Cancer Stem Cell Radioresistance and Enrichment: Where Frontline Radiation Therapy May Fail in Lung and Esophageal Cancers

Abstract
: Many studies have highlighted the role cancer stem cells (CSC) play in the development and progression of various types of cancer including lung and esophageal cancer. More recently, it has been proposed that the presence of CSCs affects treatment efficacy and patient prognosis. In reviewing this new area of cancer biology, we will give an overview of the current literature regarding lung and esophageal CSCs and radioresistance of CSC, and discuss the potential therapeutic applications of these findings.1. Introduction
Cancer has become the leading cause of human death worldwide [1]. Despite the recent advances made in cancer treatment and diagnosis, cancer mortality for advanced solid malignancies remains unchanged, in part due to the development of resistance by the tumor to radiation and chemotherapy. One possible cause for therapeutic failure is that residual tumor cells are reminiscent of stem cells, which ultimately give rise to secondary tumors or distant metastasis. From a therapeutic perspective, the state of knowledge regarding cancer stem cells will help to advance the development of treatments directed against this population of cancer cells.
Stem cells are present in many different somatic tissues and have three distinctive properties: self-renewal, the ability to develop into multiple lineages, and the potential to proliferate extensively [2]. Recent studies have defined a subset of tumor cells found in breast [3], brain [4] hematopoietic [5], colon [6], pancreatic [7], stomach [8], liver [9], skin [10], ovarian [11], bladder [12], head and neck [13], and most recently lung [14], that also possess these three features which are parallel to stem cells. Although lung and esophageal cancers are among the most common lethal forms of cancer in both men and women in the world, with the overall five-year survival rate at less than 20% [1], comparatively little is known about the biology of these two types of cancer stem cells compared to other solid tumor stem cells, particularly esophageal cancer. Therefore, there is an increasing need to understand the biology of lung and esophageal cancer stem cells so that new therapeutic developments could tackle this cell population.
Both lung and esophageal cancers are considered complex tumors in terms of their origin and regionally distinct types of neoplasia. Lung cancers are comprised of two major histological types: small-cell lung cancer (SCLC) and non-small-cell lung cancer (NSCLC), which can be further divided into squamous cell carcinoma (lung SCC), adenocarcinoma (lung ADC) and large cell carcinoma. Among these different types of lung cancer, lung ADC is the most common form of lung cancer in both smokers and nonsmokers. Lung SCC has a 25% prevalence rate and is commonly associated with tobacco smoking. Compared with other types of lung cancer, large cell carcinoma belongs to a class of poorly differentiated and less aggressive tumors. SCLC accounts for more than 20% of lung tumors in the population, and despite its initial response to therapy, it has a particular poor prognosis due to its highly metastatic, potential drug-resistance, and rapidly fatal behavior [15].
Like lung cancer, esophageal cancer is composed of two main histological subtypes: squamous cell carcinoma (SCC), mainly caused by cigarette smoking, and adenocarcinoma (ADC), mainly caused by gastric reflex and obesity. The incidence of ADC has increased remarkably over the past two decades and has supplanted SCC as the dominant phenotype in Western countries [16]. ADC frequently arises from Barrett's esophagus (BE), a chronic inflammatory condition characterized by a change in the normal esophagus epithelium into intestinal metaplasia of gastroesophageal reflux [17] and genomic instability [18]. The histological and regional diversity found in lung and esophageal cancer may partly be due to the presence of diverse pools of stem cells with different biological properties, underlining a pressing need to characterize these different stem cells for direct implications on diagnostic and therapeutic outcome.
2. Cancer Stem Cell Markers
A variety of approaches has been employed to isolate CSCs, often based on markers characteristic of normal stem cells such as CD44 [19], CD133 [20], CD15 [21], CXCR4 [22], etc. Although the list of CSC markers is not yet completed, they provide a means to isolate and substantially enrich CSCs for cancer research. In this review, we chose to focus our attention to CSC markers that are relevant to lung and esophageal tumors (Table 1).
One of the most common markers found on many of the CSCs identified to date is CD133. Although its exact function remains poorly understood, the expression of CD133 has been detected in many different types of cancer cells, including lung cancer [23]. CD133 is a highly conserved antigen that is a five-transmembrane domain cell surface glycoprotein [24]. It is a ubiquitous marker of CSCs since its expression is presented on a wide spectrum of tumors including brain [4], colorectal [6], pancreatic [25], breast [26], prostate [27], ovarian [28], liver [29], hematopoietic [30] and lung cancer [23]. CD133 was first identified in hematopoeitic stem and progenitor cells [31].
Cells isolated from primary lung tumors that are CD133+ show increased tumorigenicity and expression of stemness, adhesion, and drug efflux genes compared with the corresponding CD133− tumor cells [32]. It is striking that CD133+ lung cancer cells survive cisplastin administration in either in vitro drug exposure on A549 cells or in vivo primary tumor-derived mouse xenografts [32]. This suggests that the biological differences between cancer cells and CSCs confers the CSCs with chemoresistance, which has enormous, paradigm-shifting implications regarding patient treatment since cisplatin is a first-line agent in both lung and esophageal cancers [33]. Evidence to suggest that CSCs have an impact on survival, possibly through platinum-resistance mechanisms, includes data that the five-year survival rate of CD133+ patients was significantly lower than that of CD133−patients; furthermore, the expression of CD133 is reportedly an independent prognostic factor in some studies, thus providing the first evidence of the importance of CSC markers as potential diagnostic and prognostic indicators in lung cancer patients [34].
In contrast, whether CD133 is a prognostic factor remains unclear since CD133 is not detected among all lung cancer samples [35]. Furthermore, Salnikov et al. reported that although CD133 expression is abundant in NSCLC, it is not a prognostic indicator for NSCLC patients [36]. Additional evidence has also indicated that some CD133-negative lung CSCs also possess the ability to self-renew and generate robust xenograft outgrowth [37], again suggesting that not all lung CSCs have CD133 as a marker. Unlike lung CSCs, CD133 expression was not present in cancer stem cells isolated from NOD-SCID mice that received transplantation of esophageal adenocarcinoma cells [38].
Another common marker for the identification of CSCs is CD44, which was originally described as a leukocyte-homing receptor, is involved in cell-cell interaction and is a downstream target of the Wnt/β-catenin pathway [39]. It comprises a family of glycoproteins, all encoded by a single gene, but varying in size due to alternative splicing [40]. Its expression was first identified in breast CSCs as these cells seem to express CD44 variant isoforms, rather than a CD133+ population [3]. CD44 expression has then been identified in many types of CSCs including SCLC and NSCLC and its expression is correlated with survival [41]. CD44 expression has been found in esophageal SCC and its expression is also known to correlate with poor prognosis [42-43].
An alternative method to isolate stem cells is to rely on the ability of ABC transporters expressed in stem cell populations, to efflux the fluorescent Hoechst 33342 dye [44]. This population of cells with enhanced efflux capabilities is known as Side Population cells (SP cells), which have been described in many tumor types as being rich in stem-like properties, including those of lung and esophageal cancer [45]. In both human lung and esophageal cancer, as few as 1000 isolated SP cells could produce robust xenografts in mice whereas a higher number of non-SP cells are required to generate tumors [46-47], sometimes as high as 5 × 104 or 5 × 105. Interestingly, in both lung and esophageal cancer, the SP population cells are less sensitive to chemotherapy than non-SP cells [46-47].
SP cells are also found to self-renew and express elevated levels of other so-called stemness genes such as hTERT (telomerase) [46], Oct-4 [48], SOX2 [49], BMI [47] and ZFX [47], as well as genes that are components of the Wnt and β-catenin pathways [50]. Oct-4 is known to play an important role in cell viability, functions as a stem cell survival factor, and induces an induction of pluripotency in somatic cells [51]. In addition, Oct-4 plays a crucial role in maintaining self-renewal, CSC-like and chemo- and radio-resistant properties of CD133+ NSCLC cells [52]. In squamous cell carcinoma of the esophagus, Oct-4 expression is highly elevated in the SP population compared to non-SP cells [47].
Another marker, aldehyde dehydrogenase (ALDH) is a detoxifying enzyme, known for its role in the oxidation of intracellular aldehydes, which play a role in stem cell differentiation through metabolism of retinal to retinoic acid [53]. ALDH is highly expressed in the tumorigenic cell population of various cancers including breast [54], brain [55], colon [56], head and neck [57]. The first evidence indicating ALDH is a relevant lung CSC marker is elevated ALDH protein expression discovered in putative lung stem cell niches during malignant transformation [58]. Further evidence to support ALDH as a functional marker came from Jiang's group who reported that NSCLC cancer cells with strong ALDH1 activity showed CSC features and were positive for CD133+ [59-60]. In addition, expression of ALDH1 is positively correlated with stage and grade of lung tumors and related to poor prognosis in patients with early-stage lung cancer [58]. In esophageal cancer, patients with deficient ALDH showed high risk for developing cancer, although there is no report to date about the existence of ALDH1 in esophageal CSC [61].
3. Signal Transduction Pathways in Cancer Stem Cells
WNT, Notch, Hedgehog and BMP signaling pathways constitute the stem cell signaling network which regulates the balance of self-renewal, proliferation and differentiation among stem and progenitor cell populations [62]. β-catenin is an essential component of both intracellular junctions and the canonical Wnt signaling pathway, which has been implicated in stem cell survival [63]. Earlier observations showed that overexpression of β-catenin could promote CSC survival and tumorigenesis both in vitro and in vivo [64], suggesting an important role for the Wnt/β-catenin pathway in the maintenance and regulation of CSC self-renewal. Recently, a study by Stripp et al. suggested that activated Wnt/β-catenin signaling in the developing lung coincided with an expansion of bronchioalveolar stem cells and attenuated their differentiation [65]. Additionally, evidence suggested that activated Wnt signaling in lung tumors promoted tumorigenesis [66,67]. However, the role of Wnt/β-catenin signaling in lung CSCs might be different for NSCLC than for SCLC. For example, aberrant Wnt pathway has been shown to play a role in NSCLC such as Wnt2 is overexpressed in NSCLC and inhibition of Wnt2-mediated signaling leads to apoptosis in NSCLC cell lines [68].
There are secreted Wnt antagonists that interact directly and indirectly to modulate Wnt signaling, therefore influencing tumorigeneis [69]. A secreted Wnt antagonist, Wnt inhibitory factor (WIF1), has been shown to inhibit NSCLC growth both in vitro and in vivo [70]. Interestingly, the Wnt inhibitor Dickkopf-1 (Dkk-1) is expressed in distal pulmonary epithelium and studies have shown that knocking out Dkk-1 inhibits branching morphogenesis [71]. In esophageal ADC cancer, Dkk-1 expression increases significantly starting with normal esophagus epithelium and low grade dysplasia to high grade dysplasia and esophageal adenocarcinoma [72]. Additionally, Dkk-1 expression is highly elevated in tumor samples obtained from esophageal SCC patients and its expression is a predictor of poor survival [73]. Reduced β-catenin expression level also correlates with poor prognosis in esophageal SCC patients [74].
Notch signaling plays fundamental roles in defining cell fate, self-renew, and is frequently deregulated in human malignancies [75]. In lung and esophageal CSCs it is not yet clear if Notch signaling is required for self-renewal, although several reports suggested that components of the Notch signaling network are expressed in putative lung CSC populations [32,76]. Knockout mouse studies demonstrate a requirement for Notch signaling in lung development [77,78]. Elevated Notch ligand receptor, and its transcriptional factor, HES1 levels have been demonstrated in NSCLC lines [79,80] and Notch signaling also seems to be among key downstream effectors of oncogenic RAS [81], which is a common aberration observed in lung cancer. In addition, activation of Notch-1 in A549 adenocarcinoma cells inhibits the in vitro clonogenicity and in vivo tumorigenicity growth in mice, highlighting the complexity of the Notch pathway and its varying roles in different lung tumor subtypes [82]. Notch receptor expression is rarely seen in SCLC, likely because overexpression of activated Notch receptors inhibits SCLC growth [83]. Unlike what is seen in lung cancer, Notch signaling is inactivated in esophageal SCC, acting in an anti-oncogenic manner [84].
The Hedgehog pathway is also indispensable for normal mammalian embryo and organogenesis [85]. Although it is not yet clear if both lung and esophageal CSCs require Hedgehog signaling for self-renewal, several studies have suggested that specific inhibitors targeting the Hedgehog pathway could hamper tumor growth, some of which are currently in clinical trails for lung SCLC [86,87]. Activation of this signaling pathway occurs through the binding of the Sonic Hedgehog (Shh), Indian Hedgehog (Ihh) and Desert Hedgehog (Dhh) morphogens to their receptor Patched, subsequently inhibiting the repression of Smoothened [88]. Shh-null mice exhibit hypoblastic lung buds without airway branching [89]. On the other hand, transgenic overexpression of Shh leads to the absence of functional alveoli and hyperproliferation of epithelial and mesenchymal pulmonary cells [90]. The growth of SCLC cell lines was inhibited by KAAD-cyclopamine, a small molecule inhibitor of Smoothened and therefore the Hedgehog pathway. These results, however, were not observed in NSCLC cell lines [91]. In esophageal SCC tumors, elevated expression of Hedgehog target genes was observed and treatment of esophageal cancer cells with cyclopamine reduced cell growth and induced apoptosis [92].
These are by no means a complete summary of the most current knowledge about CSC markers signal transduction pathways in lung and esophageal cancer but they support the notion that CSCs are present in lung and esophageal cancer. Many new studies are published regularly that further expand the knowledge base of this area. Even so, additional studies are still needed to understand the pivotal role of these CSC markers in order to further elucidate the mechanisms regulating the origin and maintenance of CSCs and to delineate differences between normal stem cells and CSCs that could be exploited in the treatment and diagnosis of lung and esophageal cancer.
4. Radiation Resistance of Cancer Stem Cells
Radiation therapy plays an important role in treating esophageal and lung cancers as a part of combined modalities treatment with chemotherapy and/or surgery. These treatment approaches have improved local control and survival rates with acceptable toxicity. However, recurrence is essentially seen in most patients and some patients develop radioresistance that might limit any further treatment, demonstrating a strong need for new treatment therapies and directions in the management of these two cancers.
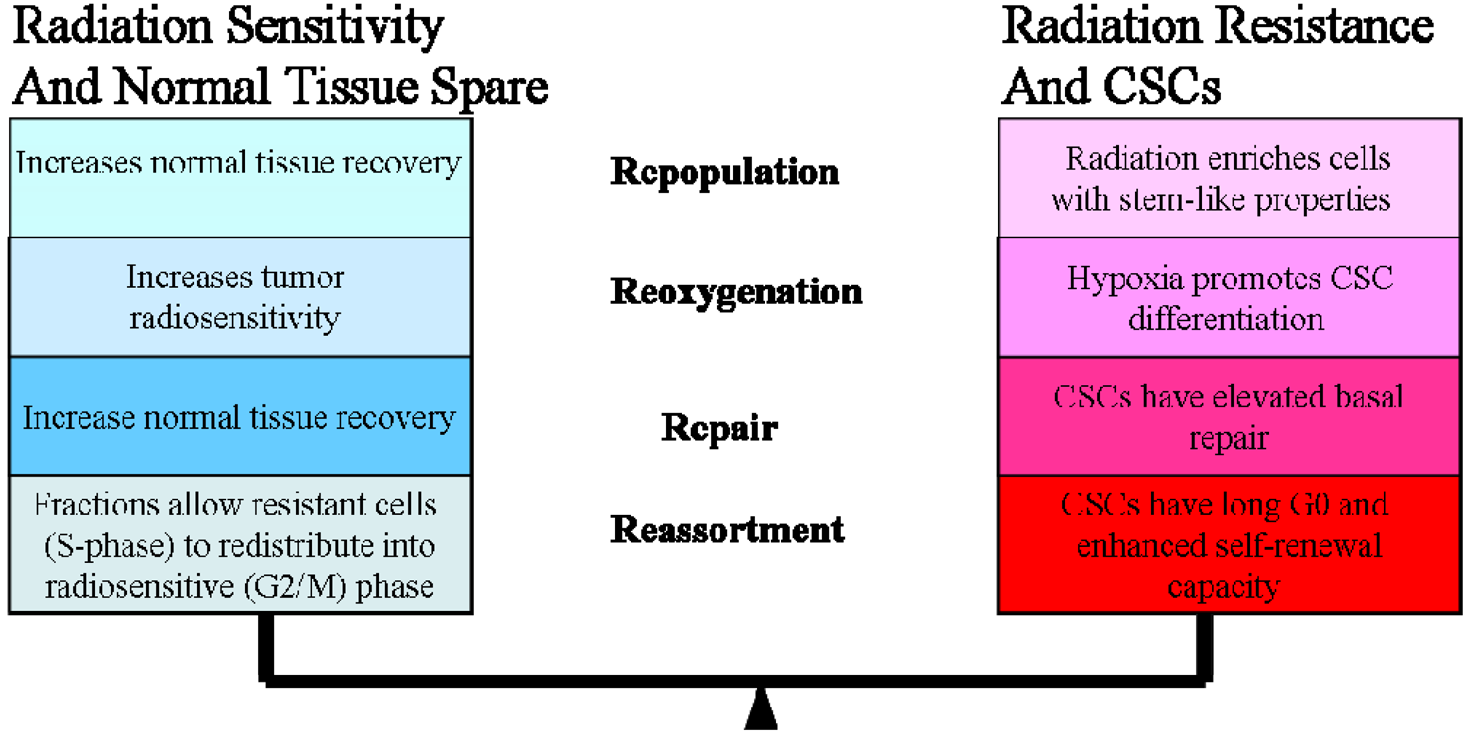
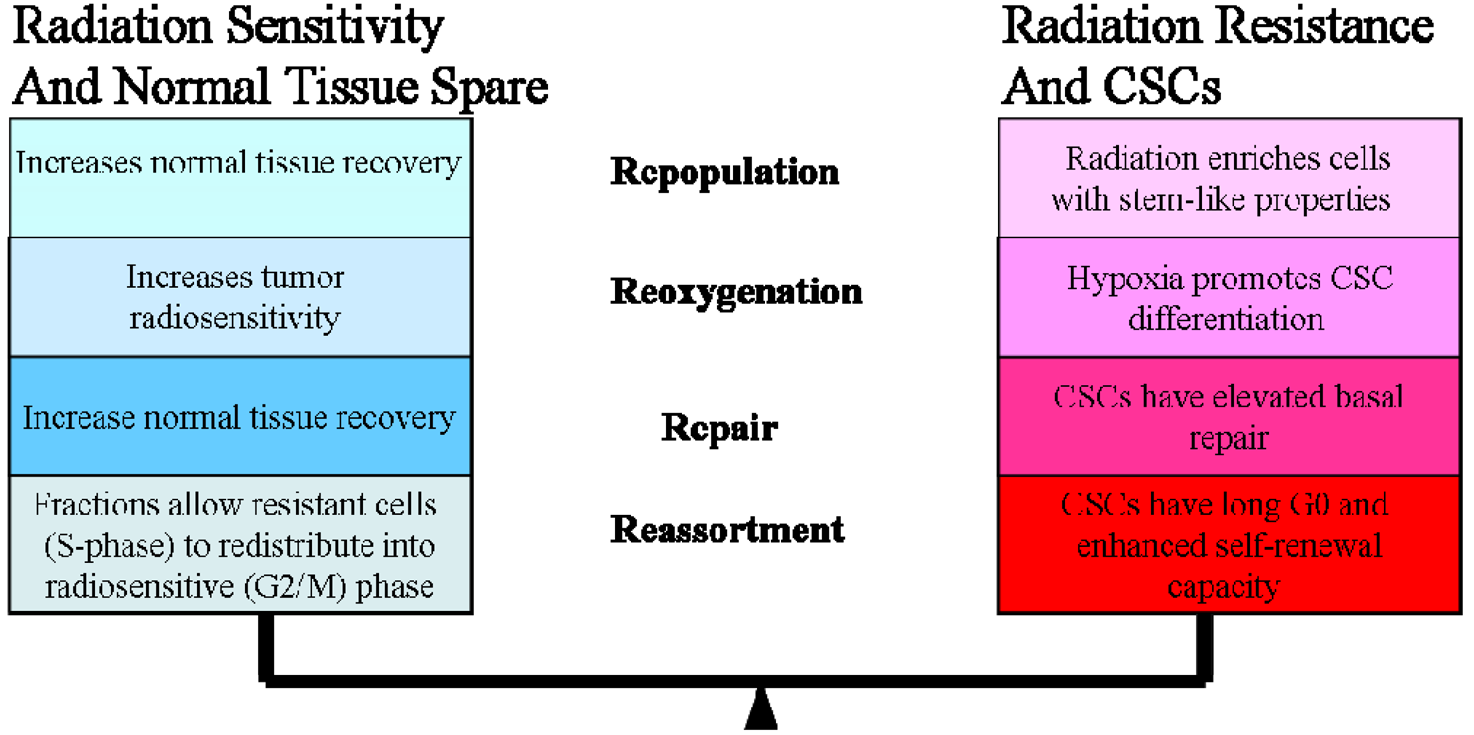
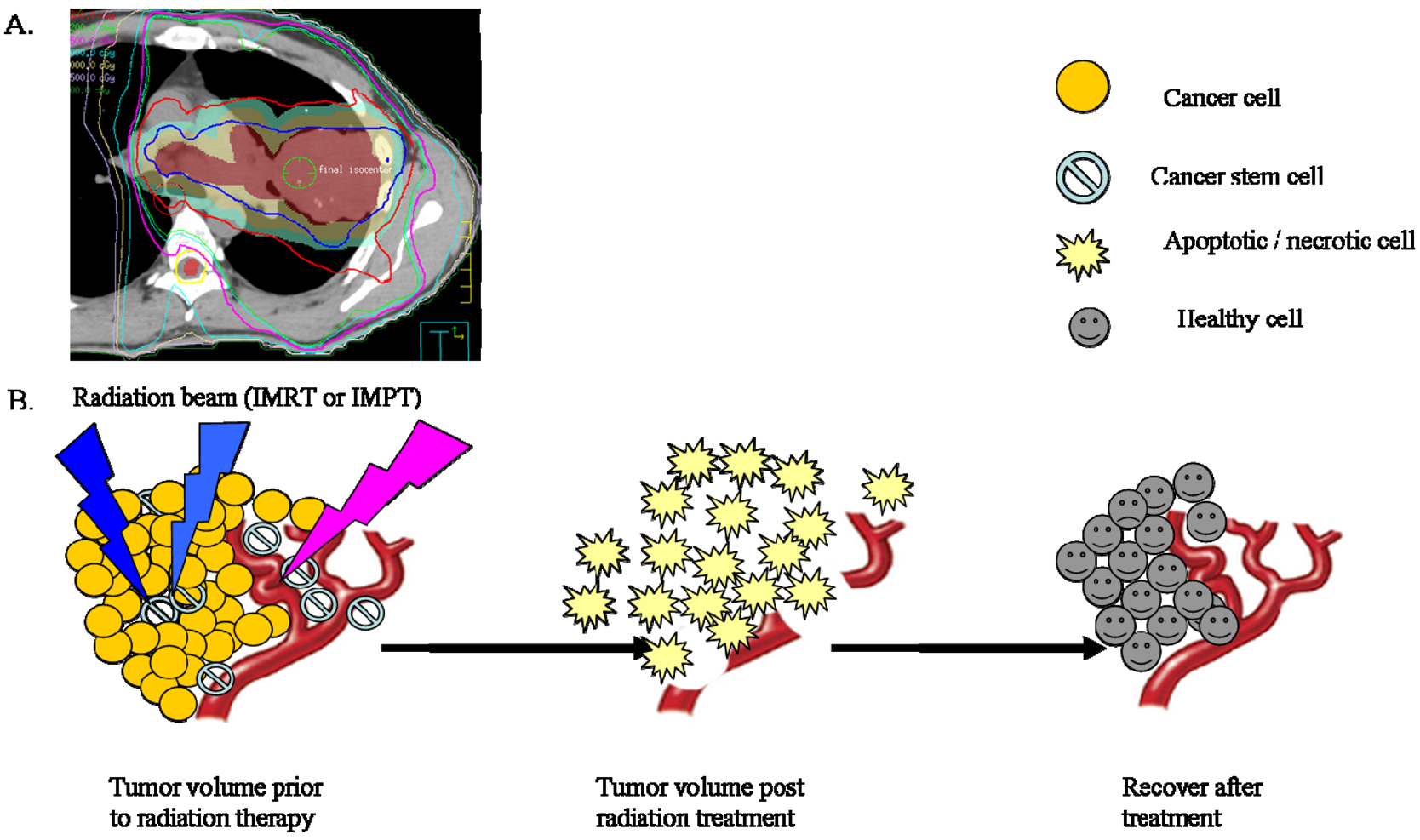
In the clinic, radiotherapy is always applied in multiple fractions, a concept that is based on the knowledge of classical “4 R's of Radiobiology” among fractionations: (1) The repair of sub-lethal damage and (2) repopulation of non-cancerous cells; (3) reoxygenation and (4) reassortment of tumor cells in radiosensitive phases [93]. In this review, we offered a systematic review of CSC research pertaining to the 4 Rs. It is difficult to summarize all the current CSC literature addressed the 4 Rs, and in many cases there are conflicting data, but we have attempted to focus on the delicate balance between CSC radiosensitivity vs. radioresistance (Figure 1).
Ionizing radiation leads to cell death by production of unrepairable DNA double-strand breaks (DSBs). Most radiation-induced DNA damage, however, is sublethal as cells are able to repair damage at lower doses. At higher dose accumulation, these sublethal lesions contribute to lethality. (The 4R's of radiation biology: Reassortment of cells within the cell-cycle, Repair of sub-lethal damage, Reoxygenation, and Repopulation.)
The ability of cells to repair the sublethal damage is different between normal and tumor cells, a concept that forms the basic foundation for using multiple fractions in radiotherapy treatment planning [94]. However, this conventional understanding of radiation-induced damage needs to be reevaluated with a specific emphasis on CSCs (Figure 1).
A hallmark of DNA DSB recognition and repair is histone H2A phosphorylation, which is thought to mark sites of DNA damage [95]. Radiation induced few or significantly fewer γ-H2AX foci in human breast CSCs, and in mouse breast CSCs these resolved faster than in non-CSCs [96-97]. Also, in glioma, radiation induced DNA damage to a similar degree in CD133+ and CD133- cells, but CD133+ cells repaired DNA damage more efficiently and rapidly than CD133- cells [98]. Interestingly, CD133+ cells showed basal activation of Rad17, a component of the DNA damage checkpoint, suggesting that cancer stem cells are primed to respond to genotoxic stresses. Inhibition of this response radiosensitized CD133+ glioma cells [98]. Although these studies focus on glioblastoma and breast CSCs, we expect that these findings may be more broadly applicable to lung and esophageal cancer types. It would be interesting to see if the checkpoint activation in CD133+ cells is biologically important in lung and esophageal CSCs.
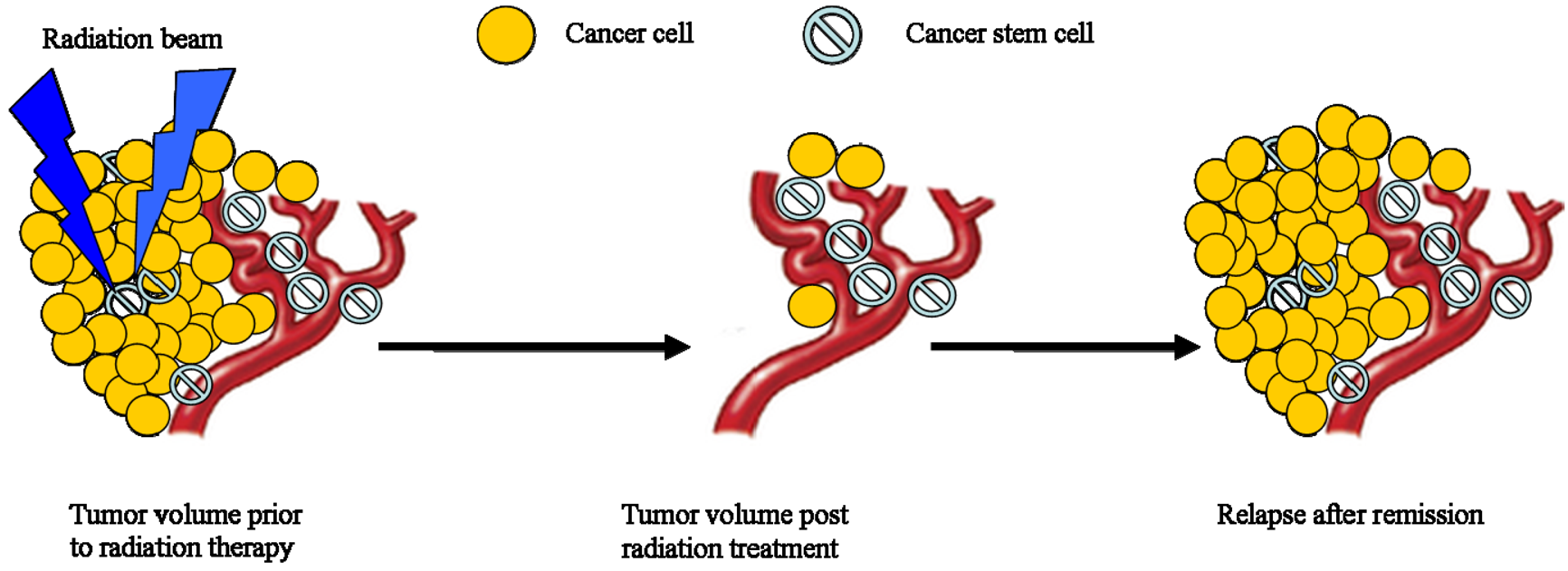
Accelerated repopulation of tumor cells during or after radiotherapy treatments are well documented as causes of treatment failure. Recent studies showed that irradiation enriches the fraction of cells expressing CSC markers [25,98]. For example, radioresistant esophageal carcinoma cells (Eca109R50Gy) obtained through fractionated irradiation (FIR) showed enhanced CSC properties and tumorigenic ability, approximately 40-times higher than that of radiosensitive cells in an in vivo xenograft tumorigenicity assay [99]. Interestingly, β-catenin expression spiked immediately with increasing irradiation doses and when the expression of β-catenin was suppressed by Wnt antagonist Dkk1, Eca109R50Gy cells displayed an enhancement of radiosensitivity [99]. In another study, Zhang et al. reported that several of the putative stem cell markers such as β-catenin, Oct3/4 and β-integrin were significantly increased in two radioresistant esophageal cell lines [100]. They also noticed that several so-called stemness genes were up-regulated while apoptotic genes were down-regulated in these cells. In addition, sorted CSCs from different types of tumors survived chemo-radiation treatments in culture much better than unsorted or negative cells [98]. This observation is supported by evidence that CSCs have high free radical scavenger levels [101], low proteasome activity [102], activated DNA checkpoints [10], slow cell-cycle progression [103-104], and efficient DNA repair machinery [98]. These factors are potential resistance mechanisms used by CSCs; highlighting the need for developing a revised radiation treatment protocol that could eliminate a small but resilient subpopulation of CSCs.
Radiation is also given on the assumption that tumors when oxygenated form highly reactive oxygen species (hROS), which rapidly interact with various biomolecules in cells, thereby causing DNA damage. There is ample evidence in the literature showing that hypoxic cells are more resistant to radiation than non-hypoxic cells [105], which would be an issue with many solid tumors. Highlighting the severity of this clinical problem is data demonstrating that human tumors containing regions of acute and chronic hypoxia are associated with poor prognosis because of local recurrence or systemic disease [106-108]. Reoxygenation between dose fractions is generally believed to improve the efficacy of radiation treatment by increasing tumor radiosensitivity. However, emerging evidence has indicated that hypoxic environment could actually trigger CSC differentiation; therefore leading to radiation resistance [109,110]. Given that CSCs have been shown to have low hROS metabolic profile, slow cycling, and express completely different survival pathways, it would be of great interest to know more about the response of CSCs to radiation under varying hypoxic conditions. Moving forward it would also have important clinical implications and therapeutic translation.
An early observation that shows CSCs are protected by hypoxia suggests this occurs through the induction of hypoxia-inducible factors (HIFs). Activation of HIF2α induces the expression of Oct-4, which is a central player in CSC self-renewal [111]. In addition, HIF2α is known to negatively interact with Notch1, therefore contributing to the maintenance of CSC undifferentiated state [112]. Another interesting point is that CSCs exist in two different states, one hypoxic for quiescent CSCs and one adjacent to endothelial cells for more activated CSCs. Transition between both states could be bidirectional, as is shown in hematopoietic stem cells [109]. These two states of CSC existence in relation to hypoxia might help to explain the variable results of clinical trials aimed at improving tumor oxygenation [113], as frequent hypoxia-reoxygenation caused by FIR could allow CSCs to differentiate and populate.
Each individual cell's position in the cell cycle influences its radiosensitivity, with cells in mitosis being most sensitive to DNA damage and cells in late S-phase most resistant [114]. Because of cell cycle progression between radiation fractions, dose fractionation allows redistribution of radioresistant S-phase tumor cells into a more sensitive phase of the cell cycle which allows maximal killing of the tumor cells while limiting toxicity to the surrounding healthy cells [115]. However, emerging evidence suggests that normal tissue stem cells and CSCs exist in the G0-phase of the cell cycle; therefore progressing slowly through the cell cycle [103,104,116] or maintaining a quiescent state for some period of time. Recent data on neural stem cells even suggest the possibility that expression of CD133 depends on the cell cycle and CD133 expression is specifically downregulated in the G0/G1 phase [98]. If this is true, it could explain why some lung cancer cells show little expression of CD133 but have the potential to self-renew [35]. Both lung cancer cell lines and human lung cancer samples contained a significant number of SP cells. These CSCs expressed a lower level of MCM7, a member of the minichromosome maintenance protein complex which is an essential component of the replication helicase complex, and is therefore a marker for S phase [46,117]. The human radioresistant esophageal cancer cell line, Eca109R50Gy had increased G0/G1 phase proportion, decreased G2/M phase proportion, and lower apoptosis rate compared with its parental cells, indicating they are relatively quiescent [99].
Another intriguing point, although it has not yet been seen in lung/esophageal samples, is that fractionated irradiation of breast CSCs induce elevated developmental signaling pathways like Notch/Jagged expression to a greater extent than single doses [96]. This data suggests that fractionated irradiation may specifically induce CSC differentiation through specific activation of different developmental pathways. If, in fact, tumors show a high percentage of slowly cycling CSCs and fractionated irradiation allows CSC differentiation, we may consider adjusting our current radiotherapy fractionation schedules to capture CSCs during their most vulnerable point in the cell cycle to induce DNA damage.
In summary, CSCs are more resistant to radiation therapy than non-stem cells. The cells that survive radiation may be enriched for more stem cells and actually exacerbate recurrence and cause refractory disease. For these important reasons, innovative radiotherapy approaches targeting CSCs might be the key to completely eradicating lung and esophageal tumors. Thus, the current understanding of radiotherapy and its treatment protocol need to be reexamined.
4. Implication for Cancer Research and Development
Despite enormous research efforts, the prognosis for both lung and esophageal cancer remains among the poorest. Surgery and radiation therapy are currently the only treatment options leading to a cancer cure for patients suffering from localized cancers. However, most current strategies may not target CSCs specifically but rather more differentiated tumor cells. Existing paradigms for cancer treatment need to be re-evaluated for relevance based upon recent findings of CSCs. One aspect that would greatly improve the current stage for treatment of lung and esophageal cancer is by further characterizing CSC markers seen in these two cancers. Although there is ample evidence of CSC markers in other solid tumors, there is little evidence on the specific CSC markers for respective subtypes of lung and esophageal cancers and the data reported sometimes conflict and can not be standardized to allow comparison across laboratories. Developing CSC markers for use in the clinic would facilitate the visualization of specific CSCs and the capture of related images used for treatment planning, which would be critical to strategically target these CSCs and reduce disease recurrence. Currently radiation doses are often prescribed on the basis of several clinical parameters including tumor volume, histology and location under the consideration of tolerance limits of the surrounding anatomical structures. Therefore, knowledge about the spatial distribution of CSCs in relation to non-stem cells within the tumor bed may become important during treatment planning. Therefore, this is an important area of research that needs to be actively examined before meaningful adjustments to current treatments can be taken.
The currently used imaging techniques for treatment planning are inadequate in discriminating between areas with active CSCs and those with non-stem cells. For instance, positron emission tomography (PET) or PET/CT with 2-deoxy-2-[18F]fluoro-D-glucose (18FDG-PET) is widely utilized for diagnosis, staging and assessment of therapeutic response during and after radiation therapy for lung/esophageal cancers [118,119]. This technology works on the basic assumption that rapidly proliferating cancer cells, which form the bulk of the tumor, rely mainly on glycolysis, a biochemical process that requires drastically increased glucose uptake; therefore, these cells will be seen on 18FDG-PET imaging of lung tumors or image-guided radiation therapy. Given what is known about CSCs, it is widely assumed that CSCs, in contrast, are mostly quiescent and metabolize glucose by oxidative phosphorylation rather than by glycolysis [120]. Therefore, areas with high numbers of quiescent CSCs with low glucose uptake may not be picked up by 18FDG-PET and therefore might not be targeted specifically during treatment planning. Thus, to improve the clinical relevance of PET/CT in tumor imaging, it is important to compliment these imaging techniques with an evaluation of CSCs. An area with enriched CSCs may in fact need a higher radiation dose to eliminate CSCs and is crucial to reduce disease progression and distant metastasis. This concept is supported by a recent clinical trial enrolling early stage lung cancer patients. These patients showed a promising clinical outcome with improved survival and local control (>95% compared with 50% using conventional radiotherapy to 60-70 Gy with 2 Gy/fraction) when hypofractionated stereotactic ablative radiotherapy, i.e., higher dose per fraction (>10 GY) to total dose of 48 to 54 Gy, was delivered [121].
Recently, intensity-modulated radiation therapy (IMRT) was developed to create more conformal radiation dose to target while minimizing dose to surrounding critical structures and allowing selectively painting the dose to subregions within the target and further customizing delivered dose distribution [121]. Given the inhomogeneity of the tumor and the presence of CSCs, it would be extremely helpful to use CSC markers as biomarkers to guide IMRT treatment when designing a comprehensive treatment plan to improve biological conformality based on CSCs. Combining these two approaches could further improve the efficacy of future radiotherapy.
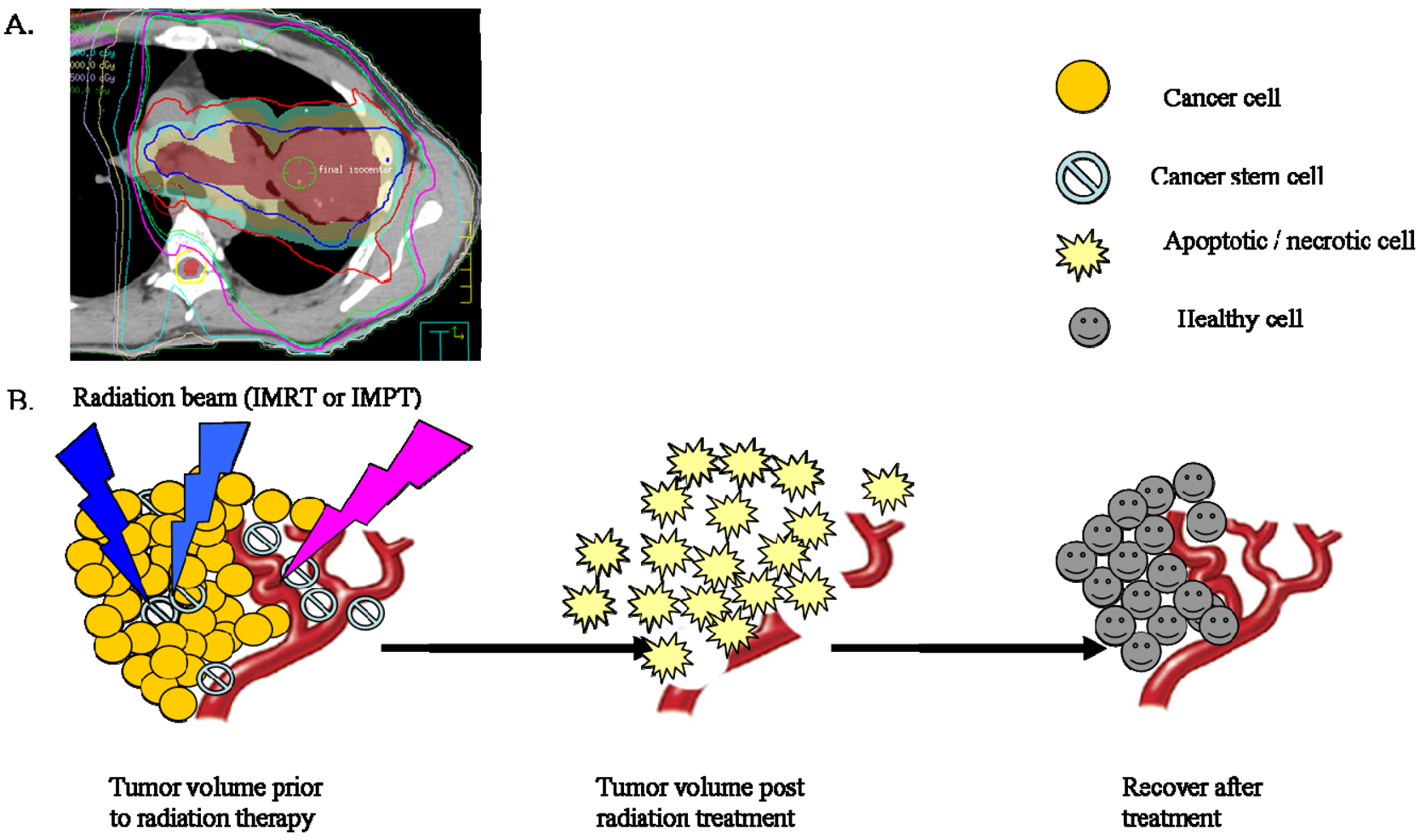
Conventional fractionated radiation therapy has evolved over more than a century as a means of delivering radiation dose over a period of time. Current standard treatment plans aim to deliver a homogenous dose over the tumor bed, assuming that CSCs are randomly distributed and therefore will be effectively removed by this method (Figure 2). Given our preliminary understanding of CSCs in their ability to recognize and repair radiation damage, to repopulate during the treatment period, and to recover from radiation damage between fractions, it is important that this concept be looked at wisely as it may change the way radiation is delivered. It might be possible that low radiation doses are given to non-CSCs while higher radiation doses are given to CSCs as this is believed to reflect their different intrinsic DNA repair efficiencies. Additionally, the time interval between radiation dose fractions might be different for CSCs vs. non-CSCs to account for the slow cycling of CSCs (Figure 3). It would be interesting to explore whether the following changes will be beneficial to cancer treatment: higher dose per fraction to overcome radiation resistance of CSCs and longer interval between fractionation to account for the slow cycling of CSCs and allow CSCs going through asymmetric division into less committed CSCs.
Radiation therapy, especially using photon energy has been a central player in the management of lung and esophageal cancer for a long time. Only recently, proton therapy has been used in lung and esophageal cancers due to its superior physical characteristics such that the proton beam can deliver a much higher therapeutic dose to certain depth without any exit dose; therefore, significantly reduce the radiation delivered to normal lung tissues and other nearby structures [122-123]. The selection of proton in cancer management becomes important because it addresses the pitfall in management of local failure in that an inadequate dose is given to the tumor bed because of toxicity concerns. Given its relatively new application in cancer management, there is little evidence regarding the applicability of proton therapy to CSCs. However, our preliminary studies have shown that proton therapy preferentially targets CSCs and increases ROS levels in treatment of resistant NSCLC to a greater extent than photons of the same dose [124]. This is a novel observation and would require further study. In addition, it would be interesting to see if similar observations are seen for other types of lung and esophageal tumors.
Given that CSCs share many of the same features as normal stem cells, there is a potential risk of killing normal stem cells while targeting CSCs. Therefore, it is important to further characterize the similarities and differences between these two types of stem cells and use new therapeutic approaches to selectively target CSC-specific pathways. One method is to target specific components of the signal transduction pathways that are highly active in CSCs, but not in normal stem cells, much like the strategy employed by therapeutics like trastuzumab (Herceptin®) in solid breast tumors [125]. An example of exploiting differences in CSC signaling is telomerase expression, which is repressed in most somatic cells but observed in stem cells as well as a high percentage of human cancers. It has recently been shown that telomerase expression is increased in radioresistant esophageal CSCs [100].
The observation that telomerase expression occurs in CSCs has important implications for developing therapeutic applications using telomerase. Since telomerase expression was believed to be restricted to CSCs, targeted strategies could be useful for therapy. Several groups have begun developing oncolytic Adenoviruses targeted to CSCs in the context of telomerase. Zhang and her colleagues engineered a telomerase-specific oncolytic Adeno vector carrying the apoptotic tumor necrosis factor-related apoptosis-inducing ligand under control of the hTERT promoter and cytomegalovirus early vector. This vector preferentially targeted radioresistant esophageal CSC-like cells and sensitized esophageal cancer to radiation [100]. However, the mechanism of tumoricidal effect of the oncolytic Adeno vector targeting the radioresistant cancer cells remains to be characterized.
Over its history, radiation oncologists have used local tumor control as an endpoint for radiotherapy with particular emphasis on the changes in tumor volume after therapy. This change in tumor volume is largely governed by the death of non-CSCs, while a subpopulation of CSCs might exist. Given what is known about the resilient growth properties of CSCs after radiation and that it only takes a very small number of CSCs, it is theoretically possible for a single surviving CSC to initiate a recurrent tumor. It is important to make a distinction that the response of the tumor cell bulk does not reflect the response of stem cells. Therefore, it might be wise to consider the absolute CSC control as the endpoint for radiotherapy. In that way, future development of new treatment planning and the assessment of local tumor control should be measured by the complete eradication of the subpopulations of CSCs.
Lastly, one cannot deny that combined-modality treatment approach has become standard and resulted in modest gain in the survival of lung and esophageal cancer patients. Therefore, the development of therapeutic agents specifically targeting CSCs coupled with a revised radiation treatment protocol will no doubt favorably alter the current poor prognosis of cancer patients. In this way, new treatments to combat lung and esophageal cancer will hopefully help to overcome the dismal outcome seen in these cancer patients.
5. Conclusions
This article gives an overview of the current literature regarding lung and esophageal CSCs and radiation resistance of CSCs. The consensus of opinion appears to be that there is little evidence of esophageal CSCs, in contrast to lung CSCs. While, at present, evidence continues to mount to support a CSC hypothesis, the extent of the remaining self-renewal capacity of lung and esophageal CSC populations after fractions of radiation is largely unknown. Further translational study to identify CSCs and analyze its behaviors will help researchers to explore CSCs targeting therapy. New novel CSC-based approaches may overcome treatment resistance and improve clinical outcome for cancer patients.


{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank Georgina Mosedale (University of Oxford) for critical reading of the manuscript and and Christine Wogan (The University of Texas M.D. Anderson Cancer Center) for editorial assistance.
References
- American Cancer Society. Global Cancer Facts and Figures. Available online: http://www5.cancer.org/downloads/STT/Cancer_Facts_and_Figures_2010.pdf (accessed on 1 March 2011).
- Weissman, I.L.; Anderson, D.J.; Gage, F. Stem and progenitor cells: Origins, phenotypes, lineage commitments, and transdifferentiations. Annu. Rev. Cell Dev. Biol. 2001, 17, 387–403. [Google Scholar]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar]
- Houghton, J.; Stoicov, C.; Nomura, S.; Rogers, A.B.; Carlson, J.; Li, H.; Cai, X.; Fox, J.G.; Goldenring, J.R.; Wang, T.C. Gastric cancer originating from bone marrow-derived cells. Science 2004, 306, 1568–1571. [Google Scholar]
- Wu, P.C.; Lai, V.C.; Fang, J.W.; Gerber, M.A.; Lai, C.L.; Lau, J.Y. Hepatocellular carcinoma expressing both hepatocellular and biliary markers also expresses cytokeratin 14, a marker of bipotential progenitor cells. J. Hepatol. 1999, 31, 965–966. [Google Scholar]
- Schatton, T.; Murphy, G.F.; Frank, N.Y.; Yamaura, K.; Waaga-Gasser, A.M.; Gasser, M.; Zhan, Q.; Jordan, S.; Duncan, L.M.; Weishaupt, C.; Fuhlbrigge, R.C.; Kupper, T.S.; Sayegh, M.H.; Frank, M.H. Identification of cells initiating human melanomas. Nature 2008, 451, 345–349. [Google Scholar]
- Szotek, P.P.; Pieretti-Vanmarcke, R.; Masiakos, P.T.; Dinulescu, D.M.; Connolly, D.; Foster, R.; Dombkowski, D.; Preffer, F.; Maclaughlin, D.T.; Donahoe, P.K. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian Inhibiting Substance responsiveness. Proc. Natl. Acad. Sci USA 2006, 103, 11154–1159. [Google Scholar]
- Chan, K.S.; Espinosa, I.; Chao, M.; Wong, D.; Ailles, L.; Diehn, M.; Gill, H.; Presti, J.; Chang, H.Y.; van de Rijn, M.; Shortliffe, L.; Weissman, I.L. Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2009, 106, 14016–14021. [Google Scholar]
- Prince, M.E.; Sivanandan, R.; Kaczorowski, A.; Wolf, G.T.; Kaplan, M.J.; Dalerba, P.; Weissman, I.L.; Clarke, M.F.; Ailles, L.E. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2007, 104, 973–978. [Google Scholar]
- Kim, C.F.; Jackson, E.L.; Woolfenden, A.E.; Lawrence, S.; Babar, I.; Vogel, S.; Crowley, D.; Bronson, R.T.; Jacks, T. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 2005, 121, 823–835. [Google Scholar]
- Govindan, R.; Page, N.; Morgensztern, D.; Read, W.; Tierney, R.; Vlahiotis, A.; Spitznagel, E.L.; Piccirillo, J. Changing epidemiology of small-cell lung cancer in the United States over the last 30 years: analysis of the surveillance, epidemiologic, and end results database. J. Clin. Oncol. 2006, 24, 4539–4544. [Google Scholar]
- Falk, J.; Carstens, H.; Lundell, L.; Albertsson, M. Incidence of carcinoma of the oesophagus and gastric cardia. Changes over time and geographical differences. Acta Oncol. 2007, 46, 1070–1074. [Google Scholar]
- Paulson, T.G.; Reid, B.J. Focus on Barrett's esophagus and esophageal adenocarcinoma. Cancer Cell 2004, 6, 11–16. [Google Scholar]
- Izzo, J.G.; Luthra, R.; Wu, T.T.; Correa, A.M.; Luthra, M.; Anandasabapathy, S.; Chao, K.S.; Hung, M.C.; Aggarwal, B.; Hittelman, W.N.; Ajani, J.A. Molecular mechanisms in Barrett's metaplasia and its progression. Semin. Oncol. 2007, 34, S2–S6. [Google Scholar]
- Krause, D.S.; Lazarides, K.; von Andrian, U.H.; Van Etten, R.A. Requirement for CD44 in homing and engraftment of BCR-ABL-expressing leukemic stem cells. Nat. Med. 2006, 12, 1175–1180. [Google Scholar]
- Wu, Y.; Wu, P.Y. CD133 as a marker for cancer stem cells: progresses and concerns. Stem Cells Dev. 2009, 18, 1127–1134. [Google Scholar]
- Capela, A.; Temple, S. LeX/ssea-1 is expressed by adult mouse CNS stem cells, identifying them as nonependymal. Neuron 2002, 35, 865–875. [Google Scholar]
- Kucia, M.; Reca, R.; Miekus, K.; Wanzeck, J.; Wojakowski, W.; Janowska-Wieczorek, A.; Ratajczak, J.; Ratajczak, M.Z. Trafficking of normal stem cells and metastasis of cancer stem cells involve similar mechanisms: pivotal role of the SDF-1-CXCR4 axis. Stem Cells 2005, 23, 879–894. [Google Scholar]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar]
- Miraglia, S.; Godfrey, W.; Yin, A.H.; Atkins, K.; Warnke, R.; Holden, J.T.; Bray, R.A.; Waller, E.K.; Buck, D.W. A novel five-transmembrane hematopoietic stem cell antigen: isolation, characterization, and molecular cloning. Blood 1997, 90, 5013–521. [Google Scholar]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar]
- Wright, M.H.; Calcagno, A.M.; Salcido, C.D.; Carlson, M.D.; Ambudkar, S.V.; Varticovski, L. Brca1 breast tumors contain distinct CD44+/CD24− and CD133+ cells with cancer stem cell characteristics. Breast Cancer Res. 2008, 10, R10. [Google Scholar]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar]
- Ferrandina, G.; Bonanno, G.; Pierelli, L.; Perillo, A.; Procoli, A.; Mariotti, A.; Corallo, M.; Martinelli, E.; Rutella, S.; Paglia, A.; Zannoni, G.; Mancuso, S.; Scambia, G. Expression of CD133-1 and CD133-2 in ovarian cancer. Int. J. Gynecol. Cancer 2008, 18, 506–14. [Google Scholar]
- Suetsugu, A.; Nagaki, M.; Aoki, H.; Motohashi, T.; Kunisada, T.; Moriwaki, H. Characterization of CD133+ hepatocellular carcinoma cells as cancer stem/progenitor cells. Biochem. Biophys. Res. Commun. 2006, 351, 820–824. [Google Scholar]
- Toren, A.; Bielorai, B.; Jacob-Hirsch, J.; Fisher, T.; Kreiser, D.; Moran, O.; Zeligson, S.; Givol, D.; Yitzhaky, A.; Itskovitz-Eldor, J.; Kventsel, I.; Rosenthal, E.; Amariglio, N.; Rechavi, G. CD133-positive hematopoietic stem cell “stemness” genes contain many genes mutated or abnormally expressed in leukemia. Stem Cells 2005, 23, 1142–1153. [Google Scholar]
- Yin, A.H.; Miraglia, S.; Zanjani, E.D.; Almeida-Porada, G.; Ogawa, M.; Leary, A.G.; Olweus, J.; Kearney, J.; Buck, D.W. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood 1997, 90, 5002–5012. [Google Scholar]
- Bertolini, G.; Roz, L.; Perego, P.; Tortoreto, M.; Fontanella, E.; Gatti, L.; Pratesi, G.; Fabbri, A.; Andriani, F.; Tinelli, S.; Roz, E.; Caserini, R.; Lo Vullo, S.; Camerini, T.; Mariani, L.; Delia, D.; Calabrò, E.; Pastorino, U.; Sozzi, G. Highly tumorigenic lung cancer CD133+ cells display stemlike features and are spared by cisplatin treatment. Proc. Natl. Acad. Sci. 2009, 106, 16281–16286. [Google Scholar]
- Chu, E.; DeVita, V.T. Physicians' Cancer Chemotherapy Drug Manual; Jones and Bartlett: Sudbury, MA, USA, 2008. [Google Scholar]
- Zhang, H.Z.; Wei, Y.P.; Wang, M.; Wu, C.; Yang, Y.Q.; Chen, J.; Cao, Y.K. Association of CD133 and endothelin-converting enzyme expressions with prognosis of non-small cell lung carcinoma. Nan Fang Yi Ke Da Xue Xue Bao 2007, 27, 696–699. [Google Scholar]
- Tirino, V.; Camerlingo, R.; Franco, R.; Malanga, D.; La Rocca, A.; Viglietto, G.; Rocco, G.; Pirozzi, G. The role of CD133 in the identification and characterisation of tumour-initiating cells in non-small-cell lung cancer. Eur. J. Cardiothorac. Surg. 2009, 36, 446–453. [Google Scholar]
- Salnikov, A.V.; Gladkich, J.; Moldenhauer, G.; Volm, M.; Mattern, J.; Herr, I. CD133 is indicative for a resistance phenotype but does not represent a prognostic marker for survival of non-small cell lung cancer patients. Int. J. Cancer 2010, 126, 950–958. [Google Scholar]
- Meng, X.; Li, M.; Wang, X.; Wang, Y.; Ma, D. Both CD133+ and CD133- subpopulations of A549 and H446 cells contain cancer-initiating cells. Cancer Sci. 2009, 100, 1040–1046. [Google Scholar]
- Grotenhuis, B.A.; Dinjens, W.N.; Wijnhoven, B.P.; Sonneveld, P.; Sacchetti, A.; Franken, P.F.; van Dekken, H.; Tilanus, H.W.; van Lanschot, J.J.; Fodde, R. Barrett's oesophageal adenocarcinoma encompasses tumour-initiating cells that do not express common cancer stem cell markers. J. Pathol. 2010, 221, 379–389. [Google Scholar]
- Takaishi, S.; Okumura, T.; Tu, S.; Wang, S.S.; Shibata, W.; Vigneshwaran, R.; Gordon, S.A.; Shimada, Y.; Wang, T.C. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells 2009, 27, 1006–1020. [Google Scholar]
- Marhaba, R.; Klingbeil, P.; Nuebel, T.; Nazarenko, I.; Buechler, M.W.; Zoeller, M. CD44 and EpCAM: cancer-initiating cell markers. Curr. Mol. Med. 2008, 8, 784–804. [Google Scholar]
- Lee, L.N.; Kuo, S.H.; Lee, Y.C.; Chang, Y.L.; Chang, H.C.; Jan, I.S.; Yang, P.C. CD44 splicing pattern is associated with disease progression in pulmonary adenocarcinoma. J. Formos. Med. Assoc. 2005, 104, 541–548. [Google Scholar]
- Chai, L.X.; Sun, K.L.; Guo, L.P.; Zhang, H.T.; Lu, S.X. Expression of ezrin and CD44-v6 in human esophageal squamous cell carcinoma and its clinical significance. Zhonghua Zhong Liu Za Zhi 2007, 29, 685–688. [Google Scholar]
- Takayama, N.; Arima, S.; Haraoka, S.; Kotho, T.; Futami, K.; Iwashita, A. Relationship between the expression of adhesion molecules in primary esophageal squamous cell carcinoma and metastatic lymph nodes. Anticancer Res. 2003, 23, 4435–4442. [Google Scholar]
- Zhou, S.; Schuetz, J.D.; Bunting, K.D.; Colapietro, A.M.; Sampath, J.; Morris, J.J.; Lagutina, I.; Grosveld, G.C.; Osawa, M.; Nakauchi, H.; Sorrentino, B.P. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat. Med. 2001, 7, 1028–1034. [Google Scholar]
- Hirschmann-Jax, C.; Foster, A.E.; Wulf, G.G.; Nuchtern, J.G.; Jax, T.W.; Gobel, U.; Goodell, M.A.; Brenner, M.K. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc. Natl. Acad. Sci. USA 2004, 101, 14228–14233. [Google Scholar]
- Ho, M.M.; Ng, A.V.; Lam, S.; Hung, J.Y. Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer Res. 2007, 67, 4827–4833. [Google Scholar]
- Huang, D.; Gao, Q.; Guo, L.; Zhang, C.; Jiang, W.; Li, H.; Wang, J.; Han, X.; Shi, Y.; Lu, S.H. Isolation and identification of cancer stem-like cells in esophageal carcinoma cell lines. Stem Cells Dev. 2009, 18, 465–473. [Google Scholar]
- Hoshi, N.; Kusakabe, T.; Taylor, B.J.; Kimura, S. Side population cells in the mouse thyroid exhibit stem/progenitor cell-like characteristics. Endocrinology 2007, 148, 4251–4258. [Google Scholar]
- Nishii, T.; Yashiro, M.; Shinto, O.; Sawada, T.; Ohira, M.; Hirakawa, K. Cancer stem cell-like SP cells have a high adhesion ability to the peritoneum in gastric carcinoma. Cancer Sci. 2009, 100, 1397–1402. [Google Scholar]
- Liu, B.Y.; McDermott, S.P.; Khwaja, S.S.; Alexander, C.M. The transforming activity of Wnt effectors correlates with their ability to induce the accumulation of mammary progenitor cells. Proc. Natl. Acad. Sci. USA 2004, 101, 4158–4163. [Google Scholar]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar]
- Chen, Y.C.; Hsu, H.S.; Chen, Y.W.; Tsai, T.H.; How, C.K.; Wang, C.Y.; Hung, S.C.; Chang, Y.L.; Tsai, M.L.; Lee, Y.Y.; Ku, H.H.; Chiou, S.H. Oct-4 expression maintained cancer stem-like properties in lung cancer-derived CD133-positive cells. PLoS One 2008, 3, e2637. [Google Scholar]
- Chute, J.P.; Muramoto, G.G.; Whitesides, J.; Colvin, M.; Safi, R.; Chao, N.J.; McDonnell, D.P. Inhibition of aldehyde dehydrogenase and retinoid signaling induces the expansion of human hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 2006, 103, 11707–11712. [Google Scholar]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; Schott, A.; Hayes, D.; Birnbaum, D.; Wicha, M.S.; Dontu, G. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar]
- Corti, S.; Locatelli, F.; Papadimitriou, D.; Donadoni, C.; Salani, S.; Del Bo, R.; Strazzer, S.; Bresolin, N.; Comi, G.P. Identification of a primitive brain-derived neural stem cell population based on aldehyde dehydrogenase activity. Stem Cells 2006, 24, 975–985. [Google Scholar]
- Huang, E.H.; Hynes, M.J.; Zhang, T.; Ginestier, C.; Dontu, G.; Appelman, H.; Fields, J.Z.; Wicha, M.S.; Boman, B.M. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res 2009, 69, 3382–3389. [Google Scholar]
- Chen, Y.C.; Chen, Y.W.; Hsu, H.S.; Tseng, L.M.; Huang, P.I.; Lu, K.H.; Chen, D.T.; Tai, L.K.; Yung, M.C.; Chang, S.C.; Ku, H.H.; Chiou, S.H.; Lo, W.L. Aldehyde dehydrogenase 1 is a putative marker for cancer stem cells in head and neck squamous cancer. Biochem. Biophys. Res Commun. 2009, 385, 307–313. [Google Scholar]
- Patel, M.; Lu, L.; Zander, D.S.; Sreerama, L.; Coco, D.; Moreb, J.S. ALDH1A1 and ALDH3A1 expression in lung cancers: correlation with histologic type and potential precursors. Lung Cancer 2008, 59, 340–349. [Google Scholar]
- Jiang, F.; Qiu, Q.; Khanna, A.; Todd, N.W.; Deepak, J.; Xing, L.; Wang, H.; Liu, Z.; Su, Y.; Stass, S.A.; Katz, R.L. Aldehyde dehydrogenase 1 is a tumor stem cell-associated marker in lung cancer. Mol. Cancer Res. 2009, 7, 330–338. [Google Scholar]
- Moreb, J.S.; Baker, H.V.; Chang, L.J.; Amaya, M.; Lopez, M.C.; Ostmark, B.; Chou, W. ALDH isozymes downregulation affects cell growth, cell motility and gene expression in lung cancer cells. Mol. Cancer 2008, 7, 87. [Google Scholar]
- Cui, R.; Kamatani, Y.; Takahashi, A.; Usami, M.; Hosono, N.; Kawaguchi, T.; Tsunoda, T.; Kamatani, N.; Kubo, M.; Nakamura, Y.; Matsuda, K. Functional variants in ADH1B and ALDH2 coupled with alcohol and smoking synergistically enhance esophageal cancer risk. Gastroenterology 2009, 137, 1768–1775. [Google Scholar]
- Pardal, R.; Clarke, M.F.; Morrison, S.J. Applying the principles of stem-cell biology to cancer. Nat. Rev. Cancer 2003, 3, 895–902. [Google Scholar]
- Polakis, P. The many ways of Wnt in cancer. Curr. Opin. Genet. Dev. 2007, 17, 45–51. [Google Scholar]
- Li, Y.; Welm, B.; Podsypanina, K.; Huang, S.; Chamorro, M.; Zhang, X.; Rowlands, T.; Egeblad, M.; Cowin, P.; Werb, Z.; Tan, L.K.; Rosen, J.M.; Varmus, H.E. Evidence that transgenes encoding components of the Wnt signaling pathway preferentially induce mammary cancers from progenitor cells. Proc. Natl. Acad. Sci. USA 2003, 100, 15853–15858. [Google Scholar]
- Reynolds, S.D.; Zemke, A.C.; Giangreco, A.; Brockway, B.L.; Teisanu, R.M.; Drake, J.A.; Mariani, T.; Di, P.Y.; Taketo, M.M.; Stripp, B.R. Conditional stabilization of beta-catenin expands the pool of lung stem cells. Stem Cells 2008, 26, 1337–146. [Google Scholar]
- Uematsu, K.; He, B.; You, L.; Xu, Z.; McCormick, F.; Jablons, D.M. Activation of the Wnt pathway in non small cell lung cancer: evidence of dishevelled overexpression. Oncogene 2003, 22, 7218–7221. [Google Scholar]
- Uematsu, K.; Kanazawa, S.; You, L.; He, B.; Xu, Z.; Li, K.; Peterlin, B.M.; McCormick, F.; Jablons, D.M. Wnt pathway activation in mesothelioma: evidence of Dishevelled overexpression and transcriptional activity of beta-catenin. Cancer Res. 2003, 63, 4547–4551. [Google Scholar]
- You, L.; He, B.; Xu, Z.; Uematsu, K.; Mazieres, J.; Mikami, I.; Reguart, N.; Moody, T.W.; Kitajewski, J.; McCormick, F.; Jablons, D.M. Inhibition of Wnt-2-mediated signaling induces programmed cell death in non-small-cell lung cancer cells. Oncogene 2004, 23, 6170–6174. [Google Scholar]
- Klaus, A.; Birchmeier, W. Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer 2008, 8, 387–398. [Google Scholar]
- Kim, J.; You, L.; Xu, Z.; Kuchenbecker, K.; Raz, D.; He, B.; Jablons, D. Wnt inhibitory factor inhibits lung cancer cell growth. J. Thorac. Cardiovasc. Surg. 2007, 133, 733–737. [Google Scholar]
- De Langhe, S.P.; Sala, F.G.; Del Moral, P.M.; Fairbanks, T.J.; Yamada, K.M.; Warburton, D.; Burns, R.C.; Bellusci, S. Dickkopf-1 (DKK1) reveals that fibronectin is a major target of Wnt signaling in branching morphogenesis of the mouse embryonic lung. Dev. Biol. 2005, 277, 316–331. [Google Scholar]
- Darlavoix, T.; Seelentag, W.; Yan, P.; Bachmann, A.; Bosman, F.T. Altered expression of CD44 and DKK1 in the progression of Barrett's esophagus to esophageal adenocarcinoma. Virchows Arch. 2009, 454, 629–637. [Google Scholar]
- Makino, T.; Yamasaki, M.; Takemasa, I.; Takeno, A.; Nakamura, Y.; Miyata, H.; Takiguchi, S.; Fujiwara, Y.; Matsuura, N.; Mori, M.; Doki, Y. Dickkopf-1 expression as a marker for predicting clinical outcome in esophageal squamous cell carcinoma. Ann. Surg. Oncol. 2009, 16, 2058–2064. [Google Scholar]
- Hsu, P.K.; Li, A.F.; Wang, Y.C.; Hsieh, C.C.; Huang, M.H.; Hsu, W.H.; Hsu, H.S. Reduced membranous beta-catenin protein expression is associated with metastasis and poor prognosis in squamous cell carcinoma of the esophagus. J. Thorac. Cardiovasc. Surg. 2008, 135, 1029–1035. [Google Scholar]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch signaling: cell fate control and signal integration in development. Science 1999, 284, 770–776. [Google Scholar]
- Levina, V.; Marrangoni, A.M.; DeMarco, R.; Gorelik, E.; Lokshin, A.E. Drug-selected human lung cancer stem cells: cytokine network, tumorigenic and metastatic properties. PLoS One 2008, 3, e3077. [Google Scholar]
- Ito, T.; Udaka, N.; Yazawa, T.; Okudela, K.; Hayashi, H.; Sudo, T.; Guillemot, F.; Kageyama, R.; Kitamura, H. Basic helix-loop-helix transcription factors regulate the neuroendocrine differentiation of fetal mouse pulmonary epithelium. Development 2000, 127, 3913–3921. [Google Scholar]
- Ito, T.; Udaka, N.; Ikeda, M.; Yazawa, T.; Kageyama, R.; Kitamura, H. Significance of proneural basic helix-loop-helix transcription factors in neuroendocrine differentiation of fetal lung epithelial cells and lung carcinoma cells. Histol. Histopathol. 2001, 16, 335–343. [Google Scholar]
- Chen, H.; Thiagalingam, A.; Chopra, H.; Borges, M.W.; Feder, J.N.; Nelkin, B.D.; Baylin, S.B.; Ball, D.W. Conservation of the Drosophila lateral inhibition pathway in human lung cancer: a hairy-related protein (HES-1) directly represses achaete-scute homolog-1 expression. Proc. Natl. Acad. Sci. USA 1997, 94, 5355–5360. [Google Scholar]
- Konishi, J.; Kawaguchi, K.S.; Vo, H.; Haruki, N.; Gonzalez, A.; Carbone, D.P.; Dang, T.P. Gamma-secretase inhibitor prevents Notch3 activation and reduces proliferation in human lung cancers. Cancer Res. 2007, 67, 8051–8057. [Google Scholar]
- Weijzen, S.; Rizzo, P.; Braid, M.; Vaishnav, R.; Jonkheer, S.M.; Zlobin, A.; Osborne, B.A.; Gottipati, S.; Aster, J.C.; Hahn, W.C.; Rudolf, M.; Siziopikou, K.; Kast, W.M.; Miele, L. Activation of Notch-1 signaling maintains the neoplastic phenotype in human Ras-transformed cells. Nat. Med. 2002, 8, 979–986. [Google Scholar]
- Zheng, Q.; Qin, H.; Zhang, H.; Li, J.; Hou, L.; Wang, H.; Zhang, X.; Zhang, S.; Feng, L.; Liang, Y.; Han, H.; Yi, D. Notch signaling inhibits growth of the human lung adenocarcinoma cell line A549. Oncol. Rep. 2007, 17, 847–852. [Google Scholar]
- Sriuranpong, V.; Borges, M.W.; Ravi, R.K.; Arnold, D.R.; Nelkin, B.D.; Baylin, S.B.; Ball, D.W. Notch signaling induces cell cycle arrest in small cell lung cancer cells. Cancer Res. 2001, 61, 3200–3205. [Google Scholar]
- Zhang, K.J.; Lu, Q.Y.; Niu, X.Q.; Zhang, P.; Zhao, J.N.; Wang, Z.; Hu, J.S.; Li, P.; Liu, W.L. Notch1 signaling inhibits growth of EC109 esophageal carcinoma cells through downmodulation of HPV18 E6/E7 gene expression. Acta Pharmacol. Sin. 2009, 30, 153–158. [Google Scholar]
- Varjosalo, M.; Taipale, J. Hedgehog: functions and mechanisms. Genes Dev. 2008, 22, 2454–72. [Google Scholar]
- Dlugosz, A.A.; Talpaz, M. Following the hedgehog to new cancer therapies. N. Engl. J. Med. 2009, 361, 1202–1205. [Google Scholar]
- Hyman, J.M.; Firestone, A.J.; Heine, V.M.; Zhao, Y.; Ocasio, C.A.; Han, K.; Sun, M.; Rack, P.G.; Sinha, S.; Wu, J.J.; Solow-Cordero, D.E.; Jiang, J.; Rowitch, D.H.; Chen, J.K. Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc. Natl. Acad. Sci. USA 2009, 106, 14132–14137. [Google Scholar]
- Varjosalo, M.; Taipale, J. Hedgehog signaling. J. Cell. Sci. 2007, 120, 3–6. [Google Scholar]
- Pepicelli, C.V.; Lewis, P.M.; McMahon, A.P. Sonic hedgehog regulates branching morphogenesis in the mammalian lung. Curr. Biol. 1998, 8, 1083–1086. [Google Scholar]
- Bellusci, S.; Furuta, Y.; Rush, M.G.; Henderson, R.; Winnier, G.; Hogan, B.L. Involvement of Sonic hedgehog (Shh) in mouse embryonic lung growth and morphogenesis. Development 1997, 124, 53–63. [Google Scholar]
- Watkins, D.N.; Berman, D.M.; Burkholder, S.G.; Wang, B.; Beachy, P.A.; Baylin, S.B. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 2003, 422, 313–317. [Google Scholar]
- Ma, X.; Sheng, T.; Zhang, Y.; Zhang, X.; He, J.; Huang, S.; Chen, K.; Sultz, J.; Adegboyega, P.A.; Zhang, H.; Xie, J. Hedgehog signaling is activated in subsets of esophageal cancers. Int. J. Cancer 2006, 118, 139–148. [Google Scholar]
- Withers, H.R. The four R's of radiotherapy. In Advances in Radiation Biology; Lett, J.T., Adler, H., Zelle, M., Eds.; Academic Press: New York, NY, USA, 1975; Volume 5, pp. 241–271. [Google Scholar]
- LAJTHA, L.G.; OLIVER, R.; ELLIS, F. Rationalisation of fractionation in radiotherapy. Br. J. Radiol. 1960, 33, 634–635. [Google Scholar]
- Olive, P.L. Detection of DNA damage in individual cells by analysis of histone H2AX phosphorylation. Methods Cell Biol. 2004, 75, 355–373. [Google Scholar]
- Phillips, T.M.; McBride, W.H.; Pajonk, F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J. Natl. Cancer Inst. 2006, 98, 1777–1785. [Google Scholar]
- Woodward, W.A.; Chen, M.S.; Behbod, F.; Alfaro, M.P.; Buchholz, T.A.; Rosen, J.M. WNT/beta-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc. Natl. Acad. Sci. USA 2007, 104, 618–623. [Google Scholar]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar]
- Che, S.M.; Zhang, X.Z.; Liu, X.L.; Chen, X.; Hou, L. The radiosensitization effect of NS398 on esophageal cancer stem cell-like radioresistant cells. Dis. Esophagus 2010. [Google Scholar] [CrossRef]
- Zhang, X.; Komaki, R.; Wang, L.; Fang, B.; Chang, J.Y. Treatment of radioresistant stem-like esophageal cancer cells by an apoptotic gene-armed, telomerase-specific oncolytic adenovirus. Clin. Cancer Res. 2008, 14, 2813–2823. [Google Scholar]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; Joshua, B.; Kaplan, M.J.; Wapnir, I.; Dirbas, F.M.; Somlo, G.; Garberoglio, C.; Paz, B.; Shen, J.; Lau, S.K.; Quake, S.R.; Brown, J.M.; Weissman, I.L.; Clarke, M.F. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar]
- Vlashi, E.; Kim, K.; Lagadec, C.; Donna, L.D.; McDonald, J.T.; Eghbali, M.; Sayre, J.W.; Stefani, E.; McBride, W.; Pajonk, F. In vivo imaging, tracking, and targeting of cancer stem cells. J. Natl. Cancer Inst. 2009, 101, 350–359. [Google Scholar]
- Arai, F.; Hirao, A.; Ohmura, M.; Sato, H.; Matsuoka, S.; Takubo, K.; Ito, K.; Koh, G.Y.; Suda, T. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell 2004, 118, 149–161. [Google Scholar]
- Wilson, A.; Laurenti, E.; Oser, G.; van der Wath, R.C.; Blanco-Bose, W.; Jaworski, M.; Offner, S.; Dunant, C.F.; Eshkind, L.; Bockamp, E.; Lió, P.; Macdonald, H.R.; Trumpp, A. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 2008, 135, 1118–1129. [Google Scholar]
- Bertout, J.A.; Patel, S.A.; Simon, M.C. The impact of O2 availability on human cancer. Nat. Rev. Cancer 2008, 8, 967–975. [Google Scholar]
- Brizel, D.M.; Scully, S.P.; Harrelson, J.M.; Layfield, L.J.; Bean, J.M.; Prosnitz, L.R.; Dewhirst, M.W. Tumor oxygenation predicts for the likelihood of distant metastases in human soft tissue sarcoma. Cancer Res. 1996, 56, 941–943. [Google Scholar]
- Hockel, M.; Schlenger, K.; Aral, B.; Mitze, M.; Schaffer, U.; Vaupel, P. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res. 1996, 56, 4509–4515. [Google Scholar]
- Nordsmark, M.; Bentzen, S.M.; Rudat, V.; Brizel, D.; Lartigau, E.; Stadler, P.; Becker, A.; Adam, M.; Molls, M.; Dunst, J.; Terris, D.J.; Overgaard, J. Prognostic value of tumor oxygenation in 397 head and neck tumors after primary radiation therapy. An international multi-center study. Radiother. Oncol. 2005, 77, 18–24. [Google Scholar]
- Jang, Y.Y.; Sharkis, S.J. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood 2007, 110, 3056–3063. [Google Scholar]
- Heddleston, J.M.; Li, Z.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009, 8, 3274–3284. [Google Scholar]
- Covello, K.L.; Kehler, J.; Yu, H.; Gordan, J.D.; Arsham, A.M.; Hu, C.J.; Labosky, P.A.; Simon, M.C.; Keith, B. HIF-2alpha regulates Oct-4: Effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006, 20, 557–570. [Google Scholar]
- Gustafsson, M.V.; Zheng, X.; Pereira, T.; Gradin, K.; Jin, S.; Lundkvist, J.; Ruas, J.L.; Poellinger, L.; Lendahl, U.; Bondesson, M. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev. Cell 2005, 9, 617–628. [Google Scholar]
- Eriksen, J.G.; Overgaard, J.; Danish Head and Neck Cancer Study Group (DAHANCA). Lack of prognostic and predictive value of CA IX in radiotherapy of squamous cell carcinoma of the head and neck with known modifiable hypoxia: an evaluation of the DAHANCA 5 study. Radiother. Oncol. 2007, 83, 383–388. [Google Scholar]
- Pawlik, T.M.; Keyomarsi, K. Role of cell cycle in mediating sensitivity to radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 928–942. [Google Scholar]
- Withers, H.R. Cell cycle redistribution as a factor in multifraction irradiation. Radiology 1975, 114, 199–202. [Google Scholar]
- Shima, H.; Takubo, K.; Tago, N.; Iwasaki, H.; Arai, F.; Takahashi, T.; Suda, T. Acquisition of G(0) state by CD34-positive cord blood cells after bone marrow transplantation. Exp. Hematol. 2010, 38, 1231–1240. [Google Scholar]
- Going, J.J.; Keith, W.N.; Neilson, L.; Stoeber, K.; Stuart, R.C.; Williams, G.H. Aberrant expression of minichromosome maintenance proteins 2 and 5, and Ki-67 in dysplastic squamous oesophageal epithelium and Barrett's mucosa. Gut 2002, 50, 373–377. [Google Scholar]
- Baum, R.P.; Swietaszczyk, C.; Prasad, V. FDG-PET/CT in lung cancer: an update. Front Radiat. Ther. Oncol. 2010, 42, 15–45. [Google Scholar]
- Chuang, H.H.; Macapinlac, H.A. The evolving role of PET-CT in the management of esophageal cancer. J. Nucl. Med. Mol. Imaging 2009, 53, 201–209. [Google Scholar]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar]
- Chang, J.Y.; Cox, J.D. Improving radiation conformality in the treatment of non-small cell lung cancer. Semin. Radiat. Oncol. 2010, 20, 171–177. [Google Scholar]
- Durante, M.; Loeffler, J.S. Charged particles in radiation oncology. Nat. Rev. Clin. Oncol. 2010, 7, 37–43. [Google Scholar]
- Olsen, D.R.; Bruland, O.S.; Frykholm, G.; Norderhaug, I.N. Proton therapy—A systematic review of clinical effectiveness. Radiother. Oncol. 2007, 83, 123–132. [Google Scholar]
- Chang, J.Y.; Zhang, X.; Gillin, M.; Fang, F.; Mohan, R. Proton therapy targets cancer stem cells in treatment-resistant non-small cell lung cancer. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, S644. [Google Scholar]
- Korkaya, H.; Paulson, A.; Iovino, F.; Wicha, M.S. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene 2008, 27, 6120–6130. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nguyen, G.H.; Murph, M.M.; Chang, J.Y. Cancer Stem Cell Radioresistance and Enrichment: Where Frontline Radiation Therapy May Fail in Lung and Esophageal Cancers. Cancers 2011, 3, 1232-1252. https://doi.org/10.3390/cancers3011232
Nguyen GH, Murph MM, Chang JY. Cancer Stem Cell Radioresistance and Enrichment: Where Frontline Radiation Therapy May Fail in Lung and Esophageal Cancers. Cancers. 2011; 3(1):1232-1252. https://doi.org/10.3390/cancers3011232
Chicago/Turabian StyleNguyen, Giang Huong, Mandi M. Murph, and Joe Y. Chang. 2011. "Cancer Stem Cell Radioresistance and Enrichment: Where Frontline Radiation Therapy May Fail in Lung and Esophageal Cancers" Cancers 3, no. 1: 1232-1252. https://doi.org/10.3390/cancers3011232