Galectin-8 binds to the Farnesylated C-terminus of K-Ras4B and Modifies Ras/ERK Signaling and Migration in Pancreatic and Lung Carcinoma Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
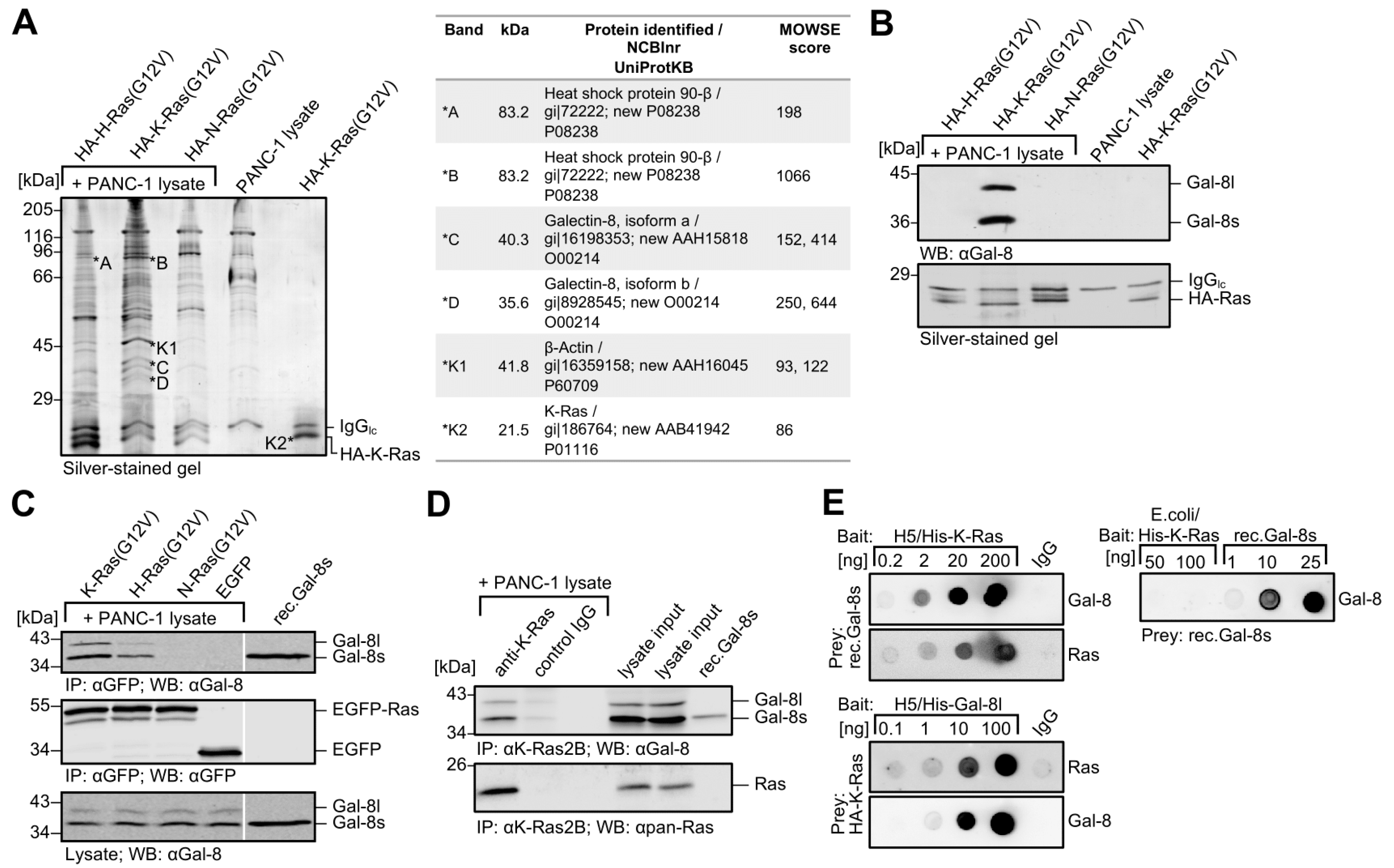
2.1. Identification of Galectin-8 as a Novel Binding Protein for K-Ras
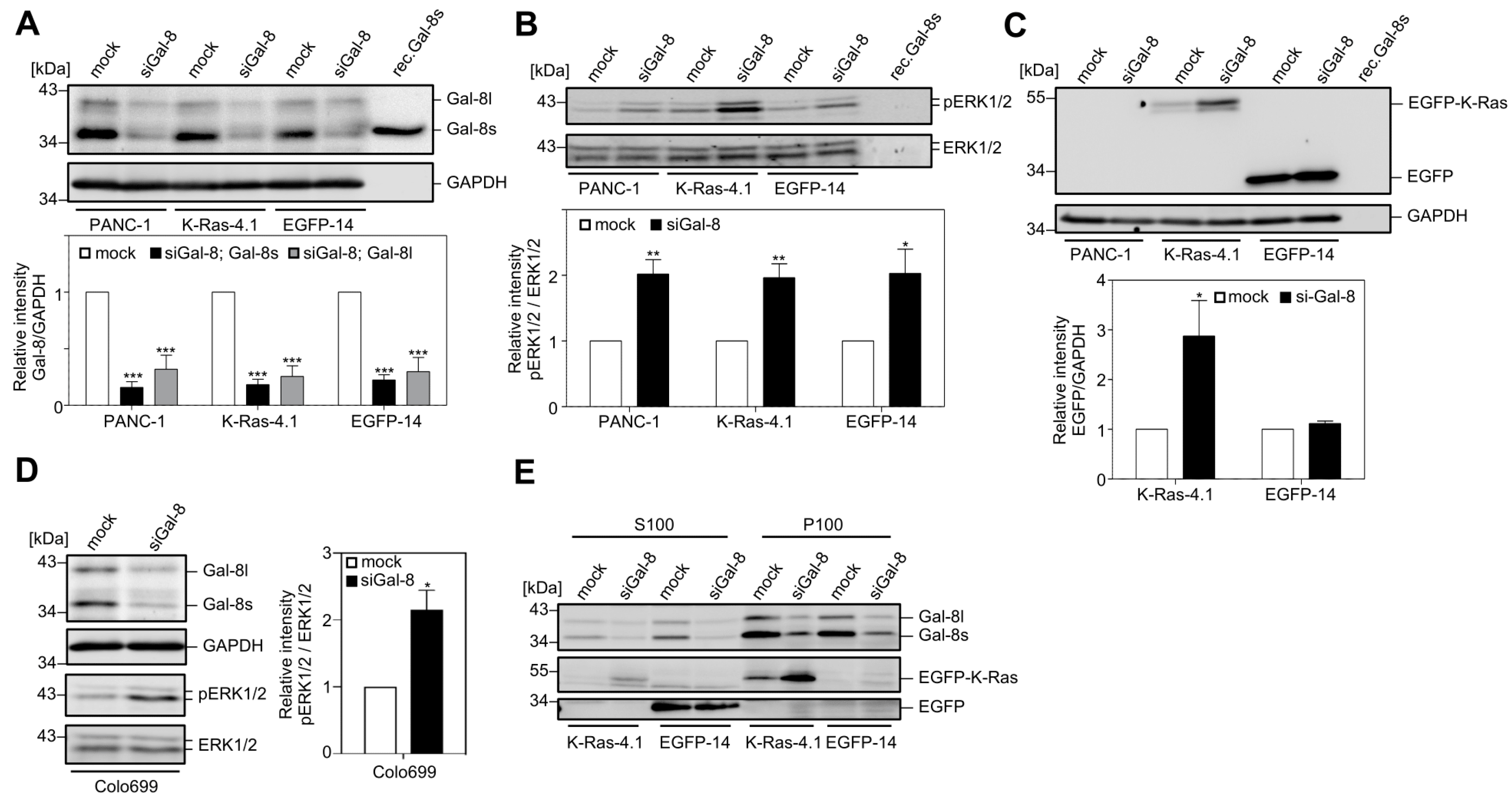
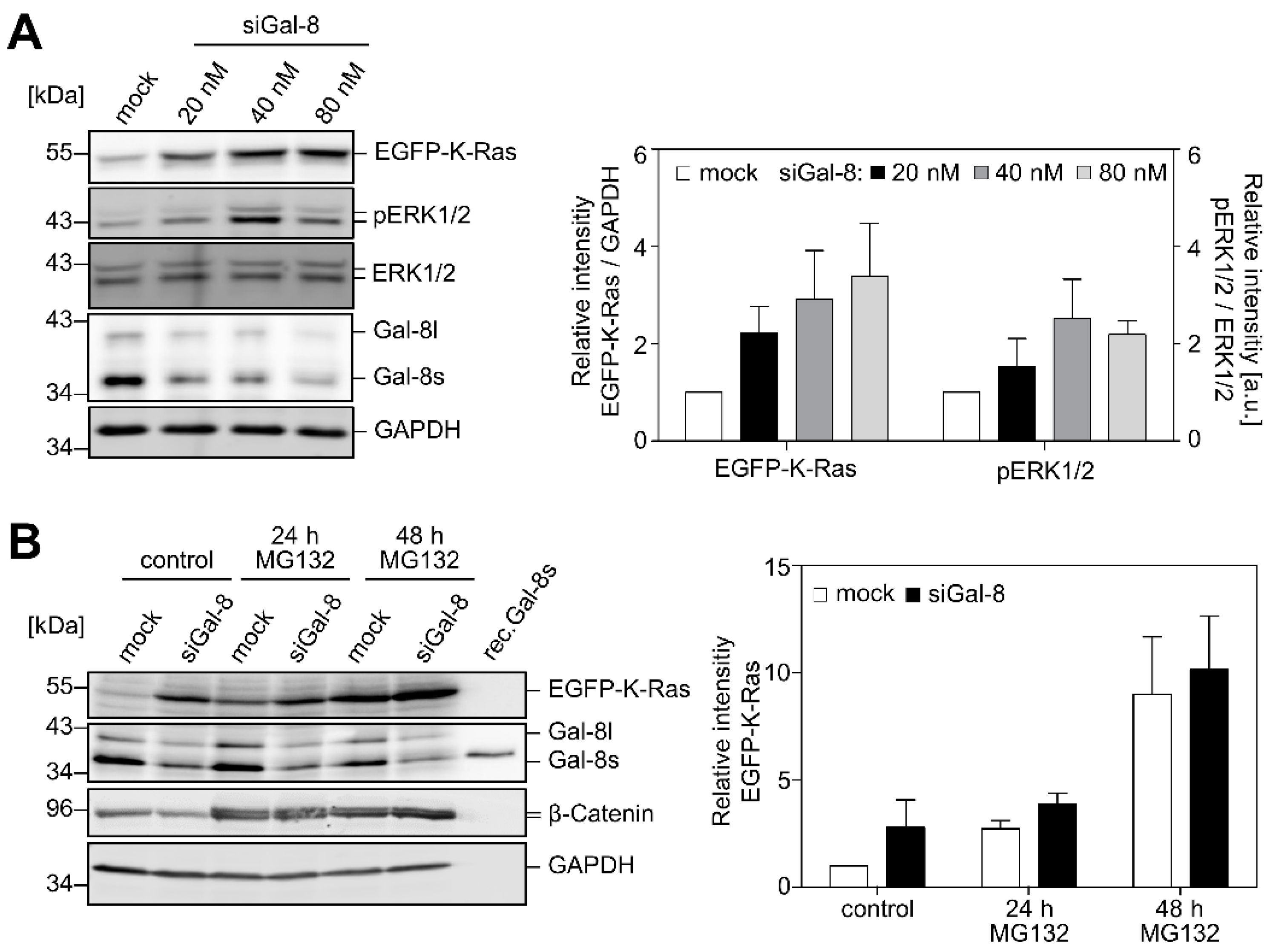
2.2. Downregulation of Galectin-8 Affects K-Ras and ERK1/2
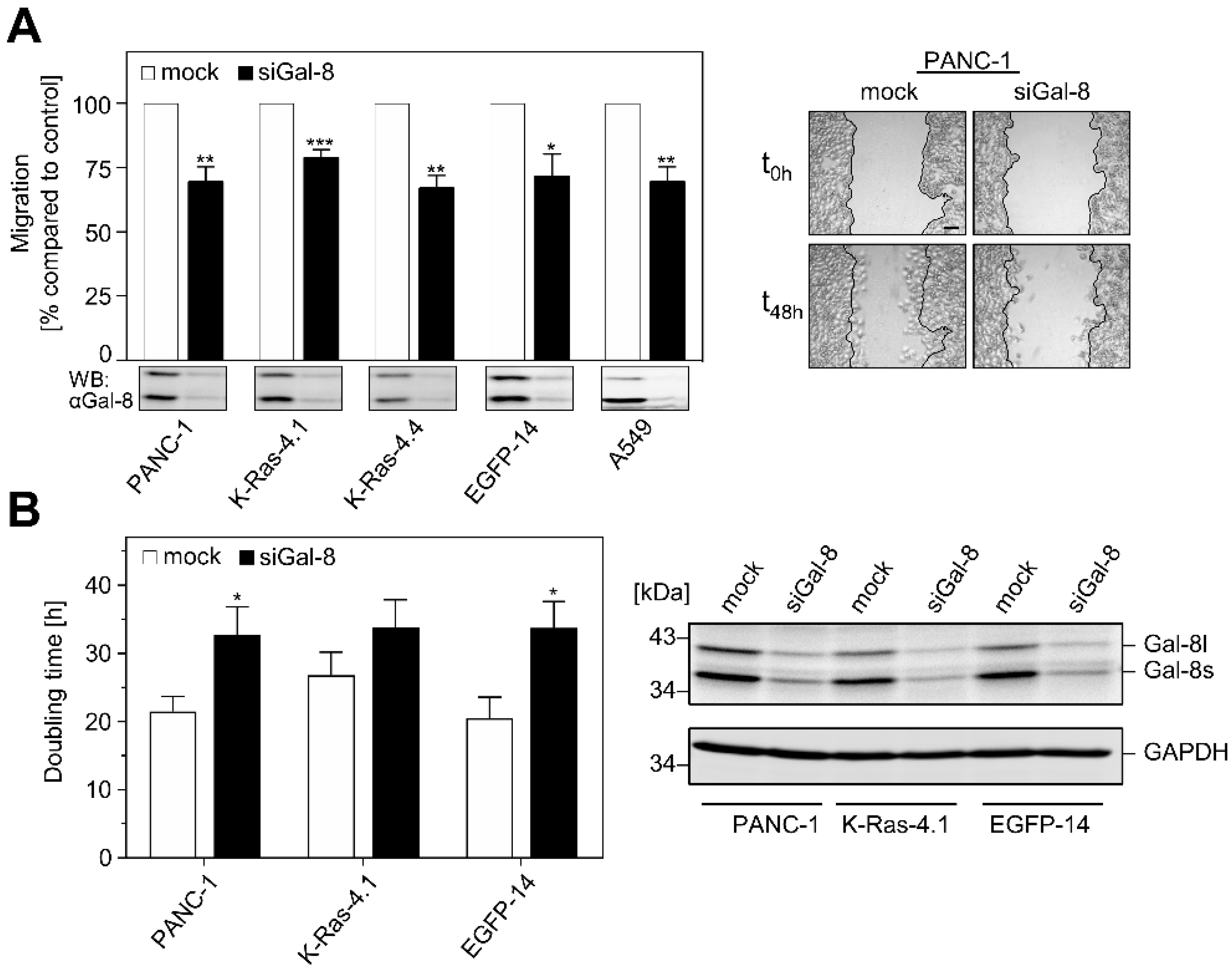
2.3. Impact of Galectin-8 Deletion on Cell Migration and Proliferation
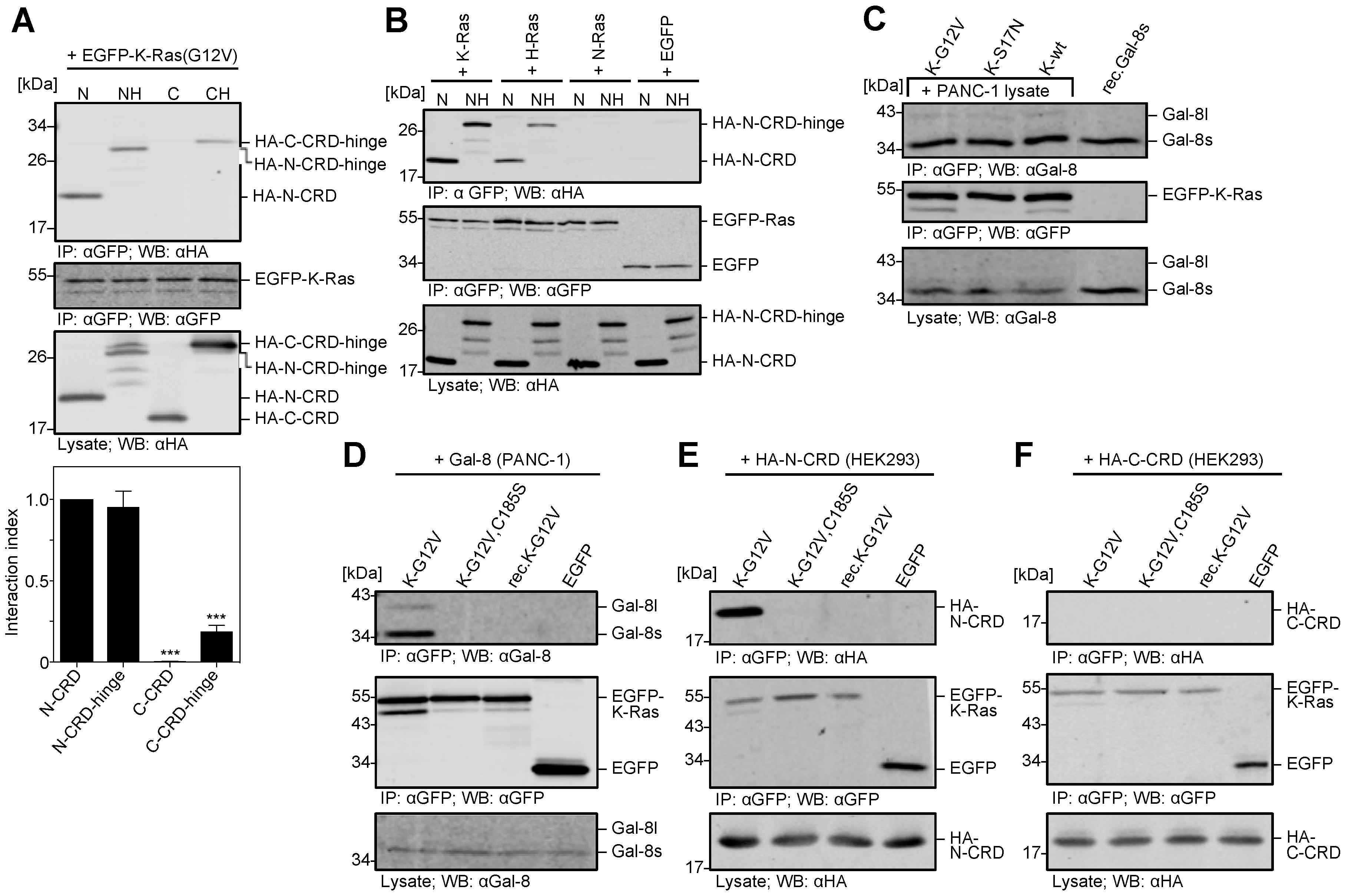
2.4. The N-CRD of Galectin-8 Interacts with Farnesylated K-Ras
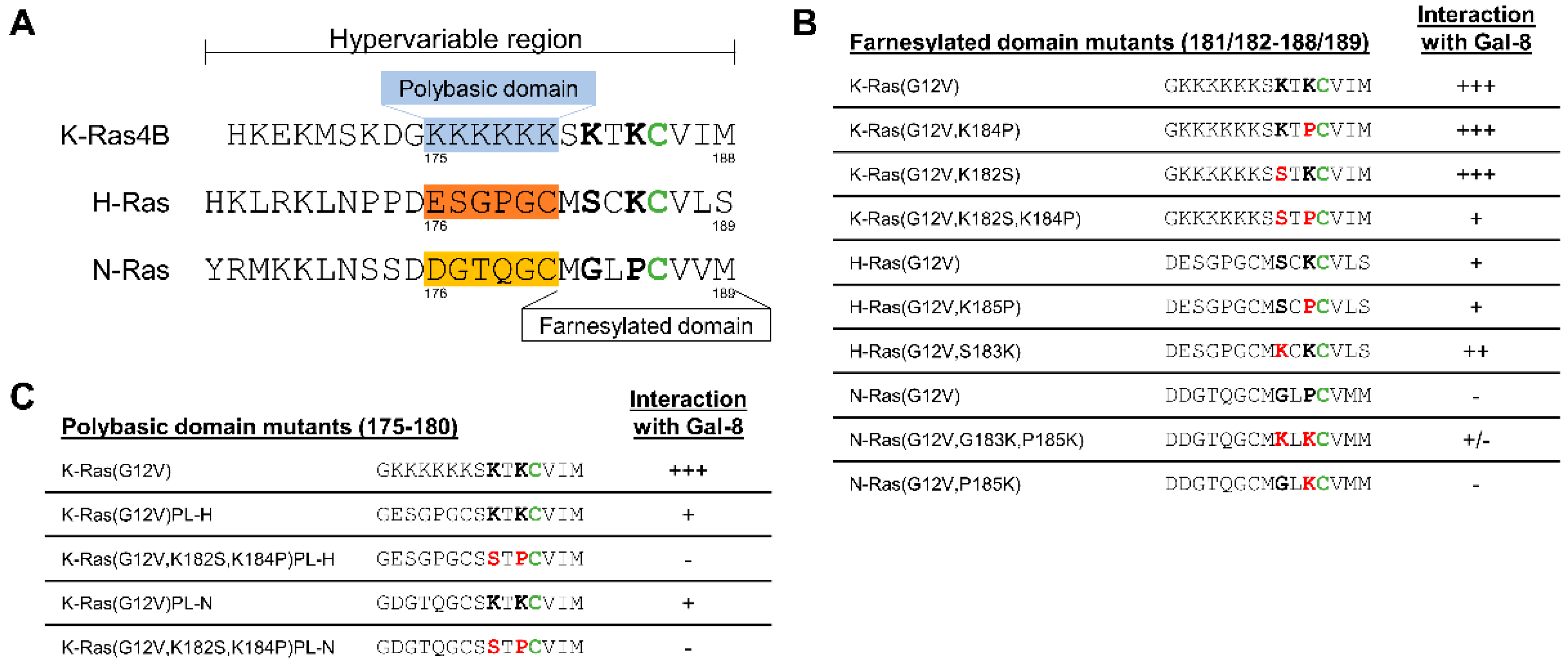
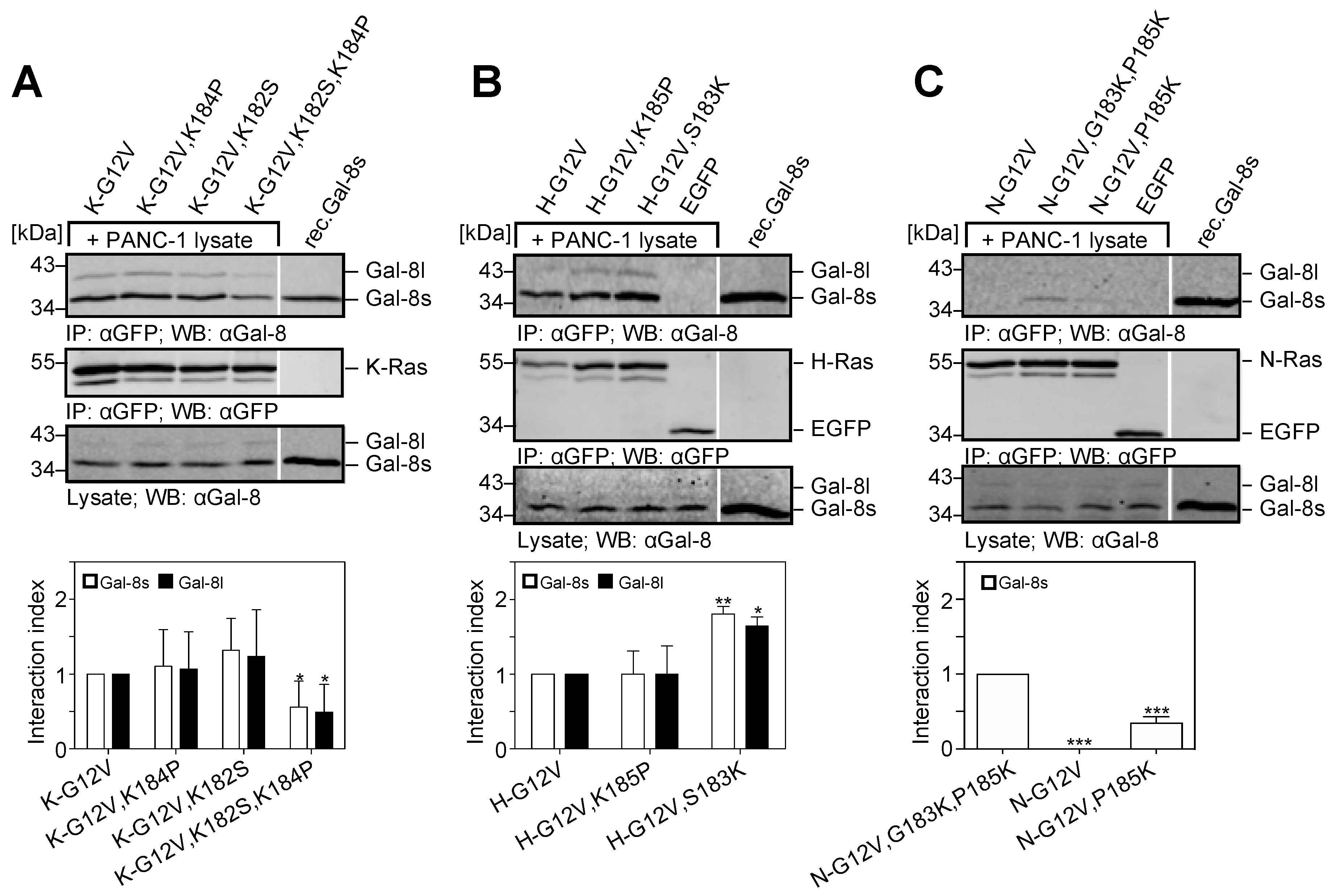
2.5. The Two Lysine Residues in the Farnesylated Domain of K-Ras Promote the Interaction with Galectin-8
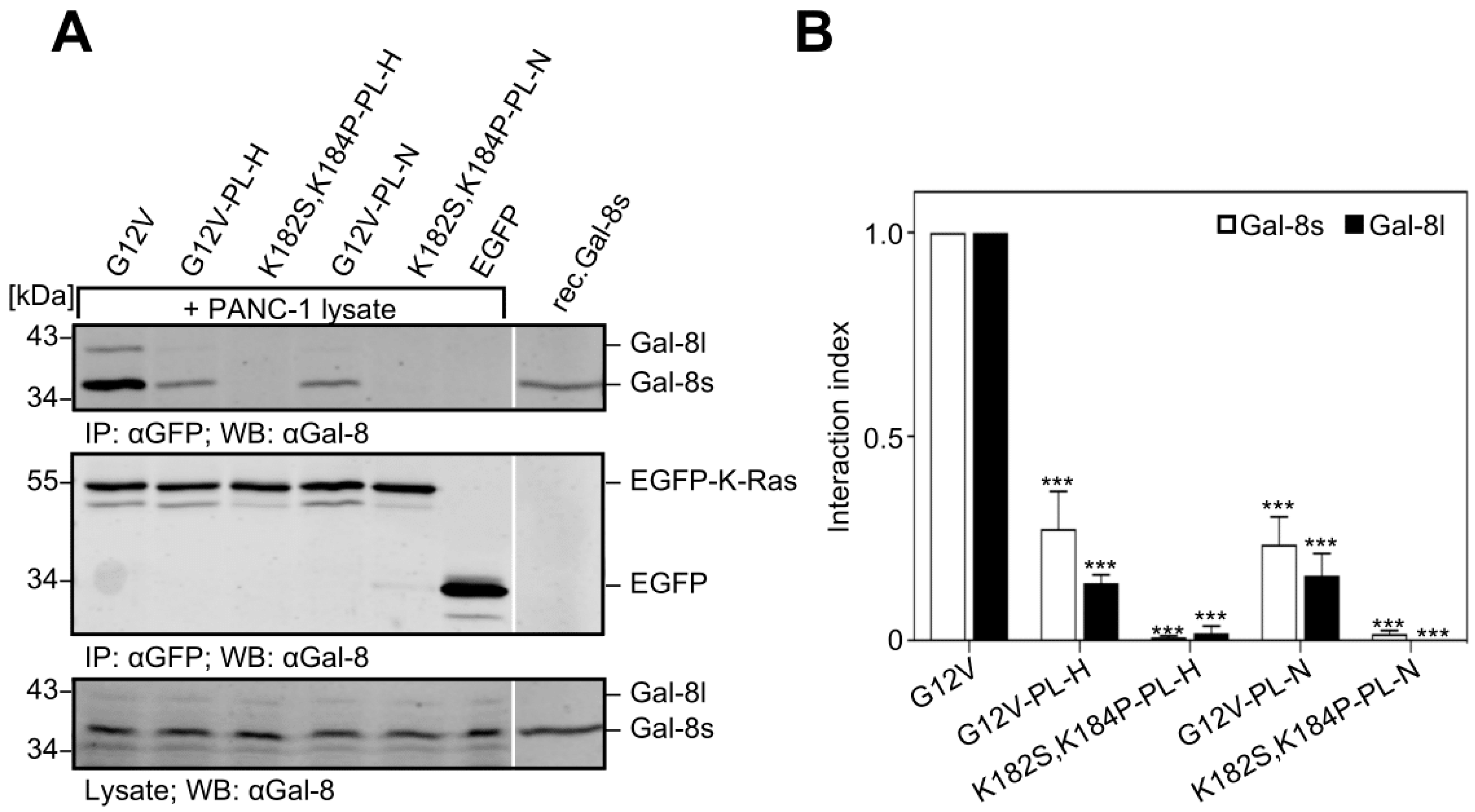
2.6. Removal of the Hexa-Lysine PBD Markedly Abrogates the K-Ras-Galectin-8 Interaction
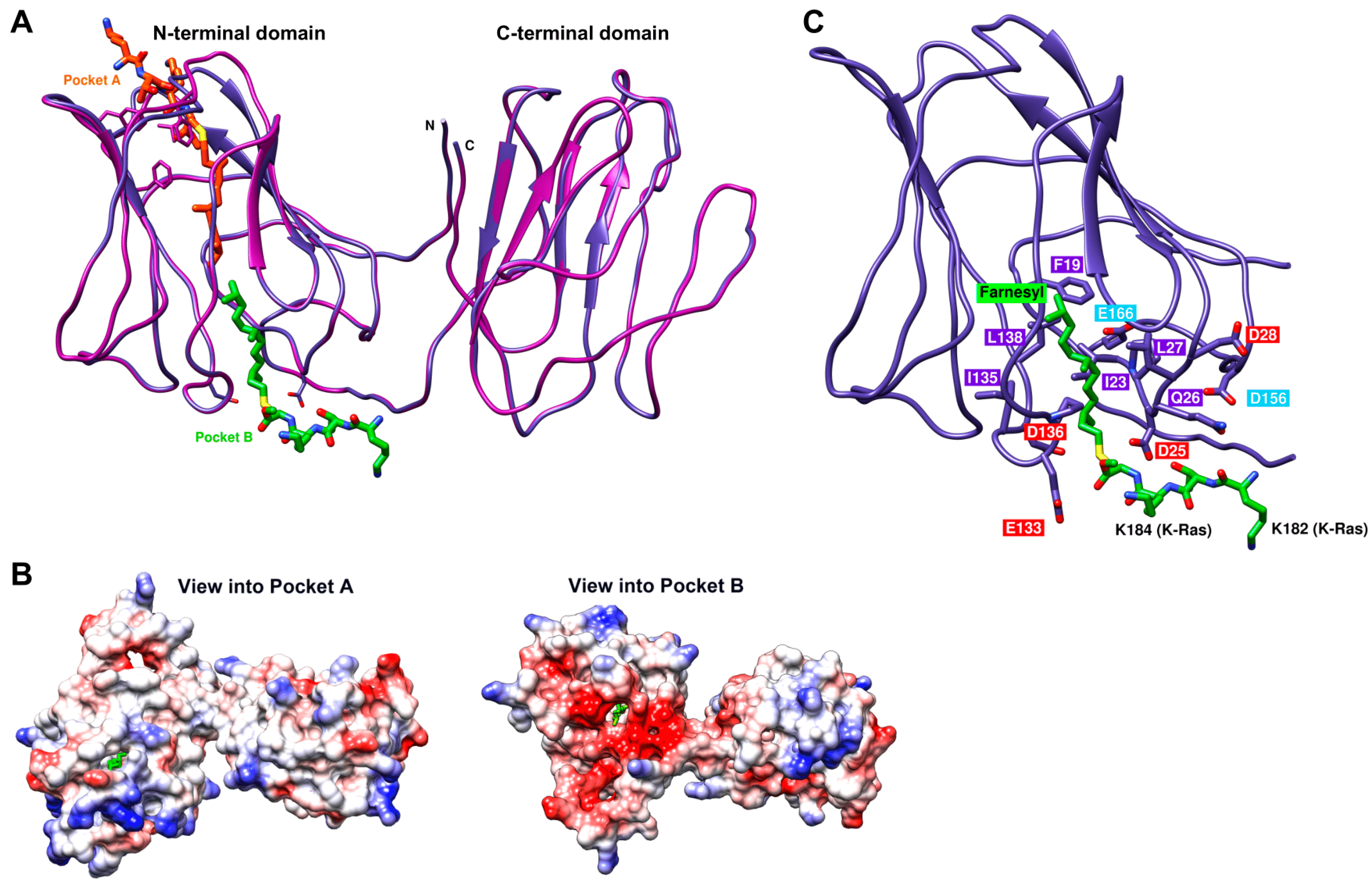
2.7. Structural Model of the Galectin-8 Binding Interface
3. Discussion
4. Materials and Methods
4.1. Antibodies, Inhibitors and Recombinant Proteins
4.2. Plasmids, Primers and Buffers
4.3. Cell Lines and Culture Conditions
4.4. Protein Lysate Preparations
4.5. Subcellular Fractionation
4.6. Purification of His-Tagged Proteins Using Ni-NTA Agarose Resin
4.7. Identification of Ras-Interacting Proteins
4.8. Protein Interaction and Co-Immunoprecipitation Assays
4.9. Ras Activity Assays
4.10. Migration and Proliferation Assays
4.11. Structural Modeling
4.12. Miscellaneous
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, F.D.; Lopes, M.S.; Zhou, M.; Court, H.; Ponce, O.; Fiordalisi, J.J.; Gierut, J.J.; Cox, A.D.; Haigis, K.M.; Philips, M.R. K-Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Proc. Natl. Acad. Sci. USA 2015, 112, 779–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nussinov, R.; Tsai, C.J.; Chakrabarti, M.; Jang, H. A New View of Ras Isoforms in Cancers. Cancer Res. 2016, 76, 18–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, T.L.; Zheng, H.; Walker, J.; Coloff, J.L.; Buss, J.E. Distinct rates of palmitate turnover on membrane-bound cellular and oncogenic H-ras. J. Biol. Chem. 2003, 278, 19292–19300. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, J.S.; Drake, K.R.; Rogers, C.; Wright, L.; Lippincott-Schwartz, J.; Philips, M.R.; Kenworthy, A.K. Depalmitoylated Ras traffics to and from the Golgi complex via a nonvesicular pathway. J. Cell Biol. 2005, 170, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Rocks, O.; Peyker, A.; Kahms, M.; Verveer, P.J.; Koerner, C.; Lumbierres, M.; Kuhlmann, J.; Waldmann, H.; Wittinghofer, A.; Bastiaens, P.I. An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science 2005, 307, 1746–1752. [Google Scholar] [CrossRef]
- Hancock, J.F. Ras proteins: Different signals from different locations. Nat. Rev. Mol. Cell Biol. 2003, 4, 373–384. [Google Scholar] [CrossRef]
- Hancock, J.F.; Paterson, H.; Marshall, C.J. A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell 1990, 63, 133–139. [Google Scholar] [CrossRef]
- Wang, M.; Casey, P.J.; Cox, A.D.; Der, C.J.; Philips, M.R. Protein prenylation: Unique fats make their mark on biology. Nat. Rev. Mol. Cell Biol. 2016, 17, 110–122. [Google Scholar] [CrossRef]
- Bivona, T.G.; Quatela, S.E.; Bodemann, B.O.; Ahearn, I.M.; Soskis, M.J.; Mor, A.; Miura, J.; Wiener, H.H.; Wright, L.; Saba, S.G.; et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol. Cell 2006, 21, 481–493. [Google Scholar] [CrossRef]
- Zimmermann, G.; Papke, B.; Ismail, S.; Vartak, N.; Chandra, A.; Hoffmann, M.; Hahn, S.A.; Triola, G.; Wittinghofer, A.; Bastiaens, P.I.; et al. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature 2013, 497, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Schmick, M.; Vartak, N.; Papke, B.; Kovacevic, M.; Truxius, D.C.; Rossmannek, L.; Bastiaens, P.I.H. KRas localizes to the plasma membrane by spatial cycles of solubilization, trapping and vesicular transport. Cell 2014, 157, 459–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenberg, S.; Beckett, A.J.; Prior, I.A.; Dekker, F.J.; Hedberg, C.; Waldmann, H.; Ehrlich, M.; Henis, Y.I. Raft protein clustering alters N-Ras membrane interactions and activation pattern. Mol. Cell Biol. 2011, 31, 3938–3952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prior, I.A.; Hancock, J.F. Ras trafficking, localization and compartmentalized signalling. Semin. Cell Dev. Biol. 2012, 23, 145–153. [Google Scholar] [CrossRef] [Green Version]
- Abankwa, D.; Gorfe, A.A.; Inder, K.; Hancock, J.F. Ras membrane orientation and nanodomain localization generate isoform diversity. Proc. Natl. Acad. Sci. USA 2010, 107, 1130–1135. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Harding, A.; Inder, K.; Plowman, S.; Parton, R.G.; Hancock, J.F. Plasma membrane nanoswitches generate high-fidelity Ras signal transduction. Nat. Cell Biol. 2007, 9, 905–914. [Google Scholar] [CrossRef]
- Zhou, Y.; Hancock, J.F.; Zhou, Y.; Liang, H.; Rodkey, T.; Ariotti, N.; Parton, R.G.; Hancock, J.F. Ras nanoclusters: Versatile lipid-based signaling platforms. Biochim. Biophys. Acta 2015, 1853, 841–849. [Google Scholar] [CrossRef] [Green Version]
- Dings, R.P.M.; Miller, M.C.; Griffin, R.J.; Mayo, K.H. Galectins as Molecular Targets for Therapeutic Intervention. Int. J. Mol. Sci. 2018, 19, 905. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.-Y.; Rabinovich, G.A.; Liu, F.-T. Galectins: Structure, function and therapeutic potential. Expert Rev. Mol. Med. 2008, 10. [Google Scholar] [CrossRef]
- Vladoiu, M.C.; Labrie, M.; St-Pierre, Y. Intracellular galectins in cancer cells: Potential new targets for therapy (Review). Int. J. Oncol. 2014, 44, 1001–1014. [Google Scholar] [CrossRef] [Green Version]
- Johannes, L.; Jacob, R.; Leffler, H. Galectins at a glance. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belanis, L.; Plowman, S.J.; Rotblat, B.; Hancock, J.F.; Kloog, Y. Galectin-1 is a novel structural component and a major regulator of h-ras nanoclusters. Mol. Biol. Cell 2008, 19, 1404–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalom-Feuerstein, R.; Plowman, S.J.; Rotblat, B.; Ariotti, N.; Tian, T.; Hancock, J.F.; Kloog, Y. K-ras nanoclustering is subverted by overexpression of the scaffold protein galectin-3. Cancer Res. 2008, 68, 6608–6616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elad-Sfadia, G.; Haklai, R.; Balan, E.; Kloog, Y. Galectin-3 augments K-Ras activation and triggers a Ras Ssignal that attenuates ERK but not Phosphoinositide 3-Kinase activity. J. Biol. Chem. 2004, 279, 34922–34930. [Google Scholar] [CrossRef] [Green Version]
- Blazevits, O.; Mideksa, Y.G.; Solman, M.; Ligabue, A.; Ariotti, N.; Nakhaeizadeh, H.; Fansa, E.K.; Papageorgiou, A.C.; Wittinghofer, A.; Ahmadian, M.R.; et al. Galectin-1 dimers can scaffold Raf-effectors to increase H-ras nanoclustering. Sci. Rep. 2016, 6, 24165. [Google Scholar] [CrossRef] [Green Version]
- Bhagatji, P.; Leventis, R.; Rich, R.; Lin, C.J.; Silvius, J.R. Multiple cellular proteins modulate the dynamics of K-Ras association with the plasma membrane. Biophys. J. 2010, 99, 3327–3335. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Ji, B.; Ramachandran, V.; Wang, H.; Hafley, M.; Logsdon, C.; Bresalier, R.S. Overexpressed galectin-3 in pancreatic cancer induces cell proliferation and invasion by binding Ras and activating Ras signaling. PLoS ONE 2012, 7, e42699. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.L.; Huang, E.Y.; Jhu, E.W.; Huang, Y.H.; Su, W.H.; Chuang, P.C.; Yang, K.D. Overexpression of galectin-3 enhances migration of colon cancer cells related to activation of the K-Ras-Raf-Erk1/2 pathway. J. Gastroenterol. 2013, 48, 350–359. [Google Scholar] [CrossRef]
- Weisz, B.; Giehl, K.; Gana-Weisz, M.; Egozi, Y.; Ben-Baruch, G.; Marciano, D.; Gierschik, P.; Kloog, Y. A new functional Ras antagonist inhibits human pancreatic tumor growth in nude mice. Oncogene 1999, 18, 2579–2588. [Google Scholar] [CrossRef] [Green Version]
- Levy, R.; Grafi-Cohen, M.; Kraiem, Z.; Kloog, Y.; Shalom-Feuerstein, R.; Plowman, S.J.; Rotblat, B.; Ariotti, N.; Tian, T.; Hancock, J.F.; et al. Galectin-3 promotes chronic activation of K-Ras and differentiation block in malignant thyroid carcinomas. Mol. Cancer Ther. 2010, 9, 2208–2219. [Google Scholar] [CrossRef] [Green Version]
- Ruvolo, P.P. Galectins as regulators of cell survival in the leukemia niche. Adv. Biol. Regul. 2019, 71, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Michael, J.V.; Goldfinger, L.E. Concepts and advances in cancer therapeutic vulnerabilities in RAS membrane targeting. Semin. Cancer Biol. 2019, 54, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Jang, H.; Zhang, J.; Nussinov, R.; Nussinov, R.; Tsai, C.J.; Jang, H. Raf-1 Cysteine-Rich Domain Increases the Affinity of K-Ras/Raf at the Membrane, Promoting MAPK Signaling. Is Nanoclustering essential for all oncogenic KRas pathways? Can it explain why wild-type KRas can inhibit its oncogenic variant? Structure 2018, 26, 513–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreissigacker, U.; Mueller, M.S.; Unger, M.; Siegert, P.; Genze, F.; Gierschik, P.; Giehl, K. Oncogenic K-Ras down-regulates Rac1 and RhoA activity and enhances migration and invasion of pancreatic carcinoma cells through activation of p38. Cell Signal. 2006, 18, 1156–1168. [Google Scholar] [CrossRef]
- Lu, A.; Tebar, F.; Alvarez-Moya, B.; Lopez-Alcala, C.; Calvo, M.; Enrich, C.; Agell, N.; Nakamura, T.; Matsuda, M.; Bachs, O. A clathrin-dependent pathway leads to KRas signaling on late endosomes en route to lysosomes. J. Cell Biol. 2009, 184, 863–879. [Google Scholar] [CrossRef] [Green Version]
- Koo, K.H.; Jeong, W.J.; Cho, Y.H.; Park, J.C.; Min do, S.; Choi, K.Y. K-Ras stabilization by estrogen via PKCdelta is involved in endometrial tumorigenesis. Oncotarget 2015, 6, 21328–21340. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.; Allam, U.S.; Ahsan, A.; Chen, G.; Krishnamurthy, P.M.; Marsh, K.; Rumschlag, M.; Shankar, S.; Whitehead, C.; Schipper, M.; et al. KRAS protein stability is regulated through SMURF2: UBCH5 complex-mediated beta-TrCP1 degradation. Neoplasia 2014, 16, 115–128. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.W.; Reid, D.; Parker, J.A.; Salter, S.; Knihtila, R.; Kuzmic, P.; Mattos, C. The small GTPases K-Ras, N-Ras, and H-Ras have distinct biochemical properties determined by allosteric effects. J. Biol. Chem. 2017, 292, 12981–12993. [Google Scholar] [CrossRef] [Green Version]
- Rotblat, B.; Niv, H.; Andre, S.; Kaltner, H.; Gabius, H.J.; Kloog, Y. Galectin-1(L11A) predicted from a computed galectin-1 farnesyl-binding pocket selectively inhibits Ras-GTP. Cancer Res. 2004, 64, 3112–3118. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, G.R.; Nassar, N.; Cerione, R.A. Structure of the Rho family GTP-binding protein Cdc42 in complex with the multifunctional regulator RhoGDI. Cell 2000, 100, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Elola, M.T.; Ferragut, F.; Cardenas Delgado, V.M.; Nugnes, L.G.; Gentilini, L.; Laderach, D.; Troncoso, M.F.; Compagno, D.; Wolfenstein-Todel, C.; Rabinovich, G.A. Expression, localization and function of galectin-8, a tandem-repeat lectin, in human tumors. Histol. Histopathol. 2014, 29, 1093–1105. [Google Scholar] [CrossRef] [PubMed]
- Satelli, A.; Rao, P.S.; Gupta, P.K.; Lockman, P.R.; Srivenugopal, K.S.; Rao, U.S. Varied expression and localization of multiple galectins in different cancer cell lines. Oncol. Rep. 2008, 19, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Zick, Y.; Eisenstein, M.; Goren, R.A.; Hadari, Y.R.; Levy, Y.; Ronen, D. Role of galectin-8 as a modulator of cell adhesion and cell growth. Glycoconj. J. 2002, 19, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Wdowiak, K.; Francuz, T.; Gallego-Colon, E.; Ruiz-Agamez, N.; Kubeczko, M.; Grochola, I.; Wojnar, J. Galectin Targeted Therapy in Oncology: Current Knowledge and Perspectives. Int. J. Mol. Sci. 2018, 19, 210. [Google Scholar] [CrossRef] [Green Version]
- Reticker-Flynn, N.E.; Malta, D.F.; Winslow, M.M.; Lamar, J.M.; Xu, M.J.; Underhill, G.H.; Hynes, R.O.; Jacks, T.E.; Bhatia, S.N. A combinatorial extracellular matrix platform identifies cell-extracellular matrix interactions that correlate with metastasis. Nat. Commun. 2012, 3, 1122. [Google Scholar] [CrossRef] [Green Version]
- Danguy, A.; Rorive, S.; Decaestecker, C.; Bronckart, Y.; Kaltner, H.; Hadari, Y.R.; Goren, R.; Zich, Y.; Petein, M.; Salmon, I.; et al. Immunohistochemical profile of galectin-8 expression in benign and malignant tumors of epithelial, mesenchymatous and adipous origins, and of the nervous system. Histol. Histopathol. 2001, 16, 861–868. [Google Scholar] [CrossRef]
- Prior, I.A.; Muncke, C.; Parton, R.G.; Hancock, J.F. Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J. Cell Biol. 2003, 160, 165–170. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.; van der Hoeven, D.; Zhou, Y.; Maekawa, M.; Ma, X.; Chen, W.; Fairn, G.D.; Hancock, J.F. Inhibition of acid Sphingomyelinase depletes cellular phosphatidylserine and mislocalizes K-Ras from the plasma membrane. Mol. Cell Biol. 2016, 36, 363–374. [Google Scholar] [CrossRef] [Green Version]
- Ashery, U.; Yizhar, O.; Rotblat, B.; Elad-Sfadia, G.; Barkan, B.; Haklai, R.; Kloog, Y. Spatiotemporal organization of Ras signaling: Rasosomes and the galectin switch. Cell Mol. Neurobiol. 2006, 26, 471–495. [Google Scholar] [CrossRef]
- Cho, K.J.; Park, J.H.; Piggott, A.M.; Salim, A.A.; Gorfe, A.A.; Parton, R.G.; Capon, R.J.; Lacey, E.; Hancock, J.F. Staurosporines disrupt phosphatidylserine trafficking and mislocalize Ras proteins. J. Biol. Chem. 2012, 287, 43573–43584. [Google Scholar] [CrossRef] [Green Version]
- van der Hoeven, D.; Cho, K.J.; Ma, X.; Chigurupati, S.; Parton, R.G.; Hancock, J.F. Fendiline inhibits K-Ras plasma membrane localization and blocks K-Ras signal transmission. Mol. Cell Biol. 2013, 33, 237–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandra, A.; Grecco, H.E.; Pisupati, V.; Perera, D.; Cassidy, L.; Skoulidis, F.; Ismail, S.A.; Hedberg, C.; Hanzal-Bayer, M.; Venkitaraman, A.R.; et al. The GDI-like solubilizing factor PDEdelta sustains the spatial organization and signalling of Ras family proteins. Nat. Cell Biol. 2011, 14, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Moepps, B.; Thelen, M. Monitoring scavenging activity of chemokine receptors. Methods Enzymol. 2016, 570, 87–118. [Google Scholar] [CrossRef] [PubMed]
- Hala, M.; Cole, R.; Synek, L.; Drdova, E.; Pecenkova, T.; Nordheim, A.; Lamkemeyer, T.; Madlung, J.; Hochholdinger, F.; Fowler, J.E.; et al. An exocyst complex functions in plant cell growth in Arabidopsis and tobacco. Plant Cell 2008, 20, 1330–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markus, G.; Birgit, M.; Anne, Z.; Kristin, W.; Stefan, H.; Stefan, P. Cell migration analysis: Segmenting scratch assay images with level sets and support vector machines. Intern. J. Pattern Recognit. 2012, 45, 3154–3165. [Google Scholar] [CrossRef]
- Kim, B.W.; Hong, S.B.; Kim, J.H.; Kwon, D.H.; Song, H.K. Structural basis for recognition of autophagic receptor NDP52 by the sugar receptor galectin-8. Nat. Commun. 2013, 4, 1613. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Yamashita, S.; Teraoka, M.; Itoh, A.; Nakakita, S.; Nishi, N.; Kamitori, S. X-ray structure of a protease-resistant mutant form of human galectin-8 with two carbohydrate recognition domains. FEBS J. 2012, 279, 3937–3951. [Google Scholar] [CrossRef]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Krissinel, E.; Henrick, K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2256–2268. [Google Scholar] [CrossRef]
- Dharmaiah, S.; Bindu, L.; Tran, T.H.; Gillette, W.K.; Frank, P.H.; Ghirlando, R.; Nissley, D.V.; Esposito, D.; McCormick, F.; Stephen, A.G.; et al. Structural basis of recognition of farnesylated and methylated KRAS4b by PDEdelta. Proc. Natl. Acad. Sci. USA 2016, 113, E6766–E6775. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meinohl, C.; Barnard, S.J.; Fritz-Wolf, K.; Unger, M.; Porr, A.; Heipel, M.; Wirth, S.; Madlung, J.; Nordheim, A.; Menke, A.; et al. Galectin-8 binds to the Farnesylated C-terminus of K-Ras4B and Modifies Ras/ERK Signaling and Migration in Pancreatic and Lung Carcinoma Cells. Cancers 2020, 12, 30. https://doi.org/10.3390/cancers12010030
Meinohl C, Barnard SJ, Fritz-Wolf K, Unger M, Porr A, Heipel M, Wirth S, Madlung J, Nordheim A, Menke A, et al. Galectin-8 binds to the Farnesylated C-terminus of K-Ras4B and Modifies Ras/ERK Signaling and Migration in Pancreatic and Lung Carcinoma Cells. Cancers. 2020; 12(1):30. https://doi.org/10.3390/cancers12010030
Chicago/Turabian StyleMeinohl, Christopher, Sarah J. Barnard, Karin Fritz-Wolf, Monika Unger, Andreea Porr, Marisa Heipel, Stefanie Wirth, Johannes Madlung, Alfred Nordheim, Andre Menke, and et al. 2020. "Galectin-8 binds to the Farnesylated C-terminus of K-Ras4B and Modifies Ras/ERK Signaling and Migration in Pancreatic and Lung Carcinoma Cells" Cancers 12, no. 1: 30. https://doi.org/10.3390/cancers12010030