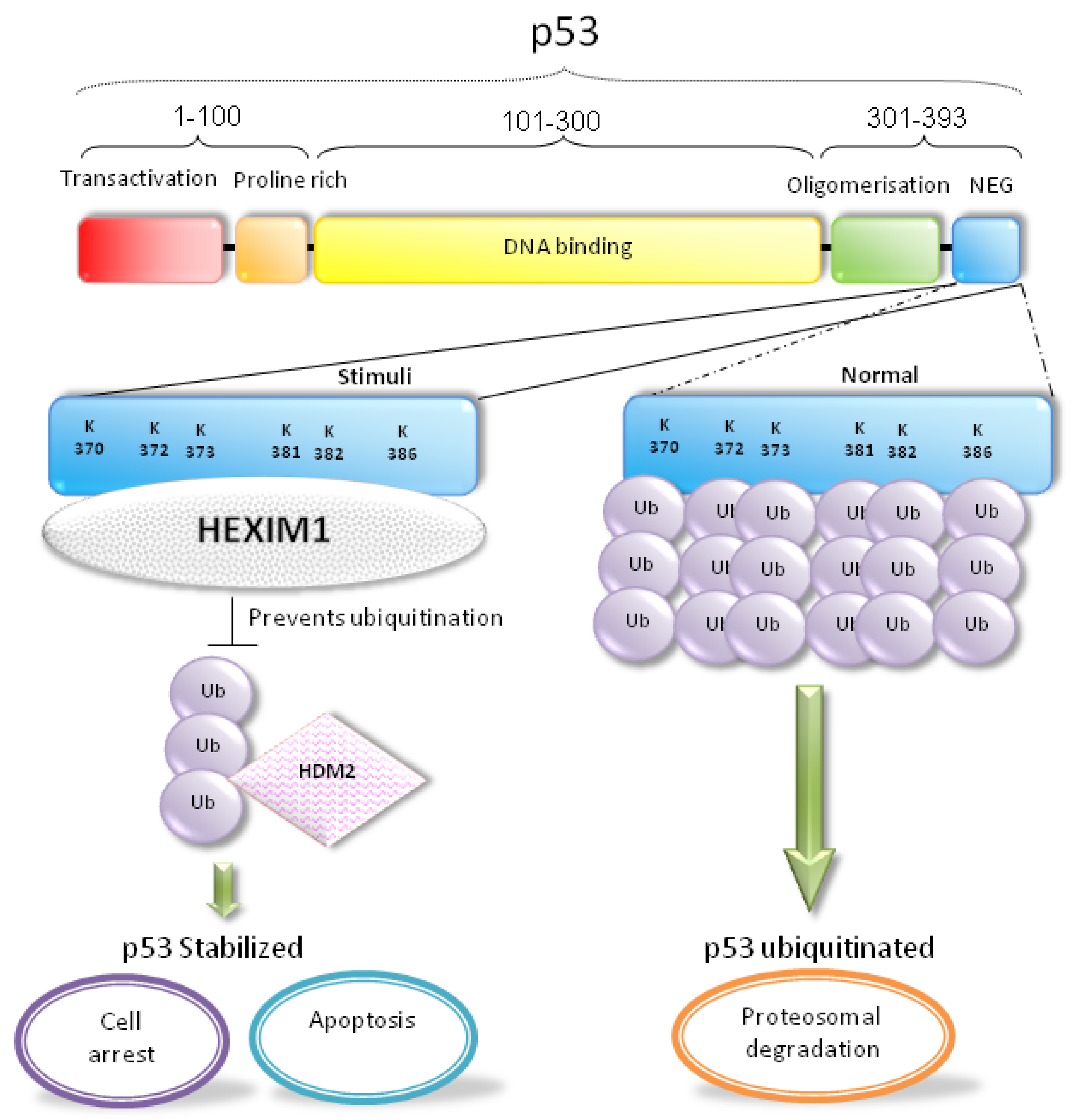
6. Conclusions
Conflict of Interest
References
- Kusuhara, M.N.K.; Kimura, K.; Maass, N.; Manabe, T.; Ishikawa, S.; Aikawa, M.; Miyazaki, K.; Yamaguchi, K. Cloning of hexamethylene-bis-acetamide-inducible transcript, HEXIM1, in human vascular smooth muscle cells. Biomed. Res. 1999, 20, 273–279. [Google Scholar]
- Ghatpande, S.; Goswami, S.; Mathew, S.; Rong, G.; Cai, L.; Shafiq, S.; Siddiqui, M.A. Identification of a novel cardiac lineage-associated protein(cCLP-1): A candidate regulator of cardiogenesis. Dev. Biol. 1999, 208, 210–221. [Google Scholar] [CrossRef]
- Huang, F.; Wagner, M.; Siddiqui, M.A. Ablation of the CLP-1 gene leads to down-regulation of the HAND1 gene and abnormality of the left ventricle of the heart and fetal death. Mech. Dev. 2004, 121, 559–572. [Google Scholar] [CrossRef]
- Huang, F.; Wagner, M.; Siddiqui, M.A. Structure, expression, and functional characterization of the mouse CLP-1 gene. Gene 2002, 292, 245–259. [Google Scholar] [CrossRef]
- Wittmann, B.M.; Wang, N.; Montano, M.M. Identification of a novel inhibitor of breast cell growth that is down-regulated by estrogens and decreased in breast tumors. Cancer Res. 2003, 63, 5151–5158. [Google Scholar]
- Michels, A.A.; Nguyen, V.T.; Fraldi, A.; Labas, V.; Edwards, M.; Bonnet, F.; Lania, L.; Bensaude, O. MAQ1 and 7SK RNA interact with CDK9/cyclin T complexes in a transcription-dependent manner. Mol. Cell Biol. 2003, 23, 4859–4869. [Google Scholar] [CrossRef]
- Yik, J.H.; Chen, R.; Nishimura, R.; Jennings, J.L.; Link, A.J.; Zhou, Q. Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol. Cell 2003, 12, 971–982. [Google Scholar] [CrossRef]
- Marshall, N.F.; Price, D.H. Control of formation of two distinct classes of RNA polymerase II elongation complexes. Mol. Cell Biol. 1992, 12, 2078–2090. [Google Scholar]
- Marshall, N.F.; Price, D.H. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J. Biol. Chem. 1995, 270, 12335–12338. [Google Scholar] [CrossRef]
- Peng, J.; Marshall, N.F.; Price, D.H. Identification of a cyclin subunit required for the function of Drosophila P-TEFb. J. Biol. Chem. 1998, 273, 13855–13860. [Google Scholar] [CrossRef]
- Peng, J.; Zhu, Y.; Milton, J.T.; Price, D.H. Identification of multiple cyclin subunits of human P-TEFb. Genes Dev. 1998, 12, 755–762. [Google Scholar] [CrossRef]
- Price, D.H. P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol. Cell Biol. 2000, 20, 2629–2634. [Google Scholar] [CrossRef]
- Peterlin, B.M.; Price, D.H. Controlling the elongation phase of transcription with P-TEFb. Mol. Cell 2006, 23, 297–305. [Google Scholar] [CrossRef]
- Chao, S.H.; Fujinagam, K.; Marion, J.E.; Taube, R.; Sausville, A.; Senderowicz, A.M.; Peterlin, B.M.; Price, D.H. Flavopiridol inhibits P-TEFb and blocks HIV-1 replication. J. Biol. Chem. 2000, 275, 28345–28348. [Google Scholar] [CrossRef]
- Chao, S.H.; Price, D.H. Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J. Biol. Chem. 2001, 276, 31793–31799. [Google Scholar] [CrossRef]
- Guenther, M.G.; Levine, S.S.; Boyer, L.A.; Jaenisch, R.; Young, R.A. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 2007, 130, 77–88. [Google Scholar] [CrossRef]
- Zeitlinger, J.; Stark, A.; Kellis, M.; Hong, J.W.; Nechaev, S.; Adelman, K.; Levine, M.; Young, R.A. RNA polymerase stalling at developmental control genes in the Drosophila melanogaster embryo. Nat. Genet. 2007, 39, 1512–1516. [Google Scholar] [CrossRef]
- Muse, G.W.; Gilchrist, D.A.; Nechaev, S.; Shah, R.; Parker, J.S.; Grissom, S.F.; Zeitlinger, J.; Adelman, K. RNA polymerase is poised for activation across the genome. Nat. Genet. 2007, 39, 1507–1511. [Google Scholar] [CrossRef]
- Wimmer, J.; Fujinaga, K.; Taube, R.; Cujec, T.P.; Zhu, Y.; Peng, J.; Price, D.H.; Peterlin, B.M. Interactions between Tat and TAR and human immunodeficiency virus replication are facilitated by human cyclin T1 but not cyclins T2a or T2b. Virology 1999, 255, 182–189. [Google Scholar] [CrossRef]
- Wei, P.; Garber, M.E.; Fang, S.M.; Fischer, W.H.; Jones, K.A. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 1998, 92, 451–462. [Google Scholar] [CrossRef]
- Zhu, Y.; Peery, T.; Peng, J.; Ramanathan, Y.; Marshall, N.; Marshall, T.; Amendt, B.; Mathews, M.B.; Price, D.H. Transcription elongation factor P-TEFb is required for HIV-1 tat transactivation in vitro. Genes Dev. 1997, 11, 2622–2632. [Google Scholar] [CrossRef]
- Ramanathan, Y.; Reza, S.M.; Young, T.M.; Mathews, M.B.; Peery, T. Human and rodent transcription elongation factor P-TEFb: Interactions with human immunodeficiency virus type 1 tat and carboxy-terminal domain substrate. J. Virol. 1999, 73, 5448–5458. [Google Scholar]
- Garriga, J.; Mayol, X.; Grana, X. The CDC2-related kinase PITALRE is the catalytic subunit of active multimeric protein complexes. Biochem. J. 1996, 319, 293–298. [Google Scholar]
- Nguyen, V.T.; Kiss, T.; Michels, A.A.; Bensaude, O. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature 2001, 414, 322–325. [Google Scholar] [CrossRef]
- Yang, Z.; Zhu, Q.; Luo, K.; Zhou, Q. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature 2001, 414, 317–322. [Google Scholar] [CrossRef]
- Michels, A.A.; Fraldi, A.; Li, Q.; Adamson, T.E.; Bonnet, F.; Nguyen, V.T.; Sedore, S.C.; Price, J.P.; Price, D.H.; Lania, L.; et al. Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor. EMBO J. 2004, 23, 2608–2619. [Google Scholar] [CrossRef]
- Krueger, B.J.; Jeronimo, C.; Roy, B.B.; Bouchard, A.; Barrandon, C.; Byers, S.A.; Searcey, C.E.; Cooper, J.J.; Bensaude, O.; Cohen, E.A.; et al. LARP7 is a stable component of the 7SK snRNP while P-TEFb, HEXIM1 and hnRNP A1 are reversibly associated. Nucleic Acids Res. 2008, 36, 2219–2229. [Google Scholar] [CrossRef]
- Jeronimo, C.; Forget, D.; Bouchard, A.; Li, Q.; Chua, G.; Poitras, C.; Therien, C.; Bergeron, D.; Bourassa, S.; Greenblatt, J.; et al. Systematic analysis of the protein interaction network for the human transcription machinery reveals the identity of the 7SK capping enzyme. Mol. Cell 2007, 27, 262–274. [Google Scholar] [CrossRef]
- Xue, Y.; Yang, Z.; Chen, R.; Zhou, Q. A capping-independent function of MePCE in stabilizing 7SK snRNA and facilitating the assembly of 7SK snRNP. Nucleic Acids Res. 2010, 38, 360–369. [Google Scholar] [CrossRef]
- Li, Q.; Price, J.P.; Byers, S.A.; Cheng, D.; Peng, J.; Price, D.H. Analysis of the large inactive P-TEFb complex indicates that it contains one 7SK molecule, a dimer of HEXIM1 or HEXIM2, and two P-TEFb molecules containing Cdk9 phosphorylated at threonine 186. J. Biol. Chem. 2005, 280, 28819–28826. [Google Scholar] [CrossRef]
- Barboric, M.; Kohoutek, J.; Price, J.P.; Blazek, D.; Price, D.H.; Peterlin, B.M. Interplay between 7SK snRNA and oppositely charged regions in HEXIM1 direct the inhibition of P-TEFb. EMBO J. 2005, 24, 4291–4303. [Google Scholar] [CrossRef]
- Blazek, D.; Barboric, M.; Kohoutek, J.; Oven, I.; Peterlin, B.M. Oligomerization of HEXIM1 via 7SK snRNA and coiled-coil region directs the inhibition of P-TEFb. Nucleic Acids Res. 2005, 33, 7000–7010. [Google Scholar] [CrossRef]
- Dulac, C.; Michels, A.A.; Fraldi, A.; Bonnet, F.; Nguyen, V.T.; Napolitano, G.; Lania, L.; Bensaude, O. Transcription-dependent association of multiple positive transcription elongation factor units to a HEXIM multimer. J. Biol. Chem. 2005, 280, 30619–30629. [Google Scholar] [CrossRef]
- Galatioto, J.; Mascareno, E.; Siddiqui, M.A. CLP-1 associates with MyoD and HDAC to restore skeletal muscle cell regeneration. J. Cell Sci. 2010, 123, 3789–3795. [Google Scholar] [CrossRef]
- Czudnochowski, N.; Vollmuth, F.; Baumann, S.; Vogel-Bachmayr, K.; Geyer, M. Specificity of Hexim1 and Hexim2 complex formation with cyclin T1/T2, importin alpha and 7SK snRNA. J. Mol. Biol. 2010, 395, 28–41. [Google Scholar] [CrossRef]
- Ali, S.; Coombes, R.C. Estrogen receptor alpha in human breast cancer: Occurrence and significance. J. Mammary Gland Biol. Neoplasia 2000, 5, 271–281. [Google Scholar] [CrossRef]
- Katzenellenbogen, B.S.; Frasor, J. Therapeutic targeting in the estrogen receptor hormonal pathway. Semin. Oncol. 2004, 31, 28–38. [Google Scholar] [CrossRef]
- Wittmann, B.M.; Fujinaga, K.; Deng, H.; Ogba, N.; Montano, M.M. The breast cell growth inhibitor, estrogen down regulated gene 1, modulates a novel functional interaction between estrogen receptor alpha and transcriptional elongation factor cyclin T1. Oncogene 2005, 24, 5576–5588. [Google Scholar] [CrossRef]
- Ouchida, R.; Kusuhara, M.; Shimizu, N.; Hisada, T.; Makino, Y.; Morimoto, C.; Handa, H.; Ohsuzu, F.; Tanaka, H. Suppression of NF-kappaB-dependent gene expression by a hexamethylene bisacetamide-inducible protein HEXIM1 in human vascular smooth muscle cells. Genes Cells 2003, 8, 95–107. [Google Scholar] [CrossRef]
- Barboric, M.; Nissen, R.M.; Kanazawa, S.; Jabrane-Ferrat, N.; Peterlin, B.M. NF-kappaB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol. Cell 2001, 8, 327–337. [Google Scholar] [CrossRef]
- Shimizu, N.; Ouchida, R.; Yoshikawa, N.; Hisada, T.; Watanabe, H.; Okamoto, K.; Kusuhara, M.; Handa, H.; Morimoto, C.; Tanaka, H. HEXIM1 forms a transcriptionally abortive complex with glucocorticoid receptor without involving 7SK RNA and positive transcription elongation factor b. Proc. Natl. Acad. Sci. USA 2005, 102, 8555–8560. [Google Scholar] [CrossRef]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- May, P.; May, E. Twenty years of p53 research: Structural and functional aspects of the p53 protein. Oncogene 1999, 18, 7621–7636. [Google Scholar] [CrossRef]
- Lu, H.; Levine, A.J. Human TAFII31 protein is a transcriptional coactivator of the p53 protein. Proc. Natl. Acad. Sci. USA 1995, 92, 5154–5158. [Google Scholar] [CrossRef]
- Thut, C.J.; Chen, J.L.; Klemm, R.; Tjian, R. p53 transcriptional activation mediated by coactivators TAFII40 and TAFII60. Science 1995, 267, 100–104. [Google Scholar]
- Sakamuro, D.; Sabbatini, P.; White, E.; Prendergast, G.C. The polyproline region of p53 is required to activate apoptosis but not growth arrest. Oncogene 1997, 15, 887–898. [Google Scholar]
- Walker, K.K.; Levine, A.J. Identification of a novel p53 functional domain that is necessary for efficient growth suppression. Proc. Natl. Acad. Sci. USA 1996, 93, 15335–15340. [Google Scholar] [CrossRef]
- Pavletich, N.P.; Chambers, K.A.; Pabo, C.O. The DNA-binding domain of p53 contains the four conserved regions and the major mutation hot spots. Genes Dev. 1993, 7, 2556–2564. [Google Scholar] [CrossRef]
- Chene, P. The role of tetramerization in p53 function. Oncogene 2001, 20, 2611–2617. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Li, M.; Luo, J.; Brooks, C.L.; Gu, W. Acetylation of p53 inhibits its ubiquitination by Mdm2. J. Biol. Chem. 2002, 277, 50607–50611. [Google Scholar]
- Ko, L.J.; Prives, C. p53: Puzzle and paradigm. Genes Dev. 1996, 10, 1054–1072. [Google Scholar] [CrossRef]
- Brooks, C.L.; Li, M.; Hu, M.; Shi, Y.; Gu, W. The p53-Mdm2-HAUSP complex is involved in p53 stabilization by HAUSP. Oncogene 2007, 26, 7262–7266. [Google Scholar] [CrossRef]
- Rodriguez, M.S.; Desterro, J.M.; Lain, S.; Lane, D.P.; Hay, R.T. Multiple C-terminal lysine residues target p53 for ubiquitin-proteasome-mediated degradation. Mol. Cell Biol. 2000, 20, 8458–8467. [Google Scholar] [CrossRef]
- Poyurovsky, M.V.; Katz, C.; Laptenko, O.; Beckerman, R.; Lokshin, M.; Ahn, J.; Byeon, I.J.; Gabizon, R.; Mattia, M.; Zupnick, A.; et al. The C-terminus of p53 binds the N-terminal domain of MDM2. Nat. Struct. Mol. Biol. 2010, 17, 982–989. [Google Scholar] [CrossRef]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiong, Y.; Yarbrough, W.G. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell 1998, 92, 725–734. [Google Scholar] [CrossRef]
- Kuo, M.L.; den Besten, W.; Bertwistle, D.; Roussel, M.F.; Sherr, C.J. N-terminal polyubiquitination and degradation of the Arf tumor suppressor. Genes Dev. 2004, 18, 1862–1874. [Google Scholar] [CrossRef]
- Bertwistle, D.; Sugimoto, M.; Sherr, C.J. Physical and functional interactions of the Arf tumor suppressor protein with nucleophosmin/B23. Mol. Cell Biol. 2004, 24, 985–996. [Google Scholar] [CrossRef]
- Kurki, S.; Peltonen, K.; Latonen, L.; Kiviharju, T.M.; Ojala, P.M.; Meek, D.; Laiho, M. Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2-mediated degradation. Cancer Cell 2004, 5, 465–475. [Google Scholar] [CrossRef]
- Savkur, R.S.; Olson, M.O. Preferential cleavage in pre-ribosomal RNA byprotein B23 endoribonuclease. Nucleic Acids Res. 1998, 26, 4508–4515. [Google Scholar] [CrossRef]
- Itahana, K.; Bhat, K.P.; Jin, A.; Itahana, Y.; Hawke, D.; Kobayashi, R.; Zhang, Y. Tumor suppressor ARF degrades B23, a nucleolar protein involved in ribosome biogenesis and cell proliferation. Mol. Cell 2003, 12, 1151–1164. [Google Scholar] [CrossRef]
- Grisendi, S.; Mecucci, C.; Falini, B.; Pandolfi, P.P. Nucleophosmin and cancer. Nat. Rev. Cancer 2006, 6, 493–505. [Google Scholar]
- Okuwaki, M.; Matsumoto, K.; Tsujimoto, M.; Nagata, K. Function of nucleophosmin/B23, a nucleolar acidic protein, as a histone chaperone. FEBS Lett. 2001, 506, 272–276. [Google Scholar] [CrossRef]
- Krause, A. Hoffmann I: Polo-like kinase 2-dependent phosphorylation of NPM/B23 on serine 4 triggers centriole duplication. PLoS One 2010, 5, e9849. [Google Scholar] [CrossRef]
- Weber, J.D.; Taylor, L.J.; Roussel, M.F.; Sherr, C.J.; Bar-Sagi, D. Nucleolar Arf sequesters Mdm2 and activates p53. Nat. Cell Biol. 1999, 1, 20–26. [Google Scholar] [CrossRef]
- Kamijo, T.; Zindy, F.; Roussel, M.F.; Quelle, D.E.; Downing, J.R.; Ashmun, R.A.; Grosveld, G.; Sherr, C.J. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell 1997, 91, 649–659. [Google Scholar] [CrossRef]
- Yun, J.P.; Chew, E.C.; Liew, C.T.; Chan, J.Y.; Jin, M.L.; Ding, M.X.; Fai, Y.H.; Li, H.K.; Liang, X.M.; Wu, Q.L. Nucleophosmin/B23 is a proliferate shuttle protein associated with nuclear matrix. J. Cell Biochem. 2003, 90, 1140–1148. [Google Scholar] [CrossRef]
- Colombo, E.; Marine, J.C.; Danovi, D.; Falini, B.; Pelicci, P.G. Nucleophosmin regulates the stability and transcriptional activity of p53. Nat. Cell Biol. 2002, 4, 529–533. [Google Scholar] [CrossRef]
- Grisendi, S.; Bernardi, R.; Rossi, M.; Cheng, K.; Khandker, L.; Manova, K.; Pandolfi, P.P. Role of nucleophosmin in embryonic development and tumorigenesis. Nature 2005, 437, 147–153. [Google Scholar] [CrossRef]
- Tanaka, M.; Sasaki, H.; Kino, I.; Sugimura, T.; Terada, M. Genes preferentially expressed in embryo stomach are predominantly expressed in gastric cancer. Cancer Res. 1992, 52, 3372–3377. [Google Scholar]
- Nozawa, Y.; van Belzen, N.; van der Made, A.C.; Dinjens, W.N.; Bosman, F.T. Expression of nucleophosmin/B23 in normal and neoplastic colorectal mucosa. J. Pathol. 1996, 178, 48–52. [Google Scholar] [CrossRef]
- Shields, L.B.; Gercel-Taylor, C.; Yashar, C.M.; Wan, T.C.; Katsanis, W.A.; Spinnato, J.A.; Taylor, D.D. Induction of immune responses to ovarian tumor antigens by multiparity. J. Soc. Gynecol. Investig. 1997, 4, 298–304. [Google Scholar] [CrossRef]
- Subong, E.N.; Shue, M.J.; Epstein, J.I.; Briggman, J.V.; Chan, P.K.; Partin, A.W. Monoclonal antibody to prostate cancer nuclear matrix protein (PRO:4–216) recognizes nucleophosmin/B23. Prostate 1999, 39, 298–304. [Google Scholar] [CrossRef]
- Tsui, K.H.; Cheng, A.J.; Chang, P.; Pan, T.L.; Yung, B.Y. Association of nucleophosmin/B23 mRNA expression with clinical outcome in patients with bladder carcinoma. Urology 2004, 64, 839–844. [Google Scholar] [CrossRef]
- Skaar, T.C.; Prasad, S.C.; Sharareh, S.; Lippman, M.E.; Brunner, N.; Clarke, R. Two-dimensional gel electrophoresis analyses identify nucleophosmin as an estrogen regulated protein associated with acquired estrogen-independence in human breast cancer cells. J. Steroid Biochem. Mol. Biol. 1998, 67, 391–402. [Google Scholar] [CrossRef]
- Li, J.; Sejas, D.P.; Rani, R.; Koretsky, T.; Bagby, G.C.; Pang, Q. Nucleophosmin regulates cell cycle progression and stress response in hematopoietic stem/progenitor cells. J. Biol. Chem. 2006, 281, 16536–16545. [Google Scholar]
- Liu, Y.; Zhang, F.; Zhang, X.F.; Qi, L.S.; Yang, L.; Guo, H.; Zhang, N. Expression of nucleophosmin/NPM1 correlates with migration and invasiveness of colon cancer cells. J. Biomed. Sci. 2012, 19, 53. [Google Scholar] [CrossRef]
- Ye, K. Nucleophosmin/B23, a multifunctional protein that can regulate apoptosis. Cancer Biol. Ther. 2005, 4, 918–923. [Google Scholar]
- Falini, B.; Mecucci, C.; Tiacci, E.; Alcalay, M.; Rosati, R.; Pasqualucci, L.; La Starza, R.; Diverio, D.; Colombo, E.; Santucci, A.; et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl. J. Med. 2005, 352, 254–266. [Google Scholar] [CrossRef]
- Quentmeier, H.; Martelli, M.P.; Dirks, W.G.; Bolli, N.; Liso, A.; Macleod, R.A.; Nicoletti, I.; Mannucci, R.; Pucciarini, A.; Bigerna, B.; et al. Cell line OCI/AML3 bears exon-12 NPM gene mutation-A and cytoplasmic expression of nucleophosmin. Leukemia 2005, 19, 1760–1767. [Google Scholar] [CrossRef]
- Falini, B.; Bolli, N.; Shan, J.; Martelli, M.P.; Liso, A.; Pucciarini, A.; Bigerna, B.; Pasqualucci, L.; Mannucci, R.; Rosati, R.; et al. Both carboxy-terminus NES motif and mutated tryptophan(s) are crucial for aberrant nuclear export of nucleophosmin leukemic mutants in NPMc+ AML. Blood 2006, 107, 4514–4523. [Google Scholar] [CrossRef]
- Falini, B.; Nicoletti, I.; Martelli, M.F.; Mecucci, C. Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc+ AML): Biologic and clinical features. Blood 2007, 109, 874–885. [Google Scholar]
- Alcalay, M.; Tiacci, E.; Bergomas, R.; Bigerna, B.; Venturini, E.; Minardi, S.P.; Meani, N.; Diverio, D.; Bernard, L.; Tizzoni, L.; et al. Acute myeloid leukemia bearing cytoplasmic nucleophosmin (NPMc+ AML) shows a distinct gene expression profile characterized by up-regulation of genes involved in stem-cell maintenance. Blood 2005, 106, 899–902. [Google Scholar] [CrossRef]
- Gurumurthy, M.; Tan, C.H.; Ng, R.; Zeiger, L.; Lau, J.; Lee, J.; Dey, A.; Philp, R.; Li, Q.; Lim, T.M.; et al. Nucleophosmin interacts with HEXIM1 and regulates RNA polymerase II transcription. J. Mol. Biol. 2008, 378, 302–317. [Google Scholar] [CrossRef]
- Lew, Q.J.; Tan, C.H.; Gurumurthy, M.; Chu, K.L.; Cheong, N.; Lane, D.P.; Chao, S.H. NPMc(+) AML cell line shows differential protein expression and lower sensitivity to DNA-damaging and p53-inducing anticancer compounds. Cell Cycle 2011, 10, 1978–1987. [Google Scholar] [CrossRef]
- Momand, J.; Zambetti, G.P.; Olson, D.C.; George, D.; Levine, A.J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992, 69, 1237–1245. [Google Scholar] [CrossRef]
- Oliner, J.D.; Pietenpol, J.A.; Thiagalingam, S.; Gyuris, J.; Kinzler, K.W.; Vogelstein, B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 1993, 362, 857–860. [Google Scholar] [CrossRef]
- Fang, S.; Jensen, J.P.; Ludwig, R.L.; Vousden, K.H.; Weissman, A.M. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J. Biol. Chem. 2000, 275, 8945–8951. [Google Scholar]
- Lai, Z.; Ferry, K.V.; Diamond, M.A.; Wee, K.E.; Kim, Y.B.; Ma, J.; Yang, T.; Benfield, P.A.; Copeland, R.A.; Auger, K.R. Human mdm2 mediates multiple mono-ubiquitination of p53 by a mechanism requiring enzyme isomerization. J. Biol. Chem. 2001, 276, 31357–31367. [Google Scholar] [CrossRef]
- Grossman, S.R.; Deato, M.E.; Brignone, C.; Chan, H.M.; Kung, A.L.; Tagami, H.; Nakatani, Y.; Livingston, D.M. Polyubiquitination of p53 by a ubiquitin ligase activity of p300. Science 2003, 300, 342–344. [Google Scholar] [CrossRef]
- Ito, A.; Lai, C.H.; Zhao, X.; Saito, S.; Hamilton, M.H.; Appella, E.; Yao, T.P. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J. 2001, 20, 1331–1340. [Google Scholar]
- Yu, Z.K.; Geyer, R.K.; Maki, C.G. MDM2-dependent ubiquitination of nuclear and cytoplasmic P53. Oncogene 2000, 19, 5892–5897. [Google Scholar] [CrossRef]
- Geyer, R.K.; Yu, Z.K.; Maki, C.G. The MDM2 RING-finger domain is required to promote p53 nuclear export. Nat. Cell Biol. 2000, 2, 569–573. [Google Scholar] [CrossRef]
- Boyd, S.D.; Tsai, K.Y.; Jacks, T. An intact HDM2 RING-finger domain is required for nuclear exclusion of p53. Nat. Cell Biol. 2000, 2, 563–568. [Google Scholar] [CrossRef]
- Palmer, A.; Mason, G.G.; Paramio, J.M.; Knecht, E.; Rivett, A.J. Changes in proteasome localization during the cell cycle. Eur. J. Cell Biol. 1994, 64, 163–175. [Google Scholar]
- Chene, P.; Fuchs, J.; Bohn, J.; Garcia-Echeverria, C.; Furet, P.; Fabbro, D. A small synthetic peptide, which inhibits the p53-hdm2 interaction, stimulates the p53 pathway in tumour cell lines. J. Mol. Biol. 2000, 299, 245–253. [Google Scholar] [CrossRef]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef]
- Issaeva, N.; Bozko, P.; Enge, M.; Protopopova, M.; Verhoef, L.G.; Masucci, M.; Pramanik, A.; Selivanova, G. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat. Med. 2004, 10, 1321–1328. [Google Scholar] [CrossRef]
- Blaydes, J.P.; Gire, V.; Rowson, J.M.; Wynford-Thomas, D. Tolerance of high levels of wild-type p53 in transformed epithelial cells dependent on auto-regulation by mdm-2. Oncogene 1997, 14, 1859–1868. [Google Scholar]
- Midgley, C.A.; Desterro, J.M.; Saville, M.K.; Howard, S.; Sparks, A.; Hay, R.T.; Lane, D.P. An N-terminal p14ARF peptide blocks Mdm2-dependent ubiquitination in vitro and can activate p53 in vivo. Oncogene 2000, 19, 2312–2323. [Google Scholar] [CrossRef]
- Lau, J.; Lew, Q.J.; Diribarne, G.; Michels, A.A.; Dey, A.; Bensaude, O.; Lane, D.P.; Chao, S.H. Ubiquitination of HEXIM1 by HDM2. Cell Cycle 2009, 8, 2247–2254. [Google Scholar] [CrossRef]
- Brown, C.J.; Lain, S.; Verma, C.S.; Fersht, A.R.; Lane, D.P. Awakening guardian angels: Drugging the p53 pathway. Nat. Rev. Cancer 2009, 9, 862–873. [Google Scholar] [CrossRef]
- Fritsche, M.; Haessler, C.; Brandner, G. Induction of nuclear accumulation of the tumor-suppressor protein p53 by DNA-damaging agents. Oncogene 1993, 8, 307–318. [Google Scholar]
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Cummins, J.M.; Vogelstein, B. HAUSP is required for p53 destabilization. Cell Cycle 2004, 3, 689–692. [Google Scholar]
- Roe, J.S.; Kim, H.; Lee, S.M.; Kim, S.T.; Cho, E.J.; Youn, H.D. p53 stabilization and transactivation by a von Hippel-Lindau protein. Mol. Cell 2006, 22, 395–405. [Google Scholar] [CrossRef]
- Thompson, T.; Tovar, C.; Yang, H.; Carvajal, D.; Vu, B.T.; Xu, Q.; Wahl, G.M.; Heimbrook, D.C.; Vassilev, L.T. Phosphorylation of p53 on key serines is dispensable for transcriptional activation and apoptosis. J. Biol. Chem. 2004, 279, 53015–53022. [Google Scholar] [CrossRef]
- An, W.G.; Kanekal, M.; Simon, M.C.; Maltepe, E.; Blagosklonny, M.V.; Neckers, L.M. Stabilization of wild-type p53 by hypoxia-inducible factor 1alpha. Nature 1998, 392, 405–408. [Google Scholar] [CrossRef]
- Hammond, E.M.; Giaccia, A.J. Hypoxia-inducible factor-1 and p53: Friends, acquaintances, or strangers? Clin. Cancer Res. 2006, 12, 5007–5009. [Google Scholar] [CrossRef]
- Yuan, Z.M.; Huang, Y.; Ishiko, T.; Nakada, S.; Utsugisawa, T.; Shioya, H.; Utsugisawa, Y.; Yokoyama, K.; Weichselbaum, R.; Shi, Y.; et al. Role for p300 in stabilization of p53 in the response to DNA damage. J. Biol. Chem. 1999, 274, 1883–1886. [Google Scholar] [CrossRef]
- Lew, Q.J.; Chia, Y.L.; Chu, K.L.; Lam, Y.T.; Gurumurthy, M.; Xu, S.; Lam, K.P.; Cheong, N.; Chao, S.H. Identification of HEXIM1 as a Positive Regulator of p53. J. Biol. Chem. 2012, 287, 36443–36454. [Google Scholar] [CrossRef]
- Byers, S.A.; Price, J.P.; Cooper, J.J.; Li, Q.; Price, D.H. HEXIM2, a HEXIM1-related protein, regulates positive transcription elongation factor b through association with 7SK. J. Biol. Chem. 2005, 280, 16360–16367. [Google Scholar]
- Amente, S.; Gargano, B.; Napolitano, G.; Lania, L.; Majello, B. Camptothecin releases P-TEFb from the inactive 7SK snRNP complex. Cell Cycle 2009, 8, 1249–1255. [Google Scholar] [CrossRef]
- Claudio, P.P.; Cui, J.; Ghafouri, M.; Mariano, C.; White, M.K.; Safak, M.; Sheffield, J.B.; Giordano, A.; Khalili, K.; Amini, S.; et al. Cdk9 phosphorylates p53 on serine 392 independently of CKII. J. Cell Physiol. 2006, 208, 602–612. [Google Scholar] [CrossRef]
- Radhakrishnan, S.K.; Gartel, A.L. CDK9 phosphorylates p53 on serine residues 33, 315 and 392. Cell Cycle 2006, 5, 519–521. [Google Scholar] [CrossRef]
- Zheng, H.; You, H.; Zhou, X.Z.; Murray, S.A.; Uchida, T.; Wulf, G.; Gu, L.; Tang, X.; Lu, K.P.; Xiao, Z.X. The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature 2002, 419, 849–853. [Google Scholar] [CrossRef]
- Zacchi, P.; Gostissa, M.; Uchida, T.; Salvagno, C.; Avolio, F.; Volinia, S.; Ronai, Z.; Blandino, G.; Schneider, C.; del Sal, G. The prolyl isomerase Pin1 reveals a mechanism to control p53 functions after genotoxic insults. Nature 2002, 419, 853–857. [Google Scholar] [CrossRef]
- Hupp, T.R.; Meek, D.W.; Midgley, C.A.; Lane, D.P. Regulation of the specific DNA binding function of p53. Cell 1992, 71, 875–886. [Google Scholar] [CrossRef]
- Jeong, J.H.; Nakajima, H.; Magae, J.; Furukawa, C.; Taki, K.; Otsuka, K.; Tomita, M.; Lee, I.S.; Kim, C.H.; Chang, H.W.; et al. Ascochlorin activates p53 in a manner distinct from DNA damaging agents. Int. J. Cancer 2009, 124, 2797–2803. [Google Scholar] [CrossRef]
- Keller, D.M.; Zeng, X.; Wang, Y.; Zhang, Q.H.; Kapoor, M.; Shu, H.; Goodman, R.; Lozano, G.; Zhao, Y.; Lu, H. A DNA damage-induced p53 serine 392 kinase complex contains CK2, hSpt16, and SSRP1. Mol. Cell 2001, 7, 283–292. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lew, Q.J.; Chu, K.L.; Chia, Y.L.; Cheong, N.; Chao, S.-H. HEXIM1, a New Player in the p53 Pathway. Cancers 2013, 5, 838-856. https://doi.org/10.3390/cancers5030838
Lew QJ, Chu KL, Chia YL, Cheong N, Chao S-H. HEXIM1, a New Player in the p53 Pathway. Cancers. 2013; 5(3):838-856. https://doi.org/10.3390/cancers5030838
Chicago/Turabian StyleLew, Qiao Jing, Kai Ling Chu, Yi Ling Chia, Nge Cheong, and Sheng-Hao Chao. 2013. "HEXIM1, a New Player in the p53 Pathway" Cancers 5, no. 3: 838-856. https://doi.org/10.3390/cancers5030838
APA StyleLew, Q. J., Chu, K. L., Chia, Y. L., Cheong, N., & Chao, S.-H. (2013). HEXIM1, a New Player in the p53 Pathway. Cancers, 5(3), 838-856. https://doi.org/10.3390/cancers5030838