Is Glioblastoma an Epigenetic Malignancy?


Abstract
:1. Nature of Epigenetic Modifications
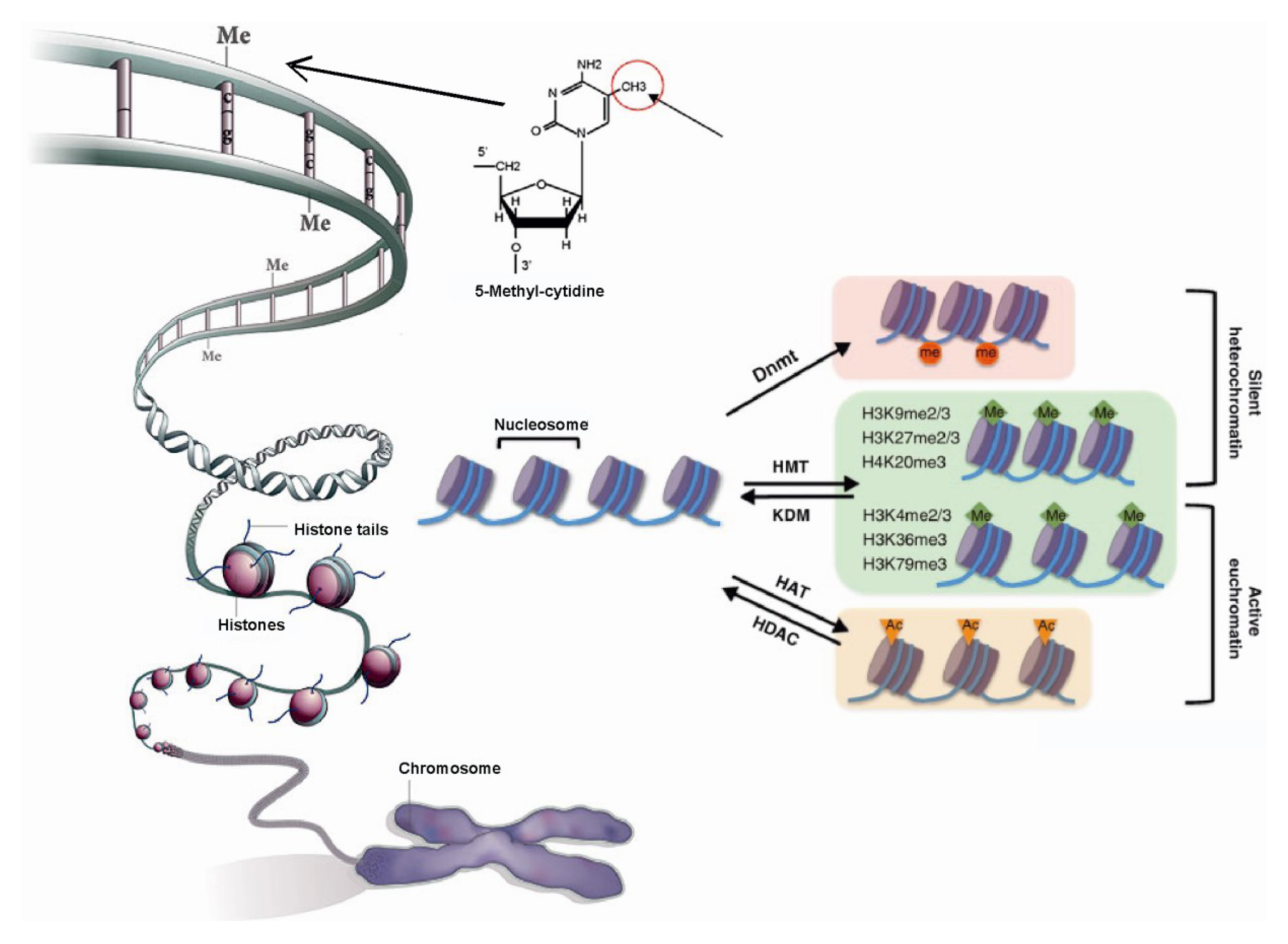
1.1. Role of Histone Modifications in Gene Regulation
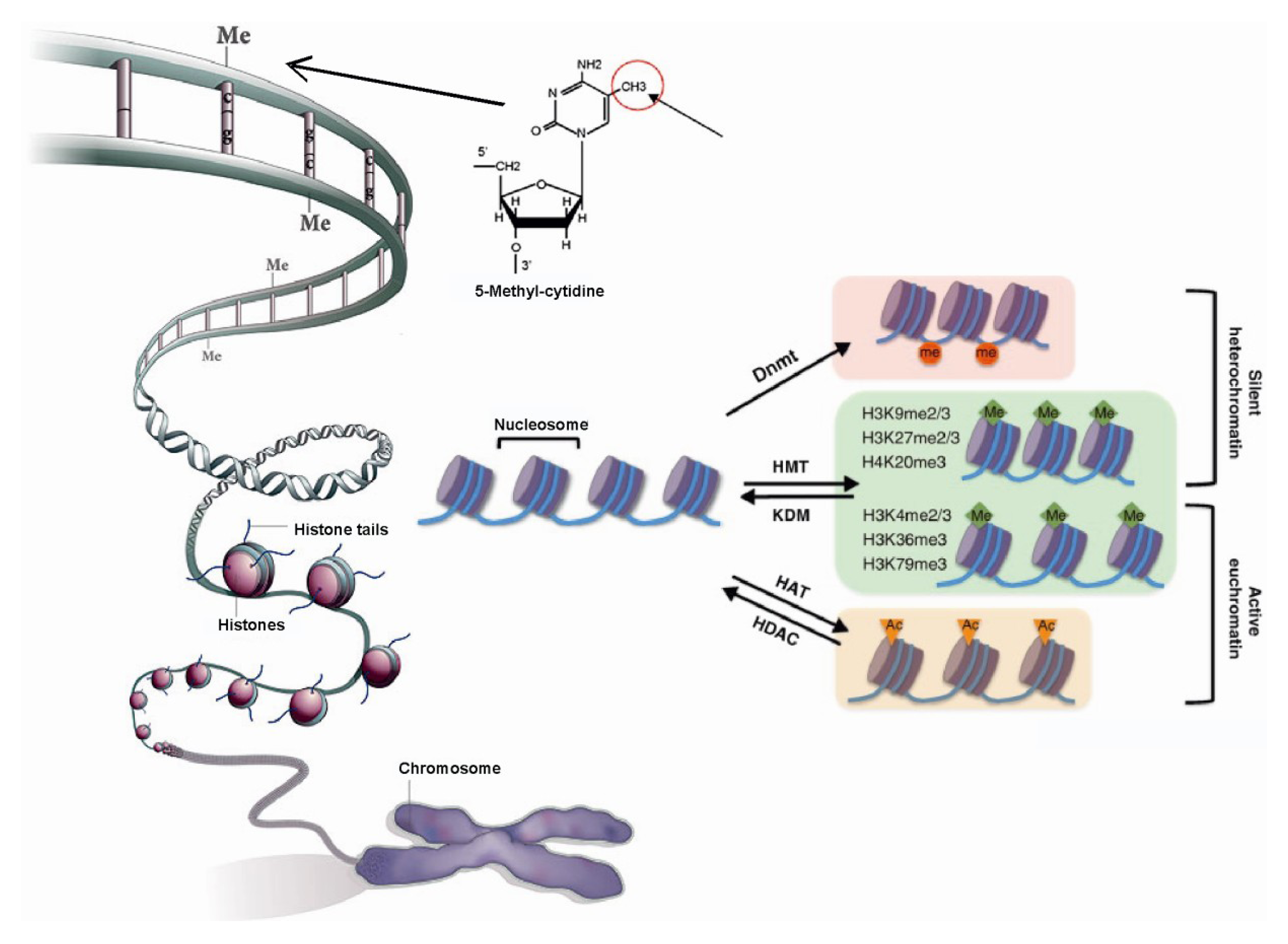
1.2. Role of DNA Methylation in Gene Regulation
2. Deregulation of the Epigenetic Landscape in Cancerogenesis
2.1. Activation or Inactivation of Epigenetic Enzymes in Cancer
2.2. Metabolic Disturbances as a Source of Epigenetic Deregulation
3. Epigenetic Modifications in Glioblastomas

{kind=link}
| DNA methylation | Function | Gene | References |
|---|---|---|---|
| Hypermethylation | DNA repair | MGMT | [57] |
| Tumor suppressors | RB, HIC1, CDKN2A, p14, p16INK4, PTEN, RRP22, TP53, TES, BEX1, BEX2, BLU | [57,59,60,61,62,63] | |
| Cell proliferation | EMP3 | 64 | |
| Apoptosis | RASFF1A, CASP8, TNFRSF10A, TMS1 | [57,62,63] | |
| Suppressors of cytokine signaling | SOCS1, SOCS2, SOCS3 | [57] | |
| Wnt signaling | SFRP1, SFRP2, NKD2 | [57] | |
| Transcription factors | GATA6, HOXA, RFX1, RUNX3 | [61,63,65] | |
| Hypomethylation | Epigenetics | DNMT3B | [66] |
| Invasiveness | MMP9 | [57] | |
| Stemness | CD133 | [67] | |
| IL8, POTEH, IGF2 | [57,68,69] |
| Tumor classification | IDH1 mutation (%) | IDH2 mutation (%) | |||
|---|---|---|---|---|---|
| Yan et al. [74] | Hartman et al. [77] | Yan et al. [74] | Hartman et al. [77] | ||
| Astrocytic tumors | Pilocytic astrocytoma (grade I) | 0.0 | - | 0.0 | - |
| Subependymal giant-cell astrocytoma (grade II) | 0.0 | - | - | - | |
| Diffuse astrocytoma (grade II) | 83.3 | 72.7 | 6.6 | 0.9 | |
| Pleomorphic xanthoastrocytoma (grade II) | 14.0 | - | - | - | |
| Anaplastic astrocytoma (grade III) | 69.2 | 64.0 | 3.8 | 0.9 | |
| Secondary glioblastoma (grade IV) | 85.0 | - | 0.0 | - | |
| Primary adult glioblastoma (grade IV) | 5.0 | - | 0.0 | - | |
| Primary pediatric glioblastoma (grade IV) | 0.0 | - | 0.0 | - | |
| Oligodendroglial tumors | Oligodendroglioma (grade II) | 80.3 | 82.0 | 3.9 | 4.7 |
| Anaplastic oligodendroglioma (grade III) | 86.1 | 69.5 | 8.3 | 5.2 | |
| Oligoastrocytic tumors | Oligoastrocytoma (grade II) | 100.0 | 81.6 | - | 1.3 |
| Anaplastic oligoastrocytoma (grade III) | 100.0 | 66.1 | - | 6.2 | |
4. Epigenetic Inhibitors as Potential Anti-Glioblastoma Therapeutics
| Experiment | Enzyme | Inhibitor | References |
|---|---|---|---|
| in vitro | HDACs | Vorinostat (SAHA), PCI-24781, TSA, VPA, Scriptaid, MS-275, AR42 | [104,105,106,107,108,109,110,111,112] |
| HAT | curcumin | [113] | |
| LSD1 | tranylcypromine | [109] | |
| DNMT | 5-azacytidine, 5-aza-2'-deoxycytidine, zebularine, psammaplin A | [114] | |
| EZH2 | 3-deazaneplanocin | [67] | |
| in vivo | HDACs | Vorinostat (SAHA), TSA, VPA, MS-275 | [104,107,111,115,116] |
| DNMT | 5-azacytidine | [115] |
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Khorasanizadeh, S. The nucleosome: From genomic organization to genomic regulation. Cell 2004, 116, 259–272. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Turner, B.M. Reading signals on the nucleosome with a new nomenclature for modified histones. Nat. Struct. Mol. Biol. 2005, 12, 110–112. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Shogren-Knaak, M.; Ishii, H.; Sun, J.M.; Pazin, M.J.; Davie, J.R.; Peterson, C.L. Histone h4-k16 acetylation controls chromatin structure and protein interactions. Science 2006, 311, 844–847. [Google Scholar] [CrossRef]
- Wei, Y.; Yu, L.; Bowen, J.; Gorovsky, M.A.; Allis, C.D. Phosphorylation of histone H3 is required for proper chromosome condensation and segregation. Cell 1999, 97, 99–109. [Google Scholar] [CrossRef]
- Waldmann, T.; Schneider, R. Targeting histone modifications—Epigenetics in cancer. Curr. Opin. Cell Biol. 2013, 25, 184–189. [Google Scholar] [CrossRef]
- Yun, M.; Wu, J.; Workman, J.L.; Li, B. Readers of histone modifications. Cell Res. 2011, 21, 564–578. [Google Scholar] [CrossRef]
- Yang, X.J.; Seto, E. HATs and HDACs: From structure, function and regulation to novel strategies for therapy and prevention. Oncogene 2007, 26, 5310–5318. [Google Scholar] [CrossRef]
- Rea, S.; Eisenhaber, F.; O’Carroll, D.; Strahl, B.D.; Sun, Z.W.; Schmid, M.; Opravil, S.; Mechtler, K.; Ponting, C.P.; Allis, C.D.; et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 2000, 406, 593–599. [Google Scholar] [CrossRef]
- Black, J.C.; van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef]
- Dillon, S.C.; Zhang, X.; Trievel, R.C.; Cheng, X. The set-domain protein superfamily: Protein lysine methyltransferases. Genome Biol. 2005. [Google Scholar] [CrossRef]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef]
- Tsukada, Y.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006, 439, 811–816. [Google Scholar]
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef]
- Bergman, Y.; Cedar, H. DNA methylation dynamics in health and disease. Nat. Struct. Mol. Biol. 2013, 20, 274–281. [Google Scholar] [CrossRef]
- Chen, T.; Li, E. Structure and function of eukaryotic DNA methyltransferases. Curr. Top. Dev. Biol. 2004, 60, 55–89. [Google Scholar] [CrossRef]
- Bourc’his, D.; Xu, G.L.; Lin, C.S.; Bollman, B.; Bestor, T.H. Dnmt3L and the establishment of maternal genomic imprints. Science 2001, 294, 2536–2539. [Google Scholar] [CrossRef]
- Hata, K.; Okano, M.; Lei, H.; Li, E. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development 2002, 129, 1983–1993. [Google Scholar]
- Goll, M.G.; Kirpekar, F.; Maggert, K.A.; Yoder, J.A.; Hsieh, C.L.; Zhang, X.; Golic, K.G.; Jacobsen, S.E.; Bestor, T.H. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 2006, 311, 395–398. [Google Scholar] [CrossRef]
- Filion, G.J.; Zhenilo, S.; Salozhin, S.; Yamada, D.; Prokhortchouk, E.; Defossez, P.A. A family of human zinc finger proteins that bind methylated DNA and repress transcription. Mol. Cell Biol. 2006, 26, 169–181. [Google Scholar] [CrossRef]
- Dawson, M.A.; Kouzarides, T.; Huntly, B.J. Targeting epigenetic readers in cancer. N. Engl. J. Med. 2012, 367, 647–657. [Google Scholar] [CrossRef]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.; et al. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 2010, 363, 2424–2433. [Google Scholar] [CrossRef]
- Yan, X.J.; Xu, J.; Gu, Z.H.; Pan, C.M.; Lu, G.; Shen, Y.; Shi, J.Y.; Zhu, Y.M.; Tang, L.; Zhang, X.W.; et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat. Genet. 2011, 43, 309–315. [Google Scholar] [CrossRef]
- Simó-Riudalbas, L.; Melo, S.A.; Esteller, M. DNMT3B gene amplification predicts resistance to dna demethylating drugs. Genes Chromosomes Cancer 2011, 50, 527–534. [Google Scholar] [CrossRef]
- Delhommeau, F.; Dupont, S.; Valle, V.D.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; le Couedic, J.P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Weissmann, S.; Alpermann, T.; Grossmann, V.; Kowarsch, A.; Nadarajah, N.; Eder, C.; Dicker, F.; Fasan, A.; Haferlach, C.; Haferlach, T.; et al. Landscape of TET2 mutations in acute myeloid leukemia. Leukemia 2012, 26, 934–942. [Google Scholar] [CrossRef]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef]
- Krivtsov, A.V.; Armstrong, S.A. MLL translocations, histone modifications and leukaemia stem-cell development. Nat. Rev. Cancer 2007, 7, 823–833. [Google Scholar] [CrossRef]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.; Otte, A.P.; Hayes, D.F.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef]
- Ernst, T.; Chase, A.J.; Score, J.; Hidalgo-Curtis, C.E.; Bryant, C.; Jones, A.V.; Waghorn, K.; Zoi, K.; Ross, F.M.; Reiter, A.; et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet. 2010, 42, 722–726. [Google Scholar] [CrossRef]
- Nikoloski, G.; Langemeijer, S.M.; Kuiper, R.P.; Knops, R.; Massop, M.; Tönnissen, E.R.; van der Heijden, A.; Scheele, T.N.; Vandenberghe, P.; de Witte, T.; et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat. Genet. 2010, 42, 665–667. [Google Scholar] [CrossRef]
- Weikert, S.; Christoph, F.; Köllermann, J.; Müller, M.; Schrader, M.; Miller, K.; Krause, H. Expression levels of the EZH2 polycomb transcriptional repressor correlate with aggressiveness and invasive potential of bladder carcinomas. Int. J. Mol. Med. 2005, 16, 349–353. [Google Scholar]
- Van Haaften, G.; Dalgliesh, G.L.; Davies, H.; Chen, L.; Bignell, G.; Greenman, C.; Edkins, S.; Hardy, C.; O’Meara, S.; Teague, J.; et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat. Genet. 2009, 41, 521–523. [Google Scholar] [CrossRef]
- Johnstone, R.W.; Licht, J.D. Histone deacetylase inhibitors in cancer therapy: Is transcription the primary target? Cancer Cell 2003, 4, 13–18. [Google Scholar] [CrossRef]
- Bereshchenko, O.R.; Gu, W.; Dalla-Favera, R. Acetylation inactivates the transcriptional repressor BCL6. Nat. Genet. 2002, 32, 606–613. [Google Scholar] [CrossRef]
- Kaelin, W.G.; McKnight, S.L. Influence of metabolism on epigenetics and disease. Cell 2013, 153, 56–69. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef]
- Baysal, B.E.; Ferrell, R.E.; Willett-Brozick, J.E.; Lawrence, E.C.; Myssiorek, D.; Bosch, A.; van der Mey, A.; Taschner, P.E.; Rubinstein, W.S.; Myers, E.N.; et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000, 287, 848–851. [Google Scholar] [CrossRef]
- Tomlinson, I.P.; Alam, N.A.; Rowan, A.J.; Barclay, E.; Jaeger, E.E.; Kelsell, D.; Leigh, I.; Gorman, P.; Lamlum, H.; Rahman, S.; et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 2002, 30, 406–410. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef]
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874. [Google Scholar] [CrossRef] [Green Version]
- Mullen, A.R.; DeBerardinis, R.J. Genetically-defined metabolic reprogramming in cancer. Trends Endocrinol. Metab. 2012, 23, 552–559. [Google Scholar] [CrossRef]
- Rakheja, D.; Konoplev, S.; Medeiros, L.J.; Chen, W. IDH mutations in acute myeloid leukemia. Hum. Pathol. 2012, 43, 1541–1551. [Google Scholar] [CrossRef]
- Letouzé, E.; Martinelli, C.; Loriot, C.; Burnichon, N.; Abermil, N.; Ottolenghi, C.; Janin, M.; Menara, M.; Nguyen, A.T.; Benit, P.; et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 2013, 23, 739–752. [Google Scholar] [CrossRef]
- Grier, J.T.; Batchelor, T. Low-grade gliomas in adults. Oncologist 2006, 11, 681–693. [Google Scholar] [CrossRef]
- Minniti, G.; Muni, R.; Lanzetta, G.; Marchetti, P.; Enrici, R.M. Chemotherapy for glioblastoma: Current treatment and future perspectives for cytotoxic and targeted agents. Anticancer Res. 2009, 29, 5171–5184. [Google Scholar]
- Sarin, H. Recent progress towards development of effective systemic chemotherapy for the treatment of malignant brain tumors. J. Transl. Med. 2009. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Li, A.; Walling, J.; Ahn, S.; Kotliarov, Y.; Su, Q.; Quezado, M.; Oberholtzer, J.C.; Park, J.; Zenklusen, J.C.; Fine, H.A. Unsupervised analysis of transcriptomic profiles reveals six glioma subtypes. Cancer Res. 2009, 69, 2091–2099. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef]
- Kim, T.M.; Huang, W.; Park, R.; Park, P.J.; Johnson, M.D. A developmental taxonomy of glioblastoma defined and maintained by micrornas. Cancer Res. 2011, 71, 3387–3399. [Google Scholar] [CrossRef]
- Dubuc, A.M.; Mack, S.; Unterberger, A.; Northcott, P.A.; Taylor, M.D. The epigenetics of brain tumors. Methods Mol. Biol. 2012, 863, 139–153. [Google Scholar] [CrossRef]
- Polivka, J.; Rohan, V.; Topolcan, O.; Ferda, J. New molecularly targeted therapies for glioblastoma multiforme. Anticancer Res. 2012, 32, 2935–2946. [Google Scholar]
- Foltz, G.; Ryu, G.Y.; Yoon, J.G.; Nelson, T.; Fahey, J.; Frakes, A.; Lee, H.; Field, L.; Zander, K.; Sibenaller, Z.; et al. Genome-wide analysis of epigenetic silencing identifies BEX1 and BEX2 as candidate tumor suppressor genes in malignant glioma. Cancer Res. 2006, 66, 6665–6674. [Google Scholar] [CrossRef]
- Schmidt, N.; Windmann, S.; Reifenberger, G.; Riemenschneider, M.J. DNA hypermethylation and histone modifications downregulate the candidate tumor suppressor gene RRP22 on 22q12 in human gliomas. Brain Pathol. 2012, 22, 17–25. [Google Scholar] [CrossRef]
- Mueller, W.; Nutt, C.L.; Ehrich, M.; Riemenschneider, M.J.; von Deimling, A.; van den Boom, D.; Louis, D.N. Downregulation of RUNX3 and TES by hypermethylation in glioblastoma. Oncogene 2007, 26, 583–593. [Google Scholar] [CrossRef]
- Hesson, L.; Bièche, I.; Krex, D.; Criniere, E.; Hoang-Xuan, K.; Maher, E.R.; Latif, F. Frequent epigenetic inactivation of RASSF1A and BLU genes located within the critical 3p21.3 region in gliomas. Oncogene 2004, 23, 2408–2419. [Google Scholar] [CrossRef]
- Martinez, R.; Martin-Subero, J.I.; Rohde, V.; Kirsch, M.; Alaminos, M.; Fernandez, A.F.; Ropero, S.; Schackert, G.; Esteller, M. A microarray-based DNA methylation study of glioblastoma multiforme. Epigenetics 2009, 4, 255–264. [Google Scholar]
- Alaminos, M.; Dávalos, V.; Ropero, S.; Setién, F.; Paz, M.F.; Herranz, M.; Fraga, M.F.; Mora, J.; Cheung, N.K.; Gerald, W.L.; et al. Emp3, a myelin-related gene located in the critical 19q13.3 region, is epigenetically silenced and exhibits features of a candidate tumor suppressor in glioma and neuroblastoma. Cancer Res. 2005, 65, 2565–2571. [Google Scholar] [CrossRef]
- Cecener, G.; Tunca, B.; Egeli, U.; Bekar, A.; Tezcan, G.; Erturk, E.; Bayram, N.; Tolunay, S. The promoter hypermethylation status of GATA6, MGMT, and FHIT in glioblastoma. Cell Mol. Neurobiol. 2012, 32, 237–244. [Google Scholar] [CrossRef]
- Rajendran, G.; Shanmuganandam, K.; Bendre, A.; Muzumdar, D.; Mujumdar, D.; Goel, A.; Shiras, A. Epigenetic regulation of dna methyltransferases: DNMT1 and DNMT3B in gliomas. J. Neurooncol. 2011, 104, 483–494. [Google Scholar] [CrossRef]
- Alimova, I.; Venkataraman, S.; Harris, P.; Marquez, V.E.; Northcott, P.A.; Dubuc, A.; Taylor, M.D.; Foreman, N.K.; Vibhakar, R. Targeting the enhancer of zeste homologue 2 in medulloblastoma. Int. J. Cancer 2012, 131, 1800–1809. [Google Scholar] [CrossRef]
- Liu, X.; Tang, H.; Zhang, Z.; Li, W.; Wang, Z.; Zheng, Y.; Wu, M.; Li, G. Poteh hypomethylation, a new epigenetic biomarker for glioma prognosis. Brain Res. 2011, 1391, 125–131. [Google Scholar] [CrossRef]
- Uyeno, S.; Aoki, Y.; Nata, M.; Sagisaka, K.; Kayama, T.; Yoshimoto, T.; Ono, T. IGF2 but not H19 shows loss of imprinting in human glioma. Cancer Res. 1996, 56, 5356–5359. [Google Scholar]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Xiao, M.T.; Liu, L.X.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef]
- Kloosterhof, N.K.; Bralten, L.B.; Dubbink, H.J.; French, P.J.; van den Bent, M.J. Isocitrate dehydrogenase-1 mutations: A fundamentally new understanding of diffuse glioma? Lancet Oncol. 2011, 12, 83–91. [Google Scholar] [CrossRef]
- Ichimura, K. Molecular pathogenesis of IDH mutations in gliomas. Brain Tumor Pathol. 2012, 29, 131–139. [Google Scholar] [CrossRef]
- Berdasco, M.; Ropero, S.; Setien, F.; Fraga, M.F.; Lapunzina, P.; Losson, R.; Alaminos, M.; Cheung, N.K.; Rahman, N.; Esteller, M. Epigenetic inactivation of the sotos overgrowth syndromegene histone methyltransferase NSD1 in human neuroblastoma and glioma. Proc. Natl. Acad. Sci. USA 2009, 106, 21830–21835. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Balss, J.; Meyer, J.; Mueller, W.; Korshunov, A.; Hartmann, C.; von Deimling, A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008, 116, 597–602. [Google Scholar] [CrossRef]
- Bleeker, F.E.; Lamba, S.; Leenstra, S.; Troost, D.; Hulsebos, T.; Vandertop, W.P.; Frattini, M.; Molinari, F.; Knowles, M.; Cerrato, A.; et al. IDH1 mutations at residue p.R132 (IDH1R132) occur frequently in high-grade gliomas but not in other solid tumors. Hum. Mutat. 2009, 30, 7–11. [Google Scholar] [CrossRef]
- Hartmann, C.; Meyer, J.; Balss, J.; Capper, D.; Mueller, W.; Christians, A.; Felsberg, J.; Wolter, M.; Mawrin, C.; Wick, W.; et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: A study of 1,010 diffuse gliomas. Acta Neuropathol. 2009, 118, 469–474. [Google Scholar] [CrossRef] [Green Version]
- Mukasa, A.; Takayanagi, S.; Saito, K.; Shibahara, J.; Tabei, Y.; Furuya, K.; Ide, T.; Narita, Y.; Nishikawa, R.; Ueki, K.; et al. Significance of IDH mutations varies with tumor histology, grade, and genetics in Japanese glioma patients. Cancer Sci. 2012, 103, 587–592. [Google Scholar] [CrossRef]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef]
- Lai, A.; Kharbanda, S.; Pope, W.B.; Tran, A.; Solis, O.E.; Peale, F.; Forrest, W.F.; Pujara, K.; Carrillo, J.A.; Pandita, A.; et al. Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J. Clin. Oncol. 2011, 29, 4482–4490. [Google Scholar] [CrossRef]
- Laffaire, J.; Everhard, S.; Idbaih, A.; Crinière, E.; Marie, Y.; de Reyniès, A.; Schiappa, R.; Mokhtari, K.; Hoang-Xuan, K.; Sanson, M.; et al. Methylation profiling identifies 2 groups of gliomas according to their tumorigenesis. Neurooncology 2011, 13, 84–98. [Google Scholar]
- Liu, B.L.; Cheng, J.X.; Zhang, X.; Wang, R.; Zhang, W.; Lin, H.; Xiao, X.; Cai, S.; Chen, X.Y.; Cheng, H. Global histone modification patterns as prognostic markers to classify glioma patients. Cancer Epidemiol. Biomarkers Prev. 2010, 19, 2888–2896. [Google Scholar] [CrossRef]
- Venneti, S.; Felicella, M.M.; Coyne, T.; Phillips, J.J.; Gorovets, D.; Huse, J.T.; Kofler, J.; Lu, C.; Tihan, T.; Sullivan, L.M.; et al. Histone 3 lysine 9 trimethylation is differentially associated with isocitrate dehydrogenase mutations in oligodendrogliomas and high-grade astrocytomas. J. Neuropathol. Exp. Neurol. 2013, 72, 298–306. [Google Scholar] [CrossRef]
- Korur, S.; Huber, R.M.; Sivasankaran, B.; Petrich, M.; Morin, P.; Hemmings, B.A.; Merlo, A.; Lino, M.M. GSK3β regulates differentiation and growth arrest in glioblastoma. PLoS One 2009, 4, e7443. [Google Scholar] [CrossRef]
- Häyry, V.; Tanner, M.; Blom, T.; Tynninen, O.; Roselli, A.; Ollikainen, M.; Sariola, H.; Wartiovaara, K.; Nupponen, N.N. Copy number alterations of the polycomb gene BMI1 in gliomas. Acta Neuropathol. 2008, 116, 97–102. [Google Scholar] [CrossRef]
- Orzan, F.; Pellegatta, S.; Poliani, P.L.; Pisati, F.; Caldera, V.; Menghi, F.; Kapetis, D.; Marras, C.; Schiffer, D.; Finocchiaro, G. Enhancer of zeste 2 (EZH2) is up-regulated in malignant gliomas and in glioma stem-like cells. Neuropathol. Appl. Neurobiol. 2011, 37, 381–394. [Google Scholar] [CrossRef]
- Lee, J.; Son, M.J.; Woolard, K.; Donin, N.M.; Li, A.; Cheng, C.H.; Kotliarova, S.; Kotliarov, Y.; Walling, J.; Ahn, S.; et al. Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer Cell 2008, 13, 69–80. [Google Scholar] [CrossRef]
- Lucio-Eterovic, A.K.; Cortez, M.A.; Valera, E.T.; Motta, F.J.; Queiroz, R.G.; Machado, H.R.; Carlotti, C.G.; Neder, L.; Scrideli, C.A.; Tone, L.G. Differential expression of 12 histone deacetylase (HDAC) genes in astrocytomas and normal brain tissue: Class II and IV are hypoexpressed in glioblastomas. BMC Cancer 2008, 8, e243. [Google Scholar] [CrossRef]
- Sparmann, A.; van Lohuizen, M. Polycomb silencers control cell fate, development and cancer. Nat. Rev. Cancer 2006, 6, 846–856. [Google Scholar] [CrossRef]
- Suvà, M.L.; Riggi, N.; Janiszewska, M.; Radovanovic, I.; Provero, P.; Stehle, J.C.; Baumer, K.; le Bitoux, M.A.; Marino, D.; Cironi, L.; et al. EZH2 is essential for glioblastoma cancer stem cell maintenance. Cancer Res. 2009, 69, 9211–9218. [Google Scholar] [CrossRef]
- Natsume, A.; Ito, M.; Katsushima, K.; Ohka, F.; Hatanaka, A.; Shinjo, K.; Sato, S.; Takahashi, S.; Ishikawa, Y.; Takeuchi, I.; et al. Chromatin regulator PRC2 is a key regulator of epigenetic plasticity in glioblastoma. Cancer Res. 2013. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [CrossRef]
- Lewis, P.W.; Müller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef]
- Fontebasso, A.M.; Liu, X.Y.; Sturm, D.; Jabado, N. Chromatin remodeling defects in pediatric and young adult glioblastoma: A tale of a variant histone 3 tail. Brain Pathol. 2013, 23, 210–216. [Google Scholar] [CrossRef]
- Kannan, K.; Inagaki, A.; Silber, J.; Gorovets, D.; Zhang, J.; Kastenhuber, E.R.; Heguy, A.; Petrini, J.H.; Chan, T.A.; Huse, J.T. Whole-exome sequencing identifies ATRX mutation as a key molecular determinant in lower-grade glioma. Oncotarget 2012, 3, 1194–1203. [Google Scholar]
- Hsiao, H.H.; Nath, A.; Lin, C.Y.; Folta-Stogniew, E.J.; Rhoades, E.; Braddock, D.T. Quantitative characterization of the interactions among c-myc transcriptional regulators FUSE, FBP, and FIR. Biochemistry 2010, 49, 4620–4634. [Google Scholar] [CrossRef]
- Jiménez, G.; Shvartsman, S.Y.; Paroush, Z. The capicua repressor—A general sensor of RTK signaling in development and disease. J. Cell Sci. 2012, 125, 1383–1391. [Google Scholar] [CrossRef]
- Jiao, Y.; Killela, P.J.; Reitman, Z.J.; Rasheed, A.B.; Heaphy, C.M.; de Wilde, R.F.; Rodriguez, F.J.; Rosemberg, S.; Oba-Shinjo, S.M.; Nagahashi Marie, S.K.; et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 2012, 3, 709–722. [Google Scholar]
- Khan, O.; la Thangue, N.B. HDAC inhibitors in cancer biology: Emerging mechanisms and clinical applications. Immunol. Cell Biol. 2012, 90, 85–94. [Google Scholar] [CrossRef]
- Song, S.H.; Han, S.W.; Bang, Y.J. Epigenetic-based therapies in cancer: Progress to date. Drugs 2011, 71, 2391–2403. [Google Scholar] [CrossRef]
- Harrison, S.J.; Bishton, M.; Bates, S.E.; Grant, S.; Piekarz, R.L.; Johnstone, R.W.; Dai, Y.; Lee, B.; Araujo, M.E.; Prince, H.M. A focus on the preclinical development and clinical status of the histone deacetylase inhibitor, romidepsin (depsipeptide, istodax®). Epigenomics 2012, 4, 571–589. [Google Scholar] [CrossRef]
- Lansigan, F.; Foss, F.M. Current and emerging treatment strategies for cutaneous T-cell lymphoma. Drugs 2010, 70, 273–286. [Google Scholar] [CrossRef]
- Berendsen, S.; Broekman, M.; Seute, T.; Snijders, T.; van Es, C.; de Vos, F.; Regli, L.; Robe, P. Valproic acid for the treatment of malignant gliomas: Review of the preclinical rationale and published clinical results. Expert Opin. Investig. Drugs 2012, 21, 1391–1415. [Google Scholar] [CrossRef]
- Bajbouj, K.; Mawrin, C.; Hartig, R.; Schulze-Luehrmann, J.; Wilisch-Neumann, A.; Roessner, A.; Schneider-Stock, R. P53-dependent antiproliferative and pro-apoptotic effects of trichostatin A (TSA) in glioblastoma cells. J. Neurooncol. 2012, 107, 503–516. [Google Scholar] [CrossRef]
- Her, S.; Lee, M.S.; Morita, K. Trichostatin A stimulates steroid 5α-reductase gene expression in rat C6 glioma cells via a mechanism involving Sp1 and Sp3 transcription factors. J. Mol. Neurosci. 2010, 41, 252–262. [Google Scholar] [CrossRef]
- Höring, E.; Podlech, O.; Silkenstedt, B.; Rota, I.A.; Adamopoulou, E.; Naumann, U. The histone deacetylase inhibitor trichostatin a promotes apoptosis and antitumor immunity in glioblastoma cells. Anticancer Res. 2013, 33, 1351–1360. [Google Scholar]
- Hsu, Y.F.; Sheu, J.R.; Hsiao, G.; Lin, C.H.; Chang, T.H.; Chiu, P.T.; Wang, C.Y.; Hsu, M.J. P53 in trichostatin a induced C6 glioma cell death. Biochim. Biophys. Acta 2010, 1810, 504–513. [Google Scholar]
- Singh, M.M.; Manton, C.A.; Bhat, K.P.; Tsai, W.W.; Aldape, K.; Barton, M.C.; Chandra, J. Inhibition of LSD1 sensitizes glioblastoma cells to histone deacetylase inhibitors. Neurooncology 2011, 13, 894–903. [Google Scholar]
- Sharma, V.; Koul, N.; Joseph, C.; Dixit, D.; Ghosh, S.; Sen, E. HDAC inhibitor, scriptaid, induces glioma cell apoptosis through JNK activation and inhibits telomerase activity. J. Cell Mol. Med. 2010, 14, 2151–2161. [Google Scholar]
- Eyüpoglu, I.Y.; Hahnen, E.; Tränkle, C.; Savaskan, N.E.; Siebzehnrübl, F.A.; Buslei, R.; Lemke, D.; Wick, W.; Fahlbusch, R.; Blümcke, I. Experimental therapy of malignant gliomas using the inhibitor of histone deacetylase MS-275. Mol. Cancer Ther. 2006, 5, 1248–1255. [Google Scholar] [CrossRef]
- Yang, Y.L.; Huang, P.H.; Chiu, H.C.; Kulp, S.K.; Chen, C.S.; Kuo, C.J.; Chen, H.D. Histone deacetylase inhibitor AR42 regulates telomerase activity in human glioma cells via an Akt-dependent mechanism. Biochem. Biophys. Res. Commun. 2013, 435, 107–112. [Google Scholar] [CrossRef]
- Kang, S.K.; Cha, S.H.; Jeon, H.G. Curcumin-induced histone hypoacetylation enhances caspase-3-dependent glioma cell death and neurogenesis of neural progenitor cells. Stem Cells Dev. 2006, 15, 165–174. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, J.H.; Chie, E.K.; Young, P.D.; Kim, I.A.; Kim, I.H. DNMT (DNA methyltransferase) inhibitors radiosensitize human cancer cells by suppressing DNA repair activity. Radiat. Oncol 2012, 7, e39. [Google Scholar] [CrossRef]
- Ecke, I.; Petry, F.; Rosenberger, A.; Tauber, S.; Mönkemeyer, S.; Hess, I.; Dullin, C.; Kimmina, S.; Pirngruber, J.; Johnsen, S.A.; et al. Antitumor effects of a combined 5-aza-2'deoxycytidine and valproic acid treatment on rhabdomyosarcoma and medulloblastoma in Ptch mutant mice. Cancer Res. 2009, 69, 887–895. [Google Scholar] [CrossRef]
- Gao, J.; Chen, T.; Liu, J.; Liu, W.; Hu, G.; Guo, X.; Yin, B.; Gong, Y.; Zhao, J.; Qiang, B.; et al. Loss of NECL1, a novel tumor suppressor, can be restored in glioma by HDAC inhibitor-trichostatin a through Sp1 binding site. Glia 2009, 57, 989–999. [Google Scholar] [CrossRef]
- Spiller, S.E.; Ravanpay, A.C.; Hahn, A.W.; Olson, J.M. Suberoylanilide hydroxamic acid is effective in preclinical studies of medulloblastoma. J. Neurooncol. 2006, 79, 259–270. [Google Scholar] [CrossRef]
- Rohle, D.; Popovici-Muller, J.; Palaskas, N.; Turcan, S.; Grommes, C.; Campos, C.; Tsoi, J.; Clark, O.; Oldrini, B.; Komisopoulou, E.; et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013, 340, 626–630. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Maleszewska, M.; Kaminska, B. Is Glioblastoma an Epigenetic Malignancy? Cancers 2013, 5, 1120-1139. https://doi.org/10.3390/cancers5031120
Maleszewska M, Kaminska B. Is Glioblastoma an Epigenetic Malignancy? Cancers. 2013; 5(3):1120-1139. https://doi.org/10.3390/cancers5031120
Chicago/Turabian StyleMaleszewska, Marta, and Bozena Kaminska. 2013. "Is Glioblastoma an Epigenetic Malignancy?" Cancers 5, no. 3: 1120-1139. https://doi.org/10.3390/cancers5031120