Mouse Models of Gastric Cancer

Abstract
:1. Introduction
2. Chemical Carcinogenesis Models of Gastric Cancer
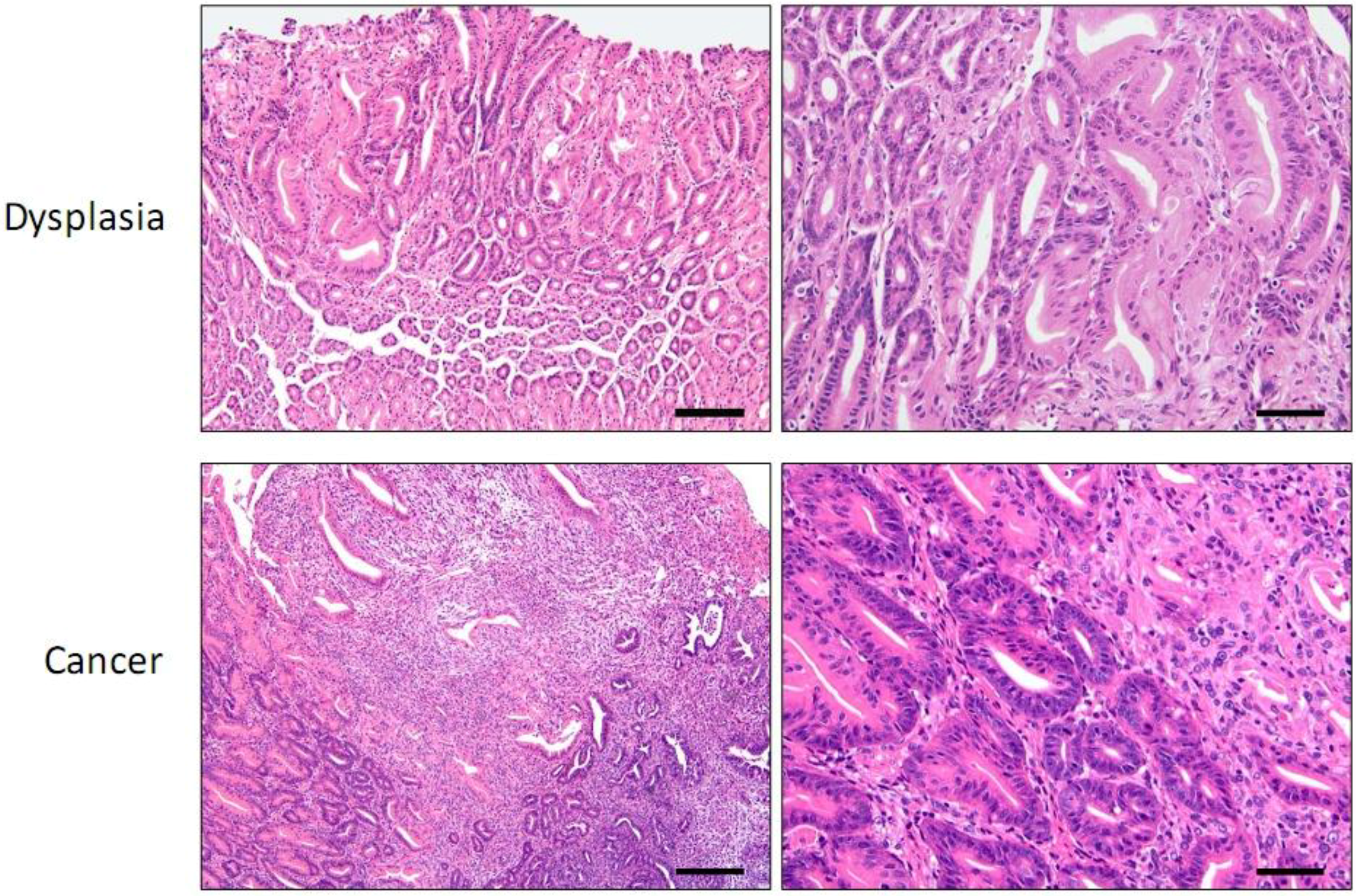
3. Helicobacter Infection Models
4. Genetically Engineered Mouse Models
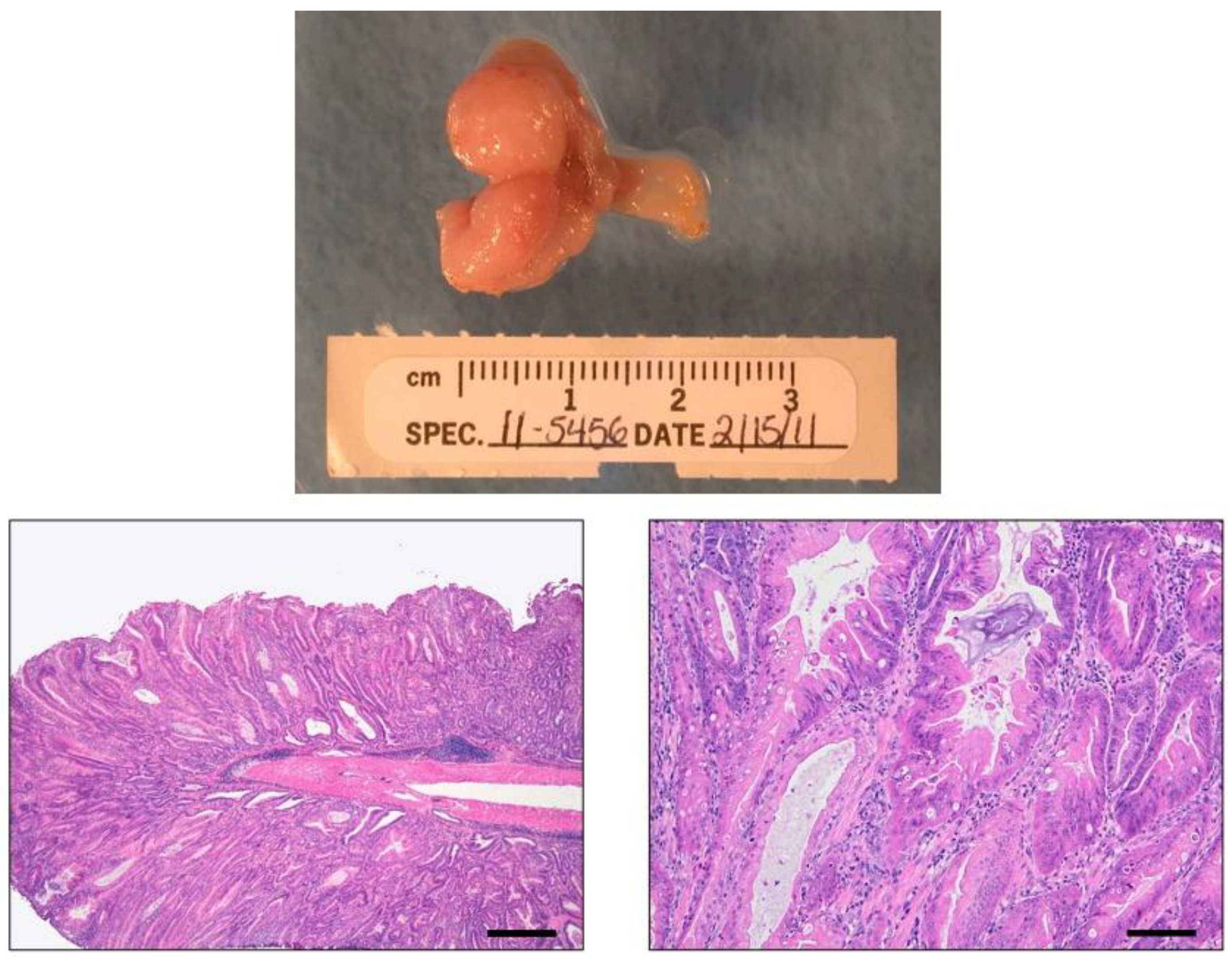
4.1. INS-GAS Mice
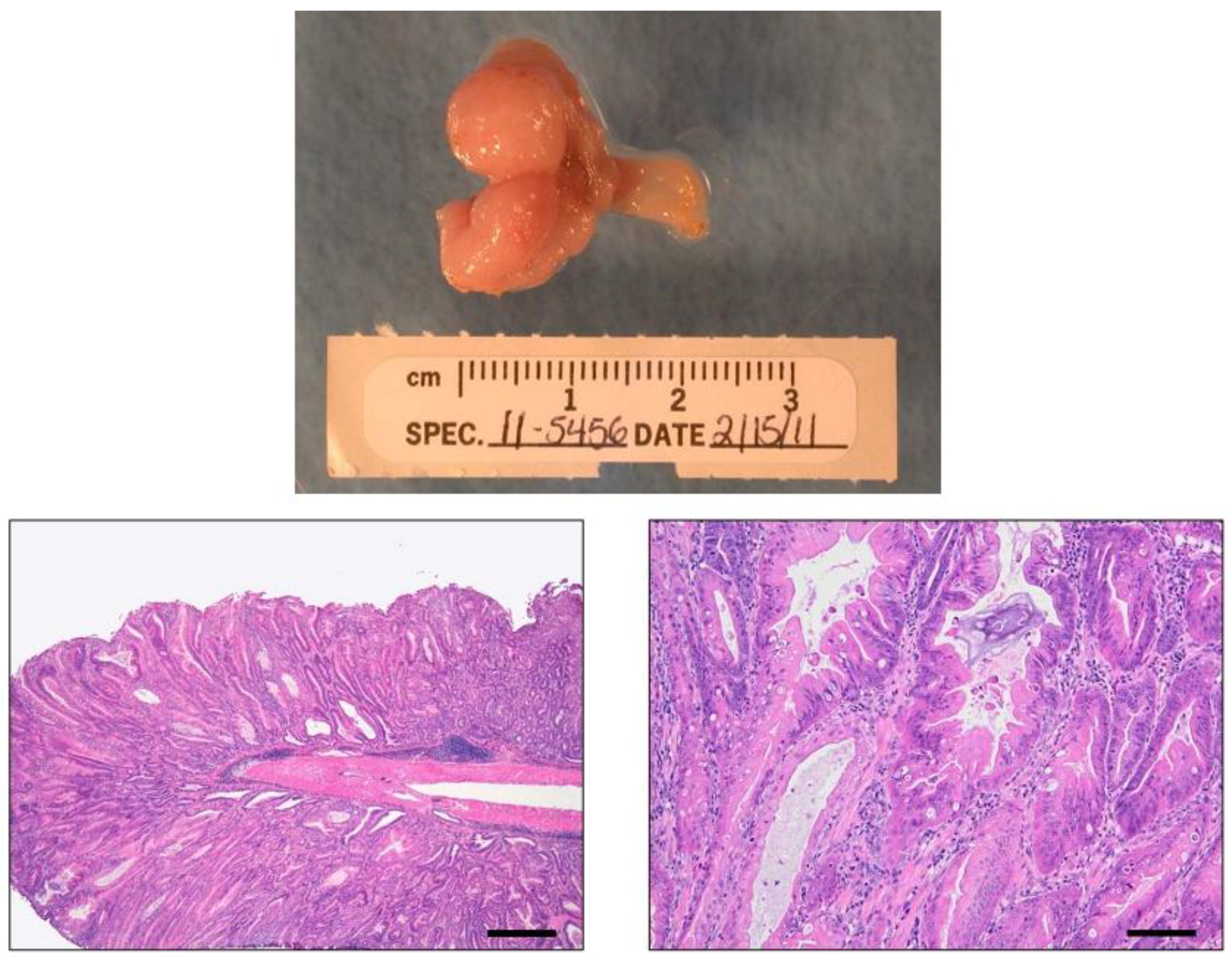

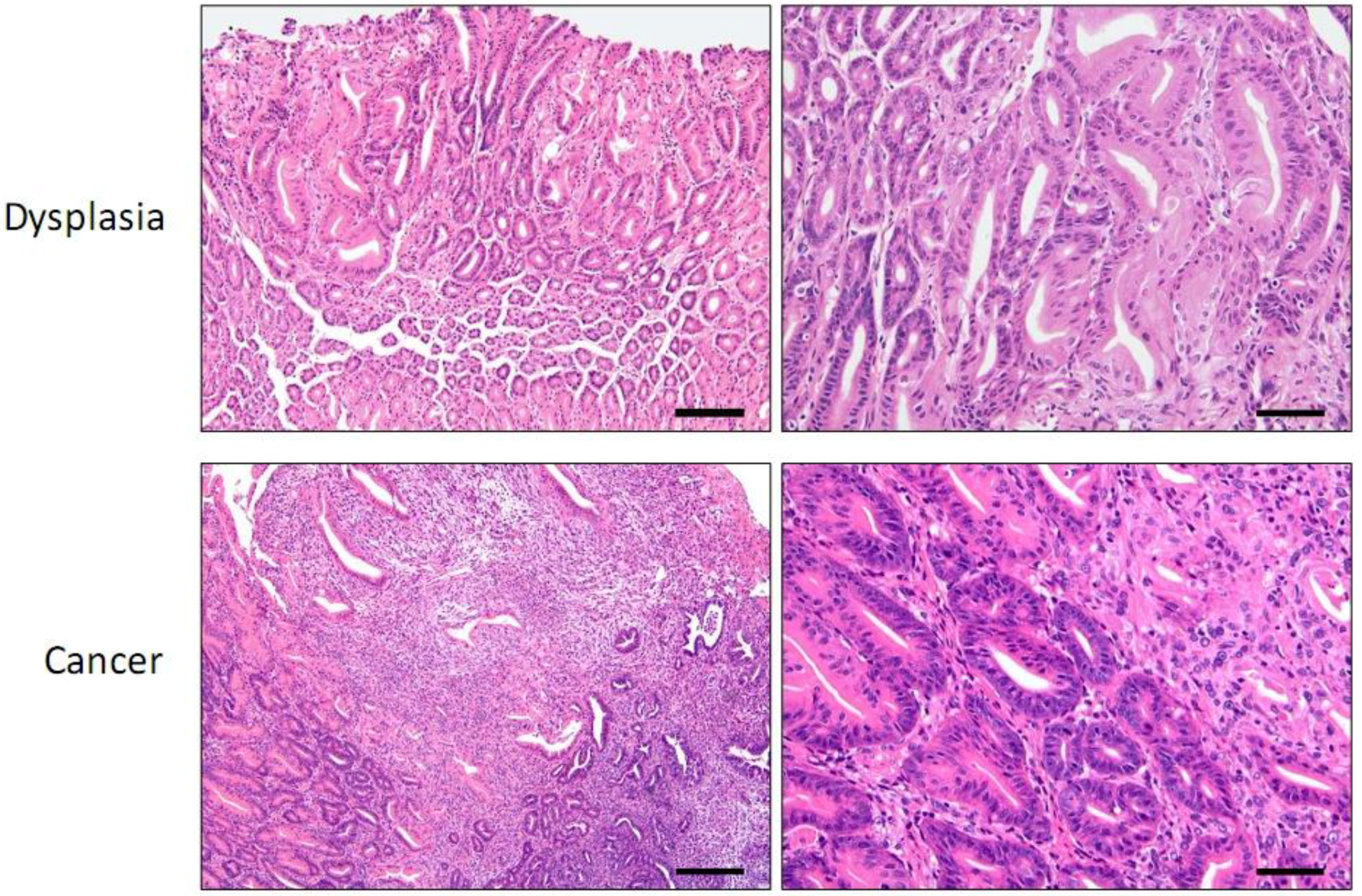{kind=link}
{kind=link}
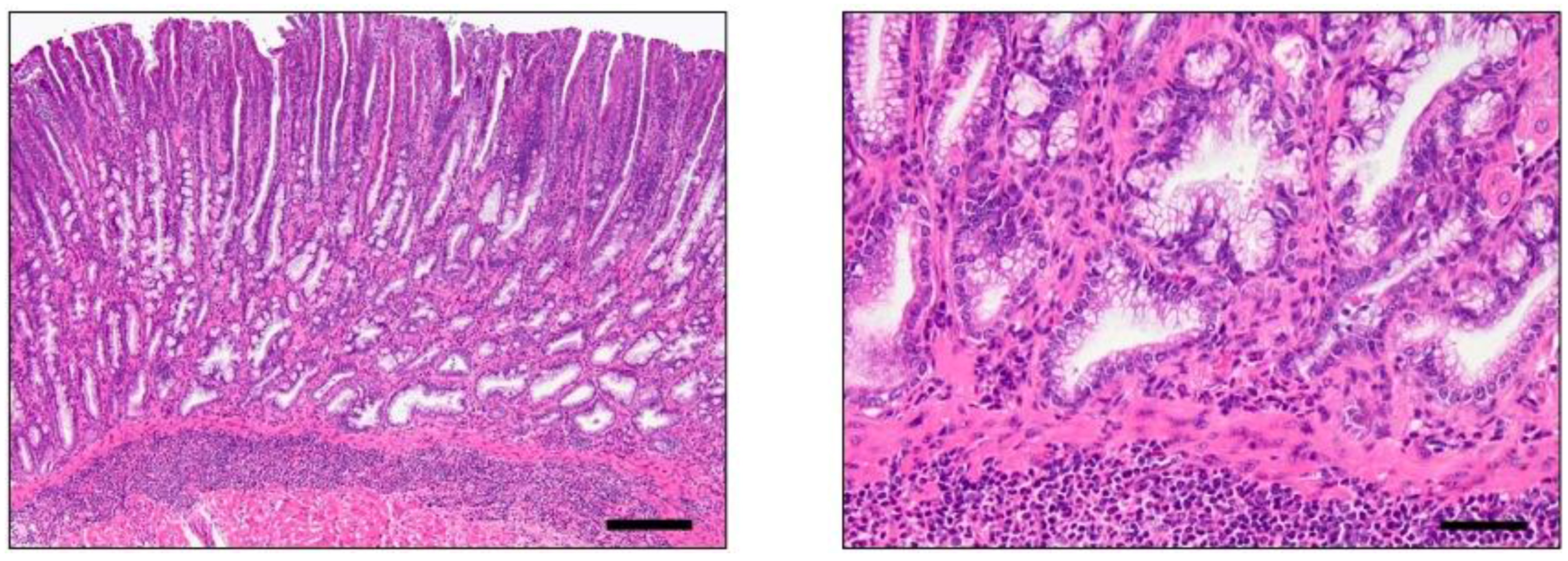{kind=link}
| Purpose of analysis | Results |
|---|---|
| Pancreatic islet cell formation [120] | gastrin and TGF-α synergistically stimulate islet growth |
| H. pylori and gastric mucosa [29] | progression to gastric atrophy, metaplasia, dysplasia, and cancer |
| Colonic carcinogenesis by AOM [137,138] | progastrin, not gastrin, promotes colon carcinogenesis |
| Gender differences [85,106] | greater gastric carcinogenesis in male INS-GAS mice with H. pylori |
| Importance of CagE [85] | loss of cagE temporally retards but does not abrogate cancer progression |
| Interaction with G-Gly [124] | G-gly synergizes with amidated gastrin to stimulate acid secretion and inhibits atrophy |
| CCK2R and Histamine receptor inhibitors [129] | CCK2R and H2R antagonists synergistically inhibit gastric atrophy and cancer |
| Intestinal crypt regeneration [139] | hypergastrinemia increases regeneration of intestinal injury |
| Apoptosis of gastric epithelium [135,136] | gastrin induces apoptosis and contribute to gastric carcinogenesis |
| Gene expression profiling [125] | identify up- and down-regulating genes among 12,000 cDNA |
| TFF2 expression [126] | TFF2 expression in the gastric fundus was elevated in INS-GAS mice |
| Swedish variant of moist oral smokeless tobacco [134] | tobacco promotes cancer formation in H. pylori-infected INS-GAS mice |
| Reg-1 expression [127] | Reg1 is increased in the stomachs of H. felis-infected INS-GAS mice |
| Role of 17-beta-estradiol [131,132,133] | 17beta-estradiol has protective effects on gastric cancer development |
| Eradication of Helicobacter [79,80,83] | eradication inhibits mouse gastric carcinogenesis |
| Commensal bacterial flora in the stomach [113] | SPF mice are more susceptible to gastric cancer than germ-free mice |
| Antral carcinogenesis [60,123] | gastrin suppresses antral carcinogenesis |
| HB-EGF, MMP-7, EMT protein [128] | neutralisation of gastrin in INS-GAS mice reduced MMP-7, HB-EGF and EMT proteins |
| Acetic acid and cytoreduction [130] | acetic acid could be a potent cytoreductive treatment of gastric cancer |
| Effect of IL-8 [34] | IL-8 promotes gastric carcinogenesis in INS-GAS mice |
| H. pylori-induced iron deficiency [140] | marked changes in expression of gastric iron transporters in H. felis-infected INS-GAS mice |
4.2. Gastrin Knockout Mice
4.3. TFF1 Knockout Mice and Gp130 Mutant Mice
4.4. H/K-ATPase-IL-1β Transgenic Mice

4.5. K-ras Transgenic Mice
4.6. Wnt1 and COX-2 Transgenic Mice
4.7. Other Mouse Models of Gastric Cancer
| Model | Incidence | Duration | Location | Phenotype |
|---|---|---|---|---|
| MNU | <70% | 12 months | Antrum | AdenoCa, Dysplasia [11,47,48,49] |
| H. felis | 80% | 18 months | SCJ/Transition | AdenoCa, Dysplasia, Metaplasia, Atrophy [21] |
| MNU + H. pylori | 80% | 12 months | Antrum | AdenoCa, Dysplasia, Metaplasia, Atrophy [110] |
| MNU + H. felis | 100% | 36 weeks | Antrum | AdenoCa, Dysplasia, Metaplasia, Atrophy [60] |
| CEA/SV40 | 100% | 50 days | Antrum | AdenoCa, Dysplasia, Invasion to submucosa and duodenum [24] |
| MMTV/Ad12 | 82%(male) | 3–4 months | SCJ | AdenoCa, AdenosquamousCa [26] |
| HPV-16 | 100% | 246–352 days | Antrum | Carcinoid, Metastasis to lymph node and liver [27] |
| MTH1−/− | 13% | 18 months | Antrum | AdenoCa, Dysplasia, Hyperplasia, Lung and liver tumors [199] |
| TFF1−/− | 30% | 5 months | Antrum | Intramucosal carcinoma, Hyperplasia, Activation of NF-kB [144,145] |
| Smad4+/− | 100% | 12–18 months | Corpus/Antrum | AdenoCa, Dysplasia, Hyperplasia, Duodenal tumor [195,196] |
| GB-Smad4F/F | 100% | 12–18 months | Antrum | Dysplasia, Hyperplasia [197] |
| INS-GAS | 75% | 20 months | Corpus | AdenoCa, Dysplasia, Metaplasia, Atrophy, Synergized with H. felis [29] |
| GAS−/− | 60% | 12 months | Antrum | Dysplasia, Metaplasia, Atrophy, Susceptible to MNU [60,143] |
| Gp130F/F | 100% | 6 months | Antrum | Adenoma, Decreased TFF1 expression [158,159] |
| IL-1β | <70% | 12 months | Transition | AdenoCa, Dysplasia, Metaplasia, Atrophy, Synergized with H. felis [163] |
| K19/K-ras | 37.5% | 16 months | Corpus | Dysplasia, Metaplasia, Atrophy [172] |
| Wnt1/C2me | 100% | 20 weeks | SCJ | AdenoCa, Dysplasia, Metaplasia, Attenuated by CD44 ablation [179,189] |
| CDH1+/− + MNU | 45.8% | 40 weeks | Antrum | Signet-ring cell Ca, Adenoma [56] |
| CDH1/p53 | 100% | 12 months | Corpus | Poorly differentiated AdenoCa, Signet-ring cell Ca [200] |
| RUNX3−/− + MNU | 71% | 52 weeks | Corpus/Antrum | AdenoCa, Metaplasia, Hyperplasia [201] |
| Villin-KLF4F/F | 29% | 80 weeks | Antrum | Adenoma, Susceptible to MNU [57] |
5. Models of Precancerous Change
| Model | Duration | Phenotype |
|---|---|---|
| H.pylori (SS-1) | 6–9 months | Atrophy, SPEM [20] |
| TGF-α transgenic | 3 months | Atrophy [122,213] |
| H/K-ATPase/DT | 28–80 days | Atrophy [214] |
| H/K-ATPase/Tk | Ganciclovir treatment | Atrophy [215] |
| H/K-ATPase-α−/− | 10 weeks | Atrophy [216] |
| H/K-ATPase-β−/− | 17 days | Atrophy [217,218] |
| NHE2−/− | 17 days | Atrophy [219] |
| Car9−/− | 4 weeks | Atrophy [220] |
| CCK2R−/− | 18 weeks | Atrophy [221,222] |
| H/K-ATPase/Shh−/− | 3–8 months | Pit cell hyperplasia, loss of parietal cell function [223] |
| DMP-777 | 7–14 days | Atrophy, SPEM [224,225] |
| L-635 | 7 days | Atrophy, SPEM [226] |
| Cdx2 transgenic | 120 days | Intestinal metaplasia [227,228] |
| Cdx1 transgenic | 120 days | Intestinal metaplasia [229] |
5.1. Models of Gastric Atrophy
5.2. Models of Metaplasia
6. Conclusions and Future Perspectives
| Gene | Location | Lineage tracing in the stomach |
|---|---|---|
| TFF1 | Surface of stomach (pit cell area) | |
| TFF2 | Isthmus of corpus & base of antrum | Give rise to parietal, mucous neck, and chief cells [242] |
| H/K-ATPase | Corpus (parietal cell) | Give rise to all gastric lineages of the corpus glands with Notch activation [243] |
| Foxa3 | Whole stomach, other organ from endoderm | |
| K19 | Whole stomach, intestine, colon, etc. | |
| Lgr5 | Cardia, Antrum, intestine, colon, etc. | Give rise to all major cell types in the cardia, antrum and transition zone [240] |
| Sox2 | Corpus, Antrum, Esophagus, Forestomach, etc. | Give rise to all major cell types in the corpus and the antrum [236] |
| Mist1 | Corpus (chief cell), Brunner gland, pancreas | Give rise to chief cell and drug-induced SPEM [226] |
| Villin | Antrum, intestine, colon | Give rise to all gastric lineages of the antral glands with IFN-γ treatment [241] |
References
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef]
- Kawase, S.; Ishikura, H. Female-predominant occurrence of spontaneous gastric adenocarcinoma in cotton rats. Lab. Anim. Sci. 1995, 45, 244–248. [Google Scholar]
- Waldum, H.L.; Rørvik, H.; Falkmer, S.; Kawase, S. Neuroendocrine (ECL cell) differentiation of spontaneous gastric carcinomas of cotton rats (Sigmodon hispidus). Lab. Anim. Sci. 1999, 49, 241–247. [Google Scholar]
- Cui, G.; Qvigstad, G.; Falkmer, S.; Sandvik, A.K.; Kawase, S.; Waldum, H.L. Spontaneous ECLomas in cotton rats (Sigmodon hispidus): Tumours occurring in hypoacidic/hypergastrinaemic animals with normal parietal cells. Carcinogenesis 2000, 21, 23–27. [Google Scholar] [CrossRef]
- Koga, T.; Takahashi, K.; Sato, K.; Kikuchi, I.; Okazaki, Y.; Miura, T.; Katsuta, M.; Narita, T. The effect of colonisation by Helicobacter pylori in Praomys (Mastomys) natalensis on the incidence of carcinoids. J. Med. Microbiol. 2002, 51, 777–785. [Google Scholar]
- Kumazawa, H.; Takagi, H.; Sudo, K.; Nakamura, W.; Hosoda, S. Adenocarcinoma and carcinoid developing spontaneously in the stomach of mutant strains of Mastomys natalensis. Virchows Arch. A Pathol. Anat. Histopathol. 1989, 416, 141–151. [Google Scholar] [CrossRef]
- Correa, P.; Haenszel, W.; Cuello, C.; Tannenbaum, S.; Archer, M. A model for gastric cancer epidemiology. Lancet 1975, 2, 58–60. [Google Scholar]
- Saito, T.; Sugimura, T. Biochemical studies on carcinogenesis in the glandular stomach of rats with N-methyl-N'-nitro-N-nitrosoguanidine. Gann 1973, 64, 373–381. [Google Scholar]
- Sugimura, T.; Fujimura, S. Tumour production in glandular stomach of rat by N-methyl-N'-nitro-N-nitrosoguanidine. Nature 1967, 216, 943–944. [Google Scholar] [CrossRef]
- Tatematsu, M.; Ogawa, K.; Hoshiya, T.; Shichino, Y.; Kato, T.; Imaida, K.; Ito, N. Induction of adenocarcinomas in the glandular stomach of BALB/c mice treated with N-methyl-N-nitrosourea. Jpn. J. Cancer Res. 1992, 83, 915–918. [Google Scholar] [CrossRef]
- Marshall, B.J.; Warren, J.R. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1984, 1, 1311–1315. [Google Scholar] [CrossRef]
- Forman, D.; Newell, D.G.; Fullerton, F.; Yarnell, J.W.; Stacey, A.R.; Wald, N.; Sitas, F. Association between infection with Helicobacter pylori and risk of gastric cancer: Evidence from a prospective investigation. BMJ 1991, 302, 1302–1305. [Google Scholar] [CrossRef]
- Nomura, A.; Stemmermann, G.N.; Chyou, P.H.; Kato, I.; Perez-Perez, G.I.; Blaser, M.J. Helicobacter pylori infection and gastric carcinoma among Japanese Americans in Hawaii. N. Engl. J. Med. 1991, 325, 1132–1136. [Google Scholar] [CrossRef]
- Parsonnet, J.; Friedman, G.D.; Vandersteen, D.P.; Chang, Y.; Vogelman, J.H.; Orentreich, N.; Sibley, R.K. Helicobacter pylori infection and the risk of gastric carcinoma. N. Engl. J. Med. 1991, 325, 1127–1131. [Google Scholar] [CrossRef]
- Uemura, N.; Okamoto, S.; Yamamoto, S.; Matsumura, N.; Yamaguchi, S.; Yamakido, M.; Taniyama, K.; Sasaki, N.; Schlemper, R.J. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 2001, 345, 784–789. [Google Scholar] [CrossRef]
- Fox, J.G.; Wang, T.C. Helicobacter pylori—Not a good bug after all! N. Engl. J. Med. 2001, 345, 829–832. [Google Scholar] [CrossRef]
- Lee, A.; Hazell, S.L.; O’Rourke, J.; Kouprach, S. Isolation of a spiral-shaped bacterium from the cat stomach. Infect. Immun. 1988, 56, 2843–2850. [Google Scholar]
- Rogers, A.B.; Taylor, N.S.; Whary, M.T.; Stefanich, E.D.; Wang, T.C.; Fox, J.G. Helicobacter pylori but not high salt induces gastric intraepithelial neoplasia in B6129 mice. Cancer Res. 2005, 65, 10709–10715. [Google Scholar] [CrossRef]
- Lee, A.; O'Rourke, J.; de Ungria, M.C.; Robertson, B.; Daskalopoulos, G.; Dixon, M.F. A standardized mouse model of Helicobacter pylori infection: Introducing the Sydney strain. Gastroenterology 1997, 112, 1386–1397. [Google Scholar] [CrossRef]
- Fox, J.G.; Sheppard, B.J.; Dangler, C.A.; Whary, M.T.; Ihrig, M.; Wang, T.C. Germ-line p53-targeted disruption inhibits helicobacter-induced premalignant lesions and invasive gastric carcinoma through down-regulation of Th1 proinflammatory responses. Cancer Res. 2002, 62, 696–702. [Google Scholar]
- Fanning, E.; Knippers, R. Structure and function of simian virus 40 large tumor antigen. Annu. Rev. Biochem. 1992, 61, 55–85. [Google Scholar] [CrossRef]
- Montag, A.G.; Oka, T.; Baek, K.H.; Choi, C.S.; Jay, G.; Agarwal, K. Tumors in hepatobiliary tract and pancreatic islet tissues of transgenic mice harboring gastrin simian virus 40 large tumor antigen fusion gene. Proc. Natl. Acad. Sci. USA 1993, 90, 6696–6700. [Google Scholar]
- Thompson, J.; Epting, T.; Schwarzkopf, G.; Singhofen, A.; Eades-Perner, A.M.; van der Putten, H.; Zimmermann, W. A transgenic mouse line that develops early-onset invasive gastric carcinoma provides a model for carcinoembryonic antigen-targeted tumor therapy. Int. J. Cancer 2000, 86, 863–869. [Google Scholar] [CrossRef]
- Li, Q.; Karam, S.M.; Gordon, J.I. Simian virus 40 T antigen-induced amplification of pre-parietal cells in transgenic mice. Effects on other gastric epithelial cell lineages and evidence for a p53-independent apoptotic mechanism that operates in a committed progenitor. J. Biol. Chem. 1995, 270, 15777–15788. [Google Scholar]
- Koike, K.; Hinrichs, S.H.; Isselbacher, K.J.; Jay, G. Transgenic mouse model for human gastric carcinoma. Proc. Natl. Acad. Sci. USA 1989, 86, 5615–5619. [Google Scholar] [CrossRef]
- Searle, P.F.; Thomas, D.P.; Faulkner, K.B.; Tinsley, J.M. Stomach cancer in transgenic mice expressing human papillomavirus type 16 early region genes from a keratin promoter. J. Gen. Virol. 1994, 75, 1125–1137. [Google Scholar] [CrossRef]
- Correa, P. Human gastric carcinogenesis: A multistep and multifactorial process—First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992, 52, 6735–6740. [Google Scholar]
- Wang, T.C.; Dangler, C.A.; Chen, D.; Goldenring, J.R.; Koh, T.; Raychowdhury, R.; Coffey, R.J.; Ito, S.; Varro, A.; Dockray, G.J.; et al. Synergistic interaction between hypergastrinemia and Helicobacter infection in a mouse model of gastric cancer. Gastroenterology 2000, 118, 36–47. [Google Scholar]
- El-Omar, E.M.; Carrington, M.; Chow, W.H.; McColl, K.E.; Bream, J.H.; Young, H.A.; Herrera, J.; Lissowska, J.; Yuan, C.C.; Rothman, N.; et al. The role of interleukin-1 polymorphisms in the pathogenesis of gastric cancer. Nature 2001, 412, 99. [Google Scholar]
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.; Takashi, S.; Baik, G.H.; Shibata, W.; Diprete, B.; Betz, K.S.; et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 2011, 19, 257–272. [Google Scholar] [CrossRef]
- Tu, S.P.; Quante, M.; Bhagat, G.; Takaishi, S.; Cui, G.; Yang, X.D.; Muthuplani, S.; Shibata, W.; Fox, J.G.; Pritchard, D.M.; et al. IFN-γ inhibits gastric carcinogenesis by inducing epithelial cell autophagy and T-cell apoptosis. Cancer Res. 2011, 71, 4247–4259. [Google Scholar] [CrossRef] [Green Version]
- Shibata, W.; Ariyama, H.; Westphalen, C.B.; Worthley, D.L.; Muthupalani, S.; Asfaha, S.; Dubeykovskaya, Z.; Quante, M.; Fox, J.G.; Wang, T.C. Stromal cell-derived factor-1 overexpression induces gastric dysplasia through expansion of stromal myofibroblasts and epithelial progenitors. Gut 2012, 62, 192–200. [Google Scholar]
- Asfaha, S.; Dubeykovskiy, A.N.; Tomita, H.; Yang, X.; Stokes, S.; Shibata, W.; Friedman, R.A.; Ariyama, H.; Dubeykovskaya, Z.A.; Muthupalani, S.; et al. Mice that Express Human Interleukin-8 Have Increased Mobilization of Immature Myeloid Cells, which Exacerbates Inflammation and Accelerates Colon Carcinogenesis. Gastroenterology 2012, 144, 155–166. [Google Scholar]
- Rogers, A.B.; Fox, J.G. Inflammation and Cancer. I. Rodent models of infectious gastrointestinal and liver cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G361–G366. [Google Scholar] [CrossRef]
- Lemke, L.B.; Ge, Z.; Whary, M.T.; Feng, Y.; Rogers, A.B.; Muthupalani, S.; Fox, J.G. Concurrent Helicobacter bilis infection in C57BL/6 mice attenuates proinflammatory H. pylori-induced gastric pathology. Infect. Immun. 2009, 77, 2147–2158. [Google Scholar] [CrossRef]
- Ge, Z.; Feng, Y.; Muthupalani, S.; Eurell, L.L.; Taylor, N.S.; Whary, M.T.; Fox, J.G. Coinfection with Enterohepatic Helicobacter species can ameliorate or promote Helicobacter pylori-induced gastric pathology in C57BL/6 mice. Infect. Immun. 2011, 79, 3861–3871. [Google Scholar] [CrossRef]
- Schoental, R. Carcinogenic activity of N-methyl-N-nitroso-N'-nitroguanidine. Nature 1966, 209, 726–727. [Google Scholar] [CrossRef]
- Ohgaki, H.; Kawachi, T.; Matsukura, N.; Morino, K.; Miyamoto, M.; Sugimura, T. Genetic control of susceptibility of rats to gastric carcinoma. Cancer Res. 1983, 43, 3663–3667. [Google Scholar]
- Tatematsu, M.; Yamamoto, M.; Shimizu, N.; Yoshikawa, A.; Fukami, H.; Kaminishi, M.; Oohara, T.; Sugiyama, A.; Ikeno, T. Induction of glandular stomach cancers in Helicobacter pylori-sensitive Mongolian gerbils treated with N-methyl-N-nitrosourea and N-methyl-N'-nitro-N-nitrosoguanidine in drinking water. Jpn. J. Cancer Res. 1998, 89, 97–104. [Google Scholar] [CrossRef]
- Takahashi, M.; Kokubo, T.; Furukawa, F.; Kurokawa, Y.; Tatematsu, M.; Hayashi, Y. Effect of high salt diet on rat gastric carcinogenesis induced by N-methyl-N'-nitro-N-nitrosoguanidine. Gann 1983, 74, 28–34. [Google Scholar]
- Tatematsu, M.; Takahashi, M.; Fukushima, S.; Hananouchi, M.; Shirai, T. Effects in rats of sodium chloride on experimental gastric cancers induced by N-methyl-N-nitro-N-nitrosoguanidine or 4-nitroquinoline-1-oxide. J. Natl. Cancer Inst. 1975, 55, 101–106. [Google Scholar]
- Tatsuta, M.; Iishi, H.; Baba, M.; Uehara, H.; Nakaizumi, A.; Taniguchi, H. Enhancing effects of calcium-deficient diet on gastric carcinogenesis by N-methyl-N'-nitro-N-nitrosoguanidine in Wistar rats. Jpn. J. Cancer Res. 1993, 84, 945–950. [Google Scholar] [CrossRef]
- Wada, S.; Hirose, M.; Shichino, Y.; Ozaki, K.; Hoshiya, T.; Kato, K.; Shirai, T. Effects of catechol, sodium chloride and ethanol either alone or in combination on gastric carcinogenesis in rats pretreated with N-methyl-N'-nitro-N-nitrosoguanidine. Cancer Lett. 1998, 123, 127–134. [Google Scholar] [CrossRef]
- Uedo, N.; Tatsuta, M.; Iishi, H.; Baba, M.; Yano, H.; Ishihara, R.; Higashino, K.; Ishiguro, S. Enhancement by interleukin-1 beta of gastric carcinogenesis induced by N-methyl-N'-nitro-N-nitrosoguanidine in Wistar rats: A possible mechanism for Helicobacter pylori-associated gastric carcinogenesis. Cancer Lett. 2003, 198, 161–168. [Google Scholar] [CrossRef]
- Danon, S.J.; Eaton, K.A. The role of gastric Helicobacter and N-methyl-N'-nitro-N-nitrosoguanidine in carcinogenesis of mice. Helicobacter 1998, 3, 260–268. [Google Scholar] [CrossRef]
- Tatematsu, M.; Yamamoto, M.; Iwata, H.; Fukami, H.; Yuasa, H.; Tezuka, N.; Masui, T.; Nakanishi, H. Induction of glandular stomach cancers in C3H mice treated with N-methyl-N-nitrosourea in the drinking water. Jpn. J. Cancer Res. 1993, 84, 1258–1264. [Google Scholar] [CrossRef]
- Yamachika, T.; Nakanishi, H.; Inada, K.; Tsukamoto, T.; Shimizu, N.; Kobayashi, K.; Fukushima, S.; Tatematsu, M. N-methyl-N-nitrosourea concentration-dependent, rather than total intake-dependent, induction of adenocarcinomas in the glandular stomach of BALB/c mice. Jpn. J. Cancer Res. 1998, 89, 385–391. [Google Scholar] [CrossRef]
- Yamamoto, M.; Furihata, C.; Ogiu, T.; Tsukamoto, T.; Inada, K.; Hirano, K.; Tatematsu, M. Independent variation in susceptibilities of six different mouse strains to induction of pepsinogen-altered pyloric glands and gastric tumor intestinalization by N-methyl-N-nitrosourea. Cancer Lett. 2002, 179, 121–132. [Google Scholar] [CrossRef]
- Yamamoto, M.; Tsukamoto, T.; Sakai, H.; Shirai, N.; Ohgaki, H.; Furihata, C.; Donehower, L.A.; Yoshida, K.; Tatematsu, M. p53 knockout mice (−/−) are more susceptible than (+/−) or (+/+) mice to N-methyl-N-nitrosourea stomach carcinogenesis. Carcinogenesis 2000, 21, 1891–1897. [Google Scholar] [CrossRef]
- Sakamoto, K.; Hikiba, Y.; Nakagawa, H.; Hayakawa, Y.; Yanai, A.; Akanuma, M.; Ogura, K.; Hirata, Y.; Kaestner, K.H.; Omata, M.; et al. Inhibitor of kappaB Kinase Beta Regulates Gastric Carcinogenesis via Interleukin-1alpha Expression. Gastroenterology 2010, 139, 226–238. [Google Scholar] [CrossRef]
- Shibata, W.; Maeda, S.; Hikiba, Y.; Yanai, A.; Sakamoto, K.; Nakagawa, H.; Ogura, K.; Karin, M.; Omata, M. c-Jun NH2-terminal kinase 1 is a critical regulator for the development of gastric cancer in mice. Cancer Res. 2008, 68, 5031–5039. [Google Scholar]
- Hayakawa, Y.; Hirata, Y.; Nakagawa, H.; Sakamoto, K.; Hikiba, Y.; Kinoshita, H.; Nakata, W.; Takahashi, R.; Tateishi, K.; Tada, M.; et al. Apoptosis signal-regulating kinase 1 and cyclin D1 compose a positive feedback loop contributing to tumor growth in gastric cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 780–785. [Google Scholar]
- Takasu, S.; Tsukamoto, T.; Cao, X.Y.; Toyoda, T.; Hirata, A.; Ban, H.; Yamamoto, M.; Sakai, H.; Yanai, T.; Masegi, T.; et al. Roles of cyclooxygenase-2 and microsomal prostaglandin E synthase-1 expression and beta-catenin activation in gastric carcinogenesis in N-methyl-N-nitrosourea-treated K19-C2mE transgenic mice. Cancer Sci. 2008, 99, 2356–2364. [Google Scholar] [CrossRef]
- Leung, W.K.; Wu, K.C.; Wong, C.Y.; Cheng, A.S.; Ching, A.K.; Chan, A.W.; Chong, W.W.; Go, M.Y.; Yu, J.; To, K.F.; et al. Transgenic cyclooxygenase-2 expression and high salt enhanced susceptibility to chemical-induced gastric cancer development in mice. Carcinogenesis 2008, 29, 1648–1654. [Google Scholar] [CrossRef]
- Humar, B.; Blair, V.; Charlton, A.; More, H.; Martin, I.; Guilford, P. E-cadherin deficiency initiates gastric signet-ring cell carcinoma in mice and man. Cancer Res. 2009, 69, 2050–2056. [Google Scholar] [CrossRef]
- Li, Q.; Jia, Z.; Wang, L.; Kong, X.; Guo, K.; Tan, D.; Le, X.; Wei, D.; Huang, S.; Mishra, L.; et al. Disruption of Klf4 in villin-positive gastric progenitor cells promotes formation and progression of tumors of the antrum in mice. Gastroenterology 2012, 142, 531–542. [Google Scholar] [CrossRef]
- Tsukamoto, T.; Mizoshita, T.; Tatematsu, M. Animal models of stomach carcinogenesis. Toxicol. Pathol. 2007, 35, 636–648. [Google Scholar] [CrossRef]
- Boffa, L.C.; Bolognesi, C. Methylating agents: Their target amino acids in nuclear proteins. Carcinogenesis 1985, 6, 1399–1401. [Google Scholar] [CrossRef]
- Tomita, H.; Takaishi, S.; Menheniott, T.R.; Yang, X.; Shibata, W.; Jin, G.; Betz, K.S.; Kawakami, K.; Minamoto, T.; Tomasetto, C.; et al. Inhibition of gastric carcinogenesis by the hormone gastrin is mediated by suppression of TFF1 epigenetic silencing. Gastroenterology 2011, 140, 879–891. [Google Scholar] [CrossRef]
- Cao, X.; Tsukamoto, T.; Nozaki, K.; Tanaka, H.; Shimizu, N.; Kaminishi, M.; Kumagai, T.; Tatematsu, M. Earlier Helicobacter pylori infection increases the risk for the N-methyl-N-nitrosourea-induced stomach carcinogenesis in Mongolian gerbils. Jpn. J. Cancer Res. 2002, 93, 1293–1298. [Google Scholar] [CrossRef]
- Maruta, F.; Sugiyama, A.; Ishida, K.; Ikeno, T.; Murakami, M.; Kawasaki, S.; Ota, H.; Tatematsu, M.; Katsuyama, T. Timing of N-methyl-N-nitrosourea administration affects gastric carcinogenesis in Mongolian gerbils infected with Helicobacter pylori. Cancer Lett. 2000, 160, 99–105. [Google Scholar] [CrossRef]
- Fox, J.G.; Wishnok, J.S.; Murphy, J.C.; Tannenbaum, S.R.; Correa, P. MNNG-induced gastric carcinoma in ferrets infected with Helicobacter mustelae. Carcinogenesis 1993, 14, 1957–1961. [Google Scholar] [CrossRef]
- Fox, J.G. Gastric disease in ferrets: Effects of Helicobacter mustelae, nitrosamines and reconstructive gastric surgery. Eur. J. Gastroenterol. Hepatol. 1994, 6, S57–S65. [Google Scholar]
- Fox, J.G.; Dangler, C.A.; Sager, W.; Borkowski, R.; Gliatto, J.M. Helicobacter mustelae-associated gastric adenocarcinoma in ferrets (Mustela putorius furo). Vet. Pathol. 1997, 34, 225–229. [Google Scholar] [CrossRef]
- Fox, J.G.; Correa, P.; Taylor, N.S.; Lee, A.; Otto, G.; Murphy, J.C.; Rose, R. Helicobacter mustelae-associated gastritis in ferrets. An animal model of Helicobacter pylori gastritis in humans. Gastroenterology 1990, 99, 352–361. [Google Scholar]
- Wirth, H.P.; Beins, M.H.; Yang, M.; Tham, K.T.; Blaser, M.J. Experimental infection of Mongolian gerbils with wild-type and mutant Helicobacter pylori strains. Infect. Immun. 1998, 66, 4856–4866. [Google Scholar]
- Ogura, K.; Maeda, S.; Nakao, M.; Watanabe, T.; Tada, M.; Kyutoku, T.; Yoshida, H.; Shiratori, Y.; Omata, M. Virulence factors of Helicobacter pylori responsible for gastric diseases in Mongolian gerbil. J. Exp. Med. 2000, 192, 1601–1610. [Google Scholar] [CrossRef]
- Israel, D.A.; Salama, N.; Arnold, C.N.; Moss, S.F.; Ando, T.; Wirth, H.P.; Tham, K.T.; Camorlinga, M.; Blaser, M.J.; Falkow, S.; et al. Helicobacter pylori strain-specific differences in genetic content, identified by microarray, influence host inflammatory responses. J. Clin. Invest. 2001, 107, 611–620. [Google Scholar] [CrossRef]
- Marchetti, M.; Rappuoli, R. Isogenic mutants of the cag pathogenicity island of Helicobacter pylori in the mouse model of infection: Effects on colonization efficiency. Microbiology 2002, 148, 1447–1456. [Google Scholar]
- Ehlers, S.; Warrelmann, M.; Hahn, H. In search of an animal model for experimental Campylobacter pylori infection: Administration of Campylobacter pylori to rodents. Zentralbl. Bakteriol. Mikrobiol. Hyg. A 1988, 268, 341–346. [Google Scholar]
- Cantorna, M.T.; Balish, E. Inability of human clinical strains of Helicobacter pylori to colonize the alimentary tract of germfree rodents. Can. J. Microbiol. 1990, 36, 237–241. [Google Scholar] [CrossRef]
- Lee, A.; Fox, J.G.; Otto, G.; Murphy, J. A small animal model of human Helicobacter pylori active chronic gastritis. Gastroenterology 1990, 99, 1315–1323. [Google Scholar]
- Lee, A.; Chen, M.; Coltro, N.; O'Rourke, J.; Hazell, S.; Hu, P.; Li, Y. Long term infection of the gastric mucosa with Helicobacter species does induce atrophic gastritis in an animal model of Helicobacter pylori infection. Zentralbl Bakteriol 1993, 280, 38–50. [Google Scholar] [CrossRef]
- Sakagami, T.; Dixon, M.; O'Rourke, J.; Howlett, R.; Alderuccio, F.; Vella, J.; Shimoyama, T.; Lee, A. Atrophic gastric changes in both Helicobacter felis and Helicobacter pylori infected mice are host dependent and separate from antral gastritis. Gut 1996, 39, 639–648. [Google Scholar] [CrossRef]
- Wang, T.C.; Goldenring, J.R.; Dangler, C.; Ito, S.; Mueller, A.; Jeon, W.K.; Koh, T.J.; Fox, J.G. Mice lacking secretory phospholipase A2 show altered apoptosis and differentiation with Helicobacter felis infection. Gastroenterology 1998, 114, 675–689. [Google Scholar]
- Stoicov, C.; Saffari, R.; Cai, X.; Hasyagar, C.; Houghton, J. Molecular biology of gastric cancer: Helicobacter infection and gastric adenocarcinoma: Bacterial and host factors responsible for altered growth signaling. Gene 2004, 341, 1–17. [Google Scholar] [CrossRef]
- Houghton, J.; Wang, T.C. Helicobacter pylori and gastric cancer: A new paradigm for inflammation-associated epithelial cancers. Gastroenterology 2005, 128, 1567–1578. [Google Scholar] [CrossRef]
- Cai, X.; Carlson, J.; Stoicov, C.; Li, H.; Wang, T.C.; Houghton, J. Helicobacter felis eradication restores normal architecture and inhibits gastric cancer progression in C57BL/6 mice. Gastroenterology 2005, 128, 1937–1952. [Google Scholar] [CrossRef]
- Lee, C.W.; Rickman, B.; Rogers, A.B.; Ge, Z.; Wang, T.C.; Fox, J.G. Helicobacter pylori eradication prevents progression of gastric cancer in hypergastrinemic INS-GAS mice. Cancer Res. 2008, 68, 3540–3548. [Google Scholar] [CrossRef]
- Sepulveda, A.R.; Coelho, L.G. Helicobacter pylori and gastric malignancies. Helicobacter 2002, 7, 37–42. [Google Scholar] [CrossRef]
- Ley, C.; Mohar, A.; Guarner, J.; Herrera-Goepfert, R.; Figueroa, L.S.; Halperin, D.; Johnstone, I.; Parsonnet, J. Helicobacter pylori eradication and gastric preneoplastic conditions: A randomized, double-blind, placebo-controlled trial. Cancer Epidemiol. Biomarkers Prev. 2004, 13, 4–10. [Google Scholar] [CrossRef]
- Lee, C.W.; Rickman, B.; Rogers, A.B.; Muthupalani, S.; Takaishi, S.; Yang, P.; Wang, T.C.; Fox, J.G. Combination of sulindac and antimicrobial eradication of Helicobacter pylori prevents progression of gastric cancer in hypergastrinemic INS-GAS mice. Cancer Res. 2009, 69, 8166–8174. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Willén, R.; Svensson, M.; Ljungh, A.; Wadström, T. Two-year follow-up of Helicobacter pylori infection in C57BL/6 and Balb/cA mice. APMIS 2003, 111, 514–522. [Google Scholar] [CrossRef]
- Fox, J.G.; Wang, T.C.; Rogers, A.B.; Poutahidis, T.; Ge, Z.; Taylor, N.; Dangler, C.A.; Israel, D.A.; Krishna, U.; Gaus, K.; et al. Host and microbial constituents influence Helicobacter pylori-induced cancer in a murine model of hypergastrinemia. Gastroenterology 2003, 124, 1879–1890. [Google Scholar]
- Higashi, H.; Tsutsumi, R.; Muto, S.; Sugiyama, T.; Azuma, T.; Asaka, M.; Hatakeyama, M. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science 2002, 295, 683–686. [Google Scholar] [CrossRef]
- Maeda, S.; Yoshida, H.; Ogura, K.; Mitsuno, Y.; Hirata, Y.; Yamaji, Y.; Akanuma, M.; Shiratori, Y.; Omata, M. H. pylori activates NF-kappaB through a signaling pathway involving IkappaB kinases, NF-kappaB-inducing kinase, TRAF2, and TRAF6 in gastric cancer cells. Gastroenterology 2000, 119, 97–108. [Google Scholar] [CrossRef]
- Mitsuno, Y.; Yoshida, H.; Maeda, S.; Ogura, K.; Hirata, Y.; Kawabe, T.; Shiratori, Y.; Omata, M. Helicobacter pylori induced transactivation of SRE and AP-1 through the ERK signalling pathway in gastric cancer cells. Gut 2001, 49, 18–22. [Google Scholar] [CrossRef]
- Viala, J.; Chaput, C.; Boneca, I.G.; Cardona, A.; Girardin, S.E.; Moran, A.P.; Athman, R.; Memet, S.; Huerre, M.R.; Coyle, A.J.; et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat. Immunol. 2004, 5, 1166–1174. [Google Scholar] [CrossRef]
- Crabtree, J.E.; Ferrero, R.L.; Kusters, J.G. The mouse colonizing Helicobacter pylori strain SS1 may lack a functional cag pathogenicity island. Helicobacter 2002, 7, 139–140. [Google Scholar] [CrossRef]
- Ohnishi, N.; Yuasa, H.; Tanaka, S.; Sawa, H.; Miura, M.; Matsui, A.; Higashi, H.; Musashi, M.; Iwabuchi, K.; Suzuki, M.; et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc. Natl. Acad. Sci. USA 2008, 105, 1003–1008. [Google Scholar]
- Arnold, I.C.; Lee, J.Y.; Amieva, M.R.; Roers, A.; Flavell, R.A.; Sparwasser, T.; Müller, A. Tolerance rather than immunity protects from Helicobacter pylori-induced gastric preneoplasia. Gastroenterology 2011, 140, 199–209. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, M.; Redline, R.; Nedrud, J.; Czinn, S. Role of the host in pathogenesis of Helicobacter-associated gastritis: H. felis infection of inbred and congenic mouse strains. Infect. Immun. 1996, 64, 238–245. [Google Scholar]
- Ottlecz, A.; Romero, J.J.; Lichtenberger, L.M. Helicobacter infection and phospholipase A2 enzymes: Effect of Helicobacter felis-infection on the expression and activity of sPLA2 enzymes in mouse stomach. Mol. Cell. Biochem. 2001, 221, 71–77. [Google Scholar] [CrossRef]
- Roth, K.A.; Kapadia, S.B.; Martin, S.M.; Lorenz, R.G. Cellular immune responses are essential for the development of Helicobacter felis-associated gastric pathology. J. Immunol. 1999, 163, 1490–1497. [Google Scholar]
- Eaton, K.A.; Mefford, M.; Thevenot, T. The role of T cell subsets and cytokines in the pathogenesis of Helicobacter pylori gastritis in mice. J. Immunol. 2001, 166, 7456–7461. [Google Scholar]
- Smythies, L.E.; Waites, K.B.; Lindsey, J.R.; Harris, P.R.; Ghiara, P.; Smith, P.D. Helicobacter pylori-induced mucosal inflammation is Th1 mediated and exacerbated in IL-4, but not IFN-gamma, gene-deficient mice. J. Immunol. 2000, 165, 1022–1029. [Google Scholar]
- Ismail, H.F.; Fick, P.; Zhang, J.; Lynch, R.G.; Berg, D.J. Depletion of neutrophils in IL-10−/− mice delays clearance of gastric Helicobacter infection and decreases the Th1 immune response to Helicobacter. J. Immunol. 2003, 170, 3782–3789. [Google Scholar]
- Berg, D.J.; Lynch, N.A.; Lynch, R.G.; Lauricella, D.M. Rapid development of severe hyperplastic gastritis with gastric epithelial dedifferentiation in Helicobacter felis-infected IL-10−/− mice. Am. J. Pathol. 1998, 152, 1377–1386. [Google Scholar]
- Ohana, M.; Okazaki, K.; Oshima, C.; Andra’s, D.; Nishi, T.; Uchida, K.; Uose, S.; Nakase, H.; Matsushima, Y.; Chiba, T. A critical role for IL-7R signaling in the development of Helicobacter felis-induced gastritis in mice. Gastroenterology 2001, 121, 329–336. [Google Scholar] [CrossRef]
- Sayi, A.; Kohler, E.; Hitzler, I.; Arnold, I.; Schwendener, R.; Rehrauer, H.; Müller, A. The CD4+ T cell-mediated IFN-gamma response to Helicobacter infection is essential for clearance and determines gastric cancer risk. J. Immunol. 2009, 182, 7085–7101. [Google Scholar]
- Sayi, A.; Kohler, E.; Toller, I.M.; Flavell, R.A.; Müller, W.; Roers, A.; Müller, A. TLR-2-activated B cells suppress Helicobacter-induced preneoplastic gastric immunopathology by inducing T regulatory-1 cells. J. Immunol. 2011, 186, 878–890. [Google Scholar] [CrossRef]
- Jones, N.L.; Day, A.S.; Jennings, H.; Shannon, P.T.; Galindo-Mata, E.; Sherman, P.M. Enhanced disease severity in Helicobacter pylori-infected mice deficient in Fas signaling. Infect. Immun. 2002, 70, 2591–2597. [Google Scholar] [CrossRef]
- Houghton, J.M.; Bloch, L.M.; Goldstein, M.; von Hagen, S.; Korah, R.M. In vivo disruption of the fas pathway abrogates gastric growth alterations secondary to Helicobacter infection. J. Infect. Dis. 2000, 182, 856–864. [Google Scholar] [CrossRef]
- Shibata, W.; Takaishi, S.; Muthupalani, S.; Pritchard, D.M.; Whary, M.T.; Rogers, A.B.; Fox, J.G.; Betz, K.S.; Kaestner, K.H.; Karin, M.; et al. Conditional deletion of IkappaB-kinase-beta accelerates helicobacter-dependent gastric apoptosis, proliferation, and preneoplasia. Gastroenterology 2010, 138, 1022–1034.e10. [Google Scholar]
- Fox, J.G.; Rogers, A.B.; Ihrig, M.; Taylor, N.S.; Whary, M.T.; Dockray, G.; Varro, A.; Wang, T.C. Helicobacter pylori-associated gastric cancer in INS-GAS mice is gender specific. Cancer Res. 2003, 63, 942–950. [Google Scholar]
- Sheh, A.; Lee, C.W.; Masumura, K.; Rickman, B.H.; Nohmi, T.; Wogan, G.N.; Fox, J.G.; Schauer, D.B. Mutagenic potency of Helicobacter pylori in the gastric mucosa of mice is determined by sex and duration of infection. Proc. Natl. Acad. Sci. USA 2010, 107, 15217–15222. [Google Scholar]
- Crabtree, J.E.; Court, M.; Aboshkiwa, M.A.; Jeremy, A.H.; Dixon, M.F.; Robinson, P.A. Gastric mucosal cytokine and epithelial cell responses to Helicobacter pylori infection in Mongolian gerbils. J. Pathol. 2004, 202, 197–207. [Google Scholar] [CrossRef]
- Shimizu, N.; Kaminishi, M.; Tatematsu, M.; Tsuji, E.; Yoshikawa, A.; Yamaguchi, H.; Aoki, F.; Oohara, T. Helicobacter pylori promotes development of pepsinogen-altered pyloric glands, a preneoplastic lesion of glandular stomach of BALB/c mice pretreated with N-methyl-N-nitrosourea. Cancer Lett. 1998, 123, 63–69. [Google Scholar] [CrossRef]
- Han, S.U.; Kim, Y.B.; Joo, H.J.; Hahm, K.B.; Lee, W.H.; Cho, Y.K.; Kim, D.Y.; Kim, M.W. Helicobacter pylori infection promotes gastric carcinogenesis in a mice model. J. Gastroenterol. Hepatol. 2002, 17, 253–261. [Google Scholar] [CrossRef]
- Fox, J.G.; Dangler, C.A.; Taylor, N.S.; King, A.; Koh, T.J.; Wang, T.C. High-salt diet induces gastric epithelial hyperplasia and parietal cell loss, and enhances Helicobacter pylori colonization in C57BL/6 mice. Cancer Res. 1999, 59, 4823–4828. [Google Scholar]
- Fox, J.G.; Beck, P.; Dangler, C.A.; Whary, M.T.; Wang, T.C.; Shi, H.N.; Nagler-Anderson, C. Concurrent enteric helminth infection modulates inflammation and gastric immune responses and reduces helicobacter-induced gastric atrophy. Nat. Med. 2000, 6, 536–542. [Google Scholar] [CrossRef]
- Lofgren, J.L.; Whary, M.T.; Ge, Z.; Muthupalani, S.; Taylor, N.S.; Mobley, M.; Potter, A.; Varro, A.; Eibach, D.; Suerbaum, S.; et al. Lack of commensal flora in Helicobacter pylori-infected INS-GAS mice reduces gastritis and delays intraepithelial neoplasia. Gastroenterology 2011, 140, 210–220. [Google Scholar] [CrossRef]
- Houghton, J.; Stoicov, C.; Nomura, S.; Rogers, A.B.; Carlson, J.; Li, H.; Cai, X.; Fox, J.G.; Goldenring, J.R.; Wang, T.C. Gastric cancer originating from bone marrow-derived cells. Science 2004, 306, 1568–1571. [Google Scholar]
- Varon, C.; Dubus, P.; Mazurier, F.; Asencio, C.; Chambonnier, L.; Ferrand, J.; Giese, A.; Senant-Dugot, N.; Carlotti, M.; Mégraud, F. Helicobacter pylori infection recruits bone marrow-derived cells that participate in gastric preneoplasia in mice. Gastroenterology 2012, 142, 281–291. [Google Scholar] [CrossRef]
- Worthley, D.L.; Giraud, A.S.; Wang, T.C. Stromal fibroblasts in digestive cancer. Cancer Microenviron 2010, 3, 117–125. [Google Scholar] [CrossRef]
- Rindi, G.; Bordi, C.; Rappel, S.; La Rosa, S.; Stolte, M.; Solcia, E. Gastric carcinoids and neuroendocrine carcinomas: Pathogenesis, pathology, and behavior. World J. Surg. 1996, 20, 168–172. [Google Scholar] [CrossRef]
- Ferrand, A.; Wang, T.C. Gastrin and cancer: A review. Cancer Lett. 2006, 238, 15–29. [Google Scholar] [CrossRef]
- Wang, T.C.; Brand, S.J. Function and regulation of gastrin in transgenic mice: A review. Yale J. Biol. Med. 1992, 65, 705–740. [Google Scholar]
- Wang, T.C.; Bonner-Weir, S.; Oates, P.S.; Chulak, M.; Simon, B.; Merlino, G.T.; Schmidt, E.V.; Brand, S.J. Pancreatic gastrin stimulates islet differentiation of transforming growth factor alpha-induced ductular precursor cells. J. Clin. Invest. 1993, 92, 1349–1356. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Shinomura, Y.; Tsutsui, S.; Zushi, S.; Higashimoto, Y.; Kanayama, S.; Higashiyama, S.; Taniguchi, N.; Matsuzawa, Y. Gastrin induces heparin-binding epidermal growth factor-like growth factor in rat gastric epithelial cells transfected with gastrin receptor. Gastroenterology 1999, 116, 78–89. [Google Scholar] [CrossRef]
- Goldenring, J.R.; Ray, G.S.; Soroka, C.J.; Smith, J.; Modlin, I.M.; Meise, K.S.; Coffey, R.J. Overexpression of transforming growth factor-alpha alters differentiation of gastric cell lineages. Dig. Dis. Sci. 1996, 41, 773–784. [Google Scholar] [CrossRef]
- Takaishi, S.; Tu, S.; Dubeykovskaya, Z.A.; Whary, M.T.; Muthupalani, S.; Rickman, B.H.; Rogers, A.B.; Lertkowit, N.; Varro, A.; Fox, J.G.; et al. Gastrin is an essential cofactor for helicobacter-associated gastric corpus carcinogenesis in C57BL/6 mice. Am. J. Pathol. 2009, 175, 365–375. [Google Scholar] [CrossRef]
- Cui, G.; Koh, T.J.; Chen, D.; Zhao, C.M.; Takaishi, S.; Dockray, G.J.; Varro, A.; Rogers, A.B.; Fox, J.G.; Wang, T.C. Overexpression of glycine-extended gastrin inhibits parietal cell loss and atrophy in the mouse stomach. Cancer Res. 2004, 64, 8160–8166. [Google Scholar]
- Takaishi, S.; Wang, T.C. Gene expression profiling in a mouse model of Helicobacter-induced gastric cancer. Cancer Sci. 2007, 98, 284–293. [Google Scholar] [CrossRef]
- Tu, S.; Chi, A.L.; Lim, S.; Cui, G.; Dubeykovskaya, Z.; Ai, W.; Fleming, J.V.; Takaishi, S.; Wang, T.C. Gastrin regulates the TFF2 promoter through gastrin-responsive cis-acting elements and multiple signaling pathways. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1726–G1737. [Google Scholar] [CrossRef]
- Steele, I.A.; Dimaline, R.; Pritchard, D.M.; Peek, R.M.; Wang, T.C.; Dockray, G.J.; Varro, A. Helicobacter and gastrin stimulate Reg1 expression in gastric epithelial cells through distinct promoter elements. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G347–G354. [Google Scholar] [CrossRef]
- Yin, Y.; Grabowska, A.M.; Clarke, P.A.; Whelband, E.; Robinson, K.; Argent, R.H.; Tobias, A.; Kumari, R.; Atherton, J.C.; Watson, S.A. Helicobacter pylori potentiates epithelial:mesenchymal transition in gastric cancer: Links to soluble HB-EGF, gastrin and matrix metalloproteinase-7. Gut 2010, 59, 1037–1045. [Google Scholar]
- Takaishi, S.; Cui, G.; Frederick, D.M.; Carlson, J.E.; Houghton, J.; Varro, A.; Dockray, G.J.; Ge, Z.; Whary, M.T.; Rogers, A.B.; et al. Synergistic inhibitory effects of gastrin and histamine receptor antagonists on Helicobacter-induced gastric cancer. Gastroenterology 2005, 128, 1965–1983. [Google Scholar] [CrossRef]
- Okabe, S.; Kodama, Y.; Cao, H.; Johannessen, H.; Zhao, C.M.; Wang, T.C.; Takahashi, R.; Chen, D. Topical application of acetic acid in cytoreduction of gastric cancer. A technical report using mouse model. J. Gastroenterol. Hepatol. 2012, 27, 40–48. [Google Scholar] [CrossRef]
- Ohtani, M.; García, A.; Rogers, A.B.; Ge, Z.; Taylor, N.S.; Xu, S.; Watanabe, K.; Marini, R.P.; Whary, M.T.; Wang, T.C.; et al. Protective role of 17 beta-estradiol against the development of Helicobacter pylori-induced gastric cancer in INS-GAS mice. Carcinogenesis 2007, 28, 2597–2604. [Google Scholar] [CrossRef]
- Ohtani, M.; Ge, Z.; García, A.; Rogers, A.B.; Muthupalani, S.; Taylor, N.S.; Xu, S.; Watanabe, K.; Feng, Y.; Marini, R.P.; et al. 17 β-estradiol suppresses Helicobacter pylori-induced gastric pathology in male hypergastrinemic INS-GAS mice. Carcinogenesis 2011, 32, 1244–1250. [Google Scholar] [CrossRef]
- Sheh, A.; Ge, Z.; Parry, N.M.; Muthupalani, S.; Rager, J.E.; Raczynski, A.R.; Mobley, M.W.; McCabe, A.F.; Fry, R.C.; Wang, T.C.; et al. 17β-estradiol and tamoxifen prevent gastric cancer by modulating leukocyte recruitment and oncogenic pathways in Helicobacter pylori-infected INS-GAS male mice. Cancer Prev. Res. (Phila.) 2011, 4, 1426–1435. [Google Scholar] [CrossRef]
- Stenström, B.; Zhao, C.M.; Rogers, A.B.; Nilsson, H.O.; Sturegård, E.; Lundgren, S.; Fox, J.G.; Wang, T.C.; Wadström, T.M.; Chen, D. Swedish moist snuff accelerates gastric cancer development in Helicobacter pylori-infected wild-type and gastrin transgenic mice. Carcinogenesis 2007, 28, 2041–2046. [Google Scholar] [CrossRef]
- Cui, G.; Takaishi, S.; Ai, W.; Betz, K.S.; Florholmen, J.; Koh, T.J.; Houghton, J.; Pritchard, D.M.; Wang, T.C. Gastrin-induced apoptosis contributes to carcinogenesis in the stomach. Lab. Invest. 2006, 86, 1037–1051. [Google Scholar] [CrossRef]
- Przemeck, S.M.; Varro, A.; Berry, D.; Steele, I.; Wang, T.C.; Dockray, G.J.; Pritchard, D.M. Hypergastrinemia increases gastric epithelial susceptibility to apoptosis. Regul. Pept. 2008, 146, 147–156. [Google Scholar] [CrossRef]
- Singh, P.; Velasco, M.; Given, R.; Wargovich, M.; Varro, A.; Wang, T.C. Mice overexpressing progastrin are predisposed for developing aberrant colonic crypt foci in response to AOM. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 278, G390–G399. [Google Scholar]
- Singh, P.; Velasco, M.; Given, R.; Varro, A.; Wang, T.C. Progastrin expression predisposes mice to colon carcinomas and adenomas in response to a chemical carcinogen. Gastroenterology 2000, 119, 162–171. [Google Scholar] [CrossRef]
- Ottewell, P.D.; Duckworth, C.A.; Varro, A.; Dimaline, R.; Wang, T.C.; Watson, A.J.; Dockray, G.J.; Pritchard, D.M. Gastrin increases murine intestinal crypt regeneration following injury. Gastroenterology 2006, 130, 1169–1180. [Google Scholar] [CrossRef]
- Thomson, M.J.; Pritchard, D.M.; Boxall, S.A.; Abuderman, A.A.; Williams, J.M.; Varro, A.; Crabtree, J.E. Gastric Helicobacter Infection Induces Iron Deficiency in the INS-GAS Mouse. PLoS One 2012, 7, e50194. [Google Scholar]
- Koh, T.J.; Goldenring, J.R.; Ito, S.; Mashimo, H.; Kopin, A.S.; Varro, A.; Dockray, G.J.; Wang, T.C. Gastrin deficiency results in altered gastric differentiation and decreased colonic proliferation in mice. Gastroenterology 1997, 113, 1015–1025. [Google Scholar] [CrossRef]
- Zavros, Y.; Rieder, G.; Ferguson, A.; Samuelson, L.C.; Merchant, J.L. Genetic or chemical hypochlorhydria is associated with inflammation that modulates parietal and G-cell populations in mice. Gastroenterology 2002, 122, 119–133. [Google Scholar] [CrossRef]
- Zavros, Y.; Eaton, K.A.; Kang, W.; Rathinavelu, S.; Katukuri, V.; Kao, J.Y.; Samuelson, L.C.; Merchant, J.L. Chronic gastritis in the hypochlorhydric gastrin-deficient mouse progresses to adenocarcinoma. Oncogene 2005, 24, 2354–2366. [Google Scholar]
- Lefebvre, O.; Chenard, M.P.; Masson, R.; Linares, J.; Dierich, A.; LeMeur, M.; Wendling, C.; Tomasetto, C.; Chambon, P.; Rio, M.C. Gastric mucosa abnormalities and tumorigenesis in mice lacking the pS2 trefoil protein. Science 1996, 274, 259–262. [Google Scholar] [CrossRef]
- Soutto, M.; Belkhiri, A.; Piazuelo, M.B.; Schneider, B.G.; Peng, D.; Jiang, A.; Washington, M.K.; Kokoye, Y.; Crowe, S.E.; Zaika, A.; et al. Loss of TFF1 is associated with activation of NF-κB-mediated inflammation and gastric neoplasia in mice and humans. J. Clin. Invest. 2011, 121, 1753–1767. [Google Scholar] [CrossRef]
- Beckler, A.D.; Roche, J.K.; Harper, J.C.; Petroni, G.; Frierson, H.F.; Moskaluk, C.A.; El-Rifai, W.; Powell, S.M. Decreased abundance of trefoil factor 1 transcript in the majority of gastric carcinomas. Cancer 2003, 98, 2184–2191. [Google Scholar] [CrossRef]
- Fujimoto, J.; Yasui, W.; Tahara, H.; Tahara, E.; Kudo, Y.; Yokozaki, H. DNA hypermethylation at the pS2 promoter region is associated with early stage of stomach carcinogenesis. Cancer Lett. 2000, 149, 125–134. [Google Scholar] [CrossRef]
- Carvalho, R.; Kayademir, T.; Soares, P.; Canedo, P.; Sousa, S.; Oliveira, C.; Leistenschneider, P.; Seruca, R.; Gött, P.; Blin, N.; et al. Loss of heterozygosity and promoter methylation, but not mutation, may underlie loss of TFF1 in gastric carcinoma. Lab. Invest. 2002, 82, 1319–1326. [Google Scholar]
- Khan, Z.E.; Wang, T.C.; Cui, G.; Chi, A.L.; Dimaline, R. Transcriptional regulation of the human trefoil factor, TFF1, by gastrin. Gastroenterology 2003, 125, 510–521. [Google Scholar] [CrossRef]
- Clyne, M.; Dillon, P.; Daly, S.; O'Kennedy, R.; May, F.E.; Westley, B.R.; Drumm, B. Helicobacter pylori interacts with the human single-domain trefoil protein TFF1. Proc. Natl. Acad. Sci. USA 2004, 101, 7409–7414. [Google Scholar]
- Reeves, E.P.; Ali, T.; Leonard, P.; Hearty, S.; O'Kennedy, R.; May, F.E.; Westley, B.R.; Josenhans, C.; Rust, M.; Suerbaum, S.; et al. Helicobacter pylori lipopolysaccharide interacts with TFF1 in a pH-dependent manner. Gastroenterology 2008, 135, 2043–2054.e2. [Google Scholar] [CrossRef]
- Rio, M.C.; Bellocq, J.P.; Daniel, J.Y.; Tomasetto, C.; Lathe, R.; Chenard, M.P.; Batzenschlager, A.; Chambon, P. Breast cancer-associated pS2 protein: Synthesis and secretion by normal stomach mucosa. Science 1988, 241, 705–708. [Google Scholar]
- Hanby, A.M.; Poulsom, R.; Singh, S.; Elia, G.; Jeffery, R.E.; Wright, N.A. Spasmolytic polypeptide is a major antral peptide: Distribution of the trefoil peptides human spasmolytic polypeptide and pS2 in the stomach. Gastroenterology 1993, 105, 1110–1116. [Google Scholar]
- Hanby, A.M.; Poulsom, R.; Elia, G.; Singh, S.; Longcroft, J.M.; Wright, N.A. The expression of the trefoil peptides pS2 and human spasmolytic polypeptide (hSP) in “gastric metaplasia” of the proximal duodenum: Implications for the nature of “gastric metaplasia”. J. Pathol. 1993, 169, 355–360. [Google Scholar] [CrossRef]
- Farrell, J.J.; Taupin, D.; Koh, T.J.; Chen, D.; Zhao, C.M.; Podolsky, D.K.; Wang, T.C. TFF2/SP-deficient mice show decreased gastric proliferation, increased acid secretion, and increased susceptibility to NSAID injury. J. Clin. Invest. 2002, 109, 193–204. [Google Scholar]
- Fox, J.G.; Rogers, A.B.; Whary, M.T.; Ge, Z.; Ohtani, M.; Jones, E.K.; Wang, T.C. Accelerated progression of gastritis to dysplasia in the pyloric antrum of TFF2−/− C57BL6 × Sv129 Helicobacter pylori-infected mice. Am. J. Pathol. 2007, 171, 1520–1528. [Google Scholar] [CrossRef]
- Peterson, A.J.; Menheniott, T.R.; O'Connor, L.; Walduck, A.K.; Fox, J.G.; Kawakami, K.; Minamoto, T.; Ong, E.K.; Wang, T.C.; Judd, L.M.; et al. Helicobacter pylori infection promotes methylation and silencing of trefoil factor 2, leading to gastric tumor development in mice and humans. Gastroenterology 2010, 139, 2005–2017. [Google Scholar] [CrossRef]
- Jenkins, B.J.; Grail, D.; Nheu, T.; Najdovska, M.; Wang, B.; Waring, P.; Inglese, M.; McLoughlin, R.M.; Jones, S.A.; Topley, N.; et al. Hyperactivation of Stat3 in gp130 mutant mice promotes gastric hyperproliferation and desensitizes TGF-beta signaling. Nat. Med. 2005, 11, 845–852. [Google Scholar]
- Tebbutt, N.C.; Giraud, A.S.; Inglese, M.; Jenkins, B.; Waring, P.; Clay, F.J.; Malki, S.; Alderman, B.M.; Grail, D.; Hollande, F.; et al. Reciprocal regulation of gastrointestinal homeostasis by SHP2 and STAT-mediated trefoil gene activation in gp130 mutant mice. Nat. Med. 2002, 8, 1089–1097. [Google Scholar]
- Jackson, C.B.; Judd, L.M.; Menheniott, T.R.; Kronborg, I.; Dow, C.; Yeomans, N.D.; Boussioutas, A.; Robb, L.; Giraud, A.S. Augmented gp130-mediated cytokine signalling accompanies human gastric cancer progression. J. Pathol. 2007, 213, 140–151. [Google Scholar] [CrossRef]
- Judd, L.M.; Ulaganathan, M.; Howlett, M.; Giraud, A.S. Cytokine signalling by gp130 regulates gastric mucosal healing after ulceration and, indirectly, antral tumour progression. J. Pathol. 2009, 217, 552–562. [Google Scholar] [CrossRef]
- Ernst, M.; Najdovska, M.; Grail, D.; Lundgren-May, T.; Buchert, M.; Tye, H.; Matthews, V.B.; Armes, J.; Bhathal, P.S.; Hughes, N.R.; et al. STAT3 and STAT1 mediate IL-11-dependent and inflammation-associated gastric tumorigenesis in gp130 receptor mutant mice. J. Clin. Invest. 2008, 118, 1727–1738. [Google Scholar]
- Tu, S.; Bhagat, G.; Cui, G.; Takaishi, S.; Kurt-Jones, E.A.; Rickman, B.; Betz, K.S.; Penz-Oesterreicher, M.; Bjorkdahl, O.; Fox, J.G.; et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 2008, 14, 408–419. [Google Scholar] [CrossRef]
- Song, X.; Krelin, Y.; Dvorkin, T.; Bjorkdahl, O.; Segal, S.; Dinarello, C.A.; Voronov, E.; Apte, R.N. CD11b+/Gr-1+ immature myeloid cells mediate suppression of T cells in mice bearing tumors of IL-1beta-secreting cells. J. Immunol. 2005, 175, 8200–8208. [Google Scholar]
- Waghray, M.; Zavros, Y.; Saqui-Salces, M.; El-Zaatari, M.; Alamelumangapuram, C.B.; Todisco, A.; Eaton, K.A.; Merchant, J.L. Interleukin-1beta promotes gastric atrophy through suppression of Sonic Hedgehog. Gastroenterology 2010, 138, 562–572.e2. [Google Scholar]
- Bos, J.L. Ras oncogenes in human cancer: A review. Cancer Res. 1989, 49, 4682–4689. [Google Scholar]
- Ellis, C.A.; Clark, G. The importance of being K-Ras. Cell. Signal. 2000, 12, 425–434. [Google Scholar] [CrossRef]
- Ushijima, T.; Sasako, M. Focus on gastric cancer. Cancer Cell 2004, 5, 121–125. [Google Scholar] [CrossRef]
- Frame, S.; Balmain, A. Integration of positive and negative growth signals during ras pathway activation in vivo. Curr. Opin. Genet. Dev. 2000, 10, 106–113. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef]
- Brembeck, F.H.; Schreiber, F.S.; Deramaudt, T.B.; Craig, L.; Rhoades, B.; Swain, G.; Grippo, P.; Stoffers, D.A.; Silberg, D.G.; Rustgi, A.K. The mutant K-ras oncogene causes pancreatic periductal lymphocytic infiltration and gastric mucous neck cell hyperplasia in transgenic mice. Cancer Res. 2003, 63, 2005–2009. [Google Scholar]
- Okumura, T.; Ericksen, R.E.; Takaishi, S.; Wang, S.S.; Dubeykovskiy, Z.; Shibata, W.; Betz, K.S.; Muthupalani, S.; Rogers, A.B.; Fox, J.G.; et al. K-ras mutation targeted to gastric tissue progenitor cells results in chronic inflammation, an altered microenvironment, and progression to intraepithelial neoplasia. Cancer Res. 2010, 70, 8435–8445. [Google Scholar]
- Ray, K.C.; Bell, K.M.; Yan, J.; Gu, G.; Chung, C.H.; Washington, M.K.; Means, A.L. Epithelial tissues have varying degrees of susceptibility to Kras(G12D)-initiated tumorigenesis in a mouse model. PLoS One 2011, 6, e16786. [Google Scholar]
- Matkar, S.S.; Durham, A.; Brice, A.; Wang, T.C.; Rustgi, A.K.; Hua, X. Systemic activation of K-ras rapidly induces gastric hyperplasia and metaplasia in mice. Am. J. Cancer Res. 2011, 1, 432–445. [Google Scholar]
- Park, W.S.; Oh, R.R.; Park, J.Y.; Lee, S.H.; Shin, M.S.; Kim, Y.S.; Kim, S.Y.; Lee, H.K.; Kim, P.J.; Oh, S.T.; et al. Frequent somatic mutations of the beta-catenin gene in intestinal-type gastric cancer. Cancer Res. 1999, 59, 4257–4260. [Google Scholar]
- Park, J.G.; Park, K.J.; Ahn, Y.O.; Song, I.S.; Choi, K.W.; Moon, H.Y.; Choo, S.Y.; Kim, J.P. Risk of gastric cancer among Korean familial adenomatous polyposis patients. Report of three cases. Dis. Colon Rectum. 1992, 35, 996–998. [Google Scholar] [CrossRef]
- Abraham, S.C.; Nobukawa, B.; Giardiello, F.M.; Hamilton, S.R.; Wu, T.T. Fundic gland polyps in familial adenomatous polyposis: Neoplasms with frequent somatic adenomatous polyposis coli gene alterations. Am. J. Pathol. 2000, 157, 747–754. [Google Scholar]
- Fox, J.G.; Dangler, C.A.; Whary, M.T.; Edelman, W.; Kucherlapati, R.; Wang, T.C. Mice carrying a truncated Apc gene have diminished gastric epithelial proliferation, gastric inflammation, and humoral immunity in response to Helicobacter felis infection. Cancer Res. 1997, 57, 3972–3978. [Google Scholar]
- Oshima, H.; Matsunaga, A.; Fujimura, T.; Tsukamoto, T.; Taketo, M.M.; Oshima, M. Carcinogenesis in mouse stomach by simultaneous activation of the Wnt signaling and prostaglandin E2 pathway. Gastroenterology 2006, 131, 1086–1095. [Google Scholar] [CrossRef]
- Ristimäki, A.; Honkanen, N.; Jänkälä, H.; Sipponen, P.; Härkönen, M. Expression of cyclooxygenase-2 in human gastric carcinoma. Cancer Res. 1997, 57, 1276–1280. [Google Scholar]
- Hu, P.J.; Yu, J.; Zeng, Z.R.; Leung, W.K.; Lin, H.L.; Tang, B.D.; Bai, A.H.; Sung, J.J. Chemoprevention of gastric cancer by celecoxib in rats. Gut 2004, 53, 195–200. [Google Scholar] [CrossRef]
- Lee, C.W.; Rickman, B.; Rogers, A.B.; Muthupalani, S.; Takaishi, S.; Yang, P.; Wang, T.C.; Fox, J.G. Combination of sulindac and antimicrobial eradication of Helicobacter pylori prevents progression of gastric cancer in hypergastrinemic INS-GAS mice. Cancer Res. 2009, 69, 8166–8174. [Google Scholar]
- Xiao, F.; Furuta, T.; Takashima, M.; Shirai, N.; Hanai, H. Effects of cyclooxygenase-2 inhibitor on gastric acid secretion in Helicobacter pylori-infected C57BL/6 mice. Scand. J. Gastroenterol. 2001, 36, 577–583. [Google Scholar] [CrossRef]
- Xiao, F.; Furuta, T.; Takashima, M.; Shirai, N.; Hanai, H. Involvement of cyclooxygenase-2 in hyperplastic gastritis induced by Helicobacter pylori infection in C57BL/6 mice. Aliment. Pharmacol. Ther. 2001, 15, 875–886. [Google Scholar] [CrossRef]
- Hahm, K.B.; Song, Y.J.; Oh, T.Y.; Lee, J.S.; Surh, Y.J.; Kim, Y.B.; Yoo, B.M.; Kim, J.H.; Han, S.U.; Nahm, K.T.; et al. Chemoprevention of Helicobacter pylori-associated gastric carcinogenesis in a mouse model: Is it possible? J. Biochem. Mol. Biol. 2003, 36, 82–94. [Google Scholar] [CrossRef]
- Oshima, H.; Oshima, M.; Inaba, K.; Taketo, M.M. Hyperplastic gastric tumors induced by activated macrophages in COX-2/mPGES-1 transgenic mice. EMBO J. 2004, 23, 1669–1678. [Google Scholar] [CrossRef]
- Oshima, M.; Oshima, H.; Matsunaga, A.; Taketo, M.M. Hyperplastic gastric tumors with spasmolytic polypeptide-expressing metaplasia caused by tumor necrosis factor-alpha-dependent inflammation in cyclooxygenase-2/microsomal prostaglandin E synthase-1 transgenic mice. Cancer Res. 2005, 65, 9147–9151. [Google Scholar] [CrossRef]
- Takaishi, S.; Okumura, T.; Tu, S.; Wang, S.S.; Shibata, W.; Vigneshwaran, R.; Gordon, S.A.; Shimada, Y.; Wang, T.C. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells 2009, 27, 1006–1020. [Google Scholar] [CrossRef]
- Ishimoto, T.; Nagano, O.; Yae, T.; Tamada, M.; Motohara, T.; Oshima, H.; Oshima, M.; Ikeda, T.; Asaba, R.; Yagi, H.; et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell 2011, 19, 387–400. [Google Scholar] [CrossRef]
- Markowitz, S.D.; Roberts, A.B. Tumor suppressor activity of the TGF-beta pathway in human cancers. Cytokine Growth Factor Rev. 1996, 7, 93–102. [Google Scholar]
- Yang, H.K.; Kang, S.H.; Kim, Y.S.; Won, K.; Bang, Y.J.; Kim, S.J. Truncation of the TGF-beta type II receptor gene results in insensitivity to TGF-beta in human gastric cancer cells. Oncogene 1999, 18, 2213–2219. [Google Scholar] [CrossRef]
- Wu, M.S.; Lee, C.W.; Shun, C.T.; Wang, H.P.; Lee, W.J.; Chang, M.C.; Sheu, J.C.; Lin, J.T. Distinct clinicopathologic and genetic profiles in sporadic gastric cancer with different mutator phenotypes. Genes Chromosomes Cancer 2000, 27, 403–411. [Google Scholar] [CrossRef]
- Crawford, S.E.; Stellmach, V.; Murphy-Ullrich, J.E.; Ribeiro, S.M.; Lawler, J.; Hynes, R.O.; Boivin, G.P.; Bouck, N. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell 1998, 93, 1159–1170. [Google Scholar] [CrossRef]
- Hahm, K.B.; Lee, K.M.; Kim, Y.B.; Hong, W.S.; Lee, W.H.; Han, S.U.; Kim, M.W.; Ahn, B.O.; Oh, T.Y.; Lee, M.H.; et al. Conditional loss of TGF-beta signalling leads to increased susceptibility to gastrointestinal carcinogenesis in mice. Aliment. Pharmacol. Ther. 2002, 16, 115–127. [Google Scholar]
- Xu, X.; Brodie, S.G.; Yang, X.; Im, Y.H.; Parks, W.T.; Chen, L.; Zhou, Y.X.; Weinstein, M.; Kim, S.J.; Deng, C.X. Haploid loss of the tumor suppressor Smad4/Dpc4 initiates gastric polyposis and cancer in mice. Oncogene 2000, 19, 1868–1874. [Google Scholar] [CrossRef]
- Takaku, K.; Miyoshi, H.; Matsunaga, A.; Oshima, M.; Sasaki, N.; Taketo, M.M. Gastric and duodenal polyps in Smad4 (Dpc4) knockout mice. Cancer Res. 1999, 59, 6113–6117. [Google Scholar]
- Hahn, J.N.; Falck, V.G.; Jirik, F.R. Smad4 deficiency in T cells leads to the Th17-associated development of premalignant gastroduodenal lesions in mice. J. Clin. Invest. 2011, 121, 4030–4042. [Google Scholar] [CrossRef]
- Kim, B.G.; Li, C.; Qiao, W.; Mamura, M.; Kasprzak, B.; Kasperczak, B.; Anver, M.; Wolfraim, L.; Hong, S.; Mushinski, E.; et al. Smad4 signalling in T cells is required for suppression of gastrointestinal cancer. Nature 2006, 441, 1015–1019. [Google Scholar]
- Tsuzuki, T.; Egashira, A.; Igarashi, H.; Iwakuma, T.; Nakatsuru, Y.; Tominaga, Y.; Kawate, H.; Nakao, K.; Nakamura, K.; Ide, F.; et al. Spontaneous tumorigenesis in mice defective in the MTH1 gene encoding 8-oxo-dGTPase. Proc. Natl. Acad. Sci. USA 2001, 98, 11456–11461. [Google Scholar]
- Shimada, S.; Mimata, A.; Sekine, M.; Mogushi, K.; Akiyama, Y.; Fukamachi, H.; Jonkers, J.; Tanaka, H.; Eishi, Y.; Yuasa, Y. Synergistic tumour suppressor activity of E-cadherin and p53 in a conditional mouse model for metastatic diffuse-type gastric cancer. Gut 2012, 61, 344–353. [Google Scholar]
- Ito, K.; Chuang, L.S.; Ito, T.; Chang, T.L.; Fukamachi, H.; Salto-Tellez, M.; Ito, Y. Loss of Runx3 is a key event in inducing precancerous state of the stomach. Gastroenterology 2011, 140, 1536–1546.e8. [Google Scholar] [CrossRef]
- Ito, K.; Liu, Q.; Salto-Tellez, M.; Yano, T.; Tada, K.; Ida, H.; Huang, C.; Shah, N.; Inoue, M.; Rajnakova, A.; et al. RUNX3, a novel tumor suppressor, is frequently inactivated in gastric cancer by protein mislocalization. Cancer Res. 2005, 65, 7743–7750. [Google Scholar]
- Li, Q.L.; Ito, K.; Sakakura, C.; Fukamachi, H.; Inoue, K.; Chi, X.Z.; Lee, K.Y.; Nomura, S.; Lee, C.W.; Han, S.B.; et al. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell 2002, 109, 113–124. [Google Scholar] [CrossRef]
- Desai, T.K.; Barkel, D. Syndromic colon cancer: Lynch syndrome and familial adenomatous polyposis. Gastroenterol. Clin. North. Am. 2008, 37, 47–72. [Google Scholar] [CrossRef]
- Fox, J.G.; Li, X.; Cahill, R.J.; Andrutis, K.; Rustgi, A.K.; Odze, R.; Wang, T.C. Hypertrophic gastropathy in Helicobacter felis-infected wild-type C57BL/6 mice and p53 hemizygous transgenic mice. Gastroenterology 1996, 110, 155–166. [Google Scholar] [CrossRef]
- Jenks, P.J.; Jeremy, A.H.; Robinson, P.A.; Walker, M.M.; Crabtree, J.E. Long-term infection with Helicobacter felis and inactivation of the tumour suppressor gene p53 cumulatively enhance the gastric mutation frequency in Big Blue transgenic mice. J. Pathol. 2003, 201, 596–602. [Google Scholar] [CrossRef]
- Ohgaki, H.; Fukuda, M.; Tohma, Y.; Huang, H.; Stoica, G.; Tatematsu, M.; Donehower, L.A. Effect of intragastric application of N-methylnitrosourea in p53 knockout mice. Mol. Carcinog. 2000, 28, 97–101. [Google Scholar] [CrossRef]
- Suzuki, H.; Miyazawa, M.; Kai, A.; Suzuki, M.; Suematsu, M.; Miura, S.; Ishii, H. No difference in the level of gastric mucosal cell apoptosis and proliferation in Helicobacter pylori-colonized p53 heterozygous knockout mice. Aliment. Pharmacol. Ther. 2002, 16, 158–166. [Google Scholar] [CrossRef]
- Wei, D.; Gong, W.; Kanai, M.; Schlunk, C.; Wang, L.; Yao, J.C.; Wu, T.T.; Huang, S.; Xie, K. Drastic down-regulation of Krüppel-like factor 4 expression is critical in human gastric cancer development and progression. Cancer Res. 2005, 65, 2746–2754. [Google Scholar]
- Becker, K.F.; Atkinson, M.J.; Reich, U.; Becker, I.; Nekarda, H.; Siewert, J.R.; Höfler, H. E-cadherin gene mutations provide clues to diffuse type gastric carcinomas. Cancer Res. 1994, 54, 3845–3852. [Google Scholar]
- Tamura, G.; Yin, J.; Wang, S.; Fleisher, A.S.; Zou, T.; Abraham, J.M.; Kong, D.; Smolinski, K.N.; Wilson, K.T.; James, S.P.; et al. E-Cadherin gene promoter hypermethylation in primary human gastric carcinomas. J. Natl. Cancer Inst. 2000, 92, 569–573. [Google Scholar] [CrossRef]
- Guilford, P.; Hopkins, J.; Harraway, J.; McLeod, M.; McLeod, N.; Harawira, P.; Taite, H.; Scoular, R.; Miller, A.; Reeve, A.E. E-cadherin germline mutations in familial gastric cancer. Nature 1998, 392, 402–405. [Google Scholar] [CrossRef]
- Takagi, H.; Fukusato, T.; Kawaharada, U.; Kuboyama, S.; Merlino, G.; Tsutsumi, Y. Histochemical analysis of hyperplastic stomach of TGF-alpha transgenic mice. Dig. Dis. Sci. 1997, 42, 91–98. [Google Scholar] [CrossRef]
- Li, Q.; Karam, S.M.; Gordon, J.I. Diphtheria toxin-mediated ablation of parietal cells in the stomach of transgenic mice. J. Biol. Chem. 1996, 271, 3671–3676. [Google Scholar]
- Canfield, V.; West, A.B.; Goldenring, J.R.; Levenson, R. Genetic ablation of parietal cells in transgenic mice: A new model for analyzing cell lineage relationships in the gastric mucosa. Proc. Natl. Acad. Sci. USA 1996, 93, 2431–2435. [Google Scholar]
- Spicer, Z.; Miller, M.L.; Andringa, A.; Riddle, T.M.; Duffy, J.J.; Doetschman, T.; Shull, G.E. Stomachs of mice lacking the gastric H,K-ATPase alpha -subunit have achlorhydria, abnormal parietal cells, and ciliated metaplasia. J. Biol. Chem. 2000, 275, 21555–21565. [Google Scholar]
- Scarff, K.L.; Judd, L.M.; Toh, B.H.; Gleeson, P.A.; van Driel, I.R. Gastric H(+),K(+)-adenosine triphosphatase beta subunit is required for normal function, development, and membrane structure of mouse parietal cells. Gastroenterology 1999, 117, 605–618. [Google Scholar] [CrossRef]
- Franic, T.V.; Judd, L.M.; Robinson, D.; Barrett, S.P.; Scarff, K.L.; Gleeson, P.A.; Samuelson, L.C.; van Driel, I.R. Regulation of gastric epithelial cell development revealed in H(+)/K(+)-ATPase beta-subunit- and gastrin-deficient mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G1502–G1511. [Google Scholar]
- Schultheis, P.J.; Clarke, L.L.; Meneton, P.; Harline, M.; Boivin, G.P.; Stemmermann, G.; Duffy, J.J.; Doetschman, T.; Miller, M.L.; Shull, G.E. Targeted disruption of the murine Na+/H+ exchanger isoform 2 gene causes reduced viability of gastric parietal cells and loss of net acid secretion. J. Clin. Invest. 1998, 101, 1243–1253. [Google Scholar] [CrossRef]
- Gut, M.O.; Parkkila, S.; Vernerová, Z.; Rohde, E.; Závada, J.; Höcker, M.; Pastorek, J.; Karttunen, T.; Gibadulinová, A.; Závadová, Z.; et al. Gastric hyperplasia in mice with targeted disruption of the carbonic anhydrase gene Car9. Gastroenterology 2002, 123, 1889–1903. [Google Scholar] [CrossRef]
- Nagata, A.; Ito, M.; Iwata, N.; Kuno, J.; Takano, H.; Minowa, O.; Chihara, K.; Matsui, T.; Noda, T. G protein-coupled cholecystokinin-B/gastrin receptors are responsible for physiological cell growth of the stomach mucosa in vivo. Proc. Natl. Acad. Sci. USA 1996, 93, 11825–11830. [Google Scholar]
- Langhans, N.; Rindi, G.; Chiu, M.; Rehfeld, J.F.; Ardman, B.; Beinborn, M.; Kopin, A.S. Abnormal gastric histology and decreased acid production in cholecystokinin-B/gastrin receptor-deficient mice. Gastroenterology 1997, 112, 280–286. [Google Scholar] [CrossRef]
- Xiao, C.; Ogle, S.A.; Schumacher, M.A.; Orr-Asman, M.A.; Miller, M.L.; Lertkowit, N.; Varro, A.; Hollande, F.; Zavros, Y. Loss of parietal cell expression of Sonic hedgehog induces hypergastrinemia and hyperproliferation of surface mucous cells. Gastroenterology 2010, 138, 550–561.e8. [Google Scholar]
- Goldenring, J.R.; Ray, G.S.; Coffey, R.J.; Meunier, P.C.; Haley, P.J.; Barnes, T.B.; Car, B.D. Reversible drug-induced oxyntic atrophy in rats. Gastroenterology 2000, 118, 1080–1093. [Google Scholar]
- Nomura, S.; Settle, S.H.; Leys, C.M.; Means, A.L.; Peek, R.M.; Leach, S.D.; Wright, C.V.; Coffey, R.J.; Goldenring, J.R. Evidence for repatterning of the gastric fundic epithelium associated with Ménétrier's disease and TGFalpha overexpression. Gastroenterology 2005, 128, 1292–1305. [Google Scholar] [CrossRef]
- Nam, K.T.; Lee, H.J.; Sousa, J.F.; Weis, V.G.; O'Neal, R.L.; Finke, P.E.; Romero-Gallo, J.; Shi, G.; Mills, J.C.; Peek, R.M.; et al. Mature chief cells are cryptic progenitors for metaplasia in the stomach. Gastroenterology 2010, 139, 2028–2037.e9. [Google Scholar] [CrossRef]
- Silberg, D.G.; Sullivan, J.; Kang, E.; Swain, G.P.; Moffett, J.; Sund, N.J.; Sackett, S.D.; Kaestner, K.H. Cdx2 ectopic expression induces gastric intestinal metaplasia in transgenic mice. Gastroenterology 2002, 122, 689–696. [Google Scholar] [CrossRef]
- Mutoh, H.; Hakamata, Y.; Sato, K.; Eda, A.; Yanaka, I.; Honda, S.; Osawa, H.; Kaneko, Y.; Sugano, K. Conversion of gastric mucosa to intestinal metaplasia in Cdx2-expressing transgenic mice. Biochem. Biophys. Res. Commun. 2002, 294, 470–479. [Google Scholar] [CrossRef]
- Mutoh, H.; Sakurai, S.; Satoh, K.; Osawa, H.; Hakamata, Y.; Takeuchi, T.; Sugano, K. Cdx1 induced intestinal metaplasia in the transgenic mouse stomach: Comparative study with Cdx2 transgenic mice. Gut 2004, 53, 1416–1423. [Google Scholar] [CrossRef]
- Friis-Hansen, L.; Sundler, F.; Li, Y.; Gillespie, P.J.; Saunders, T.L.; Greenson, J.K.; Owyang, C.; Rehfeld, J.F.; Samuelson, L.C. Impaired gastric acid secretion in gastrin-deficient mice. Am. J. Physiol. 1998, 274, G561–G568. [Google Scholar]
- Goldenring, J.R.; Nomura, S. Differentiation of the gastric mucosa III. Animal models of oxyntic atrophy and metaplasia. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G999–G1004. [Google Scholar] [CrossRef]
- Hattori, T. Development of adenocarcinomas in the stomach. Cancer 1986, 57, 1528–1534. [Google Scholar] [CrossRef]
- Goldenring, J.R.; Wang, T.C.; Mills, J.C.; Wright, N.A. Spasmolytic polypeptide-expressing metaplasia: Time for reevaluation of metaplasias and the origins of gastric cancer. Gastroenterology 2010, 138, 2207–2210. [Google Scholar]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef]
- Powell, A.E.; Wang, Y.; Li, Y.; Poulin, E.J.; Means, A.L.; Washington, M.K.; Higginbotham, J.N.; Juchheim, A.; Prasad, N.; Levy, S.E.; et al. The pan-ErbB negative regulator Lrig1 is an intestinal stem cell marker that functions as a tumor suppressor. Cell 2012, 149, 146–158. [Google Scholar] [CrossRef]
- Arnold, K.; Sarkar, A.; Yram, M.A.; Polo, J.M.; Bronson, R.; Sengupta, S.; Seandel, M.; Geijsen, N.; Hochedlinger, K. Sox2(+) adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell 2011, 9, 317–329. [Google Scholar] [CrossRef]
- Furuyama, K.; Kawaguchi, Y.; Akiyama, H.; Horiguchi, M.; Kodama, S.; Kuhara, T.; Hosokawa, S.; Elbahrawy, A.; Soeda, T.; Koizumi, M.; et al. Continuous cell supply from a Sox9-expressing progenitor zone in adult liver, exocrine pancreas and intestine. Nat. Genet. 2011, 43, 34–41. [Google Scholar] [CrossRef]
- Sangiorgi, E.; Capecchi, M.R. Bmi1 is expressed in vivo in intestinal stem cells. Nat. Genet. 2008, 40, 915–920. [Google Scholar] [CrossRef]
- Takeda, N.; Jain, R.; LeBoeuf, M.R.; Wang, Q.; Lu, M.M.; Epstein, J.A. Interconversion between intestinal stem cell populations in distinct niches. Science 2011, 334, 1420–1424. [Google Scholar] [CrossRef]
- Barker, N.; Huch, M.; Kujala, P.; van de Wetering, M.; Snippert, H.J.; van Es, J.H.; Sato, T.; Stange, D.E.; Begthel, H.; van den Born, M.; et al. Lgr5(+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell 2010, 6, 25–36. [Google Scholar] [CrossRef]
- Qiao, X.T.; Ziel, J.W.; McKimpson, W.; Madison, B.B.; Todisco, A.; Merchant, J.L.; Samuelson, L.C.; Gumucio, D.L. Prospective identification of a multilineage progenitor in murine stomach epithelium. Gastroenterology 2007, 133, 1989–1998. [Google Scholar] [CrossRef]
- Quante, M.; Marrache, F.; Goldenring, J.R.; Wang, T.C. TFF2 mRNA transcript expression marks a gland progenitor cell of the gastric oxyntic mucosa. Gastroenterology 2010, 139, 2018–2027.e2. [Google Scholar] [CrossRef]
- Kim, T.H.; Shivdasani, R.A. Notch signaling in stomach epithelial stem cell homeostasis. J. Exp. Med. 2011, 208, 677–688. [Google Scholar] [CrossRef]
- Zang, Z.J.; Cutcutache, I.; Poon, S.L.; Zhang, S.L.; McPherson, J.R.; Tao, J.; Rajasegaran, V.; Heng, H.L.; Deng, N.; Gan, A.; et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat. Genet. 2012, 44, 570–574. [Google Scholar] [CrossRef]
- Sakamoto, H.; Yoshimura, K.; Saeki, N.; Katai, H.; Shimoda, T.; Matsuno, Y.; Saito, D.; Sugimura, H.; Tanioka, F.; Kato, S.; et al. Genetic variation in PSCA is associated with susceptibility to diffuse-type gastric cancer. Nat. Genet. 2008, 40, 730–740. [Google Scholar] [CrossRef]
- Shi, Y.; Hu, Z.; Wu, C.; Dai, J.; Li, H.; Dong, J.; Wang, M.; Miao, X.; Zhou, Y.; Lu, F.; et al. A genome-wide association study identifies new susceptibility loci for non-cardia gastric cancer at 3q13.3.31 and 5p13.1. Nat. Genet. 2011, 43, 1215–1218. [Google Scholar] [CrossRef]
- Abnet, C.C.; Freedman, N.D.; Hu, N.; Wang, Z.; Yu, K.; Shu, X.O.; Yuan, J.M.; Zheng, W.; Dawsey, S.M.; Dong, L.M.; et al. A shared susceptibility locus in PLCE1 at 10q23 for gastric adenocarcinoma and esophageal squamous cell carcinoma. Nat. Genet. 2010, 42, 764–767. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hayakawa, Y.; Fox, J.G.; Gonda, T.; Worthley, D.L.; Muthupalani, S.; Wang, T.C. Mouse Models of Gastric Cancer. Cancers 2013, 5, 92-130. https://doi.org/10.3390/cancers5010092
Hayakawa Y, Fox JG, Gonda T, Worthley DL, Muthupalani S, Wang TC. Mouse Models of Gastric Cancer. Cancers. 2013; 5(1):92-130. https://doi.org/10.3390/cancers5010092
Chicago/Turabian StyleHayakawa, Yoku, James G. Fox, Tamas Gonda, Daniel L. Worthley, Sureshkumar Muthupalani, and Timothy C. Wang. 2013. "Mouse Models of Gastric Cancer" Cancers 5, no. 1: 92-130. https://doi.org/10.3390/cancers5010092