Ras and Rheb Signaling in Survival and Cell Death



{kind=link}
Abstract
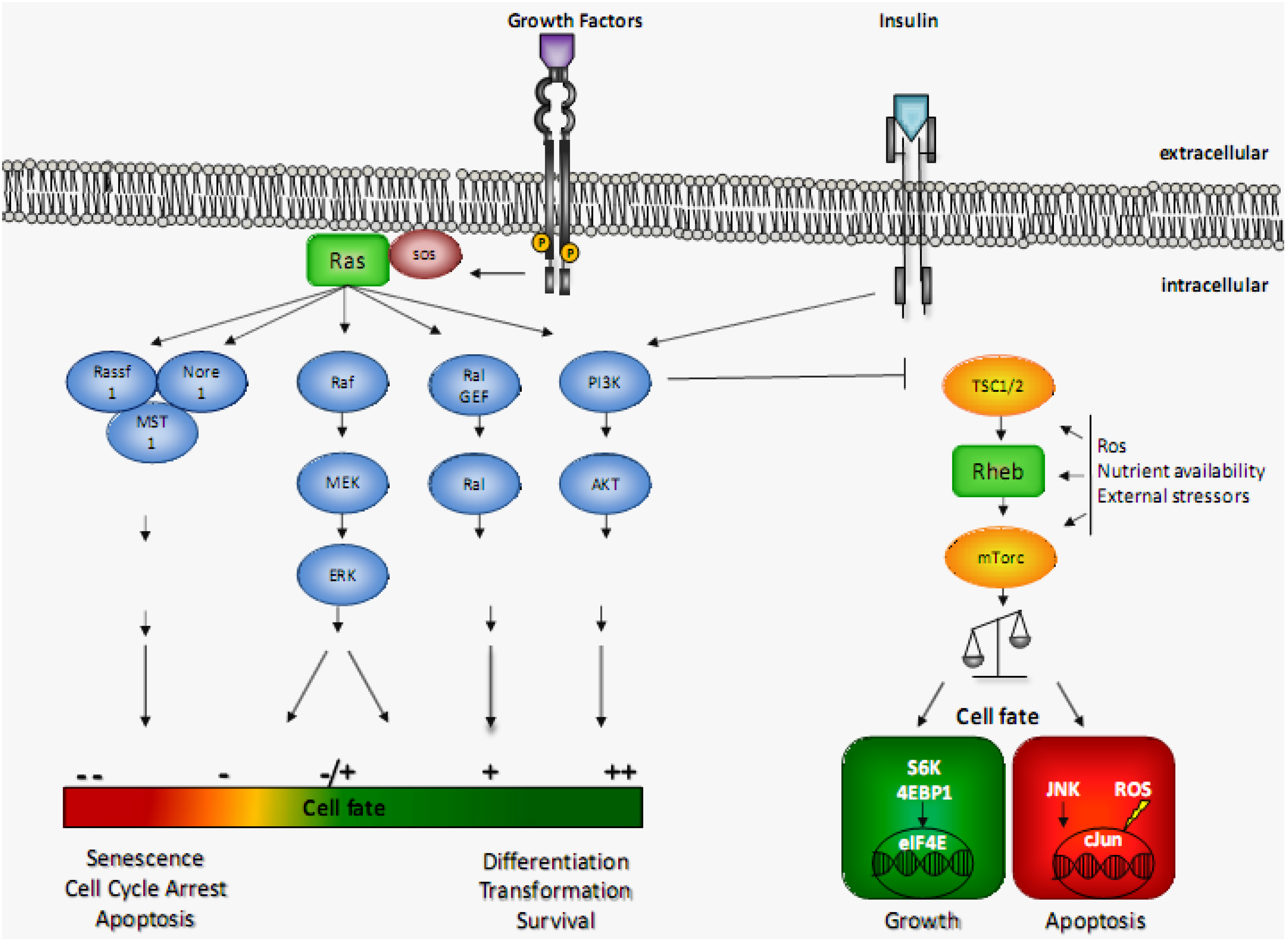
:1. Introduction
2. Ras Signaling and Induction in Apoptosis: The Pre-2000s
2.1. Growth Arrest/Senescence
2.2. Ras in Fas-Mediated Apoptosis
2.3. Role of NF-κB
2.4. Apoptosis via the Erk Pathway
2.5. Apoptosis via the Intrinsic Pathway
2.6. Apoptosis via the Extrinsic Pathway
3. Ras Signaling and Induction in Apoptosis: The Recent Findings
3.1. A trimeric Complex of Ras, Nore and MST1
3.2. Nore1a in Growth Suppression
3.3. Rassf1
4. Rheb Signaling and Induction of Apoptosis
4.1. Rheb Transcription and Posttranslational Modifications
4.2. The Two Signaling Faces of Rheb
4.3. Rheb in the Regulation of Autophagy
4.4. Tor Kinase
4.5. Tsc1/2 Complex
5. Conclusions

Acknowledgments
Conflict of Interest
References
- Karnoub, A.E.; Weinberg, R. A Ras oncogenes: Split personalities. Nat. Rev. Mol. Cell Biol. 2008, 9, 517–531. [Google Scholar] [CrossRef]
- Olson, M.F.; Marais, R. Ras protein signalling. Semin. Immunol. 2000, 12, 63–73. [Google Scholar] [CrossRef]
- Sasaki, A.T.; Firtel, R.A. Regulation of chemotaxis by the orchestrated activation of Ras, PI3K, and TOR. Eur. J. Cell. Biol 2006, 85, 873–895. [Google Scholar] [CrossRef]
- Pemberton, L.F.; Paschal, B.M. Mechanisms of receptor-mediated nuclear import and nuclear export. Traffic 2005, 6, 187–198. [Google Scholar] [CrossRef]
- Krieglstein, K.; Richter, S.; Farkas, L.; Schuster, N.; Dünker, N.; Oppenheim, R.W.; Unsicker, K. Reduction of endogenous transforming growth factors beta prevents ontogenetic neuron death. Nat. Neurosci. 2000, 3, 1085–1090. [Google Scholar] [CrossRef]
- Subramaniam, S.; Strelau, J.; Unsicker, K. Growth differentiation factor-15 prevents low potassium-induced cell death of cerebellar granule neurons by differential regulation of Akt and ERK pathways. J. Biol. Chem. 2003, 278, 8904–8912. [Google Scholar] [CrossRef]
- Borasio, G.D.; John, J.; Wittinghofer, A.; Barde, Y.A.; Sendtner, M.; Heumann, R. Ras p21 protein promotes survival and fiber outgrowth of cultured embryonic neurons. Neuron 1989, 2, 1087–1096. [Google Scholar] [CrossRef]
- Boriack-Sjodin, P.A.; Margarit, S.M.; Bar-Sagi, D.; Kuriyan, J. The structural basis of the activation of Ras by Sos. Nature 1998, 394, 337–343. [Google Scholar] [CrossRef]
- Chang, E.H.; Furth, M.E.; Scolnick, E.M.; Lowy, D.R. Tumorigenic transformation of mammalian cells induced by a normal human gene homologous to the oncogene of Harvey murine sarcoma virus. Nature 1982, 297, 479–483. [Google Scholar] [CrossRef]
- Der, C.J.; Finkel, T.; Cooper, G.M. Biological and biochemical properties of human rasH genes mutated at codon 61. Cell 1986, 44, 167–176. [Google Scholar] [CrossRef]
- Baines, A.T.; Xu, D.; Der, C.J. Inhibition of Ras for cancer treatment: The search continues. Future Med. Chem. 2011, 3, 1787–1808. [Google Scholar] [CrossRef]
- Heumann, R.; Goemans, C.; Bartsch, D.; Lingenhöhl, K.; Waldmeier, P.C.; Hengerer, B.; Allegrini, P.R.; Schellander, K.; Wagner, E.F.; Arendt, T.; et al. Transgenic activation of Ras in neurons promotes hypertrophy and protects from lesion-induced degeneration. J. Cell Biol. 2000, 151, 1537–1548. [Google Scholar] [CrossRef]
- Lowy, D.R.; Willumsen, B.M. Function and regulation of Ras. Annu. Rev. Biochem. 1993, 62, 851–891. [Google Scholar] [CrossRef]
- Kauffmann-Zeh, A.; Rodriguez-Viciana, P. Suppression of c-Myc-induced apoptosis by Ras signalling through PI (3) K and PKB. Nature 1997, 385, 544–548. [Google Scholar] [CrossRef]
- Yuspa, S.H.; Kilkenny, A.E.; Stanley, J.; Lichti, U. Keratinocytes blocked in phorbol ester-responsive early stage of terminal differentiation by sarcoma viruses. Nature 1985, 314, 459–462. [Google Scholar] [CrossRef]
- Yoshimura, T.; Arimura, N.; Kawano, Y.; Kawabata, S.; Wang, S.; Kaibuchi, K. Ras regulates neuronal polarity via the PI3-kinase/Akt/GSK-3beta/CRMP-2 pathway. Biochem. Biophys. Res. Commun. 2006, 340, 62–68. [Google Scholar] [CrossRef]
- Vavvas, D.; Li, X.; Avruch, J.; Zhang, X.F. Identification of Nore1 as a potential Ras effector. J. Biol. Chem. 1998, 273, 5439–5442. [Google Scholar] [CrossRef]
- Tommasi, S.; Dammann, R.; Jin, S.; Zhang, X.; Avruch, J.; Pfeifer, G.P. RASSF3 and NORE1: identification and cloning of two human homologues of the putative tumor suppressor gene RASSF1. Oncogene 2002, 21, 2713–2720. [Google Scholar] [CrossRef]
- Aspuria, P.-J.; Tamanoi, F. The Rheb family of GTP-binding proteins. Cell. Signal. 2004, 16, 1105–1112. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. Rheb fills a GAP between TSC and TOR. Trends Biochem. Sci. 2003, 28, 573–576. [Google Scholar] [CrossRef]
- Wataya-Kaneda, M.; Kaneda, Y.; Hino, O.; Adachi, H.; Hirayama, Y.; Seyama, K.; Satou, T.; Yoshikawa, K. Cells derived from tuberous sclerosis show a prolonged S phase of the cell cycle and increased apoptosis. Arch. Dermatol. Res. 2001, 293, 460–469. [Google Scholar] [CrossRef]
- Nardella, C.; Chen, Z.; Salmena, L.; Carracedo, A.; Alimonti, A.; Egia, A.; Carver, B.; Gerald, W.; Cordon-Cardo, C.; Pandolfi, P.P. Aberrant Rheb-mediated mTORC1 activation and Pten haploinsufficiency are cooperative oncogenic events. Gene Dev. 2008, 22, 2172–2177. [Google Scholar] [CrossRef]
- Sancak, Y.; Bar-peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to lysosomal surface and is necessary for its activation by amino acids. Cell 2011, 141, 290–303. [Google Scholar]
- Karassek, S.; Berghaus, C.; Schwarten, M.; Goemans, C.G.; Ohse, N.; Kock, G.; Jockers, K.; Neumann, S.; Gottfried, S.; Herrmann, C.; et al. Ras homolog enriched in brain (Rheb) enhances apoptotic signaling. J. Biol. Chem. 2010, 285, 33979–33991. [Google Scholar] [CrossRef]
- Cox, A.D.; Der, C.J. Ras history: The saga continues. Small GTPases 2010, 1, 2–27. [Google Scholar] [CrossRef]
- Heumann, R. Neurotrophin signalling. Curr. Opin. Neuro. Biol. 1994, 4, 668–679. [Google Scholar] [CrossRef]
- Xue, L.; Murray, J.H.; Tolkovsky, A.M. The Ras/phosphatidylinositol 3-kinase and Ras/ERK pathways function as independent survival modules each of which inhibits a distinct apoptotic signaling pathway in sympathetic neurons. J. Biol. Chem. 2000, 275, 8817–8824. [Google Scholar] [CrossRef]
- Cox, A.D.; Der, C.J. The dark side of Ras: regulation of apoptosis. Oncogene 2003, 22, 8999–9006. [Google Scholar] [CrossRef]
- Oppenheim, R. Cell Death During Development Of The Nervous System. Annu. Rev. Neurosci. 1991, 14, 453–501. [Google Scholar] [CrossRef]
- Conradt, B. Genetic control of programmed cell death during animal development. Annu. Rev. Genet. 2009, 43, 493–523. [Google Scholar] [CrossRef]
- Newbold, R.F.; Overell, R.W. Fibroblast immortality is a prerequisite for transformation by EJ c-Ha-ras oncogene. Nature 1983, 304, 648–651. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras Provokes Premature Cell Senescence Associated with Accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Gulbins, E.; Brenner, B.; Koppenhoefer, U.; Linderkamp, O.; Lang, F. Fas or ceramide induce apoptosis by Ras-regulated phosphoinositide-3-kinase activation. J. Leukocyte Biol. 1998, 63, 253–263. [Google Scholar]
- Gulbins, E.; Coggeshall, K.M.; Brenner, B.; Schlottmann, K.; Linderkamp, O.; Lang, F. Fas-induced apoptosis is mediated by activation of a Ras and Rac protein-regulated signaling pathway. J. Biol. Chem. 1996, 271, 26389–26394. [Google Scholar]
- Wajant, H. The Fas signaling pathway: More than a paradigm. Science 2002, 296, 1635–1636. [Google Scholar] [CrossRef]
- Karim, F.D.; Rubin, G.M. Ectopic expression of activated Ras1 induces hyperplastic growth and increased cell death in Drosophila imaginal tissues. Development 1998, 125, 1–9. [Google Scholar]
- Kurada, P.; White, K. Ras promotes cell survival in Drosophila by downregulating hid expression. Cell 1998, 95, 319–329. [Google Scholar] [CrossRef]
- Perona, R.; Montaner, S.; Saniger, L.; Sánchez-Pérez, I.; Bravo, R.; Lacal, J.C. Activation of the nuclear factor-kappaB by Rho, CDC42, and Rac-1 proteins. Gene Dev. 1997, 11, 463–475. [Google Scholar] [CrossRef]
- Meyer, M.; Schreck, R.; Baeuerle, P.A. H2O2 and antioxidants have opposite effects on activation of NF-kappa B and AP-1 in intact cells: AP-1 as secondary antioxidant-responsive factor. EMBO J. 1993, 12, 2005–2015. [Google Scholar]
- Sulciner, D.J.; Irani, K.; Yu, Z.X.; Ferrans, V.J.; Goldschmidt-Clermont, P.; Finkel, T. Rac1 regulates a cytokine-stimulated, redox-dependent pathway necessary for NF-kappaB activation. Mol. Cell Biol. 1996, 16, 7115–7121. [Google Scholar]
- Kaltschmidt, B.; Heinrich, M.; Kaltschmidt, C. Stimulus-dependent activation of nf-κb specifies apoptosis or neuroprotection in cerebellar granule cells. Neuromol Med. 2002, 2, 299–309. [Google Scholar] [CrossRef]
- Joneson, T.; Bar-Sagi, D. Suppression of Ras-induced apoptosis by the Rac GTPase. Mol. Cell. Biol. 1999, 19, 5892–5901. [Google Scholar]
- Irani, K. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 1997, 275, 1649–1652. [Google Scholar] [CrossRef]
- Lambert, J.M.; Lambert, Q.T.; Reuther, G.W.; Malliri, A.; Siderovski, D.P.; Sondek, J.; Collard, J.G.; Der, C.J. Tiam1 mediates Ras activation of Rac by a PI(3)K-independent mechanism. Nat. Cell Biol. 2002, 4, 621–625. [Google Scholar]
- Overmeyer, J.H.; Kaul, A.; Johnson, E.E.; Maltese, A.W. Active ras triggers death in glioblastoma cells through hyperstimulation of macropinocytosis. Mol. Cancer Res. 2008, 6, 965–977. [Google Scholar] [CrossRef]
- Bonni, A.; Brunet, A.; West, A.E.; Datta, S.R.; Takasu, M.A.; Greenberg, M.E. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 1999, 286, 1358–1362. [Google Scholar] [CrossRef]
- Hetman, M.; Xia, Z. Signaling pathways mediating anti-apoptotic action of neurotrophins. Acta Neurobiol. Exp. 2000, 60, 531–545. [Google Scholar]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef]
- Zhuang, S.; Schnellmann, R.G. A death-promoting role for extracellular signal-regulated kinase. J. Pharmacol. Exp. Ther. 2006, 319, 991–997. [Google Scholar] [CrossRef]
- Cagnol, S.; Chambard, J.-C. ERK and cell death: Mechanisms of ERK-induced cell death—apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef]
- Nowak, G. Protein kinase C-alpha and ERK1/2 mediate mitochondrial dysfunction, decreases in active Na+ transport, and cisplatin-induced apoptosis in renal cells. J. Biol. Chem. 2002, 277, 43377–43388. [Google Scholar] [CrossRef]
- Nowak, G.; Clifton, G.L.; Godwin, M.L.; Bakajsova, D. Activation of ERK1/2 pathway mediates oxidant-induced decreases in mitochondrial function in renal cells. Am. J. Physiol. RenalPhysiol. 2006, 291, 840–855. [Google Scholar]
- Rao, G.N. Hydrogen peroxide induces complex formation of SHC-Grb2-SOS with receptor tyrosine kinase and activates Ras and extracellular signal-regulated protein kinases group of mitogen-activated protein kinases. Oncogene 1996, 13, 713–719. [Google Scholar]
- Chen, W.; Martindale, J.L.; Holbrook, N.J.; Liu, Y. Tumor promoter arsenite activates extracellular signal-regulated kinase through a signaling pathway mediated by epidermal growth factor receptor and Shc. Mol. Cell. Biol. 1998, 18, 5178–5188. [Google Scholar]
- Guyton, K.Z.; Liu, Y.; Gorospe, M.; Xu, Q.; Holbrook, N.J. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J. Biol. Chem. 1996, 271, 4138–4142. [Google Scholar]
- Asai, A.; Qiu, J.H.; Narita, Y.; Chi, S.; Saito, N.; Shinoura, N.; Hamada, H.; Kuchino, Y.; Kirino, T. High level calcineurin activity predisposes neuronal cells to apoptosis. J. Biol. Chem. 1999, 274, 34450–34458. [Google Scholar] [CrossRef]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef]
- Li, D.W.; Liu, J.; Mao, Y.; Xiang, H.; Wang, J.; Ma, W.; Dong, Z.; Pike, H.M.; Brown, R.E.; Reed, J.C. Calcium-activated RAF/MEK/ERK Signaling Pathway Mediates p53-dependent apoptosis and is abrogated by B-crystallin through inhibition of RAS activation. Mol. Biol. Cell 2005, 16, 4437–4453. [Google Scholar] [CrossRef]
- Mammucari, C.; Rizzuto, R. Signaling pathways in mitochondrial dysfunction and aging. Mech. Ageing Dev. 2010, 131, 536–543. [Google Scholar] [CrossRef]
- Franklin, J.L. Redox regulation of the intrinsic pathway in neuronal apoptosis. Antioxid. Redox Sign. 2011, 14, 1437–1448. [Google Scholar] [CrossRef]
- Chen, C.Y.; Faller, D.V. Phosphorylation of Bcl-2 protein and association with p21Ras in Ras-induced apoptosis. J. Biol. Chem. 1996, 271, 2376–2379. [Google Scholar] [CrossRef]
- Denis, G.V.; Yu, Q.; Ma, P.; Deeds, L.; Faller, D.V.; Chen, C.-Y. Bcl-2, via its BH4 domain, blocks apoptotic signaling mediated by mitochondrial Ras. J. Biol. Chem. 2003, 278, 5775–5785. [Google Scholar]
- Kang, J.; Pervaiz, S. Crosstalk between Bcl-2 family and Ras family small GTPases: Potential cell fate regulation? Front. Oncol. 2012, 2, 206. [Google Scholar]
- Kuo, M.L.; Chou, Y.W.; Chau, Y.P.; Huang, T.S. Resistance to apoptosis induced by alkylating agents in v-Ha-ras-transformed cells due to defect in p53 function. Mol. Carcinog. 1997, 18, 221–231. [Google Scholar] [CrossRef]
- Davis, J.M.; Navolanic, P.M.; Weinstein-oppenheimer, C.R.; Steelman, L.S.; Hu, W.; Konopleva, M.; Blagosklonny, M.V. Raf-1 and Bcl-2 induce distinct and common pathways that contribute to breast cancer drug resistance. Clin. Cancer Res. 2003, 9, 1161–1170. [Google Scholar]
- Suzuki, M.; Abe, A.; Imagama, S.; Nomura, Y.; Tanizaki, R.; Minami, Y.; Hayakawa, F.; Ito, Y.; Katsumi, A.; Yamamoto, K.; et al. BCR-ABL-independent and RAS/MAPK pathway-dependent form of imatinib resistance in Ph-positive acute lymphoblastic leukemia cell line with activation of EphB4. Eur. J. Haematol. 2010, 84, 229–238. [Google Scholar] [CrossRef]
- Sinha, B.K.; Yamazaki, H.; Eliot, H.M.; Schneider, E.; Borner, M.M.; O’Connor, P.M. Relationships between proto-oncogene expression and apoptosis induced by anticancer drugs in human prostate tumor cells. Biochim. Biophys. Acta 1995, 1270, 12–18. [Google Scholar] [CrossRef]
- Brown, L.; Benchimol, S. The involvement of MAPK signaling pathways in determining the cellular response to p53 activation: Cell cycle arrest or apoptosis. J. Biol. Chem. 2006, 281, 3832–3840. [Google Scholar] [CrossRef]
- Avruch, J.; Hara, K.; Lin, Y.; Liu, M.; Long, X.; Ortiz-Vega, S.; Yonezawa, K. Insulin and amino-acid regulation of mTOR signaling and kinase activity through the Rheb GTPase. Oncogene 2006, 25, 6361–6372. [Google Scholar] [CrossRef]
- Renganathan, H.; Vaidyanathan, H.; Knapinska, A.; Ramos, J.W. Phosphorylation of PEA-15 switches its binding specificity from ERK/MAPK to FADD. Biochem. J. 2005, 390, 729–735. [Google Scholar] [CrossRef]
- Drosopoulos, K.G.; Roberts, M.L.; Cermak, L.; Sasazuki, T.; Shirasawa, S.; Andera, L.; Pintzas, A. Transformation by oncogenic RAS sensitizes human colon cells to TRAIL-induced apoptosis by up-regulating death receptor 4 and death receptor 5 through a MEK-dependent pathway. J. Biol. Chem. 2005, 280, 22856–22867. [Google Scholar]
- Zhou, D.; Medoff, B.D.; Chen, L.; Li, L.; Zhang, X.; Praskova, M.; Liu, M.; Landry, A.; Blumberg, R.S.; Boussiotis, V.A.; et al. The Nore1B/Mst1 complex restrains antigen receptor-induced proliferation of naïve T cells. Proc. Natl. Acad. Sci. USA 2008, 105, 20321–20326. [Google Scholar] [CrossRef]
- Hesson, L.; Dallol, A.; Minna, J.D.; Maher, E.R.; Latif, F. NORE1A, a homologue of RASSF1A tumour suppressor gene is inactivated in human cancers. Oncogene 2003, 22, 947–954. [Google Scholar] [CrossRef]
- Ortiz-vega, S.; Khokhlatchev, A.; Nedwidek, M.; Zhang, X.; Dammann, R.; Pfeifer, G.P.; Avruch, J. The putative tumor suppressor RASSF1A homodimerizes and heterodimerizes with the Ras-GTP binding protein Nore1. Oncogene 2002, 21, 1381–1390. [Google Scholar] [CrossRef]
- Stieglitz, B.; Bee, C.; Schwarz, D.; Yildiz, O.; Moshnikova, A.; Khokhlatchev, A.; Herrmann, C. Novel type of Ras effector interaction established between tumour suppressor NORE1A and Ras switch II. EMBO J. 2008, 27, 1995–2005. [Google Scholar] [CrossRef]
- Khokhlatchev, A.; Rabizadeh, S.; Xavier, R.; Nedwidek, M.; Chen, T.; Zhang, X.; Seed, B.; Avruch, J. Identification of a novel Ras-regulated proapoptotic pathway. Curr. Biol. 2002, 12, 253–265. [Google Scholar] [CrossRef]
- Park, J.; Kang, S.I.; Lee, S.-Y.; Zhang, X.F.; Kim, M.S.; Beers, L.F.; Lim, D.-S.; Avruch, J.; Kim, H.-S.; Lee, S.B. Tumor suppressor ras association domain family 5 (RASSF5/NORE1) mediates death receptor ligand-induced apoptosis. J. Biol. Chem. 2010, 285, 35029–35038. [Google Scholar] [CrossRef]
- Praskova, M.; Khoklatchev, A.; Ortiz-Vega, S.; Avruch, J. Regulation of the MST1 kinase by autophosphorylation, by the growth inhibitory proteins, RASSF1 and NORE1, and by Ras. Biochem. J. 2004, 381, 453–462. [Google Scholar] [CrossRef]
- Vos, M.D.; Martinez, A.; Ellis, C.A.; Vallecorsa, T.; Clark, G.J. The pro-apoptotic Ras effector Nore1 may serve as a Ras-regulated tumor suppressor in the lung. J. Biol. Chem. 2003, 278, 21938–21943. [Google Scholar]
- Aoyama, Y.; Avruch, J.; Zhang, X.-F. Nore1 inhibits tumor cell growth independent of Ras or the MST1/2 kinases. Oncogene 2004, 23, 3426–3433. [Google Scholar] [CrossRef]
- Dallol, A.; Agathanggelou, A.; Fenton, S.L.; Ahmed-Choudhury, J.; Hesson, L.; Vos, M.D.; Clark, G.J.; Downward, J.; Maher, E.R.; Latif, F. RASSF1A interacts with microtubule-associated proteins and modulates microtubule dynamics. Cancer Res. 2004, 64, 4112–4116. [Google Scholar] [CrossRef]
- Shao, J.; Sheng, H.; DuBois, R.N.; Beauchamp, R.D. Oncogenic Ras-mediated cell growth arrest and apoptosis are associated with increased ubiquitin-dependent cyclin D1 degradation. J. Biol. Chem. 2000, 275, 22916–22924. [Google Scholar] [CrossRef]
- Benanti, J.A.; Galloway, D.A. The normal response to RAS: Senescence or transformation? Cell Cycle 2004, 3, 715–717. [Google Scholar]
- Moshnikova, A.; Frye, J.; Shay, J.W.; Minna, J.D.; Khokhlatchev, A.V. The Growth and Tumor Suppressor NORE1A Is a Cytoskeletal Protein That Suppresses Growth by Inhibition of the ERK Pathway. J. Biol. Chem. 2006, 281, 8143–8152. [Google Scholar]
- Bee, C.; Moshnikova, A.; Mellor, C.D.; Molloy, J.E.; Koryakina, Y.; Stieglitz, B.; Khokhlatchev, A.; Herrmann, C. Growth and tumor suppressor NORE1A is a regulatory node between Ras signaling and microtubule nucleation. J. Biol. Chem. 2010, 285, 16258–16266. [Google Scholar] [CrossRef]
- Vos, M.D.; Ellis, C.A.; Bell, A.; Birrer, M.J.; Clark, G.J. Ras uses the novel tumor suppressor RASSF1 as an effector to mediate apoptosis. J. Biol. Chem. 2000, 275, 35669–35672. [Google Scholar]
- Vos, M.D.; Dallol, A.; Eckfeld, K.; Allen, N.P.C.; Donninger, H.; Hesson, L.B.; Calvisi, D.; Latif, F.; Clark, G.J. The RASSF1A tumor suppressor activates Bax via MOAP-1. J. Biol. Chem. 2006, 281, 4557–4563. [Google Scholar]
- Calvisi, D.F.; Ladu, S.; Gorden, A.; Farina, M.; Conner, E.A.; Lee, J.-S.; Factor, V.M.; Thorgeirsson, S.S. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology 2006, 130, 1117–1128. [Google Scholar] [CrossRef]
- Matallanas, D.; Romano, D.; Al-Mulla, F.; O’Neill, E.; Al-Ali, W.; Crespo, P.; Doyle, B.; Nixon, C.; Sansom, O.; Drosten, M.; et al. Mutant K-Ras activation of the proapoptotic MST2 pathway is antagonized by wild-type K-Ras. Mol. Cell 2011, 44, 893–906. [Google Scholar] [CrossRef]
- Van der Weyden, L.; Adams, D.J. The Ras-association domain family (RASSF) members and their role in human tumourigenesis. Biochim. Biophys. Acta 2007, 1776, 58–85. [Google Scholar]
- Donninger, H.; Vos, M.D.; Clark, G.J. The RASSF1A tumor suppressor. J. Cell Sci. 2007, 120, 3163–3172. [Google Scholar] [CrossRef]
- Dammann, R.; Li, C.; Yoon, J.H.; Chin, P.L.; Bates, S.; Pfeifer, G.P. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat. Genet. 2000, 25, 315–319. [Google Scholar] [CrossRef]
- Burbee, D.G.; Forgacs, E.; Zöchbauer-Müller, S.; Shivakumar, L.; Fong, K.; Gao, B.; Randle, D.; Kondo, M.; Virmani, A.; Bader, S.; et al. Epigenetic inactivation of RASSF1A in lung and breast cancers and malignant phenotype suppression. J. Nat. Cancer Inst. 2001, 93, 691–699. [Google Scholar] [CrossRef]
- Dreijerink, K.; Braga, E.; Kuzmin, I.; Geil, L.; Duh, F.M.; Angeloni, D.; Zbar, B.; Lerman, M.I.; Stanbridge, E.J.; Minna, J.D.; Protopopov, A.; et al. The candidate tumor suppressor gene, RASSF1A, from human chromosome 3p21.3 is involved in kidney tumorigenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 7504–7509. [Google Scholar] [CrossRef]
- Kuzmin, I.; Gillespie, J.W.; Protopopov, A.; Geil, L.; Dreijerink, K.; Yang, Y.; Vocke, C.D.; Duh, F.; Zabarovsky, E.; Minna, J.D.; et al. The RASSF1A tumor suppressor gene is inactivated in prostate tumors and suppresses growth of prostate carcinoma cells. Cancer Res. 2002, 62, 3498–3502. [Google Scholar]
- Hesson, L.; Bièche, I.; Krex, D.; Criniere, E.; Hoang-Xuan, K.; Maher, E.R.; Latif, F. Frequent epigenetic inactivation of RASSF1A and BLU genes located within the critical 3p21.3 region in gliomas. Oncogene 2004, 23, 2408–2419. [Google Scholar]
- Li, J.; Wang, F.; Protopopov, A.; Malyukova, A.; Kashuba, V.; Minna, J.D.; Lerman, M.I.; Klein, G.; Zabarovsky, E. Inactivation of RASSF1C during in vivo tumor growth identifies it as a tumor suppressor gene. Oncogene 2004, 23, 5941–5949. [Google Scholar] [CrossRef]
- Ji, L.; Nishizaki, M.; Gao, B.; Burbee, D.; Kondo, M.; Kamibayashi, C.; Xu, K.; Yen, N.; Atkinson, E.N.; Fang, B.; et al. Expression of several genes in the human chromosome 3p21. 3 Homozygous deletion region by an adenovirus vector results in tumor suppressor activities in vitro and in vivo. Cancer Res. 2002, 62, 2715–2720. [Google Scholar]
- Amaar, Y.G.; Minera, M.G.; Hatran, L.K.; Strong, D.D.; Mohan, S.; Reeves, M.E. Ras association domain family 1C protein stimulates human lung cancer cell proliferation. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 291, 1185–1190. [Google Scholar] [CrossRef]
- Kitagawa, D.; Kajiho, H.; Negishi, T.; Ura, S.; Watanabe, T.; Wada, T.; Ichijo, H.; Katada, T.; Nishina, H. Release of RASSF1C from the nucleus by Daxx degradation links DNA damage and SAPK/JNK activation. EMBO J. 2006, 25, 3286–3297. [Google Scholar] [CrossRef]
- Avruch, J.; Xavier, R.; Bardeesy, N.; Zhang, X.-F.; Praskova, M.; Zhou, D.; Xia, F. Rassf family of tumor suppressor polypeptides. J. Biol. Chem. 2009, 284, 11001–11005. [Google Scholar]
- Yamagata, K.; Sanders, L.K.; Kaufmann, W.E.; Yee, W.; Barnes, C.A.; Nathans, D.; Worley, P.F. Rheb, a growth factor- and synaptic activity-regulated gene, encodes a novel Ras-related protein. J. Biol. Chem. 1994, 269, 16333–16339. [Google Scholar]
- Pan, D.; Dong, J.; Zhang, Y.; Gao, X. Tuberous sclerosis complex: From Drosophila to human disease. Trends Cell. Biol. 2004, 14, 78–85. [Google Scholar] [CrossRef]
- Li, Y.-H.; Werner, H.; Püschel, A.W. Rheb and mTOR regulate neuronal polarity through Rap1B. J. Biol. Chem. 2008, 283, 33784–33792. [Google Scholar] [CrossRef]
- Zhou, X.; Ikenoue, T.; Chen, X.; Li, L.; Inoki, K.; Guan, K.-L. Rheb controls misfolded protein metabolism by inhibiting aggresome formation and autophagy. Proc. Natl. Acad. Sci. USA 2009, 106, 8923–8928. [Google Scholar]
- Wang, X.; Proud, C.G. The mTOR pathway in the control of protein synthesis. Physiology 2006, 21, 362–369. [Google Scholar] [CrossRef]
- Kita, K.; Wu, Y.P.; Sugaya, S.; Moriya, T.; Nomura, J.; Takahashi, S.; Yamamori, H.; Nakajima, N.; Suzuki, N. Search for UV-responsive genes in human cells by differential mRNA display: Involvement of human ras-related GTP-binding protein, Rheb, in UV susceptibility. Biochem. Biophys. Res. Commun. 2000, 274, 859–864. [Google Scholar] [CrossRef]
- Cao, M.; Tan, X.; Jin, W.; Zheng, H.; Xu, W.; Rui, Y.; Li, L.; Cao, J.; Wu, X.; Cui, G.; et al. Upregulation of Ras homolog enriched in the brain (Rheb) in lipopolysaccharide-induced neuroinflammation. Neurochem. Int. 2013, 62, 406–417. [Google Scholar] [CrossRef]
- Norsted Gregory, E.; Codeluppi, S.; Gregory, J.A.; Steinauer, J.; Svensson, C.I. Mammalian target of rapamycin in spinal cord neurons mediates hypersensitivity induced by peripheral inflammation. Neuroscience 2010, 169, 1392–402. [Google Scholar] [CrossRef]
- Im, E.; von Lintig, F.C.; Chen, J.; Zhuang, S.; Qui, W.; Chowdhury, S.; Worley, P.F.; Boss, G.R.; Pilz, R.B. Rheb is in a high activation state and inhibits B-Raf kinase in mammalian cells. Oncogene 2002, 21, 6356–6365. [Google Scholar] [CrossRef]
- Zheng, M.; Wang, Y.-H.; Wu, X.-N.; Wu, S.-Q.; Lu, B.-J.; Dong, M.-Q.; Zhang, H.; Sun, P.; Lin, S.-C.; Guan, K.-L.; Han, J. Inactivation of Rheb by PRAK-mediated phosphorylation is essential for energy-depletion-induced suppression of mTORC1. Nat. Cell Biol. 2011, 13, 263–272. [Google Scholar] [CrossRef]
- Yoshida, S.; Hong, S.; Suzuki, T.; Nada, S.; Mannan, A.M.; Wang, J.; Okada, M.; Guan, K.-L.; Inoki, K. Redox regulates mammalian target of rapamycin complex 1 (mTORC1) activity by modulating the TSC1/TSC2-Rheb GTPase pathway. J. Biol. Chem. 2011, 286, 32651–32660. [Google Scholar] [CrossRef]
- Ma, D.; Bai, X.; Zou, H.; Lai, Y.; Jiang, Y. Rheb GTPase controls apoptosis by regulating interaction of FKBP38 with Bcl-2 and Bcl-XL. J. Biol. Chem. 2010, 285, 8621–8627. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Shirane, M.; Nakayama, K.I. Inherent calcineurin inhibitor FKBP38 targets Bcl-2 to mitochondria and inhibits apoptosis. Nat. Cell Biol. 2003, 5, 28–37. [Google Scholar] [CrossRef]
- Edlich, F.; Weiwad, M.; Erdmann, F.; Fanghänel, J.; Jarczowski, F.; Rahfeld, J.-U.; Fischer, G. Bcl-2 regulator FKBP38 is activated by Ca2+/calmodulin. EMBO J. 2005, 24, 2688–2699. [Google Scholar] [CrossRef]
- Germain, M.; Shore, G.C. Cellular distribution of Bcl-2 family proteins. Sci STKE 2003, 2003, 1–3. [Google Scholar]
- Honjoh, S.; Yamamoto, T.; Uno, M.; Nishida, E. Signalling through RHEB-1 mediates intermittent fasting-induced longevity in C. elegans. Nature 2009, 457, 726–730. [Google Scholar] [CrossRef]
- Levine, B.; Yuan, J. Review series Autophagy in cell death: An innocent convict? J. Clin. Invest. 2005, 115, 2679–2688. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Alpha-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar]
- Iwata, A.; Riley, B.E.; Johnston, J.A.; Kopito, R.R. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 2005, 280, 40282–40292. [Google Scholar] [CrossRef]
- White, E.; Karp, C.; Strohecker, A.M.; Guo, Y.; Mathew, R. Role of autophagy in suppression of inflammation and cancer. Curr. Opin. Cell Biol. 2010, 22, 212–217. [Google Scholar] [CrossRef]
- Mavrakis, K.J.; Zhu, H.; Silva, R.L.A.; Mills, J.R.; Teruya-Feldstein, J.; Lowe, S.W.; Tam, W.; Pelletier, J.; Wendel, H. Tumorigenic activity and therapeutic inhibition of Rheb GTPase. Gene Dev. 2008, 22, 2178–2188. [Google Scholar] [CrossRef]
- Lassman, A.B.; Rossi, M.R.; Raizer, J.J.; Razier, J.R.; Abrey, L.E.; Lieberman, F.S.; Grefe, C.N.; Lamborn, K.; Pao, W.; Shih, A.H.; et al. Molecular study of malignant gliomas treated with epidermal growth factor receptor inhibitors: Tissue analysis from North American Brain Tumor Consortium Trials 01-03 and 00-01. Clin. Cancer Res. 2005, 11, 7841–7850. [Google Scholar] [CrossRef]
- White, E.; DiPaola, R.S. The double-edged sword of autophagy modulation in cancer. Clin. Cancer Res. 2009, 15, 5308–5316. [Google Scholar] [CrossRef]
- Karar, J.; Maity, A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front. Mol. Neurosci. 2011, 4, 1–8. [Google Scholar]
- Hsieh, A.C.; Costa, M.; Zollo, O.; Davis, C.; Feldman, M.E.; Testa, J.R.; Meyuhas, O.; Shokat, K.M.; Ruggero, D. Genetic Disscetion of the Oncogenic mTOR Pathway reveals Druggable Addiction of Translational Control via 4EBP-eIF4E. Cancer Cell 2010, 17, 249–261. [Google Scholar] [CrossRef]
- Wendel, H.-G.; Silva, R.L.A; Malina, A.; Mills, J.R.; Zhu, H.; Ueda, T.; Watanabe-Fukunaga, R.; Fukunaga, R.; Teruya-Feldstein, J.; Pelletier, J.; et al. Dissecting eIF4E action in tumorigenesis. Gene Dev. 2007, 21, 3232–3237. [Google Scholar] [CrossRef]
- Chen, L.; Liu, L.; Luo, Y.; Huang, S. MAPK and mTOR pathways are involved in cadmium-induced neuronal apoptosis. J. Neurochem. 2008, 105, 251–261. [Google Scholar] [CrossRef]
- Kim, J.H.; Chu, S.C.; Gramlich, J.L.; Pride, Y.B.; Babendreier, E.; Chauhan, D.; Salgia, R.; Podar, K.; Griffin, J.D.; Sattler, M. Activation of the PI3K/mTOR pathway by BCR-ABL contributes to increased production of reactive oxygen species. Blood 2005, 105, 1717–1723. [Google Scholar] [CrossRef]
- Yalcin, S.; Marinkovic, D.; Mungamuri, S.K.; Zhang, X.; Tong, W.; Sellers, R.; Ghaffari, S. ROS-mediated amplification of AKT/mTOR signalling pathway leads to myeloproliferative syndrome in Foxo3(−/−) mice. EMBO J. 2010, 29, 4118–4131. [Google Scholar] [CrossRef]
- Sattler, M.; Liang, H.; Nettesheim, D.; Meadows, R.P.; Harlan, J.E.; Eberstadt, M.; Yoon, H.S.; Shuker, S.B.; Chang, B.S.; Minn, A.J.; et al. Structure of Bcl-xL-Bak peptide complex: Recognition between regulators of apoptosis. Science 1997, 275, 983–986. [Google Scholar] [CrossRef]
- Suh, Y.A.; Arnold, R.S.; Lassegue, B.; Shi, J.; Xu, X.; Sorescu, D.; Chung, A.B.; Griendling, K.K.; Lambeth, J.D. Cell transformation by the superoxide-generating oxidase Mox1. Nature 1999, 401, 79–82. [Google Scholar] [CrossRef]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. A. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar]
- Iglesias-Bartolome, R.; Patel, V.; Cotrim, A.; Leelahavanichkul, K.; Molinolo, A.A.; Mitchell, J.B.; Gutkind, J.S. mTOR inhibition prevents epithelial stem cell senescence and protects from radiation-induced mucositis. Cell Stem Cell 2012, 11, 401–414. [Google Scholar] [CrossRef]
- Lee, J.Y.; Chang, J.W.; Yang, W.S.; Kim, S.B.; Park, S.K.; Park, J.S.; Lee, S.K. Albumin-induced epithelial-mesenchymal transition and ER stress are regulated through a common ROS-c-Src kinase-mTOR pathway: Effect of imatinib mesylate. Am. J. Physiol. Renal Physicol. 2011, 300, F1214–F1222. [Google Scholar] [CrossRef]
- Schieke, S.M.; Finkel, T. Mitochondrial signaling, TOR, and life span. Biol. Chem. 2006, 387, 1357–1361. [Google Scholar]
- Miller, A.-F. Superoxide dismutases: Ancient enzymes and new insights. FEBS Lett. 2012, 586, 585–595. [Google Scholar] [CrossRef]
- Young, J.; Povey, S. The genetic basis of tuberous sclerosis. Mol. Med. Today 1998, 4, 313–319. [Google Scholar] [CrossRef]
- Neuman, N.A.; Henske, E.P. Non-canonical functions of the tuberous sclerosis complx-Rheb signalling axis. EMBO Mol. Med. 2011, 3, 189–200. [Google Scholar] [CrossRef]
- Van Slegtenhorst, M.; Nellist, M.; Nagelkerken, B.; Cheadle, J.; Snell, R.; van den Ouweland, A.; Reuser, A.; Sampson, J.; Halley, D.; van der Sluijs, P. Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum. Mol. Genet. 1998, 7, 1053–1057. [Google Scholar] [CrossRef]
- Kang, Y.J.; Lu, M.-K.; Guan, K.-L. The TSC1 and TSC2 tumor suppressors are required for proper ER stress response and protect cells from ER stress-induced apoptosis. Cell Death Differ. 2011, 18, 133–144. [Google Scholar] [CrossRef]
- Jaeschke, A.; Hartkamp, J.; Saitoh, M.; Roworth, W.; Nobukuni, T.; Hodges, A.; Sampson, J.; Thomas, G.; Lamb, R. Tuberous sclerosis complex tumor suppressor-mediated S6 kinase inhibition by phosphatidylinositide-3-OH kinase is mTOR independent. J. Cell. Biol. 2002, 159, 217–224. [Google Scholar] [CrossRef]
- Radimerski, T.; Montagne, J.; Hemmings-Mieszczak, M.; Thomas, G. Lethality of Drosophila lacking TSC tumor suppressor function rescued by reducing dS6K signaling. Gene Dev. 2002, 16, 2627–2632. [Google Scholar] [CrossRef]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Goncharova, E.A.; Goncharov, D.A.; Eszterhas, A.; Hunter, D.S.; Glassberg, M.K.; Yeung, R.S.; Walker, C.L.; Noonan, D.; Kwiatkowski, D.J.; Chou, M.M.; et al. Tuberin regulates p70 S6 kinase activation and ribosomal protein S6 phosphorylation. A role for the TSC2 tumor suppressor gene in pulmonary lymphangioleiomyomatosis (LAM). J. Biol. Chem. 2002, 277, 30958–30967. [Google Scholar] [CrossRef]
- Kenerson, H.L.; Aicher, L.D.; True, L.D.; Yeung, R.S. Activated mammalian target of rapamycin pathway in the pathogenesis of tuberous sclerosis complex renal tumors. Cancer Res. 2002, 62, 5645–5650. [Google Scholar]
- Kwiatkowski, D.J.; Zhang, H.; Bandura, J.L.; Heiberger, K.M.; Glogauer, M.; El-Hashemite, N.; Onda, H. A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in Tsc1 null cells. Hum. Mol. Genet. 2002, 11, 525–534. [Google Scholar] [CrossRef]
- Onda, H.; Crino, P.B.; Zhang, H.; Murphey, R.D.; Rastelli, L.; Gould Rothberg, B.E.; Kwiatkowski, D.J. Tsc2 null murine neuroepithelial cells are a model for human tuber giant cells, and show activation of an mTOR pathway. Mol. Cell Neurosci. 2002, 21, 561–574. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Ozcan, U.; Ozcan, L.; Yilmaz, E.; Düvel, K.; Sahin, M.; Manning, B.D.; Hotamisligil, G.S. Loss of the tuberous sclerosis complex tumor suppressors triggers the unfolded protein response to regulate insulin signaling and apoptosis. Mol. Cell 2008, 29, 541–551. [Google Scholar] [CrossRef]
- Yokouchi, M.; Hiramatsu, N.; Hayakawa, K.; Okamura, M.; Du, S.; Kasai, A.; Takano, Y.; Shitamura, A.; Shimada, T.; Yao, J.; et al. Involvement of selective reactive oxygen species upstream of proapoptotic branches of unfolded protein response. J. Biol. Chem. 2008, 283, 4252–4260. [Google Scholar]
- Di Nardo, A.; Kramvis, I.; Cho, N.; Sadowski, A.; Meikle, L.; Kwiatkowski, D.J.; Sahin, M. Tuberous sclerosis complex activity is required to control neuronal stress responses in an mTOR-dependent manner. J. Neurosci. 2009, 29, 5926–5937. [Google Scholar] [CrossRef]
- Vojtek, A.B.; Der, C.J. Increasing complexity of the Ras signaling pathway. J. Biol. Chem. 1998, 273, 19925–19928. [Google Scholar] [CrossRef]
- Katz, M.E.; McCormick, F. Signal transduction from multiple Ras effectors. Curr. Opin. Genet. Dev. 1997, 7, 75–79. [Google Scholar] [CrossRef]
- Overmeyer, J.H.; Maltese, W.A. Death pathways triggered by activated Ras in cancer cells. Front. Biosci. 2011, 16, 1693–1713. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ehrkamp, A.; Herrmann, C.; Stoll, R.; Heumann, R. Ras and Rheb Signaling in Survival and Cell Death. Cancers 2013, 5, 639-661. https://doi.org/10.3390/cancers5020639
Ehrkamp A, Herrmann C, Stoll R, Heumann R. Ras and Rheb Signaling in Survival and Cell Death. Cancers. 2013; 5(2):639-661. https://doi.org/10.3390/cancers5020639
Chicago/Turabian StyleEhrkamp, Anja, Christian Herrmann, Raphael Stoll, and Rolf Heumann. 2013. "Ras and Rheb Signaling in Survival and Cell Death" Cancers 5, no. 2: 639-661. https://doi.org/10.3390/cancers5020639