EGFR-Targeting as a Biological Therapy: Understanding Nimotuzumab’s Clinical Effects

Abstract
: Current clinical trials of epidermal growth factor receptor (EGFR)-targeted therapies are mostly guided by a classical approach coming from the cytotoxic paradigm. The predominant view is that the efficacy of EGFR antagonists correlates with skin rash toxicity and induction of objective clinical response. Clinical benefit from EGFR-targeted therapies is well documented; however, chronic use in advanced cancer patients has been limited due to cumulative and chemotherapy-enhanced toxicity. Here we analyze different pieces of data from mechanistic and clinical studies with the anti-EGFR monoclonal antibody Nimotuzumab, which provides several clues to understand how this antibody may induce a biological control of tumor growth while keeping a low toxicity profile. Based on these results and the current state of the art on EGFR-targeted therapies, we discuss the need to evaluate new therapeutic approaches using anti-EGFR agents, which would have the potential of transforming advanced cancer into a long-term controlled chronic disease.1. Introduction
Epidermal growth factor receptor (EGFR)-targeted therapies have been extensively evaluated in the clinic [1,2], and several EGFR-targeting products have been registered worldwide for the treatment of different tumor localizations [2]. However, the clinical benefit of these products in advanced cancer patients, in terms of median overall survival, has been limited so far (Table 1). Different factors may contribute to restrict the effect of anti-EGFR drugs on survival, for example, lack or escape from EGFR-addiction in tumors through mutations in other genes (e.g., K-Ras mutations) and pathways [3]. Remarkably, no K-Ras mutations have been found in colorectal cancer patients responding to Cetuximab or Panitumumab treatment in different studies [4-8]. Furthermore, studies conducted in non-small-cell lung cancer patients indicate that K-Ras mutation may be a negative response predictor to erlotinib and gefitinib [9,10].
The limited effects on survival of EGFR antagonists might nonetheless be also a consequence, at least partially, of administering EGFR-targeting therapies in a non-optimal way. Induction and maintenance phases, treatment after early progression, combination therapies and response predictor biomarkers are important issues currently in debate to optimize the clinical benefit of these therapies. On the other hand, we are lacking in differentiation strategies for individual EGFR antagonists, which could eventually improve their clinical benefit in different patient niches. In this paper, taking the antibody Nimotuzumab as a case study, we would like to share our views regarding clinical implementations of EGFR-targeted therapies that aim to a long-term control of the advanced cancer disease.
2. The Current Paradigm: Clinical Efficacy Is Bound to Cytotoxicity
There is an increasing understanding within the clinical researchers' community that the therapeutic endpoints of molecularly targeted agents should be revised, since neither toxicity nor tumor shrinkage are necessarily adequate surrogates to evaluate their clinical efficacy [22,23]. Furthermore, in some tumors like high-grade glioma, the use of traditional, imaging-based endpoints such as overall radiographic response and progression-free survival has become problematic due to pseudoprogression, observed with different types of therapy, and pseudoresponse, reported for anti-angiogenic agents such as Bevacizumab [24].
Current clinical trials of EGFR-targeted therapies are, nevertheless, still guided by a classical approach coming from the cytotoxic paradigm. The predominant view is that the clinical efficacy of EGFR antagonists correlates with skin rash toxicity, as documented for most EGFR-targeted agents [2,25], and induction of objective clinical response [13]. This therapeutic approach, however, has shown limitations in the clinical practice, where cumulative and chemotherapy-enhanced toxicity has impaired the chronic use and combination therapies [26]. A relevant question here is whether toxicity is really welded to the clinical efficacy of EGFR-targeting drugs, assessed in terms of overall survival. If this is not the case, then EGFR-antagonists would need to be evaluated in the clinic using other criteria, different from those applied for cytotoxic drugs.
Novel criteria, designated as immune-related response criteria (irRC), have been recently proposed based on the experience with ipilimumab in patients with advanced melanoma [27]. They represent an extension of the response evaluation criteria in solid tumors (RECIST) or WHO criteria, which were developed to standardize the efficacy evaluation of cytotoxic drugs, to be applied to immunotherapeutic agents having predominantly a cytostatic effect [23]. An irRC major contribution is the definition of new clinical response patterns, involving “mixed responses”, pseudoprogression and early progression phenomena, which correlate with overall survival, thus providing a useful tool for a more accurate assessment of the efficacy of novel immunotherapeutic agents [27].
3. Diverging from the Cytotoxic Paradigm in Anti-EGFR Therapies
Relevance of EGFR-signaling for tumor biology
Self-sufficiency in growth signals is one of the six hallmarks of cancer postulated by Hanahan and Weinberg [28]. The overexpression of EGFR in human breast cancer and its relation to bad prognosis prompted us, more than twenty years ago, to extend the concept of tumor hormone-dependence to growth factors such as EGF [29]. On the other hand, HER1 (EGFR) oncogene activation results in EGFR overexpression in epithelium-derived tumors, leading to a relaxation of the growth factor dependency. Our knowledge on the role of EGFR in the cell biology has been further expanded with the emergence, in recent years, of new experimental data demonstrating an interplay between oncogene signaling pathways, tumor metabolic re-programming and cancer-related inflammation [30,31]. In consequence, our understanding of EGFR-targeting therapies should also be expanded beyond the cytotoxic paradigm, in correspondence with the pleiotropic nature of the receptor signaling network. In the following sections we discuss recent findings from mechanistic and clinical studies of Nimotuzumab (also known as h-R3), a humanized anti-EGFR monoclonal antibody (mAb) [32], which provide a different perspective on EGFR-targeting therapies.
3.1. Clinical Experience with Nimotuzumab
A low toxicity profile
The therapeutic effects observed for Nimotuzumab in patients with head and neck squamous cell carcinoma [33-35], pancreatic cancer [36], non-small cell lung cancer [37] and glioma [38-40] have been characterized by the induction of a long-term stable disease with a very low toxicity profile, in contrast to other anti-EGFR agents [32,41]. Even though Nimotuzumab produces a downstream inhibition of the EGFR signaling pathway in normal skin cells, the characteristic lymphocytic infiltrates, folliculitis or perifolliculitis induced by other EGFR inhibitors have not been observed [32].
Table 2 summarizes the published clinical results with Nimotuzumab. Although conclusive assessment of clinical efficacy is pending on completion of the currently ongoing phase III clinical trials, multiple evidences of clinical benefit have been obtained so far. Paradoxically, the increase in overall patient survival produced by Nimotuzumab is not necessarily accompanied by objective clinical responses. In fact, in some patients tumors did not regress, but became “frozen” for months and even for years. The clinical data gathered in the most recent years from more than 9,000 patients, in open populations with different ethnic characteristics [42], reinforce the conclusions extracted from the clinical trials on Nimotuzumab's safety profile.
Long-term chronic treatment
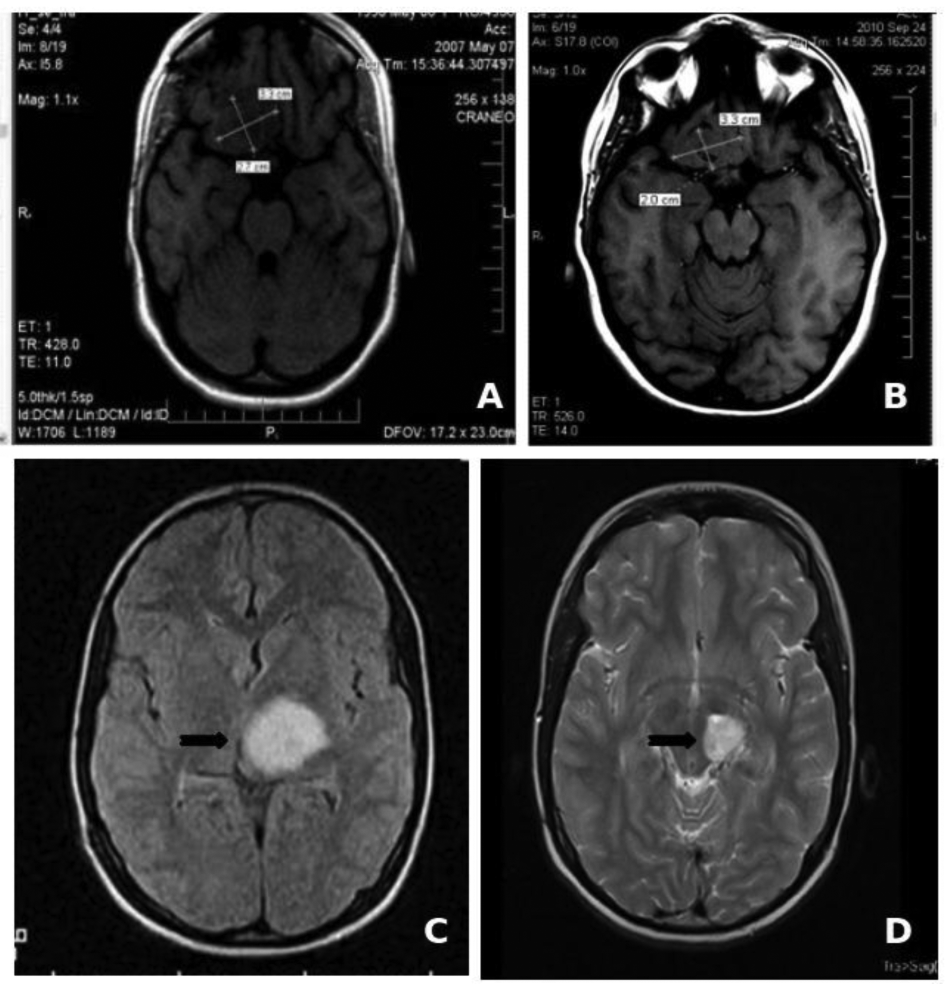
The low toxicity profile shown by Nimotuzumab has allowed its use in prolonged treatments, lasting several months, and even years in many cases. About two thirds of the 600 patients that have been treated in Cuba since 2002 have received more than six doses of the antibody (200 mg for adult, 150 mg for children), including about 50 patients that have been treated with more than 30, bi-weekly doses during more than one year [50]. In particular, two children with brain stem glioma tumors have received more than 100 doses of Nimotuzumab, continuously for more than three years, without showing adverse effects (Figure 1). It is worth noting that the frequency of adverse events (limited to grade 1 or 2) observed in these patients did not increase with drug exposure. From this clinical experience we have obtained important evidences on the impact of chronic treatment in disease stabilization and increase of overall survival in advanced cancer patients.
Perhaps such long-term treatments would be feasible also with other anti-EGFR antibodies, if the dosage and treatment schedules were adjusted to avoid a strong cumulative toxicity. As for Nimotuzumab, lowering the toxicity might result also in a reduction of the frequency of objective clinical responses. Currently, anti-EGFR antibodies are administered in large doses (hundreds of milligrams) following the paradigm developed for cytotoxic drugs, whereas biological therapies would require different evaluation criteria to define the optimal dosage and administration schedule. The relevance of chronic treatment with Nimotuzumab on disease control is currently being assessed in controlled clinical trials (e.g., NCT00561990 and NCT00753246, [51]) comparing a maintenance phase with the antibody versus the best supportive care.
3.2. Mechanisms of Action behind Nimotuzumab's Low Toxicity Profile
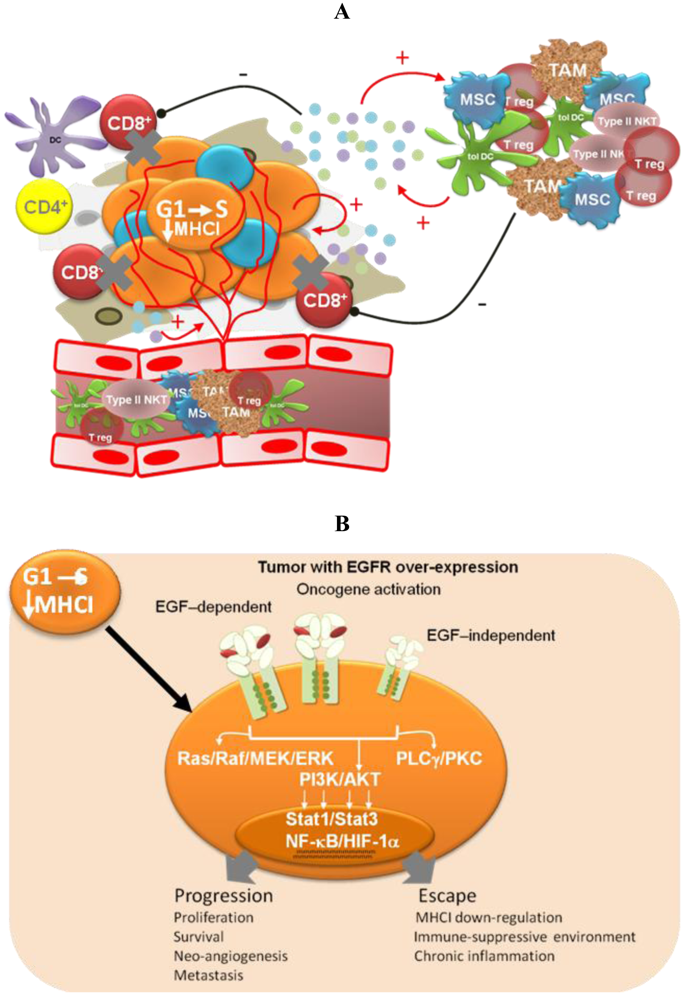
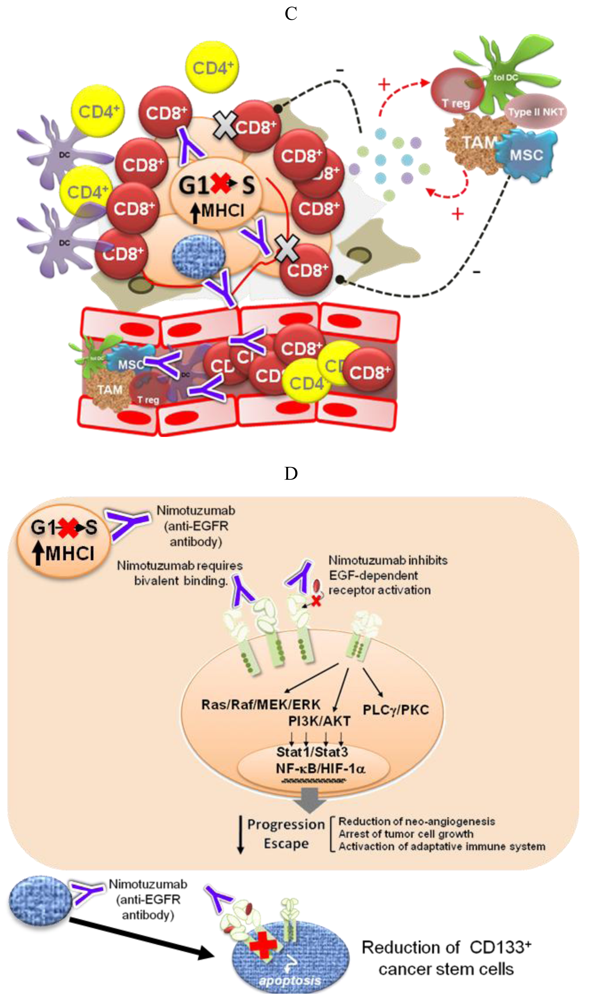
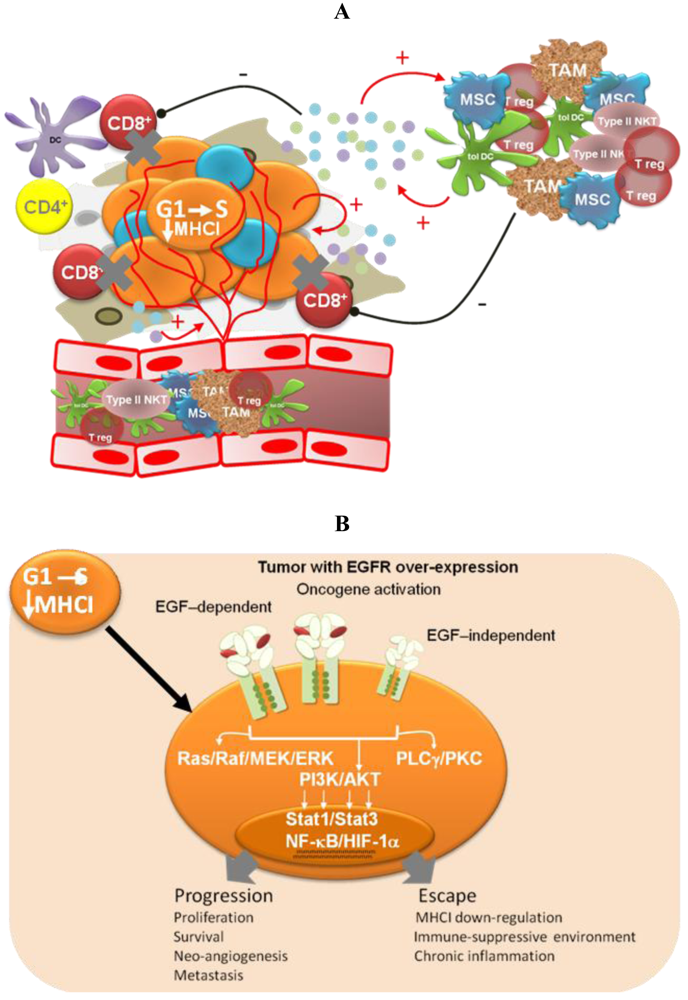
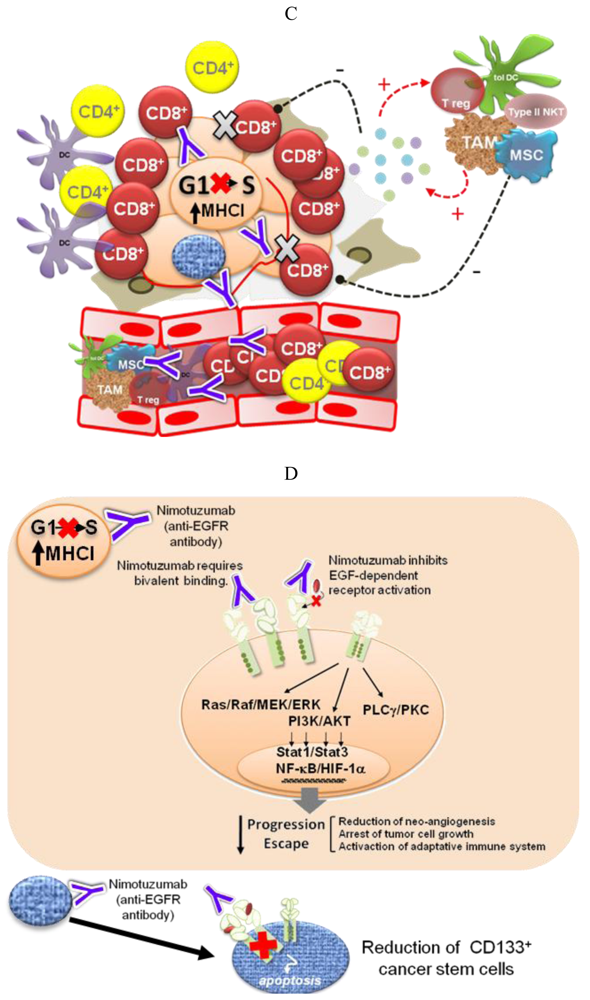
Different pieces of experimental and modeling data, gathered in recent years, support four complementary mechanisms to explain the low degree of adverse effects and the long-term disease stabilizations observed for Nimotuzumab in the clinic (Figure 2).
Intermediate affinity for EGFR and need of bivalent binding
The development of antitumor antibodies has been driven by the assumption that higher-affinity mAbs (having a dissociation constant (KD) in the nanomolar order or even lower) will have superior tumor targeting and efficacy properties. It has been shown, however, that antibodies with very high affinity have a lower penetration into solid tumors [52]. On the other hand, when the antigen targeted by the high-affinity mAb is not tumor-specific, large amounts of the antibody are retained in normal tissues. The two FDA-approved anti-EGFR antibodies, Cetuximab and Panitumumab, are high-affinity mAbs, with KD values for their monovalent Fab fragments of 2.3 × 10−9 M [53] and 5 × 10−11 M [54], respectively. Nimotuzumab, in contrast with these two antibodies, has a lower, “intermediate” affinity (KD = 2.1 × 10−8 M [55]).
Based on a mathematical model, a few years ago we put forward the hypothesis that antibodies with intermediate affinities, like Nimotuzumab, would have a higher ratio of accumulation in tumors (showing higher EGFR expression levels) with respect to normal tissues, as compared to high affinity antibodies [43]. Two recent reports [56,57] give support to this hypothesis. They show that binding of Nimotuzumab and subsequent inhibition of the EGFR phosphorylation are detected only for tumor cells lines with medium or high levels of EGFR expression (104 receptors per cell or higher). Furthermore, binding of Nimotuzumab Fab fragments was detected only for A431 cells, having the highest EGFR expression level, whereas Cetuximab Fab fragments bound also to tumor cells with lower EGFR expression levels [57]. Thus, these results sustain also the idea that Nimotuzumab requires bivalent attachment for binding to tumor cells having a surface density of EGFR molecules above certain threshold. On the other hand, Akashi and coworkers reported that the in vitro and in vivo effect of Nimotuzumab combined with radiation on human NSCLC cell lines correlated with the level of EGFR expression [56], and in a recent report of a phase II clinical trial, a significant survival improvement was observed for patients with EGFR-positive tumors that were treated with Nimotuzumab [33]. It remains to be shown whether the EGFR expression level is a predictive marker of Nimotuzumab's clinical efficacy, in contrast to high affinity antibodies like Cetuximab, for which it has been shown that the EGFR expression level is not a predictive marker of clinical benefit [58].
It might be possible that the therapeutic ratio of some of the existing high-affinity anti-EGFR antibodies could be improved by “optimizing” (in this case—lowering) the affinity, although other factors such as the location of the binding epitope on the EGFR might play an important role as well, as discussed below. Another issue to take into account is that intermediate affinity anti-EGFR antibodies might provide clinical benefit only for a subset of patients bearing EGFR-overexpressing tumors.
Inhibition of ligand-dependent receptor activation
The crystal structures of the Fab fragments of five different antibodies in complex with extracellular domains of ErbB receptors revealed that, although they show distinct modes of binding, they have one thing in common, all of them, directly or indirectly, inhibit the receptor dimerization event that triggers the proliferative signaling [59]. Cetuximab, for example, binds to domain III of the EGFR and inhibits both EGF binding and the active conformation of the receptor monomer [53]. Panitumumab also binds to domain III, to a site that overlaps with Cetuximab's epitope and with the EGF binding site on this domain. Thus, it is highly likely that Panitumumab's mechanism of EGFR inhibition is similar to that of Cetuximab [59].
Recent experimental and computer modeling data indicate that Nimotuzumab might have a different mechanism of action. The epitope recognized by this antibody on the EGFR strongly overlaps with that of Cetuximab, as demonstrated by site-directed mutagenesis, but is slightly displaced towards the C-terminus of EGFR domain III, according to a computer model [55]. In this position, Nimotuzumab sterically interferes with EGF binding while permitting the receptor to adopt its active conformation and, in consequence, form a homo—or heterodimer—with a second molecule of the EGFR family. This way, Nimotuzumab would allow a certain level of basal, ligand-independent level of EGFR activation, needed for the survival of normal epithelial cells. On the other hand, the antibody would prevent the ligand-induced shift of the equilibrium towards the active conformation, having the effect of “freezing” the tumor growth. This mechanism would explain why Nimotuzumab produces mostly a cytostatic effect [60] and induction of stable disease [32]. Interestingly, recent results show that Nimotuzumab at a high concentration is able to weaken the ligand-independent signaling in cells with a high level of EGFR expression (manuscript in preparation). Bivalent binding may account for this effect, since at high EGFR concentrations the antibody would cause a pairwise receptor cross-linking, thereby preventing the formation of active EGFR dimers, as has been shown by high resolution electron microscopy for the antibody Zalutumumab [61].
Targeting of CD133+ cancer stem cells
In a study by Díaz-Miqueli and coworkers [62] using U187 human glioma tumor xenografts in nude mice, both Nimotuzumab and Cetuximab, in spite of their differences in cytotoxicity, induced a reduction in radioresistant CD133+ tumor stem cells, impairing tumor growth progression without producing a tumor shrinkage. It has been suggested that brain tumor growth is critically dependent on the presence of an intact cancer stem cell vascular niche [63]. Therefore, the ability to target cancer stem cells may represent an important property for an anti-EGFR antibody.
Anti-angiogenic effects
The anti-tumor activity of Nimotuzumab has been associated with anti-proliferative and anti-angiogenic effects rather than direct induction of tumor cell death [60]. We demonstrated several years ago that the induction of in vivo resistance to Nimotuzumab by A431 human tumor cells was mostly due to constitutive VEGF gene overexpression [64]. In more recent experiments by Diaz-Miqueli et al. [62], Nimotuzumab combined with radiotherapy produced a reduction in the size of tumor blood vessels and the number of proliferating cells in subcutaneous tumors. The mechanism by which Nimotuzumab achieves this effect remains unknown.
3.3. Immunomodulatory Effects of EGFR Targeting
Induction of anti-tumor cellular immune responses
Analysis of the clinical data obtained in several randomized trials with Nimotuzumab evidences a delayed separation of the survival curves, indicating a non-proportional hazard ratio between treated and control patients along the follow-up time [33]. A possible explanation for these results would be a time-delayed induction of a protective immunity. Using a syngeneic mouse model, we have demonstrated that treatment with an anti-murine EGFR-antagonistic antibody, called 7A7, increases the number of various immune cells in metastatic sites, in particular T lymphocytes and dendritic cells, which might be implicated in the development of an anti-tumor specific immune response. Indeed, depletion of CD4+ and CD8+ cells in vivo totally abrogated the anti-metastatic effect produced by the 7A7 mAb [65]. Recent findings indicate that this antibody induces an immunogenic apoptotic cell death in D122-3LL murine tumor cells [66].
Active immunotherapy approaches aimed to stimulate both humoral and cellular immune responses are also being tested. One of these vaccines, consisting of a peptide enclosing the tumor-specific mutated segment of EGFRvIII, conjugated to KLH, has been shown to elicit antibody and cellular immune responses in mice and in patients [67], and is at present in phase II clinical trials for malignant glioma. We are currently developing a cancer vaccine based on the extracellular region of the EGFR, adjuvated with a proteoliposome from the outer membrane of Neisseria meningitidis bacteria [68]. Preclinical studies in animal models were recently completed, and last year the HER1 vaccine candidate entered a phase I clinical trial in Cuba, in patients with hormone-refractory prostate cancer.
It has been argued that the “vaccinal effect” of anti-tumor monoclonal antibodies may have an important weight in their clinical benefit [69], as demonstrated for the anti-CD20 antibody Rituximab, which has been shown to elicit an active T cell response specific for follicular lymphoma [70]. But for anti-EGFR mAbs, to our knowledge, there are no clinical data showing an enhancement of anti-tumor cellular immune responses as result of a passive immunotherapy. Our mechanistic studies with the 7A7 antibody prompt us to measure anti-tumor specific cytotoxic T cell (CTL) responses in patients treated with anti-EGFR antibodies.
Impairment of tumor-induced immunosuppression
Oncogene activation has been associated with a down-regulation of the antigen processing machinery, making tumor cells “less visible” to CTLs [71] More recently, oncogene activation has also been related to the induction of an immunosuppressive microenvironment by tumor cells, via up-regulation of pro-inflammatory cytokine secretion [31]. Transcription factors like Stat3, NF-Кβ and HIF-1α link oncogene activation signaling pathways with the molecular mechanisms governing the cross-talk between tumor cells and the immune system. Therefore, EGFR-targeting would have a double impact—arresting tumor cell proliferation and impairing tumor immune evasion. The experimental demonstration of this hypothesis is very appealing because of its translational impact. In particular, it becomes important to take into account the immunomodulatory effects of anti-EGFR antibodies when designing new combination therapies [72]. On the other hand, different EGFR antagonists may have different effects on cancer-related inflammation, which also needs to be translated into differentiated strategies.
4. Conclusions
4.1. EGFR-Targeted Treatment as Biological Therapy
Targeted therapies have not yet provided the expected clinical benefit in advanced cancer patients: therefore new clinical strategies are needed. We believe that targeted-therapies have the potential to transform advanced cancer into a long-term controlled chronic disease, and EGFR-targeting may represent a suitable scenario to show that this approach is feasible. Objective clinical response is a signature of cytotoxic drugs as a consequence of tumor shrinkage, but on the other hand, it has been extensively documented that in most cases there is no correlation between objective clinical response and survival time, due to tumor recurrence. Targeted-therapies may instead be aimed to stop disease progression based on biological regulatory mechanisms.
The inhibition of EGF-dependent receptor activation would stop tumor progression by at least four different mechanisms: (1) arrest of tumor cell growth; (2) homeostatic regulation of tumor population dynamics; (3) reduction of the number of cancer stem cells; and (4) enhancement of the anti-tumor cellular immune response (Box 1).
Box 1. Surpassing the cytotoxic paradigm in anti-EGFR therapies
EGFR-immunotargeting as a biological therapy
Arrest of tumor cell growth
Reduction of cancer stem cells
Homeostatic regulation of tumor population dynamics
Activation of the adaptive immune system
A tailored clinical evaluation strategy
Efficacy assessment assuming a non-proportional hazard ratio
Treatment beyond early progression
Chronic use (long-term schedule)
Combination therapies with metabolic inhibitors, anti-inflammatory drugs, immunomodulators
Because EGFR-overexpression is a hallmark of advanced epithelium-derived tumors, anti-EGFR antibodies bind preferentially to tumor cells. Nevertheless, binding to normal epithelial cells also occurs, causing toxic effects. An advantageous therapeutic ratio for a given anti-EGFR antagonistic antibody might be attained by adjusting the administration schedule based on pharmacodynamic studies. Such a treatment schedule could be used for long time periods without resulting in cumulative toxicity. Combination therapies and long-term treatment schedules, even after early disease progression, are currently being evaluated for Nimotuzumab.

{kind=link}
{kind=link}
{kind=link}
| Drug | Indication | Treatment | Clinical benefit |
|---|---|---|---|
| Cetuximab | Locally or regionally advanced SCCHN | Radiation + Cetuximab vs. Radiation | Duration of loco-regional control: 24.4 vs. 14.9 months [11] |
| Recurrent or metastatic SCCHN in progression after platinum based chemotherapy | Cetuximab monotherapy | RR: 13%. Duration of response: 5.8 months [12] | |
| Metastatic colorectal cancer refractory to Irinotecan and oxaliplatin based therapy | Cetuximab vs. BSC | MST: 6.14 vs. 4.57 months *1 [13] | |
| Metastatic colorectal cancer (Irinotecan refractory) | Cetuximab + Irinotecan vs. Cetuximab | RR: 23% vs. 11%. Duration of response: 5.7 vs. 4.1 months *,1 [14] | |
| Panitumumab | Metastatic colorectal cancer with disease progression following fluoropyrimidine, oxaliplatin, and irinotecan regimens. | Panitumumab vs. BSC | PFS: 96 vs. 60 days *,1 [15] |
| Erlotinib | Locally advanced or metastatic NSCLC refractory to first or second line chemotherapy | Erlotinib vs. BSC | MST: 6.7 vs. 4.7 months [16] |
| Locally advanced or metastatic NSCLC whose disease has not progressed after four cycles of platinum-based first-line chemotherapy | Erlotinib vs. Placebo | MST: 2.8 vs. 2.5 months [17] | |
| Locally advanced, unresectable or metastatic pancreatic cancer | Erlotinib + Gemcitabine vs. Gemcitabine | MST: 6.4 vs. 6.0 months [18] | |
| Gefitinib | NSCLC refractory to first or second line chemotherapy | Gefitinib monotherapy | RR: 10.6% Duration of response: 7 months *,2 [19] |
| NSCLC refractory to first line chemotherapy | Gefitinib vs. Docetaxel | MST: 7.6 vs. 8 months *,2 [20] | |
| Advaced NSCLC naïve for chemotherapy (Asian patients, never smoking, ADC and bronchoalveolar carcinoma) | Gefitinib vs. Carbo/Taxol | PFS: 5.7 vs. 5.8 months *,2 [21] |
Abbreviations: BSC: best supportive care; MST: median survival time; NSCLC: non-small cell lung cancer; PSF: progression free survival; RR: response rate; SCCHN: squamous cell carcinoma of the head and neck;*,1The approval of cetuximab and panitumumab in colorectal cancer was later amended to include only patients with wild-type KRAS;*,2The approval of gefitinib in NSCLC was later amended to include only patients who, in the opinion of their treating physician, are currently benefiting, or have previously benefited, from gefitinib treatment.
| Indication | Trial Design | Clinical Benefit [References] |
|---|---|---|
| Advanced SCCHN | Nimo + RTP 24 pts, 6 doses, 200 mg, weekly | MST: 45.2 months [43] |
| Nimo + RTP/CTP vs. RTP/CTP 46 patients, 6 doses, 200 mg, weekly | RR (at 24 weeks): 100% vs. 70% (p = 0.02) SV rate (30 months): 69.6% vs. 21.7% (p = 0.0011) [44] | |
| Nimo + RTP vs. RTP 46 patients, 6 doses, 200 mg, weekly | RR (at 24 weeks): 76% vs. 40% (p = 0.023) RR(at 30 months): 39.1% vs. 21.7% (p = 0.2) [44] | |
| Nimo + RTP vs. placebo + RTP 106 patients, 6 doses, 200 mg, weekly | CRR: 59.5% vs. 34.2% (p = 0.038) [33] | |
| Advanced nasopharyngeal cancer | Nimo + RTP vs. RTP 137 patients, 6 doses, 100 mg, weekly | CRR: 90.63 % vs. 51.52% (p < 0.05) [45] |
| Relapsed childhood glioma | Nimo monotherapy, 47 patients, 150 mg/m2, 6 doses weekly and maintenance bi-weekly until PD | DCR: 38.1% [46] |
| Childhood glioma | Relapsed glioma: Nimo monotherapy, 37 patients.Newly diagnosed: Nimo + RTP + vinorelbine, 10 patients 150 mg/m2, 6 doses weekly and maintenance bi-weekly until PD | MST: 11 months PFS 6 months: 90% [38] |
| High grade glioma (adults) | Nimo + RTP, 29 patients 6 doses, 200 mg, weekly | MST (GBM): 17.47 months [40] |
| Advanced or recurrent gastric cancer | Nimo + irinotecan vs. irinotecan 82 patients, 6 doses,400 mg, weekly | MST: 293 vs. 227 days, HR 0.717, (p = 0.42) EGFR 1+/2+/3+ OS HR 0.584 (p = 0.242) EGFR 2+/3+ OS HR 0.295 (p = 0.077) [47] |
| NSCLC (Unfit for radical therapy) | Nimo + palliative RTP, 17 patients 6 doses, 100–400 mg, weekly and maintenance bi-weekly until PD | DCR: 94% [48] |
| Nimo + palliative RTP, 15 pts, 6 doses, 100–400 mg, weekly and maintenance bi-weekly until PD | DCR: 100% [49] |
Abbreviations: MST: median survival time; RR: response rate (complete and partial response); SV rate: survival rate; CRR: complete response rate; DCR: disease control rate (complete and partial response plus stable disease); PFS: progression free survival; OS: overall survival; HR: hazard ratio; PD: progressive disease; RTP: radiotherapy; CTP: chemotherapy.
Acknowledgments
We are grateful to Ricardo Cabanas (Juan Manuel Márquez Hospital, Havana), as well as Patricia Piedra and Gisselle Sáurez (Center of Molecular Immunology) for providing the MRI images and data on clinical studies in pedriatic glioma patients.
References
- Mendelsohn, J.; Baselga, J. Epidermal growth factor receptor targeting in cancer. Semin. Oncol. 2006, 33, 369–385. [Google Scholar]
- Ciardiello, F.; Tortora, G. EGFR antagonists in cancer treatment. N. Engl. J. Med. 2008, 358, 1160–1174. [Google Scholar]
- Weinstein, I.B.; Joe, A. Oncogene addiction. Cancer Res. 2008, 68, 3077–3080, discussion 3080. [Google Scholar]
- Lièvre, A.; Bachet, J.; Le Corre, D.; Boige, V.; Landi, B.; Emile, J.; Côté, J.; Tomasic, G.; Penna, C.; Ducreux, M.; et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006, 66, 3992–3995. [Google Scholar]
- Di Fiore, F.; Blanchard, F.; Charbonnier, F.; Le Pessot, F.; Lamy, A.; Galais, M.P.; Bastit, L.; Killian, A.; Sesboüé, R.; Tuech, J.J.; et al. Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by Cetuximab plus chemotherapy. Br. J. Cancer 2007, 96, 1166–1169. [Google Scholar]
- De Roock, W.; Piessevaux, H.; De Schutter, J.; Janssens, M.; De Hertogh, G.; Personeni, N.; Biesmans, B.; Van Laethem, J.; Peeters, M.; Humblet, Y.; et al. KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann. Oncol. 2008, 19, 508–515. [Google Scholar]
- Lièvre, A.; Bachet, J.; Boige, V.; Cayre, A.; Le Corre, D.; Buc, E.; Ychou, M.; Bouché, O.; Landi, B.; Louvet, C.; et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J. Clin. Oncol. 2008, 26, 374–379. [Google Scholar]
- Amado, R.G.; Wolf, M.; Peeters, M.; Van Cutsem, E.; Siena, S.; Freeman, D.J.; Juan, T.; Sikorski, R.; Suggs, S.; Radinsky, R.; et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 1626–1634. [Google Scholar]
- Schneider, C.; Heigener, D.; Schott-von-Römer, K.; Gütz, S.; Laack, E.; Digel, W.; Guschall, W.; Franke, A.; Bodenstein, H.; Schmidtgen, C.; et al. Epidermal growth factor receptor-related tumor markers and clinical outcomes with erlotinib in non-small cell lung cancer: an analysis of patients from german centers in the TRUST study. J. Thorac. Oncol. 2008, 3, 1446–1453. [Google Scholar]
- Linardou, H.; Dahabreh, I.J.; Kanaloupiti, D.; Siannis, F.; Bafaloukos, D.; Kosmidis, P.; Papadimitriou, C.A.; Murray, S. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008, 9, 962–972. [Google Scholar]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Azarnia, N.; Shin, D.M.; Cohen, R.B.; Jones, C.U.; Sur, R.; Raben, D.; Jassem, J.; et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2006, 354, 567–578. [Google Scholar]
- Vermorken, J.B.; Trigo, J.; Hitt, R.; Koralewski, P.; Diaz-Rubio, E.; Rolland, F.; Knecht, R.; Amellal, N.; Schueler, A.; Baselga, J. Open-label, uncontrolled, multicenter phase II study to evaluate the efficacy and toxicity of cetuximab as a single agent in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck who failed to respond to platinum-based therapy. J. Clin. Oncol. 2007, 25, 2171–2177. [Google Scholar]
- Jonker, D.J.; O'Callaghan, C.J.; Karapetis, C.S.; Zalcberg, J.R.; Tu, D.; Au, H.; Berry, S.R.; Krahn, M.; Price, T.; Simes, R.J. Cetuximab for the treatment of colorectal cancer. N. Engl. J. Med. 2007, 357, 2040–2048. [Google Scholar]
- Cunningham, D.; Humblet, Y.; Siena, S.; Khayat, D.; Bleiberg, H.; Santoro, A.; Bets, D.; Mueser, M.; Harstrick, A.; Verslype, C. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N. Engl. J. Med. 2004, 351, 337–345. [Google Scholar]
- Giusti, R.M.; Shastri, K.; Pilaro, A.M.; Fuchs, C.; Cordoba-Rodriguez, R.; Koti, K.; Rothmann, M.; Men, A.Y.; Zhao, H.; Hughes, M. U.S. Food and Drug Administration approval: Panitumumab for epidermal growth factor receptor-expressing metastatic colorectal carcinoma with progression following fluoropyrimidine-, oxaliplatin-, and irinotecan-containing chemotherapy regimens. Clin. Cancer Res. 2008, 14, 1296–1302. [Google Scholar]
- Shepherd, F.A.; Rodrigues Pereira, J.; Ciuleanu, T.; Tan, E.H.; Hirsh, V.; Thongprasert, S.; Campos, D.; Maoleekoonpiroj, S.; Smylie, M.; Martins, R.; et al. Erlotinib in previously treated non-small-cell lung cancer. N. Engl. J. Med. 2005, 353, 123–132. [Google Scholar]
- Cappuzzo, F.; Ciuleanu, T.; Stelmakh, L.; Cicenas, S.; Szczésna, A.; Juhász, E.; Esteban, E.; Molinier, O.; Brugger, W.; Melezínek, I.; et al. Erlotinib as maintenance treatment in advanced non-small-cell lung cancer: A multicentre, randomised, placebo-controlled phase 3 study. Lancet Oncol. 2010, 11, 521–529. [Google Scholar]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar]
- Fukuoka, M.; Yano, S.; Giaccone, G.; Tamura, T.; Nakagawa, K.; Douillard, J.; Nishiwaki, Y.; Vansteenkiste, J.; Kudoh, S.; Rischin, D.; et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial) [corrected]. J. Clin. Oncol. 2003, 21, 2237–2246. [Google Scholar]
- Kim, E.S.; Hirsh, V.; Mok, T.; Socinski, M.A.; Gervais, R.; Wu, Y.; Li, L.; Watkins, C.L.; Sellers, M.V.; Lowe, E.S.; et al. Gefitinib versus docetaxel in previously treated non-small-cell lung cancer (INTEREST): A randomised phase III trial. Lancet 2008, 372, 1809–1818. [Google Scholar]
- Gridelli, C.; De Marinis, F.; Di Maio, M.; Cortinovis, D.; Cappuzzo, F.; Mok, T. Gefitinib as first-line treatment for patients with advanced non-small-cell lung cancer with activating epidermal growth factor receptor mutation: Implications for clinical practice and open issues. Lung Cancer 2011, 72, 3–8. [Google Scholar]
- Di Cosimo, S.; Baselga, J. Pharmacodynamic endpoints in primary breast cancer. Ann. Oncol 2007, 18 Suppl.9, ix21–ix23. [Google Scholar]
- Hales, R.K.; Banchereau, J.; Ribas, A.; Tarhini, A.A.; Weber, J.S.; Fox, B.A.; Drake, C.G. Assessing oncologic benefit in clinical trials of immunotherapy agents. Ann. Oncol. 2010, 21, 1944–1951. [Google Scholar]
- Reardon, D.A.; Galanis, E.; Degroot, J.F.; Cloughesy, T.F.; Wefel, J.S.; Lamborn, K.R.; Lassman, A.B.; Gilbert, M.R.; Sampson, J.H.; Wick, W.; et al. Clinical trial end points for high-grade glioma: the evolving landscape. Neuro-Oncology 2011, 13, 353–361. [Google Scholar]
- Peréz-Soler, R.; Saltz, L. Cutaneous adverse effects with HER1/EGFR-targeted agents: Is there a silver lining? J. Clin. Oncol. 2005, 23, 5235–5246. [Google Scholar]
- Lacouture, M.E. Mechanisms of cutaneous toxicities to EGFR inhibitors. Nat. Rev. Cancer 2006, 6, 803–812. [Google Scholar]
- Wolchok, J.D.; Hoos, A.; O'Day, S.; Weber, J.S.; Hamid, O.; Lebbé, C.; Maio, M.; Binder, M.; Bohnsack, O.; Nichol, G.; et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin. Cancer Res. 2009, 15, 7412–7420. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar]
- Pérez, R.; Pascual, M.; Macías, A.; Lage, A. Epidermal growth factor receptors in human breast cancer. Breast Cancer Res. Treat. 1984, 4, 189–193. [Google Scholar]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer's Achilles' heel. Cancer Cell 2008, 13, 472–482. [Google Scholar]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar]
- Boland, W.K.; Bebb, G. Nimotuzumab: A novel anti-EGFR monoclonal antibody that retains anti-EGFR activity while minimizing skin toxicity. Expert Opin. Biol. Ther. 2009, 9, 1199–1206. [Google Scholar]
- Rodríguez, M.O.; Rivero, T.C.; del Castillo Bahi, R.; Muchuli, C.R.; Bilbao, M.A.; Vinageras, E.N.; Alert, J.; Galainena, J.J.; Rodríguez, E.; Gracias, E.; et al. Nimotuzumab plus radiotherapy for unresectable squamous-cell carcinoma of the head and neck. Cancer Biol. Ther. 2010, 9, 343–349. [Google Scholar]
- Rojo, F.; Gracias, E.; Villena, N.; Cruz, T.; Corominas, J.M.; Corradino, I.; Cedeño, M.; Campas, C.; Osorio, M.; Iznaga, N.; et al. Pharmacodynamic trial of nimotuzumab in unresectable squamous cell carcinoma of the head and neck: a SENDO Foundation study. Clin. Cancer Res. 2010, 16, 2474–2482. [Google Scholar]
- Basavaraj, C.; Sierra, P.; Shivu, J.; Melarkode, R.; Montero, E.; Nair, P. Nimotuzumab with chemoradiation confers a survival advantage in treatment-naïve head and neck tumors over expressing EGFR. Cancer Biol. Ther. 2010, 10, 673–681. [Google Scholar]
- Strumberg, D.; Schultheis, B.; Scheulen, M.E.; Hilger, R.A.; Krauss, J.; Marschner, N.; Lordick, F.; Bach, F.; Reuter, D.; Edler, L.; et al. Phase II study of nimotuzumab, a humanized monoclonal anti-epidermal growth factor receptor (EGFR) antibody, in patients with locally advanced or metastatic pancreatic cancer. Invest New Drugs 2010. unpublished. [Google Scholar]
- Boland, W.; Bebb, G. The emerging role of nimotuzumab in the treatment of non-small cell lung cancer. Biologics 2010, 4, 289–298. [Google Scholar]
- Massimino, M.; Bode, U.; Biassoni, V.; Fleischhack, G. Nimotuzumab for pediatric diffuse intrinsic pontine gliomas. Expert Opin. Biol. Ther. 2011, 11, 247–256. [Google Scholar]
- Lam, C.; Bouffet, E.; Bartels, U. Nimotuzumab in pediatric glioma. Future Oncol. 2009, 5, 1349–1361. [Google Scholar]
- Ramos, T.C.; Figueredo, J.; Catala, M.; González, S.; Selva, J.C.; Cruz, T.M.; Toledo, C.; Silva, S.; Pestano, Y.; Ramos, M.; et al. Treatment of high-grade glioma patients with the humanized anti-epidermal growth factor receptor (EGFR) antibody h-R3: Report from a phase I/II trial. Cancer Biol. Ther. 2006, 5, 375–379. [Google Scholar]
- Allan, D.G.P. Nimotuzumab: evidence of clinical benefit without rash. Oncologist 2005, 10, 760–761. [Google Scholar]
- YM BioSciences. Products: Nimotuzumab: Safety Profile. http://www.ymbiosciences.com/products/nimotuzumab/safety_profile.php/ (accessed 18 March 2011).
- Crombet, T.; Osorio, M.; Cruz, T.; Roca, C.; del Castillo, R.; Mon, R.; Iznaga-Escobar, N.; Figueredo, R.; Koropatnick, J.; Renginfo, E. Use of the humanized anti-epidermal growth factor receptor monoclonal antibody h-R3 in combination with radiotherapy in the treatment of locally advanced head and neck cancer patients. J. Clin. Oncol. 2004, 22, 1646–1654. [Google Scholar]
- Ramakrishnan, M.S.; Eswaraiah, A.; Crombet, T.; Piedra, P.; Saurez, G.; Iyer, H.; Arvind, A.S. Nimotuzumab, a promising therapeutic monoclonal for treatment of tumors of epithelial origin. mAbs 2009, 1, 41–48. [Google Scholar]
- Huang, X.; Yi, J.; Gao, L.; Xu, G.; Jin, J.; Yang, W.; Lu, T.; Wu, S.; Wu, R.; Hu, W. Multi-center phase II clinical trial of humanized anti-epidermal factor receptor monoclonal antibody h-R3 combined with radiotherapy for locoregionally advanced nasopharyngeal carcinoma. Zhonghua Zhong Liu Za Zhi 2007, 29, 197–201. [Google Scholar]
- Gupta, M.; Madholia, V.; Gupta, N.; Bhowmik, K. Results from a pilot study of nimotuzumab with concurrent chemoradiation in patients with locally advanced squamous cell carcinoma of head and neck. J. Clin. Oncol. 2010, 28, 15s, Abstract 5565. [Google Scholar]
- Kim, Y.; Sasaki, Y.; Lee, K.; Rha, S.; Park, S.; Boku, N.; Komatsu, Y.; Kim, T.; KIm, S.; Sakata, Y. Randomized phase II study of nimotuzumab, an anti-EGFR antibody, plus irinotecan in patients with 5-fluorouracil-based regimen-refractory advanced or recurrent gastric cancer in Korea and Japan: Preliminary results. J. Clin. Oncol. 2011, 29, 4s, Abstract 87. [Google Scholar]
- Bebb, G.; Smith, C.; Rorke, S.; Boland, W.; Nicacio, L.; Sukhoo, R.; Brade, A. Phase I clinical trial of the anti-EGFR monoclonal antibody nimotuzumab with concurrent external thoracic radiotherapy in Canadian patients diagnosed with stage IIb, III or IV non-small cell lung cancer unsuitable for radical therapy. Cancer Chemother. Pharmacol. 2010, 67, 837–45. [Google Scholar]
- Choi, H.J.; Sohn, J.H.; Lee, C.G.; Shim, H.S.; Lee, I.; Yang, W.I.; Kwon, J.E.; Kim, S.K.; Park, M.; Lee, J.H.; et al. A phase I study of nimotuzumab in combination with radiotherapy in stages IIB-IV non-small cell lung cancer unsuitable for radical therapy: Korean results. Lung Cancer 2011, 71, 55–59. [Google Scholar]
- Crombet, T.; Cabanas, R.; Alert, J.; Valdés, J.; González, M.C.; Pedrayes, J.L.; Ríos, M.; Leyva, T.; Herrera, R.; Avila, M. Nimotuzumab and radiotherapy in children and adolescents with brain stem glioma: preliminary results from a phase II study. Eur. J. Cancer Suppl. 2009, 7, 497. [Google Scholar]
- ClinicalTrials.gov. A service from the U.S. National Institutes of Health. http://clinicaltrials.gov/ (accessed 18 March 2011).
- Weiner, L.M.; Carter, P. Tunable antibodies. Nat. Biotechnol. 2005, 23, 556–557. [Google Scholar]
- Li, S.; Schmitz, K.R.; Jeffrey, P.D.; Wiltzius, J.J.W.; Kussie, P.; Ferguson, K.M. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 2005, 7, 301–311. [Google Scholar]
- Yang, X.D.; Jia, X.C.; Corvalan, J.R.; Wang, P.; Davis, C.G. Development of ABX-EGF, a fully human anti-EGF receptor monoclonal antibody, for cancer therapy. Crit. Rev. Oncol. Hematol. 2001, 38, 17–23. [Google Scholar]
- Talavera, A.; Friemann, R.; Gómez-Puerta, S.; Martinez-Fleites, C.; Garrido, G.; Rabasa, A.; López-Requena, A.; Pupo, A.; Johansen, R.F.; Sánchez, O.; et al. Nimotuzumab, an antitumor antibody that targets the epidermal growth factor receptor, blocks ligand binding while permitting the active receptor conformation. Cancer Res. 2009, 69, 5851–5859. [Google Scholar]
- Akashi, Y.; Okamoto, I.; Iwasa, T.; Yoshida, T.; Suzuki, M.; Hatashita, E.; Yamada, Y.; Satoh, T.; Fukuoka, M.; Ono, K.; et al. Enhancement of the antitumor activity of ionising radiation by nimotuzumab, a humanised monoclonal antibody to the epidermal growth factor receptor, in non-small cell lung cancer cell lines of differing epidermal growth factor receptor status. Br. J. Cancer 2008, 98, 749–755. [Google Scholar]
- Garrido, G.; Tikhomirov, I.A.; Rabasa, A.; Yang, E.; Gracia, E.; Iznaga, N.; Fernández, L.E.; Crombet, T.; Kerbel, R.; Pérez, R. Bivalent binding by intermediate affinity of nimotuzumab: A contribution to explain antibody clinical profile. Cancer Biol. Ther. 2011, 11, 1–10. [Google Scholar]
- Khambata-Ford, S.; Harbison, C.T.; Hart, L.L.; Awad, M.; Xu, L.; Horak, C.E.; Dakhil, S.; Hermann, R.C.; Lynch, T.J.; Weber, M.R. Analysis of potential predictive markers of cetuximab benefit in BMS099, a phase III study of cetuximab and first-line taxane/carboplatin in advanced non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 918–927. [Google Scholar]
- Schmitz, K.R.; Ferguson, K.M. Interaction of antibodies with ErbB receptor extracellular regions. Exp. Cell Res. 2009, 315, 659–670. [Google Scholar]
- Crombet-Ramos, T.; Rak, J.; Pérez, R.; Viloria-Petit, A. Antiproliferative, antiangiogenic and proapoptotic activity of h-R3: A humanized anti-EGFR antibody. Int. J. Cancer 2002, 101, 567–575. [Google Scholar]
- Lammerts van Bueren, J.J.; Bleeker, W.K.; Brännström, A.; von Euler, A.; Jansson, M.; Peipp, M.; Schneider-Merck, T.; Valerius, T.; van de Winkel, J.G.J.; Parren, P.W.H.I. The antibody zalutumumab inhibits epidermal growth factor receptor signaling by limiting intra- and intermolecular flexibility. Proc. Natl. Acad. Sci. USA 2008, 105, 6109–6114. [Google Scholar]
- Diaz Miqueli, A.; Rolff, J.; Lemm, M.; Fichtner, I.; Perez, R.; Montero, E. Radiosensitisation of U87MG brain tumours by anti-epidermal growth factor receptor monoclonal antibodies. Br. J. Cancer 2009, 100, 950–958. [Google Scholar]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar]
- Viloria-Petit, A.; Crombet, T.; Jothy, S.; Hicklin, D.; Bohlen, P.; Schlaeppi, J.M.; Rak, J.; Kerbel, R.S. Acquired resistance to the antitumor effect of epidermal growth factor receptor-blocking antibodies in vivo: A role for altered tumor angiogenesis. Cancer Res. 2001, 61, 5090–5101. [Google Scholar]
- Garrido, G.; Lorenzano, P.; Sánchez, B.; Beausoleil, I.; Alonso, D.F.; Pérez, R.; Fernández, L.E. T cells are crucial for the anti-metastatic effect of anti-epidermal growth factor receptor antibodies. Cancer Immunol. Immunother. 2007, 56, 1701–1710. [Google Scholar]
- Garrido, G.; Rabasa, A.; Sánchez, B.; López, M.V.; Blanco, R.; López, A.; Hernández, D.R.; Pérez, R.; Fernández, L.E. J. Immunol. Submitted for publication. 2011.
- Heimberger, A.B.; Sampson, J.H. The PEPvIII-KLH (CDX-110) vaccine in glioblastoma multiforme patients. Expert Opin. Biol. Ther. 2009, 9, 1087–1098. [Google Scholar]
- Ramírez, B.S.; Alpízar, Y.A.; Fernández, D.R.H.; Hidalgo, G.G.; Capote, A.R.; Rodríguez, R.P.; Fernández, L.E. Anti-EGFR activation, anti-proliferative and pro-apoptotic effects of polyclonal antibodies induced by EGFR-based cancer vaccine. Vaccine 2008, 26, 4918–4926. [Google Scholar]
- Weiner, L.M.; Dhodapkar, M.V.; Ferrone, S. Monoclonal antibodies for cancer immunotherapy. Lancet 2009, 373, 1033–1040. [Google Scholar]
- Hilchey, S.P.; Hyrien, O.; Mosmann, T.R.; Livingstone, A.M.; Friedberg, J.W.; Young, F.; Fisher, R.I.; Kelleher, R.J.; Bankert, R.B.; Bernstein, S.H. Rituximab immunotherapy results in the induction of a lymphoma idiotype-specific T-cell response in patients with follicular lymphoma: support for a “vaccinal effect” of rituximab. Blood 2009, 113, 3809–3812. [Google Scholar]
- Herrmann, F.; Lehr, H.; Drexler, I.; Sutter, G.; Hengstler, J.; Wollscheid, U.; Seliger, B. HER-2/neu-mediated regulation of components of the MHC class I antigen-processing pathway. Cancer Res. 2004, 64, 215–220. [Google Scholar]
- Hoos, A.; Parmiani, G.; Hege, K.; Sznol, M.; Loibner, H.; Eggermont, A.; Urba, W.; Blumenstein, B.; Sacks, N.; Keilholz, U. A clinical development paradigm for cancer vaccines and related biologics. J. Immunother. 2007, 30, 1–15. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Perez, R.; Moreno, E.; Garrido, G.; Crombet, T. EGFR-Targeting as a Biological Therapy: Understanding Nimotuzumab’s Clinical Effects. Cancers 2011, 3, 2014-2031. https://doi.org/10.3390/cancers3022014
Perez R, Moreno E, Garrido G, Crombet T. EGFR-Targeting as a Biological Therapy: Understanding Nimotuzumab’s Clinical Effects. Cancers. 2011; 3(2):2014-2031. https://doi.org/10.3390/cancers3022014
Chicago/Turabian StylePerez, Rolando, Ernesto Moreno, Greta Garrido, and Tania Crombet. 2011. "EGFR-Targeting as a Biological Therapy: Understanding Nimotuzumab’s Clinical Effects" Cancers 3, no. 2: 2014-2031. https://doi.org/10.3390/cancers3022014