Rapid Assessment of Functional Avidity of Tumor-Specific T Cell Receptors Using an Antigen-Presenting Tumor Cell Line Electroporated with Full-Length Tumor Antigen mRNA

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Quantitation of WT1-Presenting Potential Odel APC
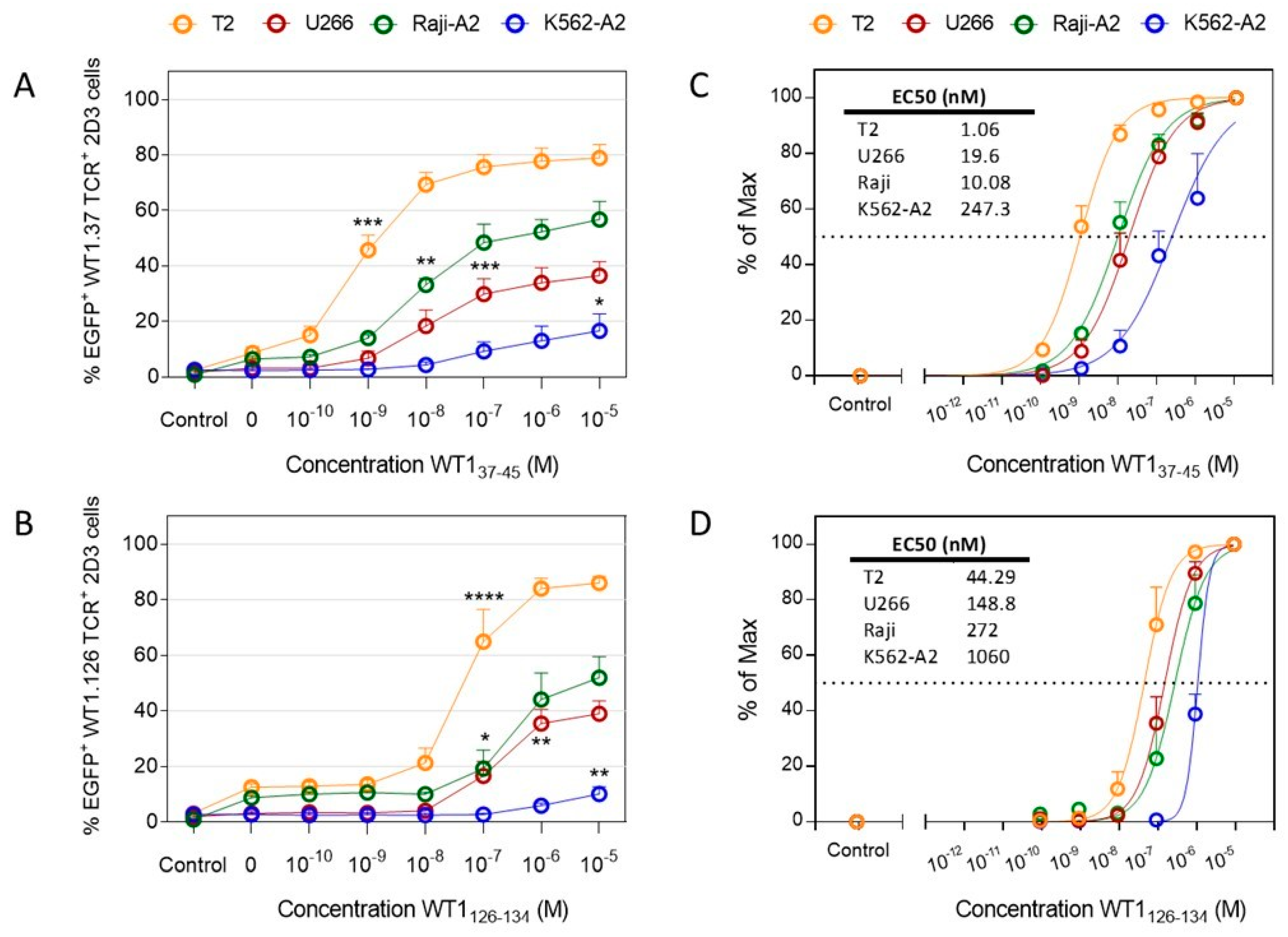
2.2. Functional Avidity of WT1-Specific T Cells Drastically Differs Depending on the APC Used
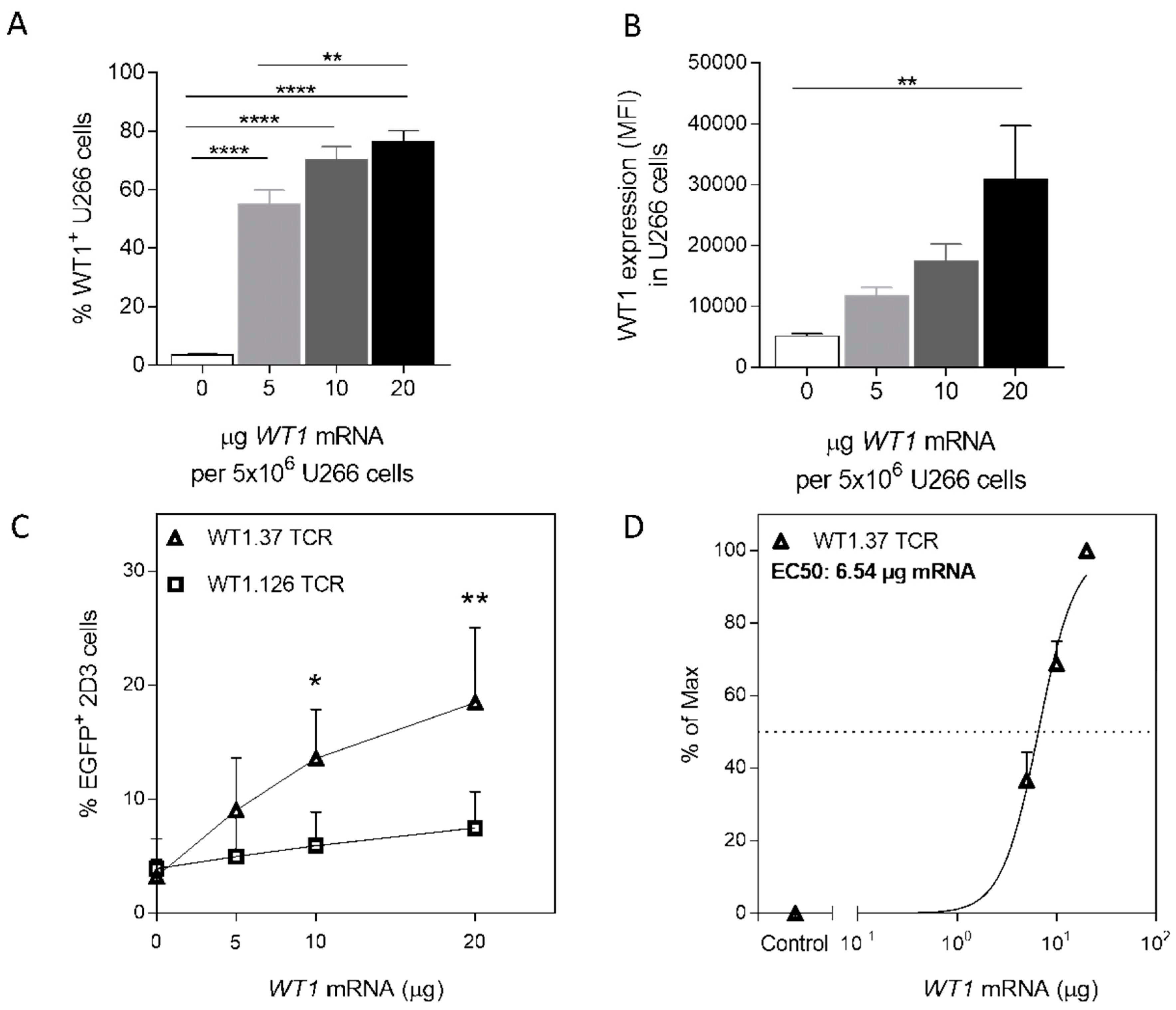
2.3. Mimicking Endogenous WT1 Expression
2.4. WT1 mRNA-Electroporated U266 Cells Activate WT1-Specific TCR-Redirected Primary Human CD8 T Cells in a Dose-Dependent Manner
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Primary Cells
4.2. In Vitro Transcription of mRNA
4.3. Electroporation
4.4. Flow Cytometry
4.5. Peptide-Pulsing of Tumor Cells
4.6. Co-Cultures
4.7. Analysis of Epitope-Specific T-Cell Activation
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tendeiro Rego, R.; Morris, E.C.; Lowdell, M.W. T-cell receptor gene-modified cells: Past promises, present methodologies and future challenges. Cytotherapy 2019, 21, 341–357. [Google Scholar] [CrossRef]
- Bullock, T.N.; Colella, T.A.; Engelhard, V.H. The density of peptides displayed by dendritic cells affects immune responses to human tyrosinase and gp100 in HLA-A2 transgenic mice. J. Immunol. 2000, 164, 2354–2361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, P.A.; Carreno, L.J.; Coombs, D.; Mora, J.E.; Palmieri, E.; Goldstein, B.; Nathenson, S.G.; Kalergis, A.M. T cell receptor binding kinetics required for T cell activation depend on the density of cognate ligand on the antigen-presenting cell. Proc. Natl. Acad. Sci. USA 2005, 102, 4824–4829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allard, M.; Hebeisen, M.; Rufer, N. Assessing T Cell Receptor Affinity and Avidity Against Tumor Antigens. In Oncoimmunology: A Practical Guide for Cancer Immunotherapy; Zitvogel, L., Kroemer, G., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 665–679. [Google Scholar] [CrossRef]
- Snyder, J.T.; Alexander-Miller, M.A.; Berzofskyl, J.A.; Belyakov, I.M. Molecular mechanisms and biological significance of CTL avidity. Curr. HIV Res. 2003, 1, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Fahmy, T.M.; Bieler, J.G.; Edidin, M.; Schneck, J.P. Increased TCR avidity after T cell activation: A mechanism for sensing low-density antigen. Immunity 2001, 14, 135–143. [Google Scholar] [CrossRef]
- Van Tendeloo, V.F.; Van de Velde, A.; Van Driessche, A.; Cools, N.; Anguille, S.; Ladell, K.; Gostick, E.; Vermeulen, K.; Pieters, K.; Nijs, G.; et al. Induction of complete and molecular remissions in acute myeloid leukemia by Wilms’ tumor 1 antigen-targeted dendritic cell vaccination. Proc. Natl. Acad. Sci. USA 2010, 107, 13824–13829. [Google Scholar] [CrossRef] [Green Version]
- Chapuis, A.G.; Ragnarsson, G.B.; Nguyen, H.N.; Chaney, C.N.; Pufnock, J.S.; Schmitt, T.M.; Duerkopp, N.; Roberts, I.M.; Pogosov, G.L.; Ho, W.Y.; et al. Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Sci. Transl. Med. 2013, 5, 174ra27. [Google Scholar] [CrossRef] [Green Version]
- Kohrt, H.E.; Shu, C.T.; Stuge, T.B.; Holmes, S.P.; Weber, J.; Lee, P.P. Rapid assessment of recognition efficiency and functional capacity of antigen-specific T-cell responses. J. Immunother. 2005, 28, 297–305. [Google Scholar] [CrossRef]
- Takagi, A.; Horiuchi, Y.; Matsui, M. Characterization of the flow cytometric assay for ex vivo monitoring of cytotoxicity mediated by antigen-specific cytotoxic T lymphocytes. Biochem. Biophys. Res. Commun. 2017, 492, 27–32. [Google Scholar] [CrossRef]
- Lovelace, P.; Maecker, H.T. Multiparameter Intracellular Cytokine Staining. Methods Mol. Biol. 2018, 1678, 151–166. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, T.; Sester, M. Detection of antigen-specific T cells based on intracellular cytokine staining using flow-cytometry. Methods Mol. Biol. 2013, 1064, 267–274. [Google Scholar] [CrossRef]
- Malyguine, A.M.; Strobl, S.; Dunham, K.; Shurin, M.R.; Sayers, T.J. ELISPOT Assay for Monitoring Cytotoxic T Lymphocytes (CTL) Activity in Cancer Vaccine Clinical Trials. Cells 2012, 1, 111–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li Pira, G.; Ivaldi, F.; Dentone, C.; Righi, E.; Del Bono, V.; Viscoli, C.; Koopman, G.; Manca, F. Evaluation of antigen-specific T-cell responses with a miniaturized and automated method. Clin. Vaccine Immunol. 2008, 15, 1811–1818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubio, V.; Stuge, T.B.; Singh, N.; Betts, M.R.; Weber, J.S.; Roederer, M.; Lee, P.P. Ex vivo identification, isolation and analysis of tumor-cytolytic T cells. Nat. Med. 2003, 9, 1377–1382. [Google Scholar] [CrossRef] [PubMed]
- Betts, M.R.; Brenchley, J.M.; Price, D.A.; De Rosa, S.C.; Douek, D.C.; Roederer, M.; Koup, R.A. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J. Immunol. Methods 2003, 281, 65–78. [Google Scholar] [CrossRef]
- Wolfl, M.; Kuball, J.; Ho, W.Y.; Nguyen, H.; Manley, T.J.; Bleakley, M.; Greenberg, P.D. Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities. Blood 2007, 110, 201–210. [Google Scholar] [CrossRef]
- Morimoto, S.; Fujiki, F.; Kondo, K.; Nakajima, H.; Kobayashi, Y.; Inatome, M.; Aoyama, N.; Nishida, Y.; Tsuboi, A.; Oka, Y.; et al. Establishment of a novel platform cell line for efficient and precise evaluation of T cell receptor functional avidity. Oncotarget 2018, 9, 34132–34141. [Google Scholar] [CrossRef]
- Rosskopf, S.; Leitner, J.; Paster, W.; Morton, L.T.; Hagedoorn, R.S.; Steinberger, P.; Heemskerk, M.H.M. A Jurkat 76 based triple parameter reporter system to evaluate TCR functions and adoptive T cell strategies. Oncotarget 2018, 9, 17608–17619. [Google Scholar] [CrossRef] [Green Version]
- Eggermont, L.J.; Paulis, L.E.; Tel, J.; Figdor, C.G. Towards efficient cancer immunotherapy: Advances in developing artificial antigen-presenting cells. Trends Biotechnol. 2014, 32, 456–465. [Google Scholar] [CrossRef] [Green Version]
- Anguille, S.; Van de Velde, A.L.; Smits, E.L.; Van Tendeloo, V.F.; Juliusson, G.; Cools, N.; Nijs, G.; Stein, B.; Lion, E.; Van Driessche, A.; et al. Dendritic cell vaccination as postremission treatment to prevent or delay relapse in acute myeloid leukemia. Blood 2017, 130, 1713–1721. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Han, Y.; Suarez Saiz, F.; Minden, M.D. A tumor suppressor and oncogene: The WT1 story. Leukemia 2007, 21, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Schmid, D.A.; Irving, M.B.; Posevitz, V.; Hebeisen, M.; Posevitz-Fejfar, A.; Sarria, J.C.; Gomez-Eerland, R.; Thome, M.; Schumacher, T.N.; Romero, P.; et al. Evidence for a TCR affinity threshold delimiting maximal CD8 T cell function. J. Immunol. 2010, 184, 4936–4946. [Google Scholar] [CrossRef] [PubMed]
- Henderson, R.A.; Michel, H.; Sakaguchi, K.; Shabanowitz, J.; Appella, E.; Hunt, D.F.; Engelhard, V.H. HLA-A2.1-associated peptides from a mutant cell line: A second pathway of antigen presentation. Science 1992, 255, 1264–1266. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, K.; Bennich, H.; Johansson, S.G.; Ponten, J. Established immunoglobulin producing myeloma (IgE) and lymphoblastoid (IgG) cell lines from an IgE myeloma patient. Clin. Exp. Immunol. 1970, 7, 477–489. [Google Scholar]
- Britten, C.M.; Meyer, R.G.; Kreer, T.; Drexler, I.; Wolfel, T.; Herr, W. The use of HLA-A*0201-transfected K562 as standard antigen-presenting cells for CD8(+) T lymphocytes in IFN-gamma ELISPOT assays. J. Immunol. Methods 2002, 259, 95–110. [Google Scholar] [CrossRef]
- Khan, M.; Carmona, S.; Sukhumalchandra, P.; Roszik, J.; Philips, A.; Perakis, A.A.; Kerros, C.; Zhang, M.; Qiao, N.; John, L.S.S.; et al. Cathepsin G Is Expressed by Acute Lymphoblastic Leukemia and Is a Potential Immunotherapeutic Target. Front. Immunol. 2017, 8, 1975. [Google Scholar] [CrossRef]
- Campillo-Davo, D.; Fujiki, F.; Van den Bergh, J.M.; De Reu, H.; Smits, E.L.; Goosens, H.; Sugiyama, H.; Lion, E.; Berneman, Z.N.; Van Tendeloo, V.F. Efficient and non-genotoxic RNA-based engineering of human T cells using tumor-specific T cell receptors with minimal TCR mispairing. Front. Immunol. 2018, 9, 2503. [Google Scholar] [CrossRef] [Green Version]
- Versteven, M.; Van den Bergh, J.M.J.; Broos, K.; Fujiki, F.; Campillo-Davo, D.; De Reu, H.; Morimoto, S.; Lecocq, Q.; Keyaerts, M.; Berneman, Z.; et al. A versatile T cell-based assay to assess therapeutic antigen-specific PD-1-targeted approaches. Oncotarget 2018, 9, 27797–27808. [Google Scholar] [CrossRef] [Green Version]
- Hasan, A.N.; Selvakumar, A.; O’Reilly, R.J. Artificial Antigen Presenting Cells: An Off the Shelf Approach for Generation of Desirable T-Cell Populations for Broad Application of Adoptive Immunotherapy. Adv. Genet. Eng. 2015, 4, 130. [Google Scholar]
- Bossi, G.; Gerry, A.B.; Paston, S.J.; Sutton, D.H.; Hassan, N.J.; Jakobsen, B.K. Examining the presentation of tumor-associated antigens on peptide-pulsed T2 cells. Oncoimmunology 2013, 2, e26840. [Google Scholar] [CrossRef] [Green Version]
- Ritz, U.; Seliger, B. The transporter associated with antigen processing (TAP): Structural integrity, expression, function, and its clinical relevance. Mol. Med. 2001, 7, 149–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweitzer, S.; Schneiders, A.M.; Langhans, B.; Kraas, W.; Jung, G.; Vidalin, O.; Inchauspe, G.; Sauerbruch, T.; Spengler, U. Flow cytometric analysis of peptide binding to major histocampatibility complex class I for hepatitis C virus core T-cell epitopes. Cytometry 2000, 41, 271–278. [Google Scholar] [CrossRef]
- Luft, T.; Rizkalla, M.; Tai, T.Y.; Chen, Q.; MacFarlan, R.I.; Davis, I.D.; Maraskovsky, E.; Cebon, J. Exogenous peptides presented by transporter associated with antigen processing (TAP)-deficient and TAP-competent cells: Intracellular loading and kinetics of presentation. J. Immunol. 2001, 167, 2529–2537. [Google Scholar] [CrossRef] [Green Version]
- Stanke, J.; Hoffmann, C.; Erben, U.; von Keyserling, H.; Stevanovic, S.; Cichon, G.; Schneider, A.; Kaufmann, A.M. A flow cytometry-based assay to assess minute frequencies of CD8+ T cells by their cytolytic function. J. Immunol. Methods 2010, 360, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Aptsiauri, N.; Doorduijn, E.M.; Garcia Lora, A.M.; van Hall, T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 2016, 39, 44–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leone, P.; Shin, E.C.; Perosa, F.; Vacca, A.; Dammacco, F.; Racanelli, V. MHC class I antigen processing and presenting machinery: Organization, function, and defects in tumor cells. J. Natl. Cancer Inst. 2013, 105, 1172–1187. [Google Scholar] [CrossRef] [Green Version]
- Van Camp, K.; Cools, N.; Stein, B.; Van de Velde, A.; Goossens, H.; Berneman, Z.N.; Van Tendeloo, V. Efficient mRNA electroporation of peripheral blood mononuclear cells to detect memory T cell responses for immunomonitoring purposes. J. Immunol. Methods 2010, 354, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Britten, C.M.; Meyer, R.G.; Frankenberg, N.; Huber, C.; Wolfel, T. The use of clonal mRNA as an antigenic format for the detection of antigen-specific T lymphocytes in IFN-gamma ELISPOT assays. J. Immunol. Methods 2004, 287, 125–136. [Google Scholar] [CrossRef]
- Etschel, J.K.; Huckelhoven, A.G.; Hofmann, C.; Zitzelsberger, K.; Maurer, K.; Bergmann, S.; Mueller-Schmucker, S.M.; Wittmann, J.; Spriewald, B.M.; Dorrie, J.; et al. HIV-1 mRNA electroporation of PBMC: A simple and efficient method to monitor T-cell responses against autologous HIV-1 in HIV-1-infected patients. J. Immunol. Methods 2012, 380, 40–55. [Google Scholar] [CrossRef]
- Shao, H.; Zhang, W.; Hu, Q.; Wu, F.; Shen, H.; Huang, S. TCR mispairing in genetically modified T cells was detected by fluorescence resonance energy transfer. Mol. Biol. Rep. 2010, 37, 3951–3956. [Google Scholar] [CrossRef]
- Al-Ramadi, B.K.; Jelonek, M.T.; Boyd, L.F.; Margulies, D.H.; Bothwell, A.L. Lack of strict correlation of functional sensitization with the apparent affinity of MHC/peptide complexes for the TCR. J. Immunol. 1995, 155, 662–673. [Google Scholar] [PubMed]
- Echchakir, H.; Dorothee, G.; Vergnon, I.; Menez, J.; Chouaib, S.; Mami-Chouaib, F. Cytotoxic T lymphocytes directed against a tumor-specific mutated antigen display similar HLA tetramer binding but distinct functional avidity and tissue distribution. Proc. Natl. Acad. Sci. USA 2002, 99, 9358–9363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, T.M.; Man, S.; Wang, E.C.; Williams, S.; Amos, N.; Gillespie, G.M.; Moss, P.A.; Borysiewicz, L.K. Functional differences between influenza A-specific cytotoxic T lymphocyte clones expressing dominant and subdominant TCR. Int. Immunol. 2001, 13, 1383–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palermo, B.; Campanelli, R.; Mantovani, S.; Lantelme, E.; Manganoni, A.M.; Carella, G.; Da Prada, G.; della Cuna, G.R.; Romagne, F.; Gauthier, L.; et al. Diverse expansion potential and heterogeneous avidity in tumor-associated antigen-specific T lymphocytes from primary melanoma patients. Eur. J. Immunol. 2001, 31, 412–420. [Google Scholar] [CrossRef] [Green Version]
- Bacher, P.; Scheffold, A. Flow-cytometric analysis of rare antigen-specific T cells. Cytom. A 2013, 83, 692–701. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campillo-Davo, D.; Versteven, M.; Roex, G.; De Reu, H.; van der Heijden, S.; Anguille, S.; Berneman, Z.N.; Van Tendeloo, V.F.I.; Lion, E. Rapid Assessment of Functional Avidity of Tumor-Specific T Cell Receptors Using an Antigen-Presenting Tumor Cell Line Electroporated with Full-Length Tumor Antigen mRNA. Cancers 2020, 12, 256. https://doi.org/10.3390/cancers12020256
Campillo-Davo D, Versteven M, Roex G, De Reu H, van der Heijden S, Anguille S, Berneman ZN, Van Tendeloo VFI, Lion E. Rapid Assessment of Functional Avidity of Tumor-Specific T Cell Receptors Using an Antigen-Presenting Tumor Cell Line Electroporated with Full-Length Tumor Antigen mRNA. Cancers. 2020; 12(2):256. https://doi.org/10.3390/cancers12020256
Chicago/Turabian StyleCampillo-Davo, Diana, Maarten Versteven, Gils Roex, Hans De Reu, Sanne van der Heijden, Sébastien Anguille, Zwi N. Berneman, Viggo F. I. Van Tendeloo, and Eva Lion. 2020. "Rapid Assessment of Functional Avidity of Tumor-Specific T Cell Receptors Using an Antigen-Presenting Tumor Cell Line Electroporated with Full-Length Tumor Antigen mRNA" Cancers 12, no. 2: 256. https://doi.org/10.3390/cancers12020256