Connecting Cancer Pathways to Tumor Engines: A Stratification Tool for Colorectal Cancer Combining Human In Vitro Tissue Models with Boolean In Silico Models

 , and
, and
Abstract
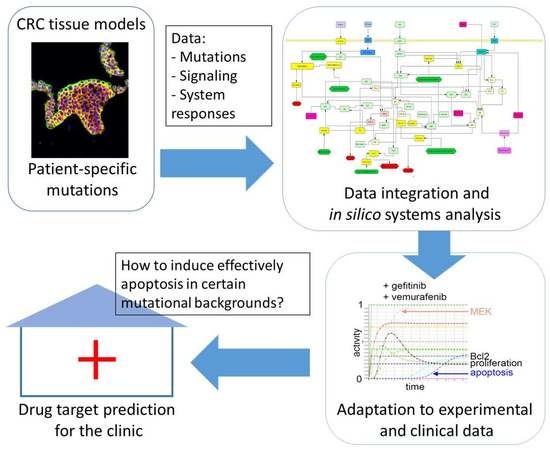
:
1. Introduction
2. Results
2.1. Characterization of Low Passage Cell Lines and Comparison of 3D Tissue Morphology with Cancerous Specimens
2.2. Proliferation and Apoptosis Responses upon Treatment
2.3. Comparison of Signaling Changes upon Treatment between Different Models
2.4. In silico System Responses Reflecting 3D In Vitro Data by Integrating HROC87 Specific Mutations and Signaling Cascades
2.5. Breaking Resistance: In Silico Mode-of-Action Analysis in p53 and BRAF Mutation Background
3. Discussion
4. Materials and Methods
4.1. SISmuc Preparation
4.2. 2D Cell Culture and Cell Lines
4.3. 3D Cell Culture of In Vitro Models
4.4. Animal Models
4.5. Fluorescence Immunohistochemistry
4.6. Total Cell Number and Proliferation Rate
4.7. Protein Lysate Preparation and WB Analysis
4.8. M30 CytoDeathTM ELISA
4.9. Statistical Analysis
4.10. In Silico Simulations
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2D | ||||||||
| Figure 5 | EGFR | Erk | pEGFR | pErk | HGFR | Akt | pHGFR | pAkt |
| Ctrl. | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| G | 70 | 96 | 49 | 32 | 44 | 76 | 1 | 227 |
| V | 75 | 77 | 80 | 18 | 52 | 73 | 86 | 130 |
| GV | 63 | 94 | 53 | 2 | 26 | 86 | 2 | 314 |
| 3D | ||||||||
| Figure 5 | EGFR | Erk | pEGFR | pErk | HGFR | Akt | pHGFR | pAkt |
| Ctrl. | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| G | 111 | 128 | 36 | 40 | 91 | 114 | 67 | 74 |
| V | 81 | 83 | 86 | 7 | 72 | 65 | 78 | 75 |
| GV | 55 | 91 | 61 | 2 | 55 | 70 | 34 | 93 |
| PDX | ||||||||
| Figure 5 | EGFR | Erk | pEGFR | pErk | HGFR | Akt | pHGFR | pAkt |
| Ctrl. | 100 | 100 | 100 | 100 | 100 | 100 | - | 100 |
| G | 70 | 102 | 37 | 38 | 55 | 134 | - | 27 |
| V | 53 | 67 | 32 | 10 | 55 | 88 | - | 17 |
| GV | 77 | 104 | 22 | 4 | 55 | 89 | - | 18 |




Appendix A.1. Ideal-Typic Transformation from a Healthy Epithelium to a Colon Carcinoma
| Stage 0 | Stage 1 | Stage 2 | Stage 3 | Stage 4 | Stage 5 | Stage 6 |
|---|---|---|---|---|---|---|
| normal epithelium | dysplastic aberrant cryptic foci | early/initial adenoma | intermediate adenoma | late adenoma | carcinoma | metastasis |
| APC (β-catenin) | COX-2 | KRAS | DCC/loss of 18q | p53 | E-cadherin/BAX |
Appendix A.2. Bioinformatics Mod$eling and Systems Biology Data
| Western Blot Data (in Comparison to Untreated Control) | Proliferation Value in 3D | Apoptosis in 3D Compared to Ctrl | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Ctrl | Gef | Vem | Gef+Vem | HROC87 | HROC87 | ||||
| pEGFR | medium | ↓↓ | ↓ | ↓↓ | Ctrl | 0.5 | Ctrl | weak | |
| pErk | strong | ↓ | ↓ | ↓↓ | Gef | 0.5 | Gef | ↑ | |
| pHGFR | strong | ↓ | 0 | ↓↓ | Vem | 0.5 | Vem | ↑ | |
| pAkt | weak | ↓ | 0 | ↑ | Gef + Vem | 0.2 | Gef+Vem | ↑ | |

Appendix A.3. Simulation of Different Treatment Scenarios

References
- World Health Organization. Globocan 2012: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012 v1.0. Available online: https://publications.iarc.fr/Databases/Iarc-Cancerbases/GLOBOCAN-2012-Estimated-Cancer-Incidence-Mortality-And-Prevalence-Worldwide-In-2012-V1.0-2012ISBN-13(Database)978-92-832-2447-1 (accessed on 23 December 2019).
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic signaling pathways in the cancer genome atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Pietrantonio, F.; Petrelli, F.; Coinu, A.; Di Bartolomeo, M.; Borgonovo, K.; Maggi, C.; Cabiddu, M.; Iacovelli, R.; Bossi, I.; Lonati, V.; et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: A meta-analysis. Eur. J. Cancer 2015, 51, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Ura, T.; Shibata, N.; Takahari, D.; Shitara, K.; Nomura, M.; Kondo, C.; Mizota, A.; Utsunomiya, S.; Muro, K.; et al. BRAF mutation is a powerful prognostic factor in advanced and recurrent colorectal cancer. Br. J. Cancer 2011, 104, 856–862. [Google Scholar] [CrossRef] [Green Version]
- Thiel, A.; Heinonen, M.; Kantonen, J.; Gylling, A.; Lahtinen, L.; Korhonen, M.; Kytölä, S.; Mecklin, J.P.; Orpana, A.; Peltomäki, P.; et al. BRAF mutation in sporadic colorectal cancer and Lynch syndrome. Virchows Arch. 2013, 463, 613–621. [Google Scholar]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [Green Version]
- Nagaraja, A.K.; Bass, A.J. Hitting the target in BRAF-mutant colorectal cancer. J. Clin. Oncol. 2015, 33, 3990–3992. [Google Scholar] [CrossRef]
- Kopetz, S.; Desai, J.; Chan, E.; Hecht, J.R.; O’Dwyer, P.J.; Lee, R.J.; Morris, V.; Janku, F.; Dasari, A.; Chung, W.; et al. PLX4032 in metastatic colorectal cancer patients with mutant BRAF tumors. J. Clin. Oncol. 2010, 28, 3534. [Google Scholar] [CrossRef]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF (V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; Della Pelle, P.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly, K.; Brungs, D.; Szeto, E.; Epstein, R.J. Anticancer activity of combination targeted therapy using cetuximab plus vemurafenib for refractory BRAF (V600E)-mutant metastatic colorectal carcinoma. Curr. Oncol. 2014, 21, e151–e154. [Google Scholar] [CrossRef] [PubMed]
- Capalbo, C.; Marchetti, P.; Coppa, A.; Calogero, A.; Anastasi, E.; Buffone, A.; Belardinilli, F.; Gulino, M.; Frati, P.; Catalano, C.; et al. Vemurafenib and panitumumab combination tailored therapy in BRAF-mutated metastatic colorectal cancer: A case report. Cancer Biol. Ther. 2014, 15, 826–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaeger, R.D.; Cercek, A.; O’Reilly, E.M.; Reidy, D.L.; Kemeny, N.E.; Wolinsky, T.; Gollub, M.J.; Lacouture, M.E.; Rosen, N.; Vakiani, E.; et al. Pilot study of vemurafenib and panitumumab combination therapy in patients with BRAF V600E mutated metastatic colorectal cancer. J. Clin. Oncol. 2015, 33, 611. [Google Scholar] [CrossRef]
- Schlatter, R.; Philippi, N.; Wangorsch, G.; Pick, R.; Sawodny, O.; Borner, C.; Timmer, J.; Ederer, M.; Dandekar, T. Integration of Boolean models exemplified on hepatocyte signal transduction. Brief Bioinform. 2012, 13, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Buiga, P.; Elson, A.; Tabernero, L.; Schwartz, J.M. Regulation of dual specificity phosphatases in breast cancer during initial treatment with Herceptin: A Boolean model analysis. BMC Syst. Biol. 2018, 12, 11. [Google Scholar] [CrossRef] [Green Version]
- Gottlich, C.; Muller, L.C.; Kunz, M.; Schmitt, F.; Walles, H.; Walles, T.; Dandekar, T.; Dandekar, G.; Nietzer, S.L. A combined 3D tissue engineered in vitro/in silico lung tumor model for predicting drug effectiveness in specific mutational backgrounds. J. Vis. Exp. 2016, 6, e53885. [Google Scholar] [CrossRef] [Green Version]
- Stratmann, A.T.; Fecher, D.; Wangorsch, G.; Gottlich, C.; Walles, T.; Walles, H.; Dandekar, T.; Dandekar, G.; Nietzer, S.L. Establishment of a human 3D lung cancer model based on a biological tissue matrix combined with a Boolean in silico model. Mol. Oncol. 2014, 8, 351–365. [Google Scholar] [CrossRef]
- Danes, B.S.; Deangelis, P.; Traganos, F.; Melamed, M.R.; Alm, T. Demonstration of altered cellular DNA content distribution in long-term colon epithelial cell lines with colon cancer genotypes. Scand. J. Gastroenterol. 1988, 23, 840–846. [Google Scholar] [CrossRef]
- Bocsi, J.; Zalatnai, A. Establishment and long-term xenografting of human pancreatic carcinomas in immunosuppressed mice: Changes and stability in morphology, DNA ploidy and proliferation activity. J. Cancer Res. Clin. Oncol. 1999, 125, 9–19. [Google Scholar] [CrossRef]
- Lee, J.M.; Mhawech-Fauceglia, P.; Lee, N.; Parsanian, L.C.; Lin, Y.G.; Gayther, S.A.; Lawrenson, K. A three-dimensional microenvironment alters protein expression and chemosensitivity of epithelial ovarian cancer cells in vitro. Lab. Invest 2013, 93, 528–542. [Google Scholar] [PubMed] [Green Version]
- Zschenker, O.; Streichert, T.; Hehlgans, S.; Cordes, N. Genome-wide gene expression analysis in cancer cells reveals 3D growth to affect ECM and processes associated with cell adhesion but not DNA repair. PLoS ONE 2012, 7, e34279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meli, L.; Jordan, E.T.; Clark, D.S.; Linhardt, R.J.; Dordick, J.S. Influence of a three-dimensional, microarray environment on human cell culture in drug screening systems. Biomaterials 2012, 33, 9087–9096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komoto, C.; Nakamura, T.; Ohmoto, N.; Kobayashi, H.; Yagami, T.; Nishiguchi, K.; Iwaki, K.; Kuwahara, A.; Yamamori, M.; Okamura, N.; et al. Three-dimensional, but not two-dimensional, culture results in tumor growth enhancement after exposure to anticancer drugs. Kobe J. Med. Sci. 2007, 53, 335–343. [Google Scholar]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Maletzki, C.; Stier, S.; Gruenert, U.; Gock, M.; Ostwald, C.; Prall, F.; Linnebacher, M. Establishment, characterization and chemosensitivity of three mismatch repair deficient cell lines from sporadic and inherited colorectal carcinomas. PLoS ONE 2012, 7, e52485. [Google Scholar] [CrossRef] [Green Version]
- Maletzki, C.; Klier, U.; Marinkovic, S.; Klar, E.; Andra, J.; Linnebacher, M. Host defense peptides for treatment of colorectal carcinoma—A comparative in vitro and in vivo analysis. Oncotarget 2014, 5, 4467–4479. [Google Scholar] [CrossRef] [Green Version]
- Mullins, C.S.; Micheel, B.; Matschos, S.; Leuchter, M.; Burtin, F.; Krohn, M.; Hühns, M.; Klar, E.; Prall, F.; Linnebacher, M. Integrated biobanking and tumor model establishment of human colorectal carcinoma provides excellent tools for preclinical research. Cancers 2019, 11, 1520. [Google Scholar] [CrossRef] [Green Version]
- Groeber, F.; Engelhardt, L.; Lange, J.; Kurdyn, S.; Schmid, F.F.; Rucker, C.; Mielke, S.; Walles, H.; Hansmann, J. A first vascularized skin equivalent for as an alternative to animal experimentation. ALTEX 2016, 33, 415–422. [Google Scholar] [CrossRef] [Green Version]
- Schanz, J.; Pusch, J.; Hansmann, J.; Walles, H. Vascularised human tissue models: A new approach for the refinement of biomedical research. J. Biotechnol. 2010, 148, 56–63. [Google Scholar] [CrossRef]
- Scheller, K.; Dally, I.; Hartmann, N.; Munst, B.; Braspenning, J.; Walles, H. Upcyte® microvascular endothelial cells repopulate decellularized scaffold. Tissue Eng. Part C Methods 2013, 19, 57–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moll, C.; Reboredo, J.; Schwarz, T.; Appelt, A.; Schurlein, S.; Walles, H.; Nietzer, S. Tissue engineering of a human 3D in vitro tumor test system. J. Vis. Exp. 2013, e50460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nietzer, S.; Baur, F.; Sieber, S.; Hansmann, J.; Schwarz, T.; Stoffer, C.; Häfner, H.; Gasser, M.; Waaga-Gasser, A.M.; Walles, H.; et al. Mimicking metastases including tumor stroma: A new technique to generate a three-dimensional colorectal cancer model based on a biological decellularized intestinal scaffold. Tissue Eng. Part C Methods 2016, 22, 621–635. [Google Scholar] [CrossRef] [PubMed]
- Gottlich, C.; Kunz, M.; Zapp, C.; Nietzer, S.L.; Walles, H.; Dandekar, T.; Dandekar, G. A combined tissue-engineered/in silico signature tool patient stratification in lung cancer. Mol. Oncol. 2018, 12. [Google Scholar] [CrossRef] [PubMed]
- Lubtow, M.M.; Nelke, L.C.; Seifert, J.; Kuhnemundt, J.; Sahay, G.; Dandekar, G.; Nietzer, S.L.; Luxenhofer, R. Drug induced micellization into ultra-high capacity and stable curcumin nanoformulations: Physico-chemical characterization and evaluation in 2D and 3D in vitro models. J. Control. Release 2019, 303, 162–180. [Google Scholar] [CrossRef]
- Wallstabe, L.; Gottlich, C.; Nelke, L.C.; Kuhnemundt, J.; Schwarz, T.; Nerreter, T.; Einsele, H.; Walles, H.; Dandekar, G.; Nietzer, S.L.; et al. ROR1-CAR T cells are effective against lung and breast cancer in advanced microphysiologic 3D tumor models. JCI Insight 2019, 4, e126345. [Google Scholar] [CrossRef] [Green Version]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Song, G.; Ouyang, G.; Bao, S. The activation of Akt/PKB signaling pathway and cell survival. J. Cell Mol. Med. 2005, 9, 59–71. [Google Scholar] [CrossRef]
- Armaghany, T.; Wilson, J.D.; Chu, Q.; Mills, G. Genetic alterations in colorectal cancer. Gastrointest. Cancer Res. 2012, 5, 19–27. [Google Scholar]
- Chubb, D.; Broderick, P.; Dobbins, S.E.; Frampton, M.; Kinnersley, B.; Penegar, S.; Price, A.; Ma, Y.P.; Sherborne, A.L.; Palles, C.; et al. Rare disruptive mutations and their contribution to the heritable risk of colorectal cancer. Nat. Commun. 2016, 7, 11883. [Google Scholar] [CrossRef]
- Chang, Y.C.; Chang, J.G.; Liu, T.C.; Lin, C.Y.; Yang, S.F.; Ho, C.M.; Chen, W.T.; Chang, Y.S. Mutation analysis of 13 driver genes of colorectal cancer-related pathways in Taiwanese patients. World J. Gastroenterol. 2016, 22, 2314–2325. [Google Scholar] [CrossRef] [PubMed]
- Di Cara, A.; Garg, A.; De Micheli, G.; Xenarios, I.; Mendoza, L. Dynamic simulation of regulatory networks using SQUAD. BMC Bioinform. 2007, 8, 462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendell, J.C.; Atreya, C.E.; André, T.; Tabernero, J.; Gordon, M.S.; Bernards, R.; Van Cutsem, E.; Tejpar, S.; Sidhu, R.; Go, W.Y.; et al. Efficacy and tolerability in an open-label phase I/II study of MEK inhibitor trametinib (T), BRAF inhibitor dabrafenib (D), and anti-EGFR antibody panitumumab (P) in combination in patients (pts) with BRAF V600E mutated colorectal cancer (CRC). J. Clin. Oncol. 2014, 32, 3515. [Google Scholar] [CrossRef]
- ClinicalTrials.Gov. BRAF/MEK/EGFR Inhibitor Combination Study in Colorectal Cancer (CRC). Available online: https://clinicaltrials.gov/show/NCT01750918 (accessed on 23 December 2019).
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, binimetinib, and cetuximab in BRAF V600E-mutated colorectal cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bissell, M.J.; Radisky, D. Putting tumours in context. Nat. Rev. Cancer 2001, 1, 46–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debnath, J.; Brugge, J.S. Modelling glandular epithelial cancers in three-dimensional cultures. Nat. Rev. Cancer 2005, 5, 675–688. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, C.; Chen, R.; Matsumoto, T.; Schmelzle, T.; Brugge, J.S.; Polverini, P.J.; Mooney, D.J. Engineering tumors with 3D scaffolds. Nat. Methods 2007, 4, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Van Es, J.H.; Clevers, H. Generation and analysis of mouse intestinal tumors and organoids harboring APC and K-Ras mutations. Methods Mol. Biol. 2015, 1267, 125–144. [Google Scholar]
- Corcoran, R.B.; Atreya, C.E.; Falchook, G.S.; Kwak, E.L.; Ryan, D.P.; Bendell, J.C.; Hamid, O.; Messersmith, W.A.; Daud, A.; Kurzrock, R.; et al. Combined BRAF and MEK Inhibition with dabrafenib and trametinib in BRAF V600-mutant colorectal cancer. J. Clin. Oncol. 2015, 33, 4023–4031. [Google Scholar] [CrossRef] [Green Version]
- Fritsche-Guenther, R.; Witzel, F.; Kempa, S.; Brummer, T.; Sers, C.; Bluthgen, N. Effects of RAF inhibitors on PI3K/AKT signalling depend on mutational status of the RAS/RAF signalling axis. Oncotarget 2016, 7, 7960–7969. [Google Scholar] [CrossRef] [Green Version]
- Puri, N.; Salgia, R. Synergism of EGFR and c-Met pathways, cross-talk and inhibition, in non-small cell lung cancer. J. Carcinog. 2008, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Bonine-Summers, A.R.; Aakre, M.E.; Brown, K.A.; Arteaga, C.L.; Pietenpol, J.A.; Moses, H.L.; Cheng, N. Epidermal growth factor receptor plays a significant role in hepatocyte growth factor mediated biological responses in mammary epithelial cells. Cancer Biol. Ther. 2007, 6, 561–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spix, J.K.; Chay, E.Y.; Block, E.R.; Klarlund, J.K. Hepatocyte growth factor induces epithelial cell motility through transactivation of the epidermal growth factor receptor. Exp. Cell Res. 2007, 313, 3319–3325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirschi, B.; Gallmeier, E.; Ziesch, A.; Marschall, M.; Kolligs, F.T. Genetic targeting of B-RafV600E affects survival and proliferation and identifies selective agents against BRAF-mutant colorectal cancer cells. Mol. Cancer. 2014, 13, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oddo, D.; Sennott, E.M.; Barault, L.; Valtorta, E.; Arena, S.; Cassingena, A.; Filiciotto, G.; Marzolla, G.; Elez, E.; van Geel, R.M.; et al. Molecular landscape of acquired resistance to targeted therapy combinations in BRAF-mutant colorectal cancer. Cancer Res. 2016, 76, 4504–4515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provencal, M.; Berger-Thibault, N.; Labbe, D.; Veitch, R.; Boivin, D.; Rivard, G.E.; Gingras, D.; Béliveau, R. Tissue factor mediates the HGF/Met-induced anti-apoptotic pathway in DAOY medulloblastoma cells. J. Neurooncol. 2010, 97, 365–372. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Gale, C.M.; Lifshits, E.; Gonzales, A.J.; Shimamura, T.; Zhao, F.; Vincent, P.W.; Naumov, G.N.; Bradner, J.E.; et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007, 67, 11924–11932. [Google Scholar] [CrossRef] [Green Version]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [Green Version]
- Jeffers, M.; Rong, S.; Vande Woude, G.F. Hepatocyte growth factor/scatter factor-Met signaling in tumorigenicity and invasion/metastasis. J. Mol. Med. 1996, 74, 505–513. [Google Scholar] [CrossRef]
- Francone, T.D.; Landmann, R.G.; Chen, C.T.; Sun, M.Y.; Kuntz, E.J.; Zeng, Z.; Dematteo, R.P.; Paty, P.B.; Weiser, M.R. Novel xenograft model expressing human hepatocyte growth factor shows ligand-dependent growth of c-Met-expressing tumors. Mol. Cancer Ther. 2007, 6, 1460–1466. [Google Scholar] [CrossRef] [Green Version]
- Hata, A.N.; Engelman, J.A.; Faber, A.C. The BCL2 family: Key mediators of the apoptotic response to targeted anticancer therapeutics. Cancer Discov. 2015, 5, 475–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nehls, O.; Okech, T.; Hsieh, C.J.; Enzinger, T.; Sarbia, M.; Borchard, F.; Gruenagel, H.H.; Gaco, V.; Hass, H.G.; Arkenau, H.T.; et al. Studies on p53, BAX and Bcl-2 protein expression and microsatellite instability in stage III (UICC) colon cancer treated by adjuvant chemotherapy: Major prognostic impact of proapoptotic BAX. Br. J. Cancer 2007, 96, 1409–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Kortlever, R.M.; Sodir, N.M.; Wilson, C.H.; Burkhart, D.L.; Pellegrinet, L.; Brown Swigart, L.; Littlewood, T.D.; Evan, G. Myc cooperates with Ras by programming inflammation and immune suppression. Cell 2017, 171, 1301–1315. [Google Scholar] [CrossRef] [PubMed]
- Linke, K.; Schanz, J.; Hansmann, J.; Walles, T.; Brunner, H.; Mertsching, H. Engineered liver-like tissue on a capillarized matrix for applied research. Tissue Eng. 2007, 13, 2699–2707. [Google Scholar] [CrossRef] [PubMed]
- Wessel, D.; Flugge, U.I. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 1984, 138, 141–143. [Google Scholar] [CrossRef]
- Weinberg, R.A. The Biology of Cancer, 1st ed.; Garland Science: New York, NY, USA, 2007. [Google Scholar]
- Pinto, D.; Clevers, H. Wnt, stem cells and cancer in the intestine. Biol. Cell 2005, 97, 185–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Dubois, R.N. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene 2010, 29, 781–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fodde, R.; Smits, R.; Clevers, H. APC, signal transduction and genetic instability in colorectal cancer. Nat. Rev. Cancer 2001, 1, 55–67. [Google Scholar] [CrossRef]
- Sun, Q.; Hua, J.; Wang, Q.; Xu, W.; Zhang, J.; Kang, J.; Li, M. Expressions of GRP78 and Bax associate with differentiation, metastasis, and apoptosis in non-small cell lung cancer. Mol. Biol. Rep. 2012, 39, 6753–6761. [Google Scholar] [CrossRef]
- Philippi, N.; Walter, D.; Schlatter, R.; Ferreira, K.; Ederer, M.; Sawodny, O.; Timmer, J.; Borner, C.; Dandekar, T. Modeling system states in liver cells: Survival, apoptosis and their modifications in response to viral infection. BMC Syst. Biol. 2009, 3, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]







| Name of Network Node | untr. | +gef | +vem | +combi | +MEK-Inh | +BCL2-Inh |
|---|---|---|---|---|---|---|
| BRAF(V600)-Act | 0.7 | 0.7 | 0.7 | 0.7 | 0.7 | 0.7 |
| EGFR | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| HGFR | 0.7 | 0.7 | 0.7 | 0.7 | 0.7 | 0.7 |
| ERK-Inh | 0.26 | 0.3 | 0.3 | 0.41 | 0.26 | 0.26 |
| p53-Mut | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 |
| Bcl2 | 0.31 | 0.3467 | 0.305 | 0.2252 | ||
| c-Myc-Inh | 0.17 | 0.097 | 0.097 | 0.17 | 0.17 | |
| AKT | 0.2 | 0.16 | 0.2 | 0.24 | 0.2 | 0.2 |
| (EGFR) * | 0.3 | 0.4 | 0.3 | |||
| (HGFR) * | 0.6 | 0.4 | ||||
| c-Myc-Act | 0.09 | |||||
| Gefitinib | 1.0 | 1.0 | ||||
| Vemurafenib | 1.0 | 1.0 | ||||
| MEK-Inhibitor | 1.0 | |||||
| Bcl2-Inhibitor | 1.0 |
| Name of Network Node | untr. | +gef | +vem | +combi | +MEK-Inh | +BCL2-Inh |
|---|---|---|---|---|---|---|
| apoptosis | 0.21 | 0.35 | 0.35 | 0.35 | 0.99 | 0.99 |
| proliferation | 0.5 | 0.5 | 0.5 | 0.2 | 0.0 | 0.5 |
| (HGFR)* | 0.97 | 0.6 | 0.97 | 0.4 | 0.97 | 0.97 |
| ERK | 0.71 | 0.59 | 0.59 | 0.3 | 0.0 | 0.71 |
| (EGFR)* | 0.5 | 0.3 | 0.4 | 0.3 | 0.5 | 0.5 |
| AKT | 0.2 | 0.16 | 0.2 | 0.24 | 0.2 | 0.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baur, F.; Nietzer, S.L.; Kunz, M.; Saal, F.; Jeromin, J.; Matschos, S.; Linnebacher, M.; Walles, H.; Dandekar, T.; Dandekar, G. Connecting Cancer Pathways to Tumor Engines: A Stratification Tool for Colorectal Cancer Combining Human In Vitro Tissue Models with Boolean In Silico Models. Cancers 2020, 12, 28. https://doi.org/10.3390/cancers12010028
Baur F, Nietzer SL, Kunz M, Saal F, Jeromin J, Matschos S, Linnebacher M, Walles H, Dandekar T, Dandekar G. Connecting Cancer Pathways to Tumor Engines: A Stratification Tool for Colorectal Cancer Combining Human In Vitro Tissue Models with Boolean In Silico Models. Cancers. 2020; 12(1):28. https://doi.org/10.3390/cancers12010028
Chicago/Turabian StyleBaur, Florentin, Sarah L. Nietzer, Meik Kunz, Fabian Saal, Julian Jeromin, Stephanie Matschos, Michael Linnebacher, Heike Walles, Thomas Dandekar, and Gudrun Dandekar. 2020. "Connecting Cancer Pathways to Tumor Engines: A Stratification Tool for Colorectal Cancer Combining Human In Vitro Tissue Models with Boolean In Silico Models" Cancers 12, no. 1: 28. https://doi.org/10.3390/cancers12010028