NO1, a New Sigma 2 Receptor/TMEM97 Fluorescent Ligand, Downregulates SOCE and Promotes Apoptosis in the Triple Negative Breast Cancer Cell Lines

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
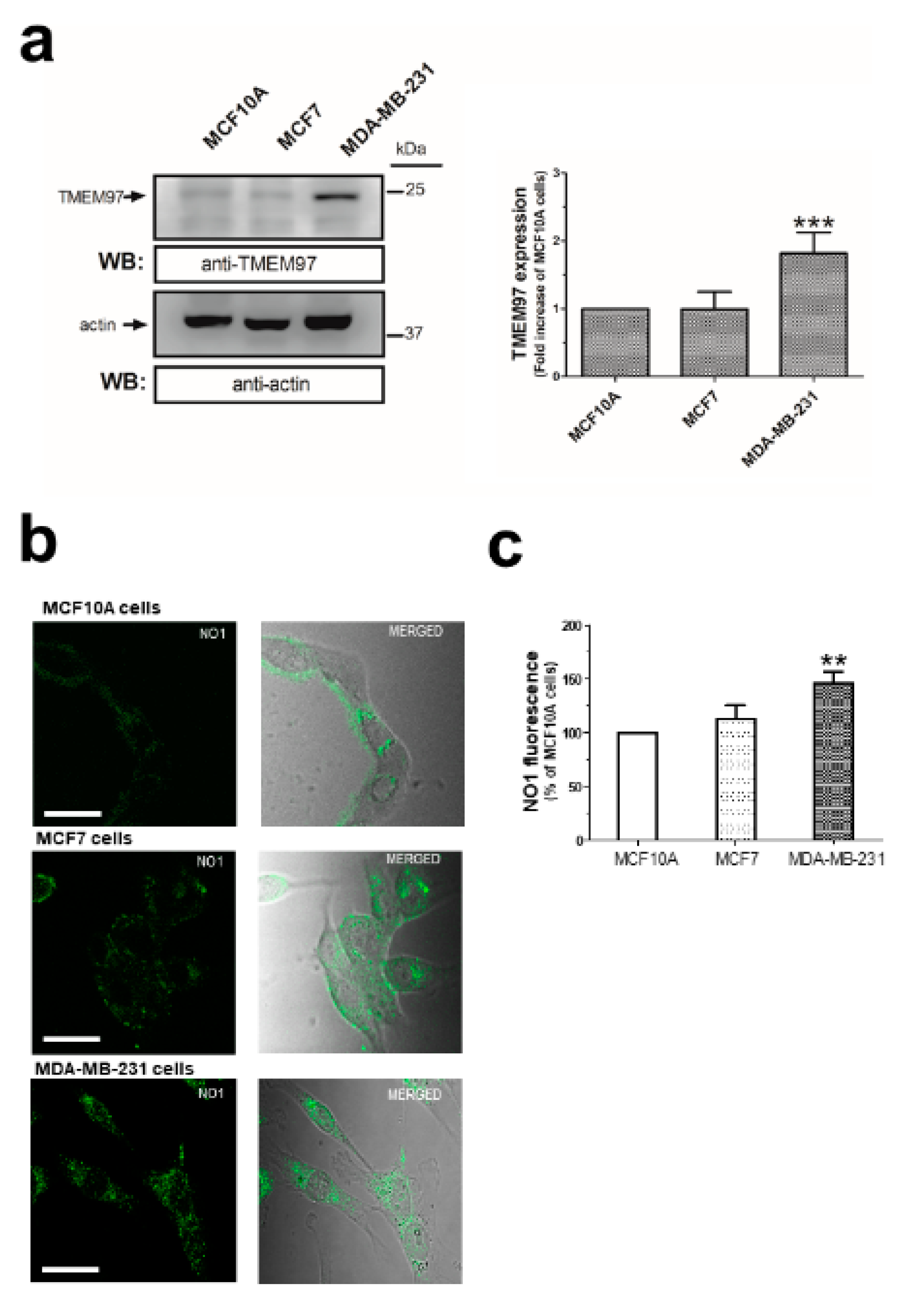
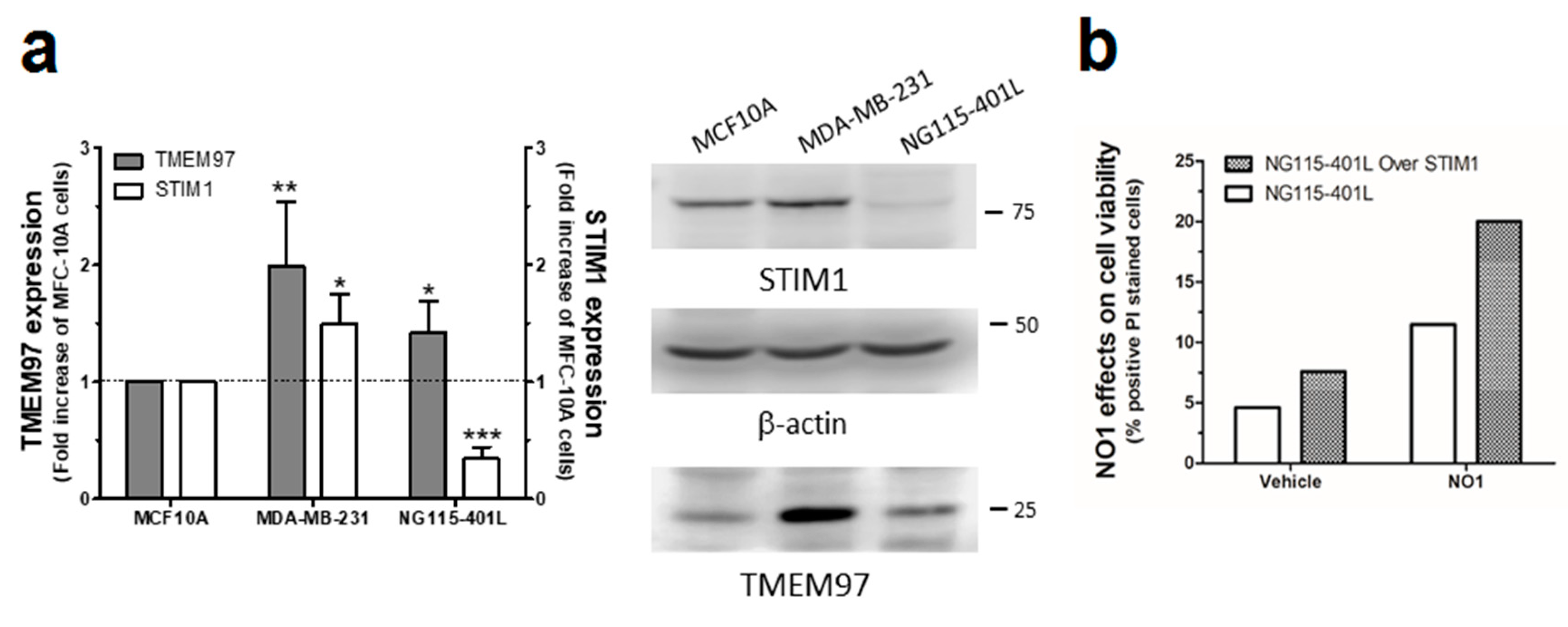
2.1. σ2R/TMEM97 Is Overexpressed in Breast Cancer Cell Lines
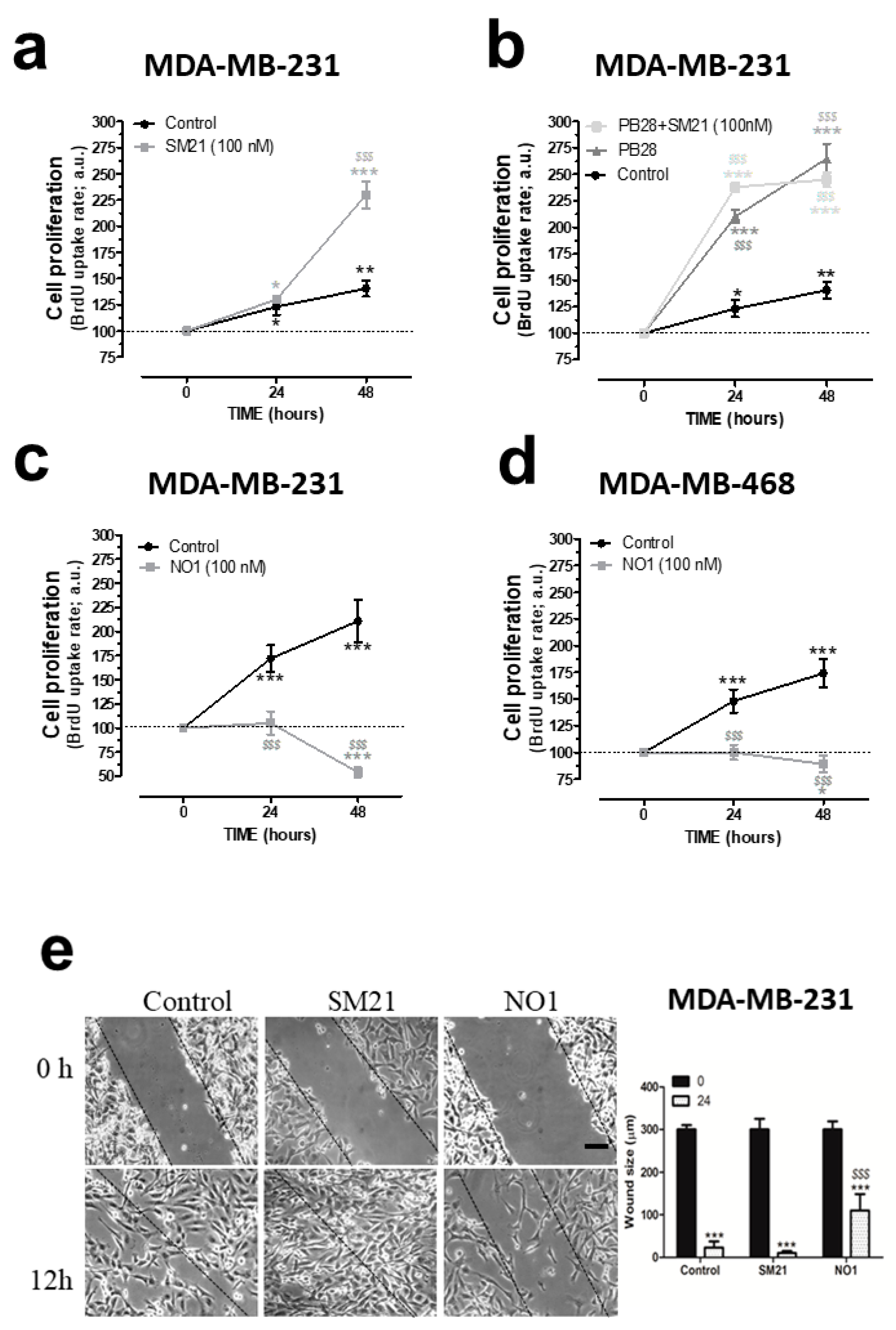
2.2. σ2R/TMEM97 Ligands Alter TNBC Cell Proliferation and Migration
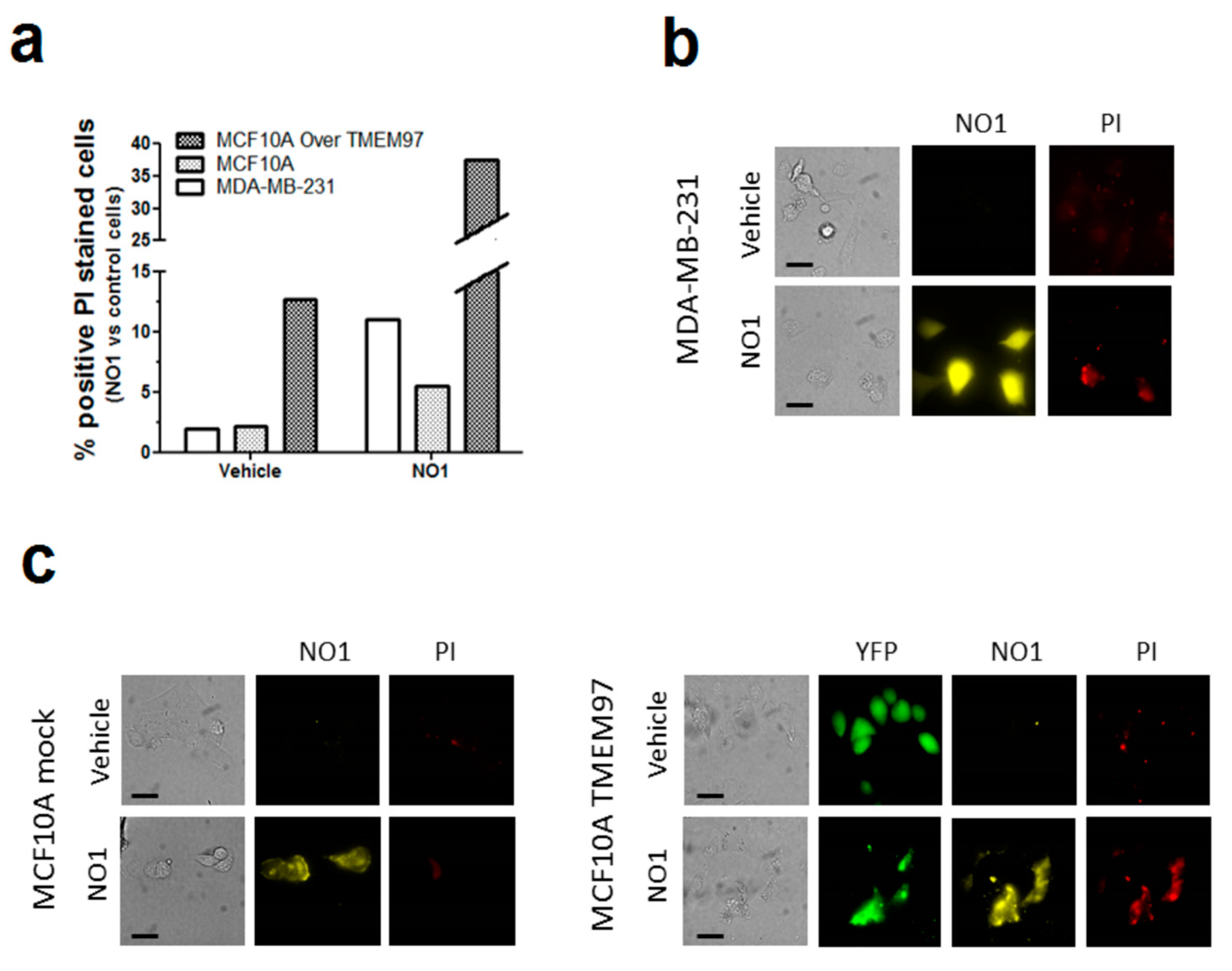
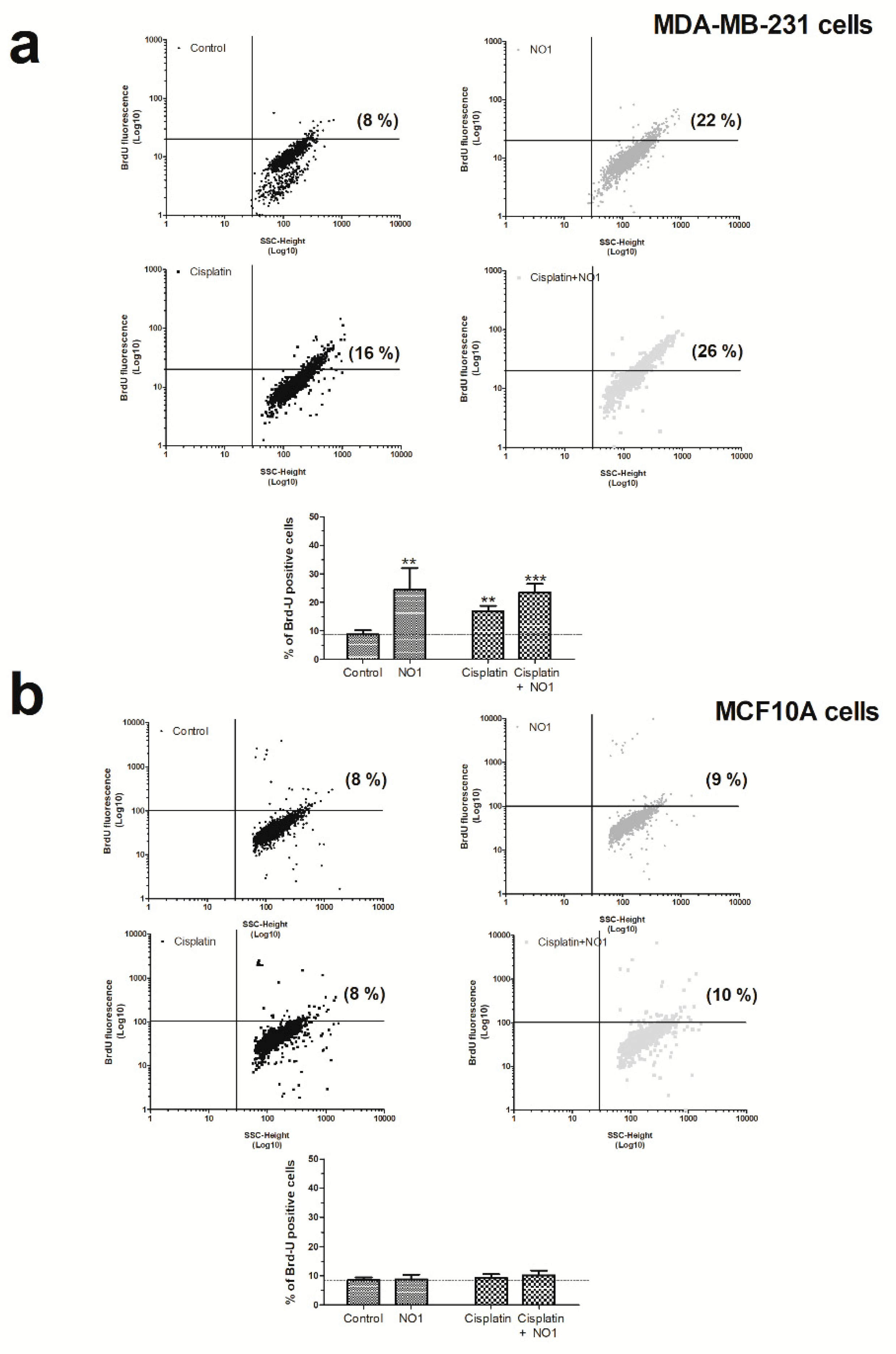
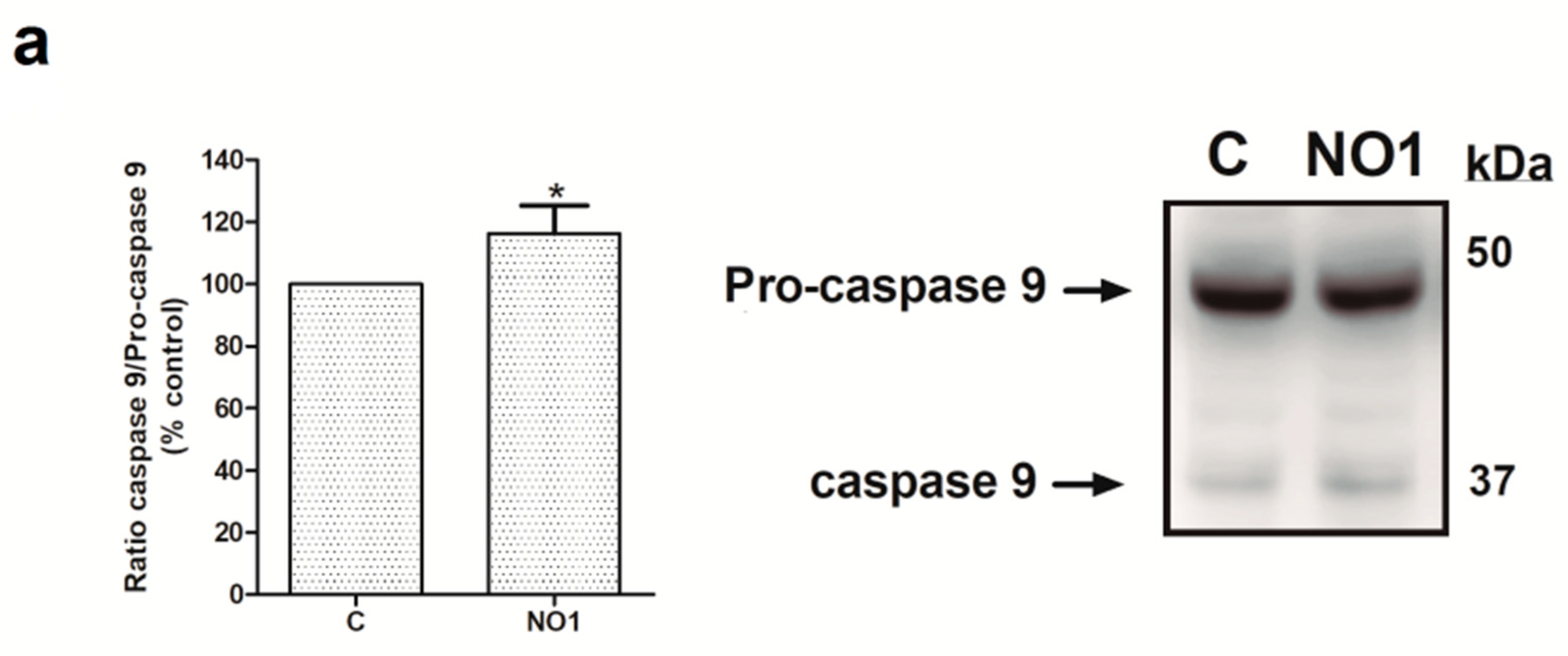
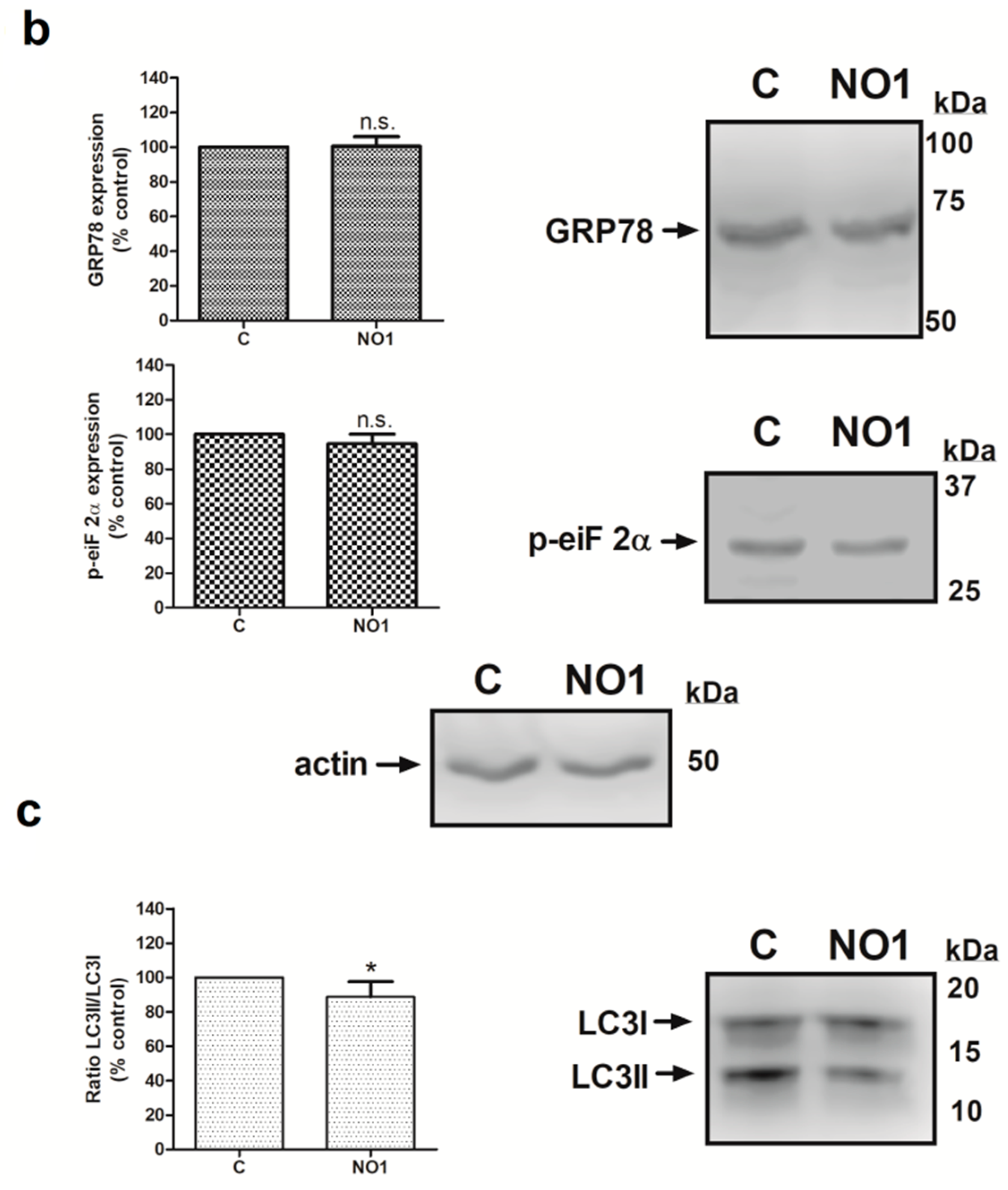
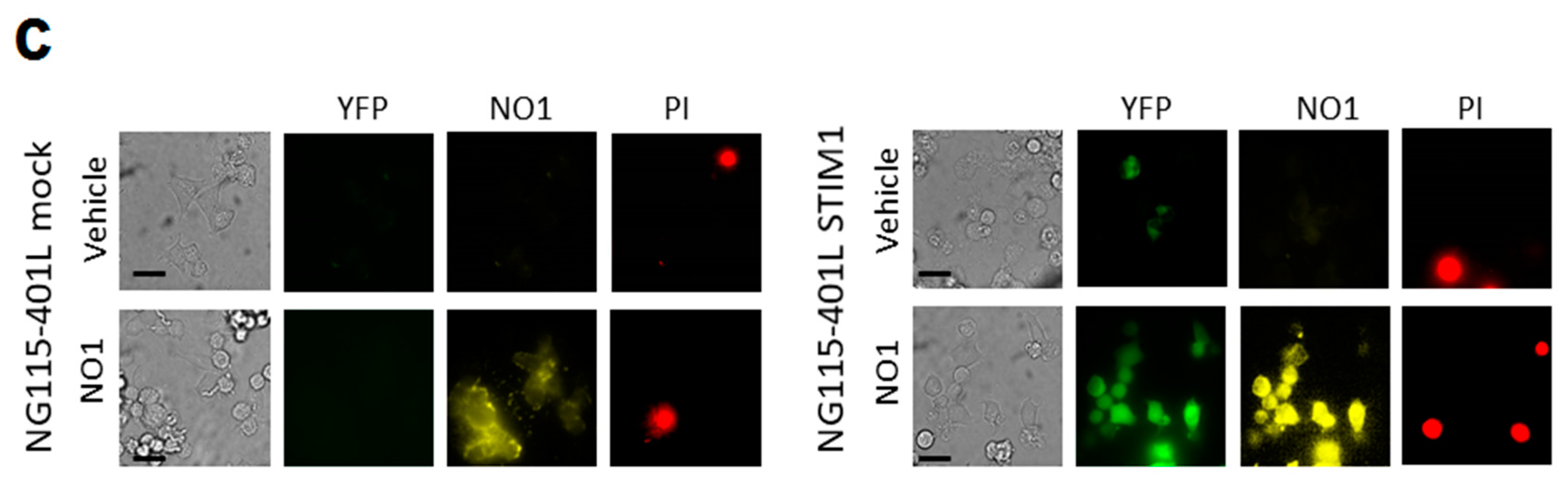
2.3. NO1 Promotes Cell Death and Apoptosis in MDA-MB-231 Cells
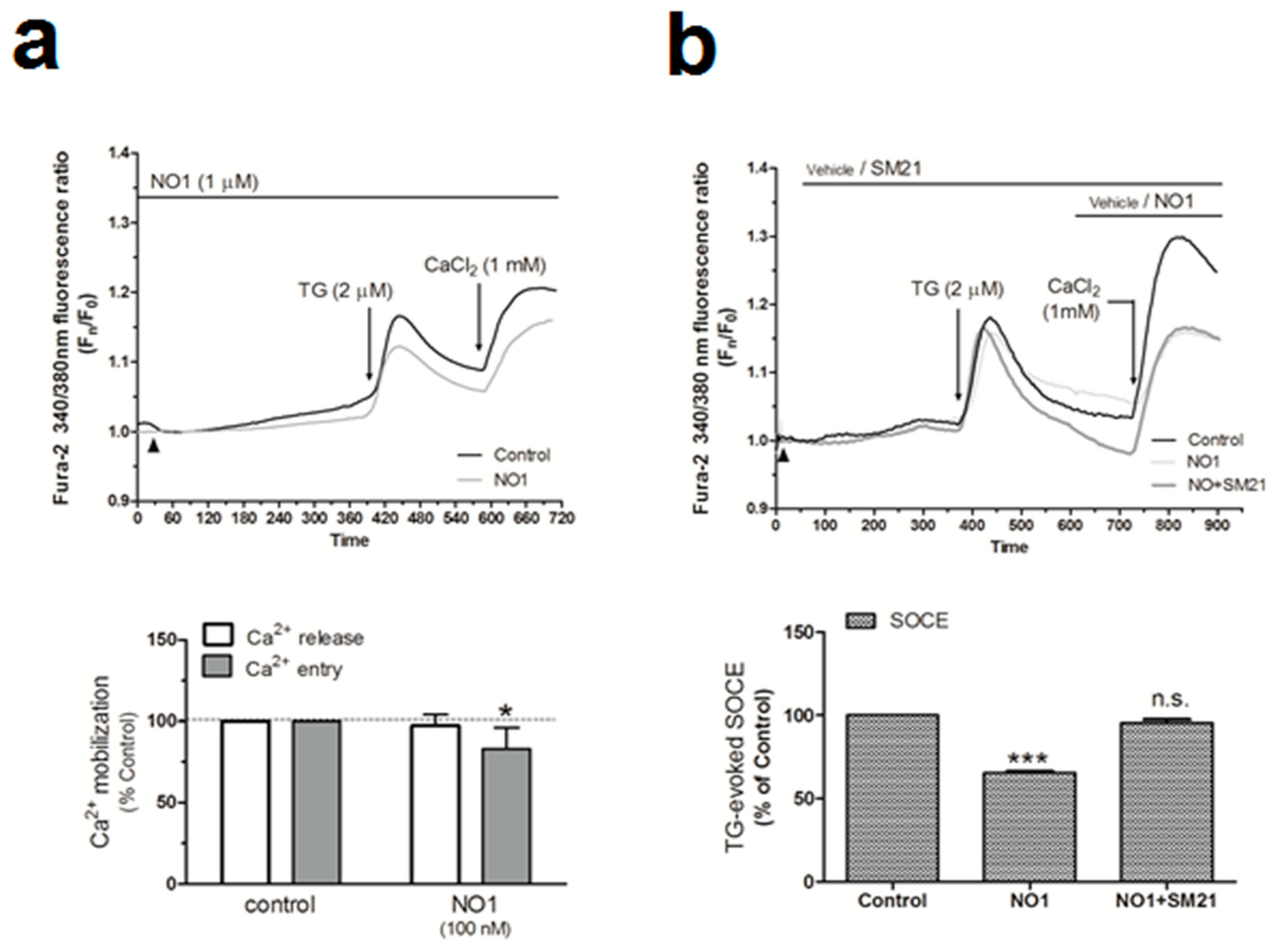
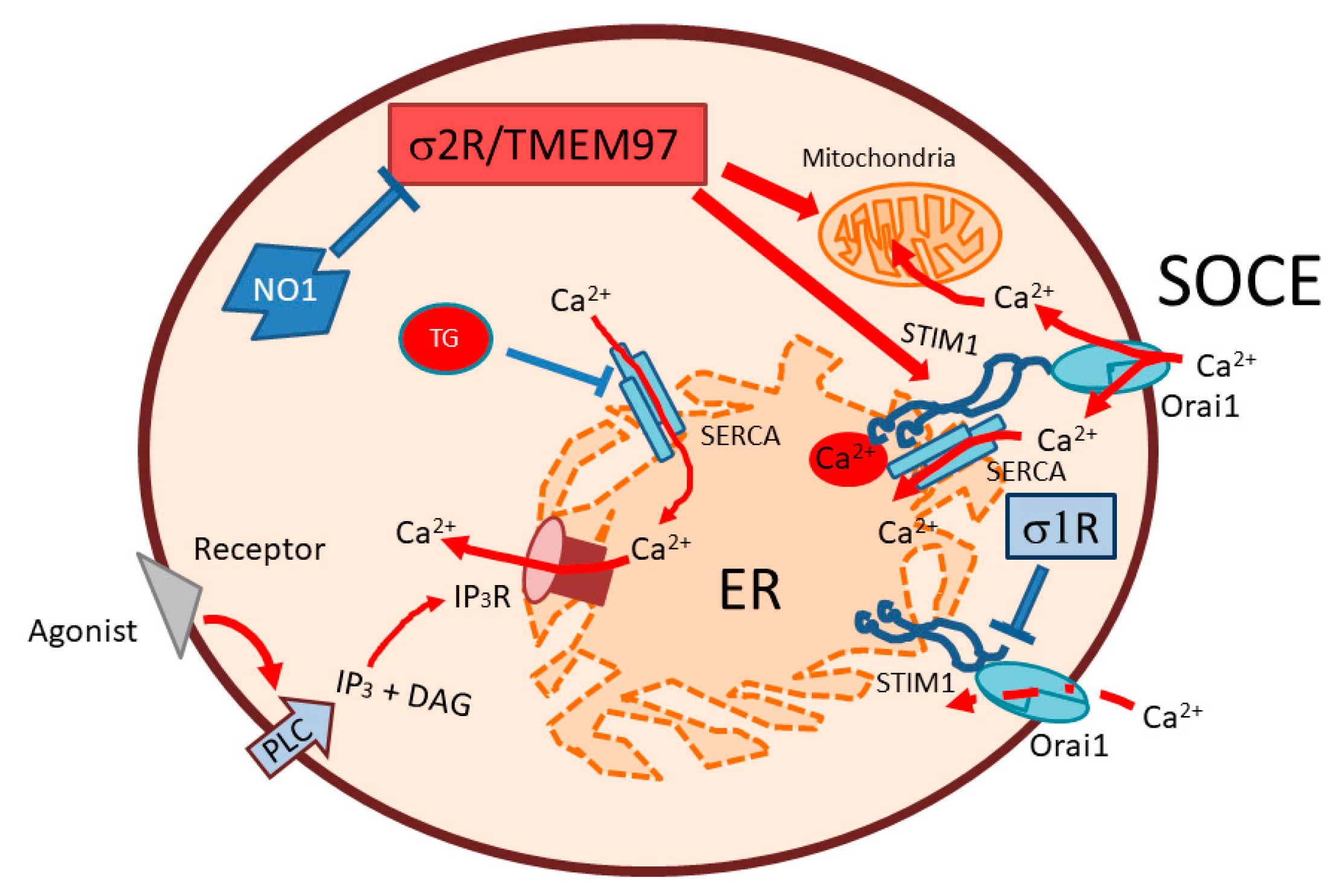
2.4. σ2R/TMEM97 Is Required for SOCE Activation in MDA-MB-231 Cells
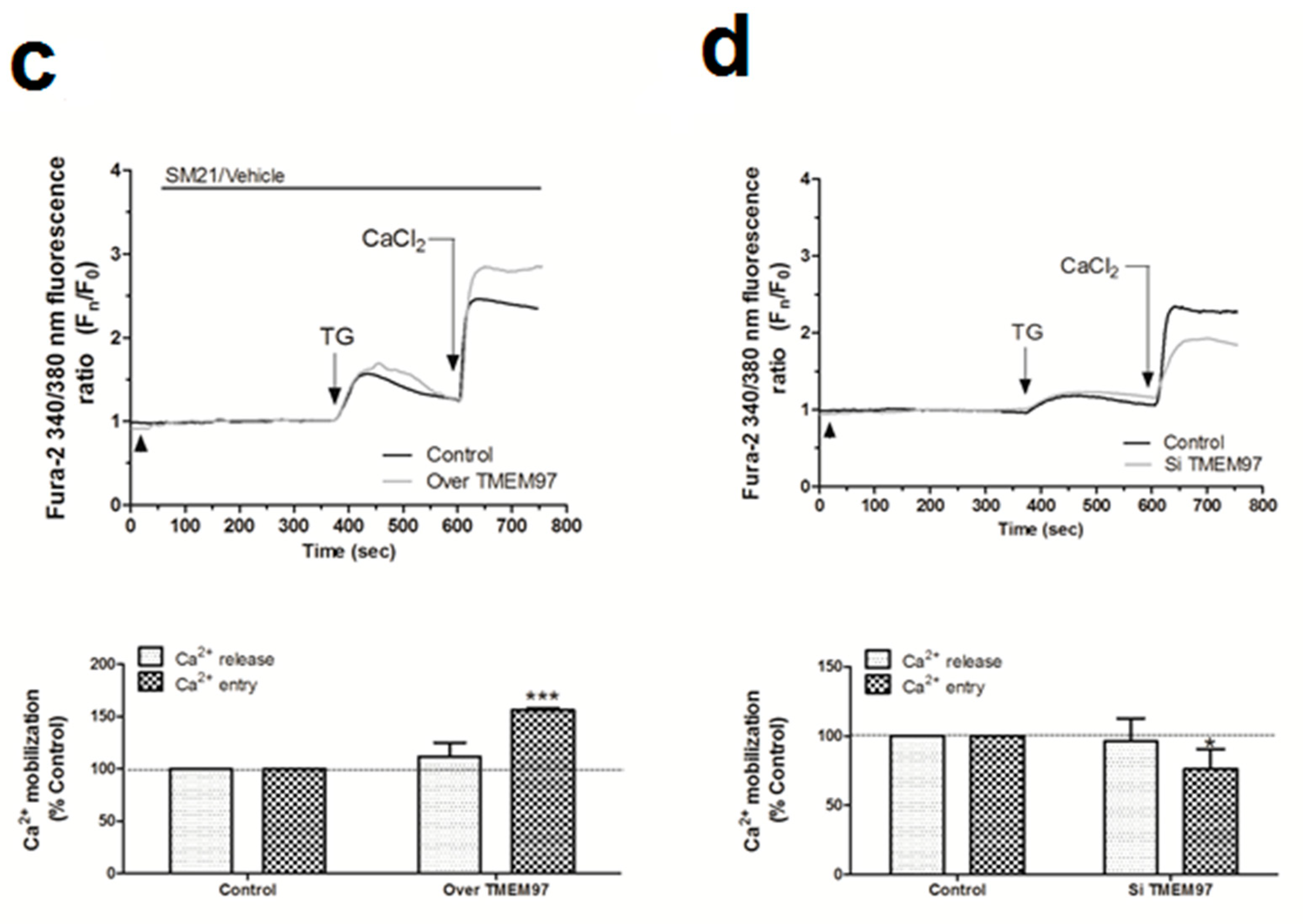
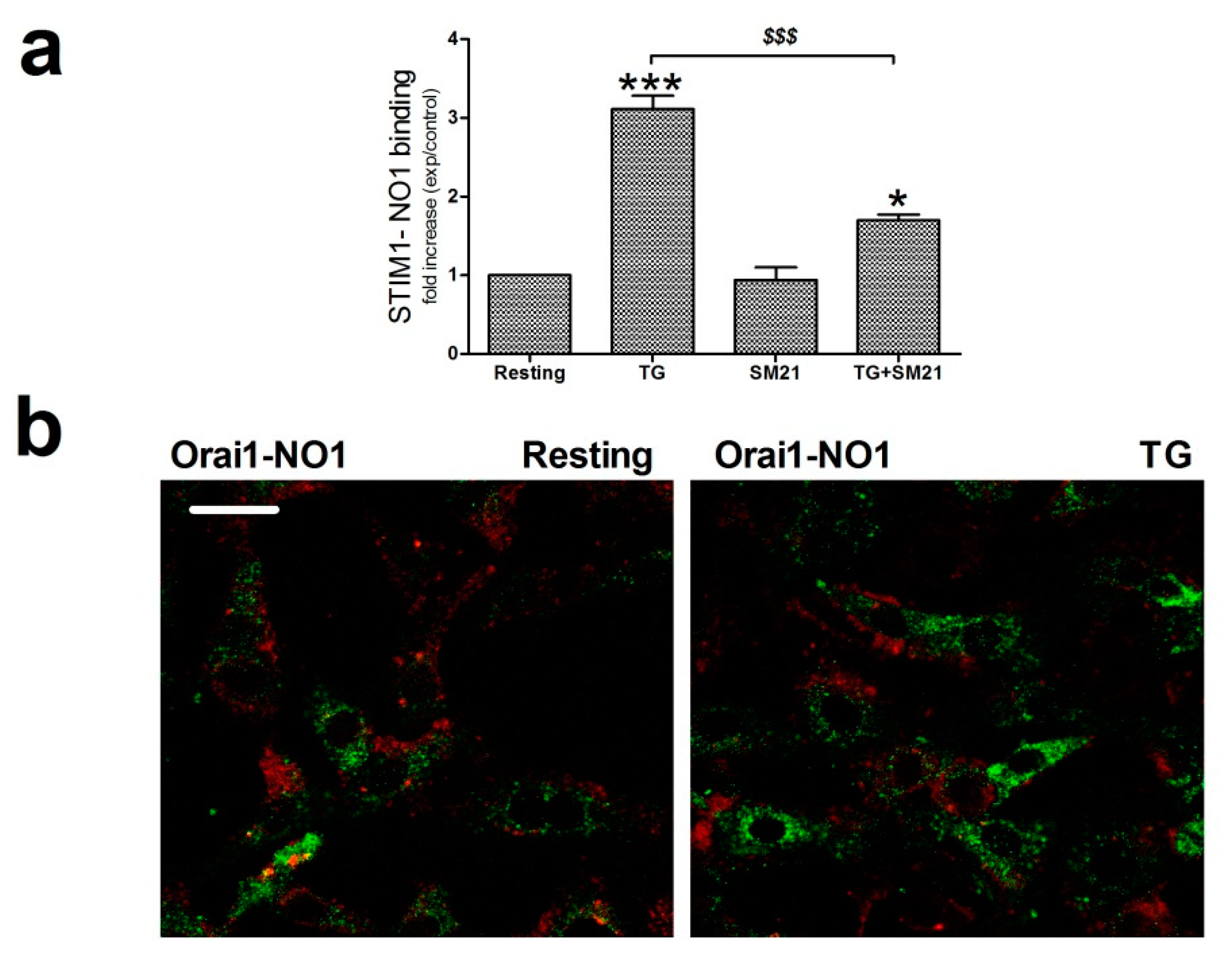
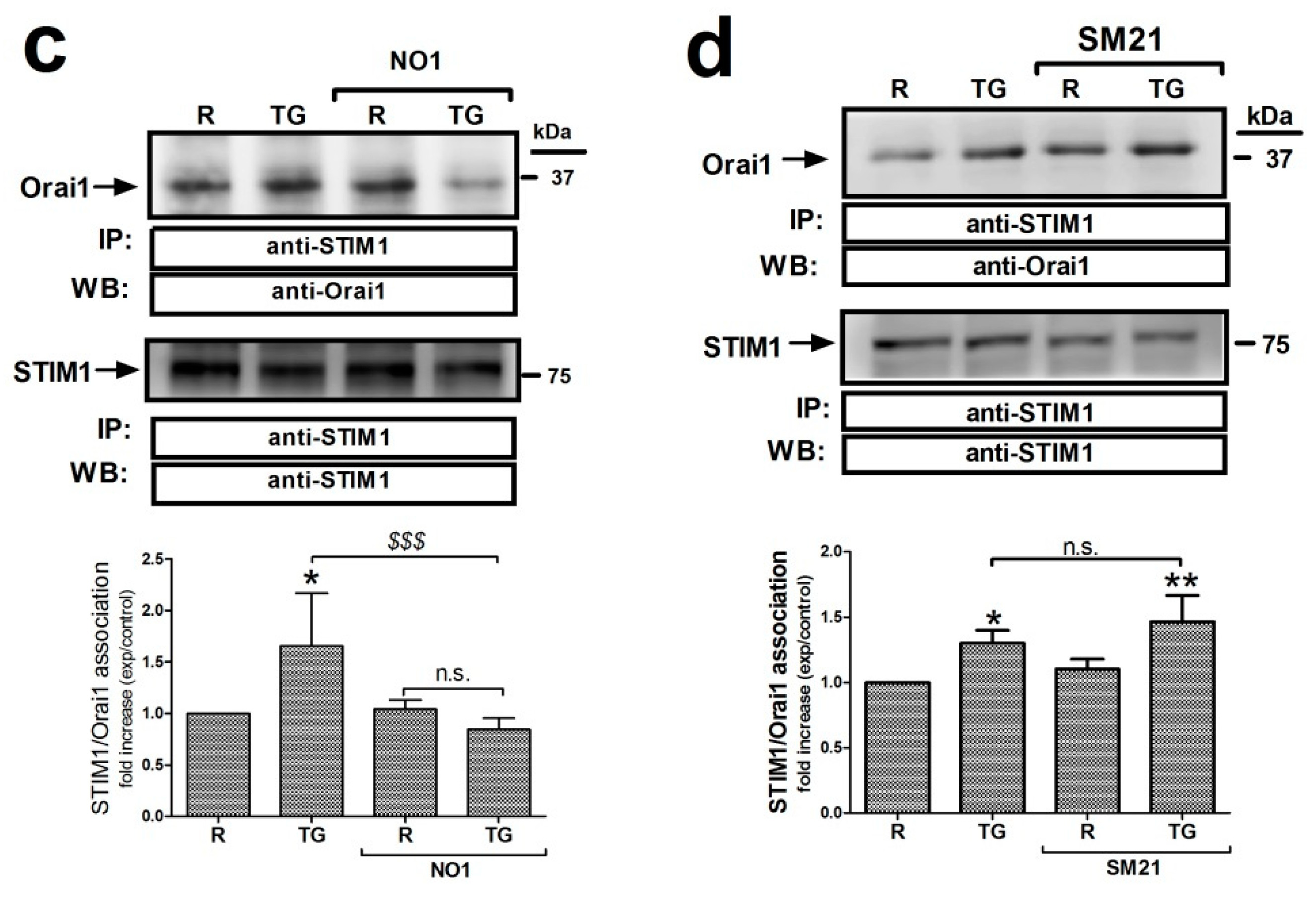
2.5. σ2R/TMEM97 Interacts with STIM1 But Not with Orai1 during SOCE Activation
3. Discussions
4. Material and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Measurement of Cytosolic Free-Ca2+Concentration
4.4. Immunoprecipitation and Western Blotting
4.5. NO1 Uptake Analysis Confocal Microscopy, and Intracellular σ2R/Orai1 Colocalization
4.6. σ2R/STIM1 Coupling Detection
4.7. Cell Migration Measurement
4.8. Cell Proliferation Assay
4.9. Analysis of the Cell Death Apoptosis Activation by Flow Cytometry (TUNEL) and Caspase Activation Using Fluorogenic Substrates
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| σR | Sigma receptor |
| TG | thapsigargin |
| NO1 | (2-{6-[2-(3-(6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)propyl)-3,4-dihydroisoquinolin-1(2H)-one-5-yloxy]hexyl}[2]-5-(dimethyllamino) bisoindoline-1,3-dione) |
| BrdU | 5-bromo-2′-desoxyuridine |
| SERCA | sarcoendoplasmic Ca2+ ATPase |
| PMCA | plasma membrane Ca2+-ATPase |
| ER | endoplasmic reticulum |
| PI | propidium iodide |
| STIM1 | stromal interaction molecule 1 |
| SOCE | store-operated calcium entry |
References
- Sun, X.; Wei, Q.; Cheng, J.; Bian, Y.; Tian, C.; Hu, Y.; Li, H. Enhanced Stim1 expression is associated with acquired chemo-resistance of cisplatin in osteosarcoma cells. Hum. Cell 2017, 30, 216–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gui, L.; Wang, Z.; Han, J.; Ma, H.; Li, Z. High Expression of Orai1 Enhances Cell Proliferation and is Associated with Poor Prognosis in Human Colorectal Cancer. Clin. Lab. 2016, 62, 1689–1698. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riesgo, E.; Gutierrez, L.G.; Ubierna, D.; Acedo, A.; Moyer, M.P.; Nunez, L.; Villalobos, C. Transcriptomic Analysis of Calcium Remodeling in Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 922. [Google Scholar] [CrossRef] [PubMed]
- Motiani, R.K.; Abdullaev, I.F.; Trebak, M. A novel native store-operated calcium channel encoded by Orai3: Selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J. Biol. Chem. 2010, 285, 19173–19183. [Google Scholar] [CrossRef] [Green Version]
- Jardin, I.; Diez-Bello, R.; Lopez, J.J.; Redondo, P.C.; Salido, G.M.; Smani, T.; Rosado, J.A. TRPC6 Channels Are Required for Proliferation, Migration and Invasion of Breast Cancer Cell Lines by Modulation of Orai1 and Orai3 Surface Exposure. Cancers 2018, 10, 331. [Google Scholar] [CrossRef] [Green Version]
- Motiani, R.K.; Zhang, X.; Harmon, K.E.; Keller, R.S.; Matrougui, K.; Bennett, J.A.; Trebak, M. Orai3 is an estrogen receptor alpha-regulated Ca2+ channel that promotes tumorigenesis. FASEB J. 2013, 27, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Miao, Y.; Zheng, X.; Gong, Y.; Zhang, J.; Zou, F.; Cai, C. STIM1 and STIM2 differently regulate endogenous Ca2+ entry and promote TGF-beta-induced EMT in breast cancer cells. Biochem. Biophys. Res. Commun. 2017, 488, 74–80. [Google Scholar] [CrossRef]
- Faouzi, M.; Hague, F.; Potier, M.; Ahidouch, A.; Sevestre, H.; Ouadid-Ahidouch, H. Down-regulation of Orai3 arrests cell-cycle progression and induces apoptosis in breast cancer cells but not in normal breast epithelial cells. J. Cell. Physiol. 2011, 226, 542–551. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, J.J.; Huang, X.Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef] [Green Version]
- Abate, C.; Niso, M.; Contino, M.; Colabufo, N.A.; Ferorelli, S.; Perrone, R.; Berardi, F. 1-Cyclohexyl-4-(4-arylcyclohexyl) piperazines: Mixed sigma and human Δ(8)-Δ(7) sterol isomerase ligands with antiproliferative and P-glycoprotein inhibitory activity. ChemMedChem 2011, 6, 73–80. [Google Scholar] [CrossRef]
- Yang, Y.; Jiang, Z.; Wang, B.; Chang, L.; Liu, J.; Zhang, L.; Gu, L. Expression of STIM1 is associated with tumor aggressiveness and poor prognosis in breast cancer. Pathol. Res. Pract. 2017, 213, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.M.; Tang, F.L.; Chang, K.J.; Russell, A. Evidence that the potential antipsychotic agent rimcazole (BW 234U) is a specific, competitive antagonist of sigma sites in brain. Life Sci. 1986, 38, 2329–2337. [Google Scholar] [CrossRef]
- Monnet, F.P. [Involvement of sigma receptors in schizophrenic syndromes. Pathophysiological approach. Presse Med. 1993, 22, 218–223. [Google Scholar] [PubMed]
- Snyder, S.H.; Largent, B.L. Receptor mechanisms in antipsychotic drug action: Focus on sigma receptors. J. Neuropsychiatry Clin. Neurosci. 1989, 1, 7–15. [Google Scholar] [CrossRef]
- Bartz, F.; Kern, L.; Erz, D.; Zhu, M.; Gilbert, D.; Meinhof, T.; Wirkner, U.; Erfle, H.; Muckenthaler, M.; Pepperkok, R.; et al. Identification of cholesterol-regulating genes by targeted RNAi screening. Cell Metab. 2009, 10, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Alon, A.; Schmidt, H.R.; Wood, M.D.; Sahn, J.J.; Martin, S.F.; Kruse, A.C. Identification of the gene that codes for the sigma2 receptor. Proc. Natl. Acad. Sci. USA 2017, 114, 7160–7165. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, S.; Lingnurkar, R.; Carey, T.S.; Pomaville, M.; Kar, P.; Feig, M.; Wilson, C.A.; Knott, J.G.; Arnosti, D.N.; Henry, R.W. The Evolutionarily Conserved C-terminal Domains in the Mammalian Retinoblastoma Tumor Suppressor Family Serve as Dual Regulators of Protein Stability and Transcriptional Potency. J. Biol. Chem. 2015, 290, 14462–14475. [Google Scholar] [CrossRef] [Green Version]
- Pati, M.L.; Hornick, J.R.; Niso, M.; Berardi, F.; Spitzer, D.; Abate, C.; Hawkins, W. Sigma-2 receptor agonist derivatives of 1-Cyclohexyl-4-[3-(5-methoxy-1,2,3,4-tetrahydronaphthalen-1-yl)propyl]piperazine (PB28) induce cell death via mitochondrial superoxide production and caspase activation in pancreatic cancer. BMC Cancer 2017, 17, 51. [Google Scholar] [CrossRef] [Green Version]
- Spruce, B.A.; Campbell, L.A.; McTavish, N.; Cooper, M.A.; Appleyard, M.V.; O’Neill, M.; Howie, J.; Samson, J.; Watt, S.; Murray, K.; et al. Small molecule antagonists of the sigma-1 receptor cause selective release of the death program in tumor and self-reliant cells and inhibit tumor growth in vitro and in vivo. Cancer Res. 2004, 64, 4875–4886. [Google Scholar] [CrossRef] [Green Version]
- van Waarde, A.; Rybczynska, A.A.; Ramakrishnan, N.K.; Ishiwata, K.; Elsinga, P.H.; Dierckx, R.A. Potential applications for sigma receptor ligands in cancer diagnosis and therapy. Biochim. Biophys. Acta 2015, 1848, 2703–2714. [Google Scholar] [CrossRef] [Green Version]
- McDonald, E.S.; Mankoff, J.; Makvandi, M.; Chu, W.; Chu, Y.; Mach, R.H.; Zeng, C. Sigma-2 ligands and PARP inhibitors synergistically trigger cell death in breast cancer cells. Biochem. Biophys. Res. Commun. 2017, 486, 788–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, C.; McDonald, E.S.; Mach, R.H. Molecular Probes for Imaging the Sigma-2 Receptor: In Vitro and In Vivo Imaging Studies. Handb. Exp. Pharmacol. 2017, 244, 309–330. [Google Scholar] [CrossRef] [PubMed]
- Vilner, B.J.; Bowen, W.D. Modulation of cellular calcium by sigma-2 receptors: Release from intracellular stores in human SK-N-SH neuroblastoma cells. J. Pharmacol. Exp. Ther. 2000, 292, 900–911. [Google Scholar] [PubMed]
- Cassano, G.; Gasparre, G.; Niso, M.; Contino, M.; Scalera, V.; Colabufo, N.A. F281, synthetic agonist of the sigma-2 receptor, induces Ca2+ efflux from the endoplasmic reticulum and mitochondria in SK-N-SH cells. Cell Calcium 2009, 45, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cuevas, J. Sigma receptors inhibit high-voltage-activated calcium channels in rat sympathetic and parasympathetic neurons. J. Neurophysiol. 2002, 87, 2867–2879. [Google Scholar] [CrossRef]
- Zeng, C.; Vangveravong, S.; Xu, J.; Chang, K.C.; Hotchkiss, R.S.; Wheeler, K.T.; Shen, D.; Zhuang, Z.P.; Kung, H.F.; Mach, R.H. Subcellular localization of sigma-2 receptors in breast cancer cells using two-photon and confocal microscopy. Cancer Res. 2007, 67, 6708–6716. [Google Scholar] [CrossRef] [Green Version]
- Chu, U.B.; Mavlyutov, T.A.; Chu, M.L.; Yang, H.; Schulman, A.; Mesangeau, C.; McCurdy, C.R.; Guo, L.W.; Ruoho, A.E. The Sigma-2 Receptor and Progesterone Receptor Membrane Component 1 are Different Binding Sites Derived from Independent Genes. EBioMedicine 2015, 2, 1806–1813. [Google Scholar] [CrossRef] [Green Version]
- Bali, N.; Arimoto, J.M.; Morgan, T.E.; Finch, C.E. Progesterone antagonism of neurite outgrowth depends on microglial activation via Pgrmc1/S2R. Endocrinology 2013, 154, 2468–2480. [Google Scholar] [CrossRef] [Green Version]
- Crudden, G.; Chitti, R.E.; Craven, R.J. Hpr6 (heme-1 domain protein) regulates the susceptibility of cancer cells to chemotherapeutic drugs. J. Pharmacol. Exp. Ther. 2006, 316, 448–455. [Google Scholar] [CrossRef]
- Niso, M.; Riganti, C.; Pati, M.L.; Ghigo, D.; Berardi, F.; Abate, C. Novel and Selective Fluorescent sigma2-Receptor Ligand with a 3,4-Dihydroisoquinolin-1-one Scaffold: A Tool to Study sigma2 Receptors in Living Cells. Chembiochem 2015, 16, 1078–1083. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, J.; Xia, X.; Yang, M.; Vangveravong, S.; Chen, J.; Mach, R.H.; Xia, Y. SV119-gold nanocage conjugates: A new platform for targeting cancer cells via sigma-2 receptors. Nanoscale 2012, 4, 421–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasparre, G.; Abate, C.; Carlucci, R.; Berardi, F.; Cassano, G. The sigma1 receptor agonist (+)-pentazocine increases store-operated Ca2+ entry in MCF7sigma1 and SK-N-SH cell lines. Pharmacol. Rep. 2017, 69, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Abate, C.; Niso, M.; Lacivita, E.; Mosier, P.D.; Toscano, A.; Perrone, R. Analogues of sigma receptor ligand 1-cyclohexyl-4-[3-(5-methoxy-1,2,3,4-tetrahydronaphthalen-1-yl)propyl]piperazine (PB28) with added polar functionality and reduced lipophilicity for potential use as positron emission tomography radiotracers. J. Med. Chem. 2011, 54, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Kopp, F.; Wagner, E.; Roidl, A. The proto-oncogene KRAS is targeted by miR-200c. Oncotarget 2014, 5, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Longhitano, L.; Castracani, C.C.; Tibullo, D.; Avola, R.; Viola, M.; Russo, G.; Prezzavento, O.; Marrazzo, A.; Amata, E.; Reibaldi, M.; et al. Sigma-1 and Sigma-2 receptor ligands induce apoptosis and autophagy but have opposite effect on cell proliferation in uveal melanoma. Oncotarget 2017, 8, 91099–91111. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, M.; Amata, E.; Vinciguerra, S.; Fiorito, J.; Giurdanella, G.; Drago, F.; Caporarello, N.; Prezzavento, O.; Arena, E.; Salerno, L.; et al. Antiangiogenic Effect of (+/−)-Haloperidol Metabolite II Valproate Ester [(+/−)-MRJF22] in Human Microvascular Retinal Endothelial Cells. J. Med. Chem. 2016, 59, 9960–9966. [Google Scholar] [CrossRef]
- Georgiadis, M.O.; Karoutzou, O.; Foscolos, A.S.; Papanastasiou, I. Sigma Receptor (sigmaR) Ligands with Antiproliferative and Anticancer Activity. Molecules 2017, 22. [Google Scholar] [CrossRef]
- Niso, M.; Abate, C.; Contino, M.; Ferorelli, S.; Azzariti, A.; Perrone, R.; Colabufo, N.A.; Berardi, F. Sigma-2 receptor agonists as possible antitumor agents in resistant tumors: Hints for collateral sensitivity. ChemMedChem 2013, 8, 2026–2035. [Google Scholar] [CrossRef]
- Gonzalez, D.; Espino, J.; Bejarano, I.; Lopez, J.J.; Rodriguez, A.B.; Pariente, J.A. Caspase-3 and −9 are activated in human myeloid HL-60 cells by calcium signal. Mol. Cell. Biochem. 2010, 333, 151–157. [Google Scholar] [CrossRef]
- Zbidi, H.; Redondo, P.C.; Lopez, J.J.; Bartegi, A.; Salido, G.M.; Rosado, J.A. Homocysteine induces caspase activation by endoplasmic reticulum stress in platelets from type 2 diabetics and healthy donors. Thromb. Haemost. 2010, 103, 1022–1032. [Google Scholar] [CrossRef]
- Cai, Y.; Zheng, Y.; Gu, J.; Wang, S.; Wang, N.; Yang, B.; Zhang, F.; Wang, D.; Fu, W.; Wang, Z. Betulinic acid chemosensitizes breast cancer by triggering ER stress-mediated apoptosis by directly targeting GRP78. Cell Death Dis. 2018, 9, 636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Yang, C.; Guo, C.; Li, X.; Yang, N.; Zhao, L.; Hang, H.; Liu, S.; Chu, P.; Sun, Z.; et al. SZC015, a synthetic oleanolic acid derivative, induces both apoptosis and autophagy in MCF-7 breast cancer cells. Chem. Biol. Interact. 2016, 244, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Rosado, J.A.; Lopez, J.J.; Gomez-Arteta, E.; Redondo, P.C.; Salido, G.M.; Pariente, J.A. Early caspase-3 activation independent of apoptosis is required for cellular function. J. Cell. Physiol. 2006, 209, 142–152. [Google Scholar] [CrossRef]
- Lopez, J.J.; Redondo, P.C.; Salido, G.M.; Pariente, J.A.; Rosado, J.A. N,N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediamine induces apoptosis through the activation of caspases-3 and -8 in human platelets. A role for endoplasmic reticulum stress. J. Thromb. Haemost. 2009, 7, 992–999. [Google Scholar] [CrossRef] [PubMed]
- McAndrew, D.; Grice, D.M.; Peters, A.A.; Davis, F.M.; Stewart, T.; Rice, M.; Smart, C.E.; Brown, M.A.; Kenny, P.A.; Roberts-Thomson, S.J.; et al. ORAI1-mediated calcium influx in lactation and in breast cancer. Mol. Cancer Ther. 2011, 10, 448–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kar, P.; Parekh, A.B. Distinct spatial Ca2+ signatures selectively activate different NFAT transcription factor isoforms. Mol. Cell 2015, 58, 232–243. [Google Scholar] [CrossRef] [Green Version]
- Albarran, L.; Lopez, J.J.; Jardin, I.; Sanchez-Collado, J.; Berna-Erro, A.; Smani, T.; Camello, P.J.; Salido, G.M.; Rosado, J.A. EFHB is a Novel Cytosolic Ca2+ Sensor That Modulates STIM1-SARAF Interaction. Cell. Physiol. Biochem. 2018, 51, 1164–1178. [Google Scholar] [CrossRef]
- Juska, A.; Redondo, P.C.; Rosado, J.A.; Salido, G.M. Dynamics of calcium fluxes in human platelets assessed in calcium-free medium. Biochem. Biophys. Res. Commun. 2005, 334, 779–786. [Google Scholar] [CrossRef]
- Redondo, P.C.; Rosado, J.A.; Pariente, J.A.; Salido, G.M. Collaborative effect of SERCA and PMCA in cytosolic calcium homeostasis in human platelets. J. Physiol. Biochem. 2005, 61, 507–516. [Google Scholar] [CrossRef]
- Azzariti, A.; Colabufo, N.A.; Berardi, F.; Porcelli, L.; Niso, M.; Simone, G.M.; Perrone, R.; Paradiso, A. Cyclohexylpiperazine derivative PB28, a sigma2 agonist and sigma1 antagonist receptor, inhibits cell growth, modulates P-glycoprotein, and synergizes with anthracyclines in breast cancer. Mol. Cancer Ther. 2006, 5, 1807–1816. [Google Scholar] [CrossRef] [Green Version]
- Aydar, E.; Onganer, P.; Perrett, R.; Djamgoz, M.B.; Palmer, C.P. The expression and functional characterization of sigma (sigma) 1 receptors in breast cancer cell lines. Cancer Lett. 2006, 242, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Srivats, S.; Balasuriya, D.; Pasche, M.; Vistal, G.; Edwardson, J.M.; Taylor, C.W.; Murrell-Lagnado, R.D. Sigma1 receptors inhibit store-operated Ca2+ entry by attenuating coupling of STIM1 to Orai1. J. Cell Biol. 2016, 213, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Redondo, P.C.; Rosado, J.A. Store-operated calcium entry: Unveiling the calcium handling signalplex. Int. Rev. Cell Mol. Biol. 2015, 316, 183–226. [Google Scholar] [CrossRef] [PubMed]
- Stathopulos, P.B.; Ikura, M. Store operated calcium entry: From concept to structural mechanisms. Cell Calcium 2017, 63, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Putney, J.W., Jr. New molecular players in capacitative Ca2+ entry. J. Cell Sci. 2007, 120, 1959–1965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riad, A.; Zeng, C.; Weng, C.C.; Winters, H.; Xu, K.; Makvandi, M.; Metz, T.; Carlin, S.; Mach, R.H. Sigma-2 Receptor/TMEM97 and PGRMC-1 Increase the Rate of Internalization of LDL by LDL Receptor through the Formation of a Ternary Complex. Sci. Rep. 2018, 8, 16845. [Google Scholar] [CrossRef]
- Ebrahimi-Fakhari, D.; Wahlster, L.; Bartz, F.; Werenbeck-Ueding, J.; Praggastis, M.; Zhang, J.; Joggerst-Thomalla, B.; Theiss, S.; Grimm, D.; Ory, D.S.; et al. Reduction of TMEM97 increases NPC1 protein levels and restores cholesterol trafficking in Niemann-pick type C1 disease cells. Hum. Mol. Genet. 2016, 25, 3588–3599. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Thomas, D.W. Stromal Interaction Molecule 1 rescues store-operated calcium entry and protects NG115-401L cells against cell death induced by endoplasmic reticulum and mitochondrial oxidative stress. Neurochem. Int. 2016, 97, 137–145. [Google Scholar] [CrossRef]
- Albarran, L.; Lopez, J.J.; Amor, N.B.; Martin-Cano, F.E.; Berna-Erro, A.; Smani, T.; Salido, G.M.; Rosado, J.A. Dynamic interaction of SARAF with STIM1 and Orai1 to modulate store-operated calcium entry. Sci. Rep. 2016, 6, 24452. [Google Scholar] [CrossRef]
- Lopez, E.; Frischauf, I.; Jardin, I.; Derler, I.; Muik, M.; Cantonero, C.; Salido, G.M.; Smani, T.; Rosado, J.A.; Redondo, P.C. STIM1 phosphorylation at Y316 modulates its interaction with SARAF and the activation of SOCE and ICRAC. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [Green Version]
- Garg, G.; Kumar, D.; Asim, M.; Husain, S.A.; Das, B.C.; Kar, P. Multiplex Reverse Transcriptase-PCR for Simultaneous Detection of Hepatitis, B., C., and E Virus. J. Clin. Exp. Hepatol. 2016, 6, 33–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, K.T.; Wang, L.M.; Wallen, C.A.; Childers, S.R.; Cline, J.M.; Keng, P.C.; Mach, R.H. Sigma-2 receptors as a biomarker of proliferation in solid tumours. Br. J. Cancer 2000, 82, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Makvandi, M.; Tilahun, E.D.; Lieberman, B.P.; Anderson, R.C.; Zeng, C.; Xu, K.; Hou, C.; McDonald, E.S.; Pryma, D.A.; Mach, R.H. The sigma-2 receptor as a therapeutic target for drug delivery in triple negative breast cancer. Biochem. Biophys. Res. Commun. 2015, 467, 1070–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, T.; Hayashi, T.; Hayashi, E.; Su, T.P. Sigma-1 receptor chaperone at the ER-mitochondrion interface mediates the mitochondrion-ER-nucleus signaling for cellular survival. PLoS ONE 2013, 8, e76941. [Google Scholar] [CrossRef] [Green Version]
- Ghelardini, C.; Galeotti, N.; Bartolini, A. Pharmacological identification of SM-21, the novel sigma(2) antagonist. Pharmacol. Biochem. Behav. 2000, 67, 659–662. [Google Scholar] [CrossRef]
- Wu, Y.; Bai, X.; Li, X.; Zhu, C.; Wu, Z.P. Overexpression of sigma-1 receptor in MCF-7 cells enhances proliferation via the classic protein kinase C subtype signaling pathway. Oncol. Lett. 2018, 16, 6763–6769. [Google Scholar] [CrossRef] [Green Version]
- Krajewski, S.; Krajewska, M.; Ellerby, L.M.; Welsh, K.; Xie, Z.; Deveraux, Q.L.; Salvesen, G.S.; Bredesen, D.E.; Rosenthal, R.E.; Fiskum, G.; et al. Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischemia. Proc. Natl. Acad. Sci. USA 1999, 96, 5752–5757. [Google Scholar] [CrossRef] [Green Version]
- Lopez, J.J.; Salido, G.M.; Gomez-Arteta, E.; Rosado, J.A.; Pariente, J.A. Thrombin induces apoptotic events through the generation of reactive oxygen species in human platelets. J. Thromb. Haemost. 2007, 5, 1283–1291. [Google Scholar] [CrossRef]
- Hernandez-Morales, M.; Sobradillo, D.; Valero, R.A.; Munoz, E.; Ubierna, D.; Moyer, M.P.; Nunez, L.; Villalobos, C. Mitochondria sustain store-operated currents in colon cancer cells but not in normal colonic cells: Reversal by non-steroidal anti-inflammatory drugs. Oncotarget 2017, 8, 55332. [Google Scholar] [CrossRef] [Green Version]
- Jardin, I.; Albarran, L.; Salido, G.M.; Lopez, J.J.; Sage, S.O.; Rosado, J.A. Fine-tuning of store-operated calcium entry by fast and slow Ca2+-dependent inactivation: Involvement of SARAF. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 463–469. [Google Scholar] [CrossRef]
- Gilabert, J.A.; Parekh, A.B. Respiring mitochondria determine the pattern of activation and inactivation of the store-operated Ca2+ current I(CRAC). EMBO J. 2000, 19, 6401–6407. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, W.; Zhang, L.; Liu, W.B.; Fei, Z. Inhibition of store-operated calcium entry attenuates MPP(+)-induced oxidative stress via preservation of mitochondrial function in PC12 cells: Involvement of Homer1a. PLoS ONE 2013, 8, e83638. [Google Scholar] [CrossRef] [PubMed]
- Rao, W.; Zhang, L.; Su, N.; Wang, K.; Hui, H.; Wang, L.; Chen, T.; Luo, P.; Yang, Y.F.; Liu, Z.B.; et al. Blockade of SOCE protects HT22 cells from hydrogen peroxide-induced apoptosis. Biochem. Biophys. Res. Commun. 2013, 441, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Shanmughapriya, S.; Rajan, S.; Hoffman, N.E.; Zhang, X.; Guo, S.; Kolesar, J.E.; Hines, K.J.; Ragheb, J.; Jog, N.R.; Caricchio, R.; et al. Ca2+ signals regulate mitochondrial metabolism by stimulating CREB-mediated expression of the mitochondrial Ca2+ uniporter gene MCU. Sci. Signal. 2015, 8, ra23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, X.; Zhang, Y.; Wang, Z.; Yu, J.; Kong, Z.; Ruzic, L. HIIT changes the expression of MuRF1 and MAFBx proteins and proteins involved in the MTOR pathway and autophagy in rat skeletal muscle. Exp. Physiol. 2019, 104, 1505–1517. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, I.; Espino, J.; Gonzalez-Flores, D.; Casado, J.G.; Redondo, P.C.; Rosado, J.A.; Barriga, C.; Pariente, J.A.; Rodriguez, A.B. Role of Calcium Signals on Hydrogen Peroxide-Induced Apoptosis in Human Myeloid HL-60 Cells. Int. J. Biomed. Sci. 2009, 5, 246–256. [Google Scholar] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cantonero, C.; Camello, P.J.; Abate, C.; Berardi, F.; Salido, G.M.; Rosado, J.A.; Redondo, P.C. NO1, a New Sigma 2 Receptor/TMEM97 Fluorescent Ligand, Downregulates SOCE and Promotes Apoptosis in the Triple Negative Breast Cancer Cell Lines. Cancers 2020, 12, 257. https://doi.org/10.3390/cancers12020257
Cantonero C, Camello PJ, Abate C, Berardi F, Salido GM, Rosado JA, Redondo PC. NO1, a New Sigma 2 Receptor/TMEM97 Fluorescent Ligand, Downregulates SOCE and Promotes Apoptosis in the Triple Negative Breast Cancer Cell Lines. Cancers. 2020; 12(2):257. https://doi.org/10.3390/cancers12020257
Chicago/Turabian StyleCantonero, Carlos, Pedro Javier Camello, Carmen Abate, Francesco Berardi, Gines Maria Salido, Juan Antonio Rosado, and Pedro C. Redondo. 2020. "NO1, a New Sigma 2 Receptor/TMEM97 Fluorescent Ligand, Downregulates SOCE and Promotes Apoptosis in the Triple Negative Breast Cancer Cell Lines" Cancers 12, no. 2: 257. https://doi.org/10.3390/cancers12020257