
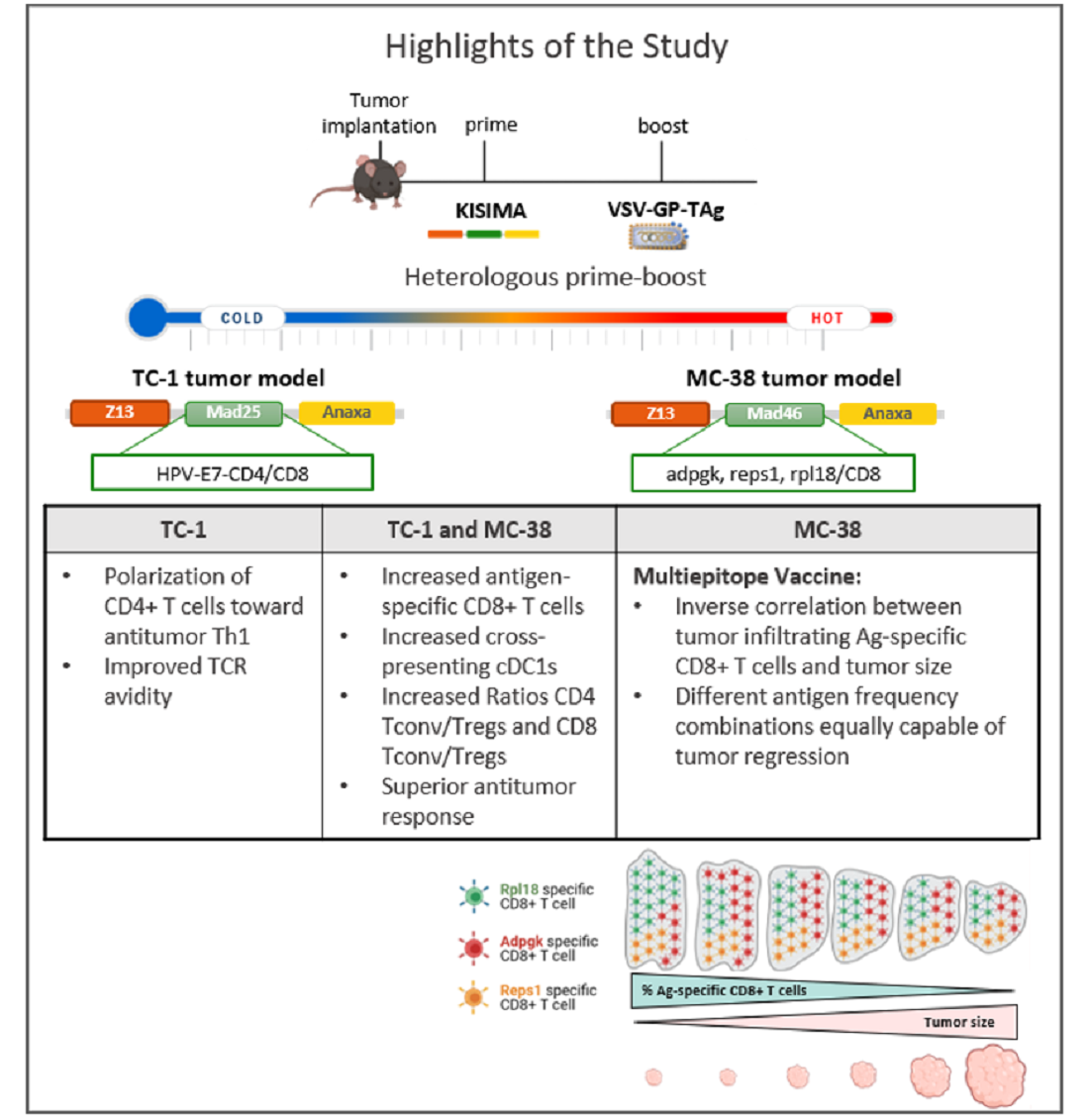
Heterologous Prime-Boost Vaccination with a Peptide-Based Vaccine and Viral Vector Reshapes Dendritic Cell, CD4+ and CD8+ T Cell Phenotypes to Improve the Antitumor Therapeutic Effect


 ,
,
Abstract
:Simple Summary
Abstract
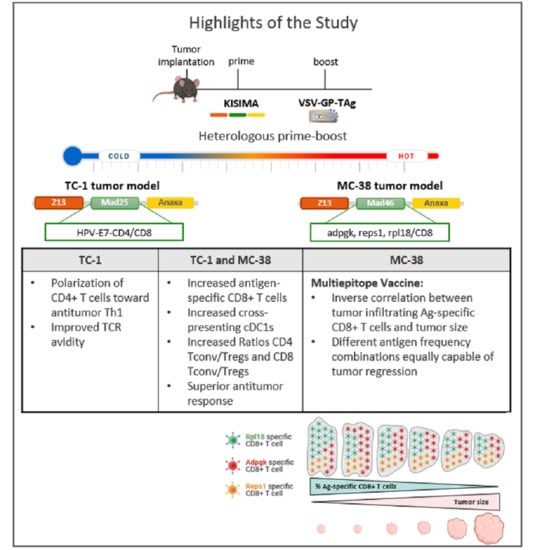
1. Introduction
2. Materials and Methods
2.1. Ethics Approval
2.2. Mice
2.3. Tumor Implantation
2.4. Generation of Vaccines
2.5. Vaccination
2.6. Tissue Processing
2.6.1. Spleen
2.6.2. Lymph Nodes
2.6.3. Tumor Tissue
2.6.4. Blood
2.6.5. Bone Marrow
2.7. FACS Analysis
2.8. TCR Avidity Assay
2.9. Cytokine Quantification
2.10. Data Analysis
3. Results
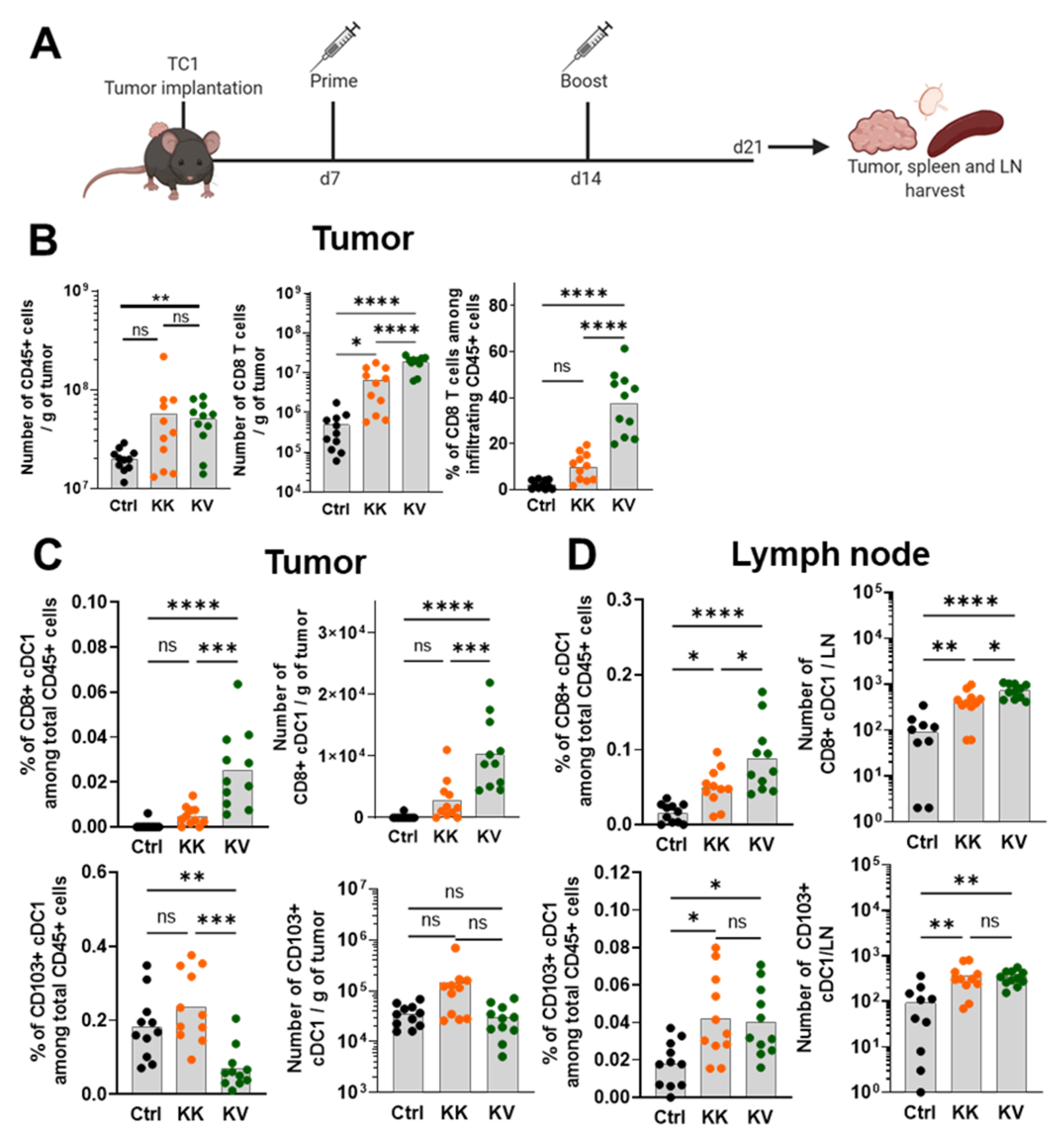
3.1. Heterologous Prime-Boost Increases the Infiltration of Cross-Presenting cDC1s in Tumor and Tumor-dLNs
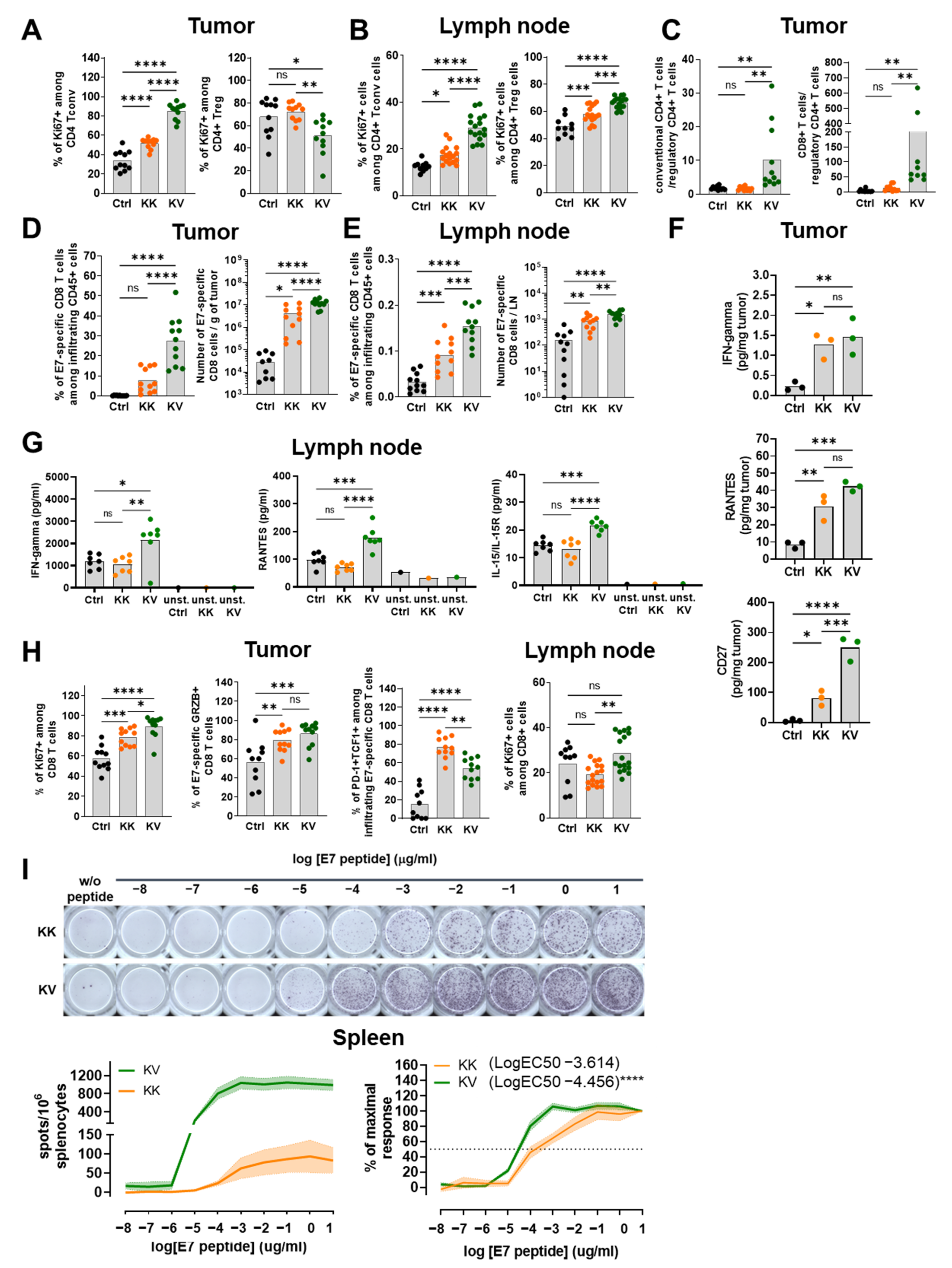
3.2. KV Induces a Th1-Biased T-Helper Cell Profile
3.3. KV Elicited CD8+ T Cells Exhibit Superior Treatment Efficacy Not Only via Increased Quantities but Also Improved Quality
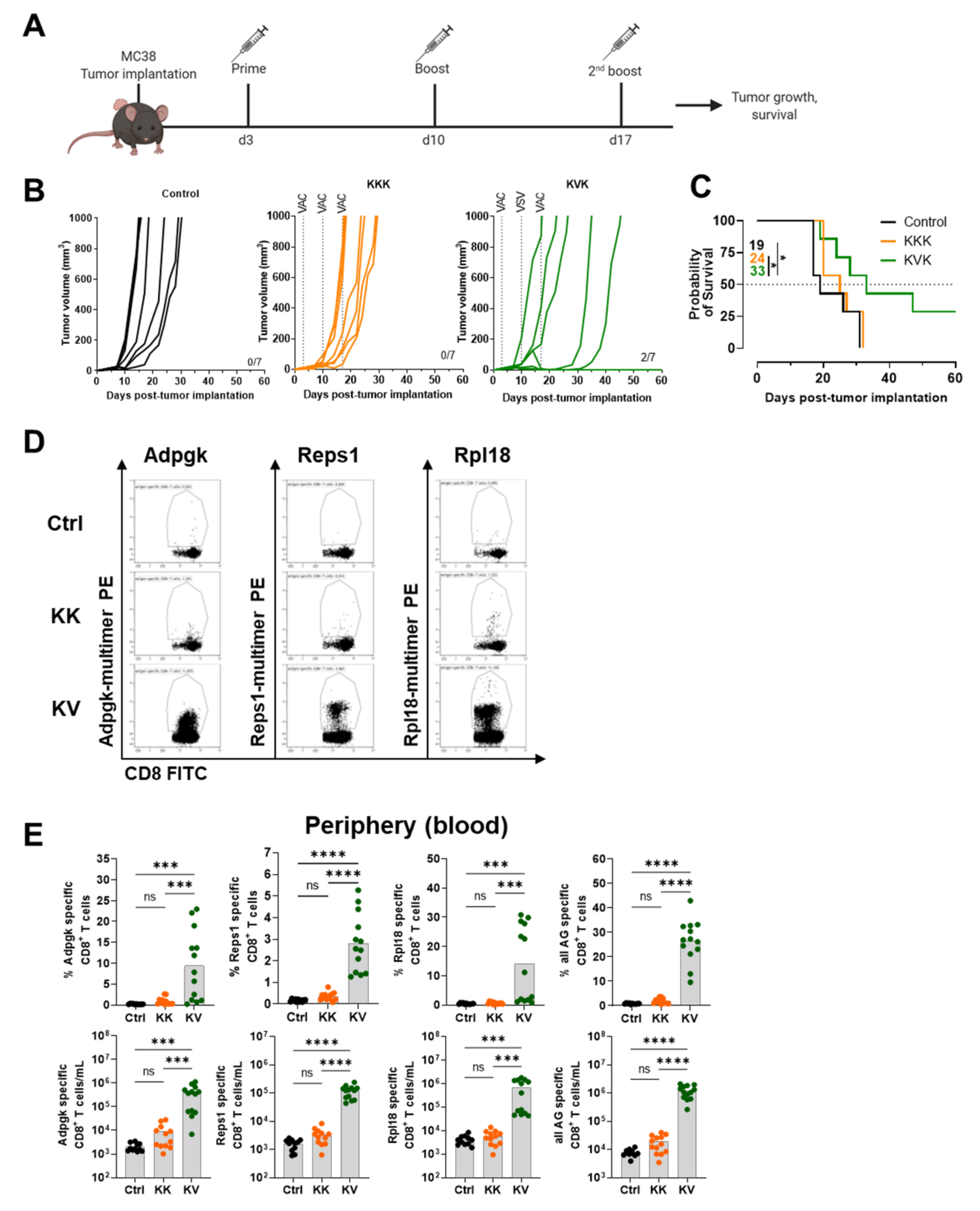
3.4. KVK-Mad46 Prolongs Overall Survival and Induces Long-Term Remission in the MC-38 Tumor Model
3.5. KV Increases the Migration of Antigen-Specific CD8+ T Cells into the Tumor and Tumor-dLNs of MC-38 Tumor-Bearing Mice
3.6. KV Reduces Tumor-Infiltrating CD4+ Tregs Whilst Increasing CD8+ cDC1s and pDCs in Tumor-dLNs
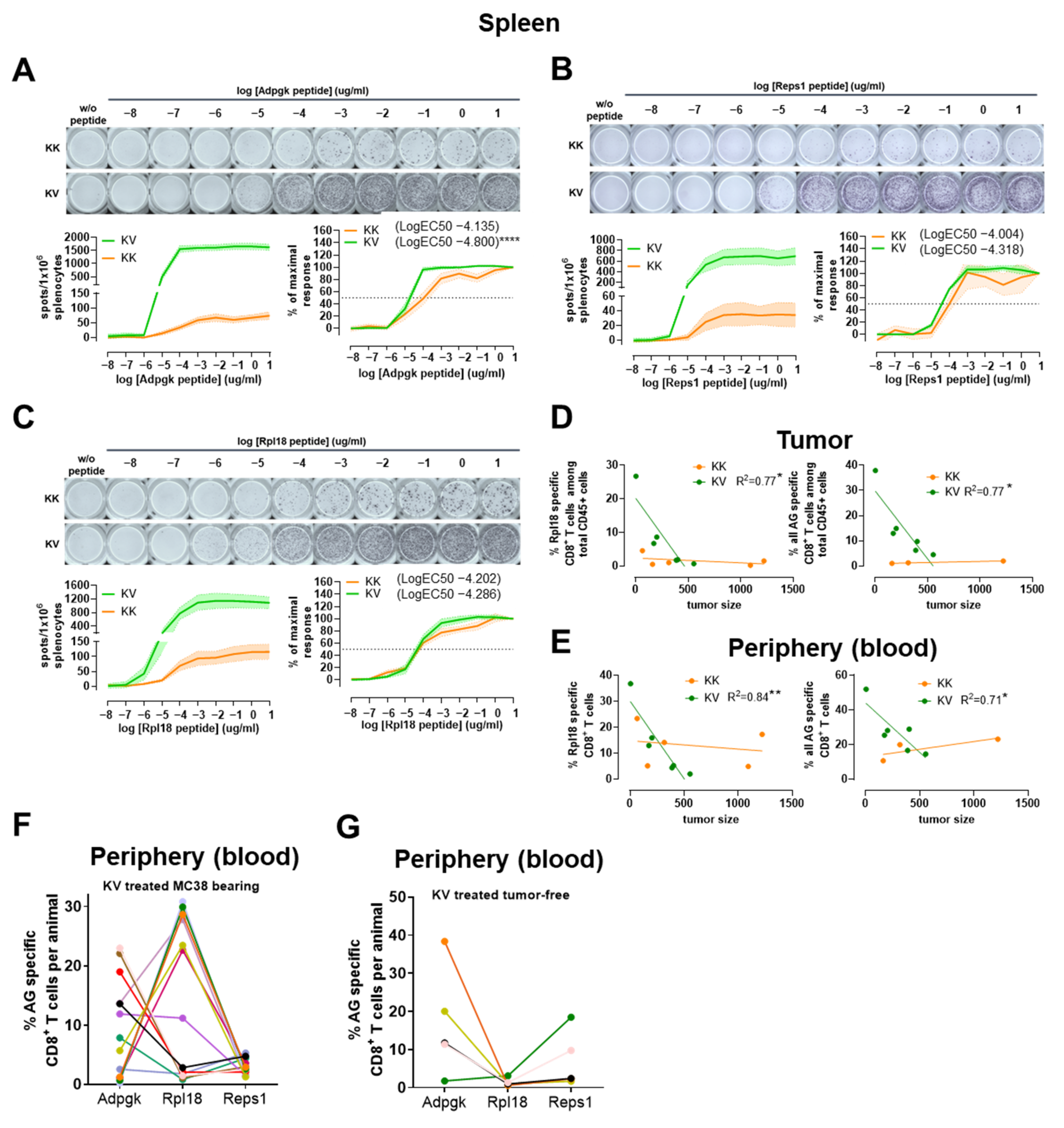
3.7. Polyfunctionality, Exhaustion Status and TCR Avidity Differ between Adpgk-, Rpl18- and Reps1-Specific CD8+ T Cells
3.8. The Presence of MC-38 Tumor Impacts the Relationship between Adpgk-, Rpl18- and Reps1-specific CD8+ T-Cell Frequencies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Derouazi, M.; Berardino-Besson, W.D.; Belnoue, E.; Hoepner, S.; Walther, R.; Benkhoucha, M.; Teta, P.; Dufour, Y.; Maroun, C.Y.; Salazar, A.M.; et al. Novel Cell-Penetrating Peptide-Based Vaccine Induces Robust CD4+ and CD8+ T Cell-Mediated Antitumor Immunity. Cancer Res. 2015, 75, 3020–3031. [Google Scholar] [CrossRef] [Green Version]
- Belnoue, E.; Mayol, J.F.; Carboni, S.; Besson, W.D.B.; Dupuychaffray, E.; Nelde, A.; Stevanovic, S.; Santiago-Raber, M.L.; Walker, P.R.; Derouazi, M. Targeting Self and Neo-Epitopes with a Modular Self-Adjuvanting Cancer Vaccine. JCI Insight 2019, 5, e127305. [Google Scholar] [CrossRef] [Green Version]
- Vasconcelos, J.R.; Dominguez, M.R.; Araújo, A.F.; Ersching, J.; Tararam, C.A.; Bruna-Romero, O.; Rodrigues, M.M. Relevance of Long-Lived CD8+ T Effector Memory Cells for Protective Immunity Elicited by Heterologous Prime-Boost Vaccination. Front. Immunol. 2012, 3, 358. [Google Scholar] [CrossRef] [Green Version]
- Jou, J.; Harrington, K.J.; Zocca, M.-B.; Ehrnrooth, E.; Cohen, E.E.W. The Changing Landscape of Therapeutic Cancer Vaccines—Novel Platforms and Neoantigen Identification. Clin. Cancer Res. 2021, 27, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Muik, A.; Stubbert, L.J.; Jahedi, R.Z.; Geibeta, Y.; Kimpel, J.; Dold, C.; Tober, R.; Volk, A.; Klein, S.; Dietrich, U.; et al. Re-Engineering Vesicular Stomatitis Virus to Abrogate Neurotoxicity, Circumvent Humoral Immunity, and Enhance Oncolytic Potency. Cancer Res. 2014, 74, 3567–3578. [Google Scholar] [CrossRef] [Green Version]
- Tober, R.; Banki, Z.; Egerer, L.; Muik, A.; Behmuller, S.; Kreppel, F.; Greczmiel, U.; Oxenius, A.; von Laer, D.; Kimpel, J. VSV-GP: A Potent Viral Vaccine Vector That Boosts the Immune Response upon Repeated Applications. J. Virol. 2014, 88, 4897–4907. [Google Scholar] [CrossRef] [Green Version]
- Wilmschen, S.; Schneider, S.; Peters, F.; Bayer, L.; Issmail, L.; Banki, Z.; Grunwald, T.; von Laer, D.; Kimpel, J. RSV Vaccine Based on Rhabdoviral Vector Protects after Single Immunization. Vaccines 2019, 7, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bresk, C.A.; Hofer, T.; Wilmschen, S.; Krismer, M.; Beierfuß, A.; Effantin, G.; Weissenhorn, W.; Hogan, M.J.; Jordan, A.P.O.; Gelman, R.S.; et al. Induction of Tier 1 HIV Neutralizing Antibodies by Envelope Trimers Incorporated into a Replication Competent Vesicular Stomatitis Virus Vector. Viruses 2019, 11, 159. [Google Scholar] [CrossRef] [Green Version]
- Huttner, A.; Dayer, J.-A.; Yerly, S.; Combescure, C.; Auderset, F.; Desmeules, J.; Eickmann, M.; Finckh, A.; Goncalves, A.R.; Hooper, J.W.; et al. The Effect of Dose on the Safety and Immunogenicity of the VSV Ebola Candidate Vaccine: A Randomised Double-Blind, Placebo-Controlled Phase 1/2 Trial. Lancet Infect. Dis. 2015, 15, 1156–1166. [Google Scholar] [CrossRef]
- Schreiber, L.M.; Urbiola, C.; Das, K.; Spiesschaert, B.; Kimpel, J.; Heinemann, F.; Stierstorfer, B.; Muller, P.; Petersson, M.; Erlmann, P.; et al. The Lytic Activity of VSV-GP Treatment Dominates the Therapeutic Effects in a Syngeneic Model of Lung Cancer. Br. J. Cancer 2019, 121, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Stojdl, D.F.; Lichty, B.D.; ten Oever, B.R.; Paterson, J.M.; Power, A.T.; Knowles, S.; Marius, R.; Reynard, J.; Poliquin, L.; Atkins, H.; et al. VSV Strains with Defects in Their Ability to Shutdown Innate Immunity Are Potent Systemic Anti-Cancer Agents. Cancer Cell 2003, 4, 263–275. [Google Scholar] [CrossRef] [Green Version]
- Das, K.; Belnoue, E.; Rossi, M.; Hofer, T.; Danklmaier, S.; Nolden, T.; Schreiber, L.-M.; Angerer, K.; Kimpel, J.; Hoegler, S.; et al. A Modular Self-Adjuvanting Cancer Vaccine Combined with an Oncolytic Vaccine Induces Potent Antitumor Immunity. Nat. Commun. 2021, 12, 5195. [Google Scholar] [CrossRef]
- Yadav, M.; Jhunjhunwala, S.; Phung, Q.T.; Lupardus, P.; Tanguay, J.; Bumbaca, S.; Franci, C.; Cheung, T.K.; Fritsche, J.; Weinschenk, T.; et al. Predicting Immunogenic Tumour Mutations by Combining Mass Spectrometry and Exome Sequencing. Nature 2014, 515, 572–576. [Google Scholar] [CrossRef]
- Hos, B.J.; Camps, M.G.M.; van den Bulk, J.; Tondini, E.; van den Ende, T.C.; Ruano, D.; Franken, K.; Janssen, G.M.C.; Ru, A.; Filippov, D.V.; et al. Identification of a Neo-Epitope Dominating Endogenous CD8 T Cell Responses to MC-38 Colorectal Cancer. Oncoimmunology 2019, 9, 1673125. [Google Scholar] [CrossRef] [Green Version]
- Santiago-Raber, M.-L.; Baccala, R.; Haraldsson, K.M.; Choubey, D.; Stewart, T.A.; Kono, D.H.; Theofilopoulos, A.N. Type-I Interferon Receptor Deficiency Reduces Lupus-like Disease in NZB Mice. J. Exp. Med. 2003, 197, 777–788. [Google Scholar] [CrossRef] [Green Version]
- Roberts, E.W.; Broz, M.L.; Binnewies, M.; Headley, M.B.; Nelson, A.E.; Wolf, D.M.; Kaisho, T.; Bogunovic, D.; Bhardwaj, N.; Krummel, M.F. Critical Role for CD103+/CD141+ Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 2016, 30, 324–336. [Google Scholar] [CrossRef] [Green Version]
- Borst, J.; Ahrends, T.; Bąbała, N.; Melief, C.J.M.; Kastenmüller, W. CD4+ T Cell Help in Cancer Immunology and Immunotherapy. Nat. Rev. Immunol. 2018, 18, 635–647. [Google Scholar] [CrossRef]
- Colonna, M.; Krug, A.; Cella, M. Interferon-Producing Cells: On the Front Line in Immune Responses against Pathogens. Curr. Opin. Immunol. 2002, 14, 373–379. [Google Scholar] [CrossRef]
- McKenna, K.; Beignon, A.-S.; Bhardwaj, N. Plasmacytoid Dendritic Cells: Linking Innate and Adaptive Immunity. J. Virol. 2005, 79, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic Cells in Cancer Immunology and Immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, H.; Zhao, J. The Role of CD4 T Cell Help for CD8 CTL Activation. Biochem. Biophys. Res. Commun. 2009, 384, 405–408. [Google Scholar] [CrossRef]
- Ostroumov, D.; Fekete-Drimusz, N.; Saborowski, M.; Kühnel, F.; Woller, N. CD4 and CD8 T Lymphocyte Interplay in Controlling Tumor Growth. Cell. Mol. Life Sci. 2018, 75, 689–713. [Google Scholar] [CrossRef] [Green Version]
- Weng, N.-P.; Liu, K.; Catalfamo, M.; Li, Y.; Henkart, P.A. IL-15 Is a Growth Factor and an Activator of CD8 Memory T Cells. Ann. N. Y. Acad. Sci. 2002, 975, 46–56. [Google Scholar] [CrossRef]
- Van de Ven, K.; Borst, J. Targeting the T-Cell Co-Stimulatory CD27/CD70 Pathway in Cancer Immunotherapy: Rationale and Potential. Immunotherapy 2015, 7, 655–667. [Google Scholar] [CrossRef] [Green Version]
- Peperzak, V.; Veraar, E.A.M.; Keller, A.M.; Xiao, Y.; Borst, J. The Pim Kinase Pathway Contributes to Survival Signaling in Primed CD8+ T Cells upon CD27 Costimulation. J. Immunol. 2010, 185, 6670–6678. [Google Scholar] [CrossRef] [Green Version]
- Zhong, W.; Myers, J.S.; Wang, F.; Wang, K.; Lucas, J.; Rosfjord, E.; Lucas, J.; Hooper, A.T.; Yang, S.; Lemon, L.A.; et al. Comparison of the Molecular and Cellular Phenotypes of Common Mouse Syngeneic Models with Human Tumors. BMC Genom. 2020, 21, 2. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.H.; Wang, T.L.; Hung, C.F.; Pardoll, D.M.; Wu, T.C. Boosting with Recombinant Vaccinia Increases HPV-16 E7-Specific T Cell Precursor Frequencies of HPV-16 E7-Expressing DNA Vaccines. Vaccine 2000, 18, 2015–2022. [Google Scholar] [CrossRef]
- Hallermalm, K.; Johansson, S.; Brave, A.; Ek, M.; Engstrom, G.; Boberg, A.; Gudmundsdotter, L.; Blomberg, P.; Mellstedt, H.; Stout, R.; et al. Pre-Clinical Evaluation of a CEA DNA Prime/Protein Boost Vaccination Strategy against Colorectal Cancer. Scand. J. Immunol. 2007, 66, 43–51. [Google Scholar] [CrossRef]
- Pol, J.G.; Zhang, L.; Bridle, B.W.; Stephenson, K.B.; Resseguier, J.; Hanson, S.; Chen, L.; Kazdhan, N.; Bramson, J.L.; Stojdl, D.F.; et al. Maraba Virus as a Potent Oncolytic Vaccine Vector. Mol. Ther. 2014, 22, 420–429. [Google Scholar] [CrossRef] [Green Version]
- Bonilla, W.V.; Kirchhammer, N.; Marx, A.-F.; Kallert, S.M.; Krzyzaniak, M.A.; Lu, M.; Darbre, S.; Schmidt, S.; Raguz, J.; Berka, U.; et al. Heterologous Arenavirus Vector Prime-Boost Overrules Self-Tolerance for Efficient Tumor-Specific CD8 T Cell Attack. Cell Rep. Med. 2021, 2, 100209. [Google Scholar] [CrossRef] [PubMed]
- Zander, R.; Schauder, D.; Xin, G.; Nguyen, C.; Wu, X.; Zajac, A.; Cui, W. CD4+ T Cell Help Is Required for the Formation of a Cytolytic CD8+ T Cell Subset That Protects against Chronic Infection and Cancer. Immunity 2019, 51, 1028-1042.e4. [Google Scholar] [CrossRef]
- Galluzzi, L.; Chan, T.A.; Kroemer, G.; Wolchok, J.D.; López-Soto, A. The Hallmarks of Successful Anticancer Immunotherapy. Sci. Transl. Med. 2018, 10, eaat7807. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage Plasticity and Polarization: In Vivo Veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Roychoudhuri, R.; Eil, R.L.; Restifo, N.P. The Interplay of Effector and Regulatory T Cells in Cancer. Curr. Opin. Immunol. 2015, 33, 101–111. [Google Scholar] [CrossRef]
- Saleh, R.; Elkord, E. FoxP3+ T Regulatory Cells in Cancer: Prognostic Biomarkers and Therapeutic Targets. Cancer Lett. 2020, 490, 174–185. [Google Scholar] [CrossRef]
- Ferris, S.T.; Durai, V.; Wu, R.; Theisen, D.J.; Ward, J.P.; Bern, M.D.; Davidson, J.T.; Bagadia, P.; Liu, T.; Briseño, C.G.; et al. CDC1 Prime and Are Licensed by CD4+ T Cells to Induce Anti-Tumour Immunity. Nature 2020, 584, 624–629. [Google Scholar] [CrossRef]
- Hildner, K.; Edelson, B.T.; Purtha, W.E.; Diamond, M.; Matsushita, H.; Kohyama, M.; Calderon, B.; Schraml, B.U.; Unanue, E.R.; Diamond, M.S.; et al. Batf3 Deficiency Reveals a Critical Role for CD8a+ Dendritic Cells in Cytotoxic T Cell Immunity. Science 2014, 322, 1097–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103+ Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef] [Green Version]
- Binnewies, M.; Mujal, A.M.; Pollack, J.L.; Combes, A.J.; Hardison, E.A.; Barry, K.C.; Tsui, J.; Ruhland, M.K.; Kersten, K.; Abushawish, M.A.; et al. Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4+ T Cell Immunity. Cell 2019, 177, 556–571.e16. [Google Scholar] [CrossRef]
- Heath, W.R.; Belz, G.T.; Behrens, G.M.N.; Smith, C.M.; Forehan, S.P.; Parish, I.A.; Davey, G.M.; Wilson, N.S.; Carbone, F.R.; Villadangos, J.A. Cross-Presentation, Dendritic Cell Subsets, and the Generation of Immunity to Cellular Antigens. Immunol. Rev. 2004, 199, 9–26. [Google Scholar] [CrossRef]
- Pinto, A.; Rega, A.; Crother, T.R.; Sorrentino, R. Plasmacytoid Dendritic Cells and Their Therapeutic Activity in Cancer. Oncoimmunology 2012, 1, 726–734. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, S.; Yang, J.; Ronchese, F. Monocyte-Derived Dendritic Cells Are Essential for CD8+ T Cell Activation and Antitumor Responses After Local Immunotherapy. Front. Immunol. 2015, 6, 584. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Cuss, S.M.; Singh, V.; Gurusamy, D.; Shoe, J.L.; Leighty, R.; Bronte, V.; Hurwitz, A.A. CD4+ T Cell Help Selectively Enhances High-Avidity Tumor Antigen-Specific CD8+ T Cells. J. Immunol. 2015, 195, 3482–3489. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-T.; Chang, T.-C.; Shaw, S.-W.; Cheng, P.-J.; Huang, C.-T.; Chao, A.; Soong, Y.-K.; Lai, C.-H. Maintenance of CD8 Effector T Cells by CD4 Helper T Cells Eradicates Growing Tumors and Promotes Long-Term Tumor Immunity. Vaccine 2006, 24, 6199–6207. [Google Scholar] [CrossRef]
- Greene, J.M.; Wiseman, R.W.; Lank, S.M.; Bimber, B.N.; Karl, J.A.; Burwitz, B.J.; Lhost, J.J.; Hawkins, O.E.; Kunstman, K.J.; Broman, K.W.; et al. Differential MHC Class I Expression in Distinct Leukocyte Subsets. BMC Immunol. 2011, 12, 39. [Google Scholar] [CrossRef] [Green Version]
- Bucks, C.M.; Norton, J.A.; Boesteanu, A.C.; Mueller, Y.M.; Katsikis, P.D. Chronic Antigen Stimulation Alone Is Sufficient to Drive CD8+ T Cell Exhaustion. J. Immunol. 2009, 182, 6697–6708. [Google Scholar] [CrossRef] [Green Version]
- Burger, M.L.; Cruz, A.M.; Crossland, G.E.; Gaglia, G.; Ritch, C.C.; Blatt, S.E.; Bhutkar, A.; Canner, D.; Kienka, T.; Tavana, S.Z.; et al. Antigen Dominance Hierarchies Shape TCF1+ Progenitor CD8 T Cell Phenotypes in Tumors. Cell 2021, 184, 4996-5014.e26. [Google Scholar] [CrossRef]
- Badovinac, V.P.; Haring, J.S.; Harty, J.T. Initial T Cell Receptor Transgenic Cell Precursor Frequency Dictates Critical Aspects of the CD8+ T Cell Response to Infection. Immunity 2007, 26, 827–841. [Google Scholar] [CrossRef] [Green Version]
- Philip, M.; Schietinger, A. CD8+ T Cell Differentiation and Dysfunction in Cancer. Nat. Rev. Immunol. 2021, 21, 1–15. [Google Scholar] [CrossRef]
- Yu, A.I.; Zhao, L.; Eaton, K.A.; Ho, S.; Chen, J.; Poe, S.; Becker, J.; Gonzalez, A.; McKinstry, D.; Hasso, M.; et al. Gut Microbiota Modulate CD8 T Cell Responses to Influence Colitis-Associated Tumorigenesis. Cell Rep. 2020, 31, 107471. [Google Scholar] [CrossRef]
- Kuai, R.; Ochyl, L.J.; Bahjat, K.S.; Schwendeman, A.; Moon, J.J. Designer Vaccine Nanodiscs for Personalized Cancer Immunotherapy. Nat. Mater. 2017, 16, 489–496. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor | Control | KK | KV | p-Value | ||
|---|---|---|---|---|---|---|
| Mean (±SD) | Ctrl vs. KK | Ctrl vs. KV | KK vs. KV | |||
| CD4 (%) | 0.86 (0.79) | 1.38 (0.59) | 2.07 (0.53) | 0.16 | 0.0003 | 0.04 |
| CD4 (×105) | 1.33 (1.04) | 8.67 (12.42) | 10.7 (7.25) | 0.11 | 0.03 | 0.83 |
| Th1 (%) | 14.84 (8.64) | 31.22 (10.59) | 51.62 (7.31) | 0.0005 | <0.0001 | <0.0001 |
| Th1 (×105) | 0.23 (0.28) | 2.89 (3.61) | 5.90 (4.21) | 0.14 | 0.0007 | 0.09 |
| Th2 (%) | 23.95 (15.74) | 21.98 (3.83) | 10.15 (8.12) | 0.74 | <0.0001 | 0.0003 |
| Th2 (×105) | 0.33 (0.26) | 2.01 (2.67) | 1.15 (1.08) | 0.06 | 0.49 | 0.46 |
| Th17 (%) | 5.66 (3.72) | 3.98 (2.16) | 0.63 (0.64) | 0.27 | 0.0002 | 0.01 |
| Th17 (×105) | 0.07 (0.05) | 0.44 (0.89) | 0.07 (0.06) | 0.24 | 1.00 | 0.23 |
| Treg (%) | 36.32 (6.16) | 38.93 (7.83) | 14.88 (8.38) | 0.70 | <0.0001 | <0.0001 |
| Treg (×105) | 0.58 (0.43) | 3.48 (4.43) | 1.78 (1.51) | 0.054 | 0.56 | 0.32 |
| Lymph Nodes | Control | KK | KV | p-Value | ||
| Mean (±SD) | Ctrl vs. KK | Ctrl vs. KV | KK vs. KV | |||
| CD4 (%) | 46.83 (2.94) | 43.64 (4.14) | 43.25 (3.31) | 0.09 | 0.04 | 0.95 |
| CD4 (×105) | 2.42 (1.23) | 1.75 (1.02) | 1.56 (0.61) | 0.20 | 0.07 | 0.82 |
| Th1 (%) | 2.24 (1.68) | 3.30 (1.97) | 6.56 (3.38) | 0.56 | 0.0004 | 0.002 |
| Th1 (×105) | 0.03 (0.04) | 0.06 (0.04) | 0.10 (0.08) | 0.34 | 0.01 | 0.11 |
| Th2 (%) | 2.01 (0.82) | 1.90 (0.68) | 1.16 (0.58) | 0.91 | 0.01 | 0.01 |
| Th2 (×105) | 0.03 (0.02) | 0.04 (0.02) | 0.02 (0.01 | 0.55 | 0.23 | 0.01 |
| Th17 (%) | 4.42 (6.53) | 6.82 (5.64) | 3.48 (2.42) | 0.45 | 0.89 | 0.14 |
| Th17 (×105) | 0.07 (0.13) | 0.13 (0.11) | 0.05 (0.05) | 0.21 | 0.94 | 0.06 |
| Treg (%) | 7.49 (2.93) | 10.23 (4.52) | 11.85 (5.30) | 0.30 | 0.054 | 0.56 |
| Treg (×105) | 0,16 (0.07) | 0,15 (0.05) | 0,16 (0.07) | 0.93 | 0.97 | 0.75 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hofer, T.; Rossi, M.; Carboni, S.; Di Berardino Besson, W.; von Laer, D.; Wollmann, G.; Derouazi, M.; Santiago-Raber, M.-L. Heterologous Prime-Boost Vaccination with a Peptide-Based Vaccine and Viral Vector Reshapes Dendritic Cell, CD4+ and CD8+ T Cell Phenotypes to Improve the Antitumor Therapeutic Effect. Cancers 2021, 13, 6107. https://doi.org/10.3390/cancers13236107
Hofer T, Rossi M, Carboni S, Di Berardino Besson W, von Laer D, Wollmann G, Derouazi M, Santiago-Raber M-L. Heterologous Prime-Boost Vaccination with a Peptide-Based Vaccine and Viral Vector Reshapes Dendritic Cell, CD4+ and CD8+ T Cell Phenotypes to Improve the Antitumor Therapeutic Effect. Cancers. 2021; 13(23):6107. https://doi.org/10.3390/cancers13236107
Chicago/Turabian StyleHofer, Tamara, Matteo Rossi, Susanna Carboni, Wilma Di Berardino Besson, Dorothee von Laer, Guido Wollmann, Madiha Derouazi, and Marie-Laure Santiago-Raber. 2021. "Heterologous Prime-Boost Vaccination with a Peptide-Based Vaccine and Viral Vector Reshapes Dendritic Cell, CD4+ and CD8+ T Cell Phenotypes to Improve the Antitumor Therapeutic Effect" Cancers 13, no. 23: 6107. https://doi.org/10.3390/cancers13236107