Manoalide Preferentially Provides Antiproliferation of Oral Cancer Cells by Oxidative Stress-Mediated Apoptosis and DNA Damage

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
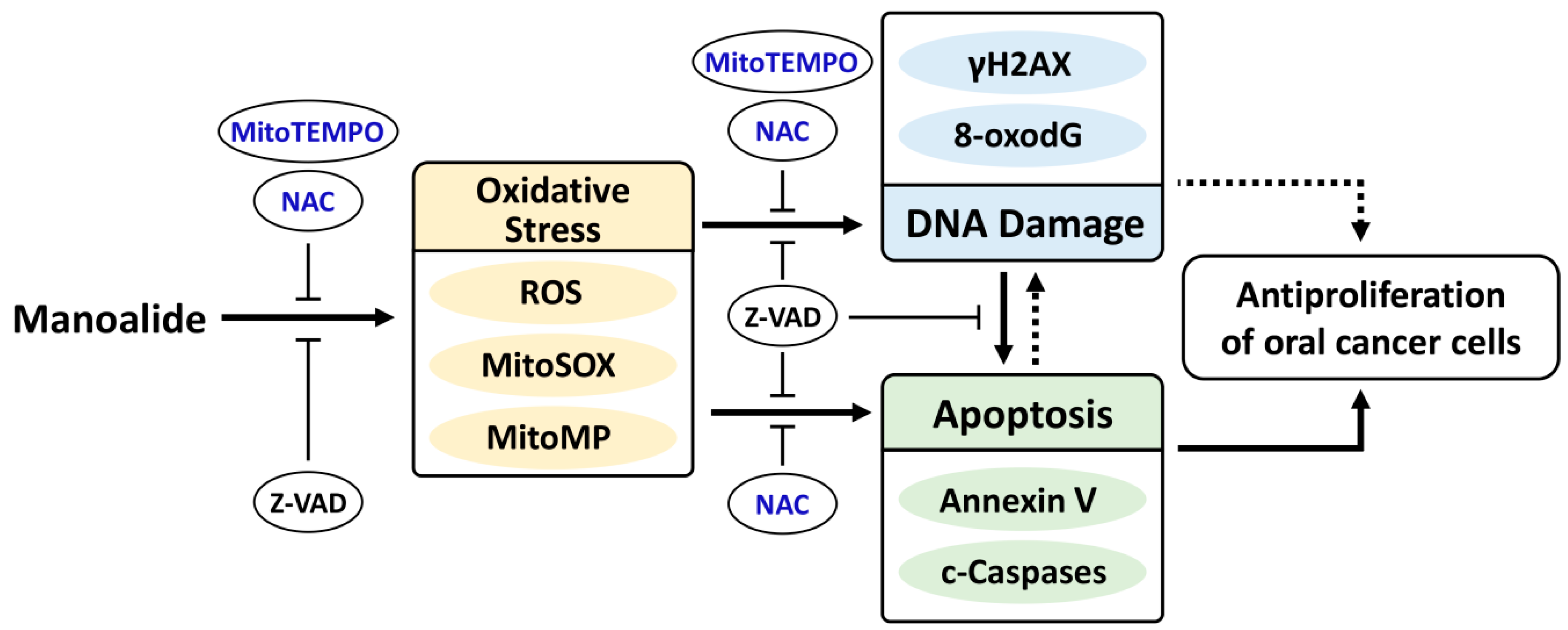
2. Results
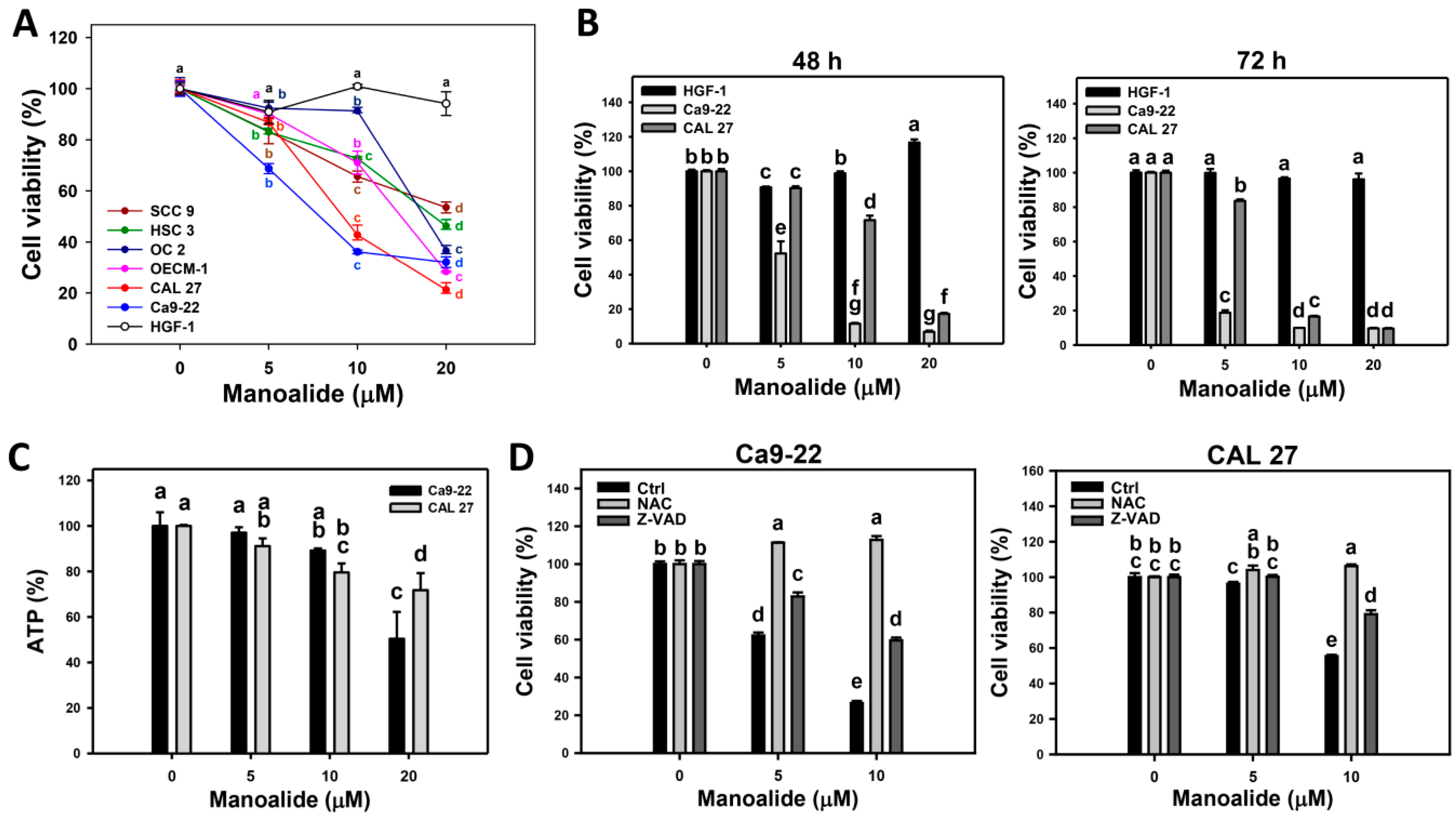
2.1. Cell Viability of Manoalide-Treated Oral Cancer and Normal Oral Cells with or Without Pretreatments of NAC or Z-VAD
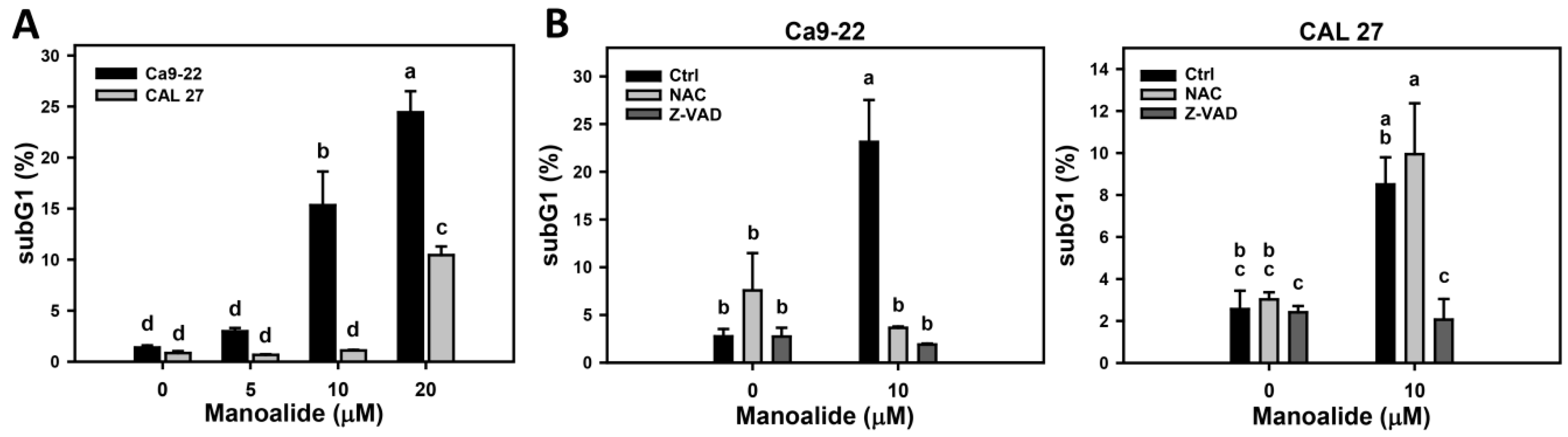
2.2. Cell Cycle Changes of Manoalide-Treated Oral Cancer Cells with or Without Pretreatments of NAC or Z-VAD
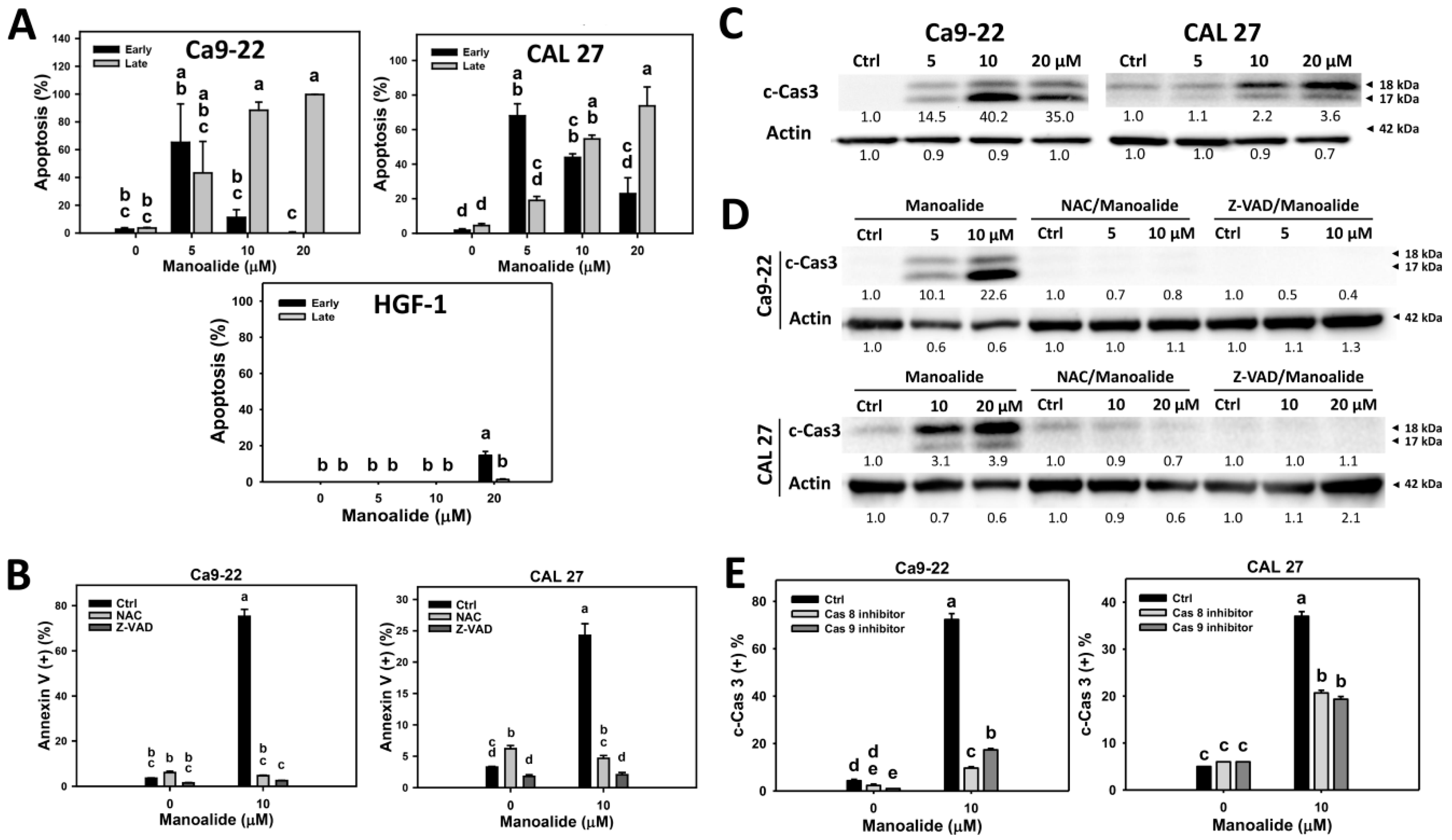
2.3. Apoptosis of Manoalide-Treated Oral Cancer Cells with or Without Pretreatments of NAC or Z-VAD
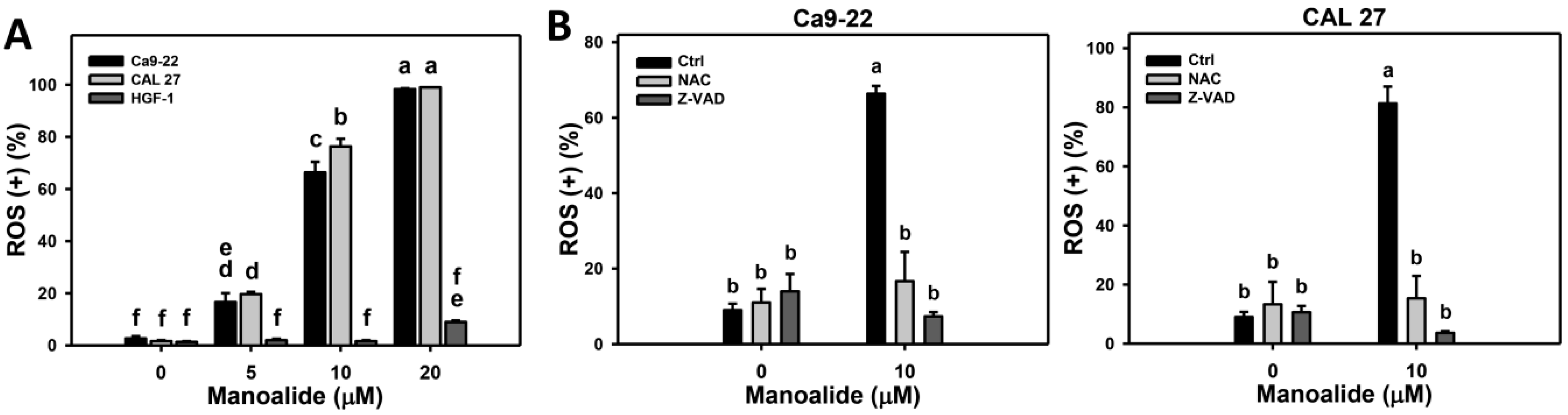
2.4. ROS Production of Manoalide-Treated Oral Cancer and Normal Oral Cells
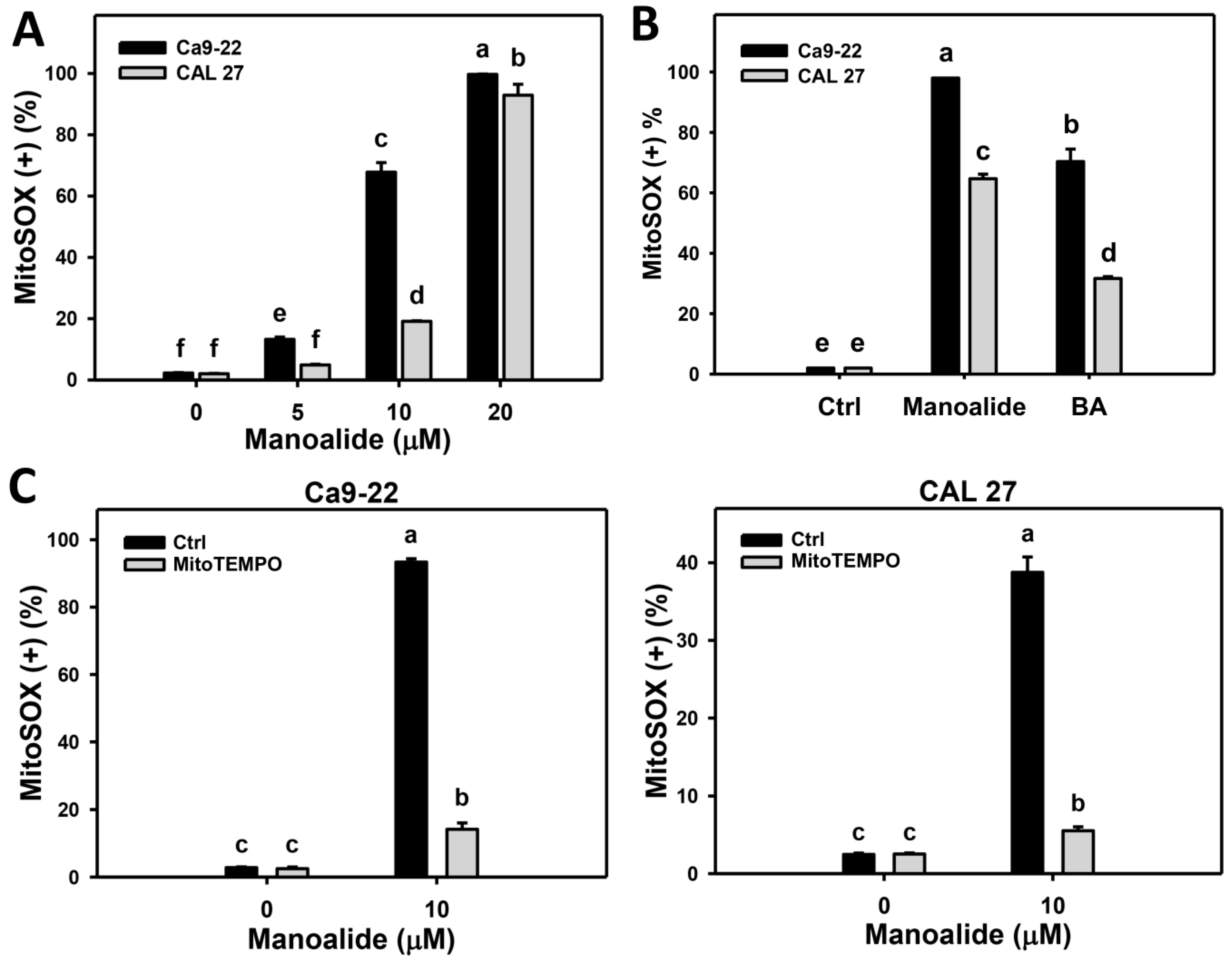
2.5. Mitochondrial Superoxide (MitoSOX) Production of Manoalide-Treated Oral Cancer Cells with or Without Pretreatment of MitoTEMPO
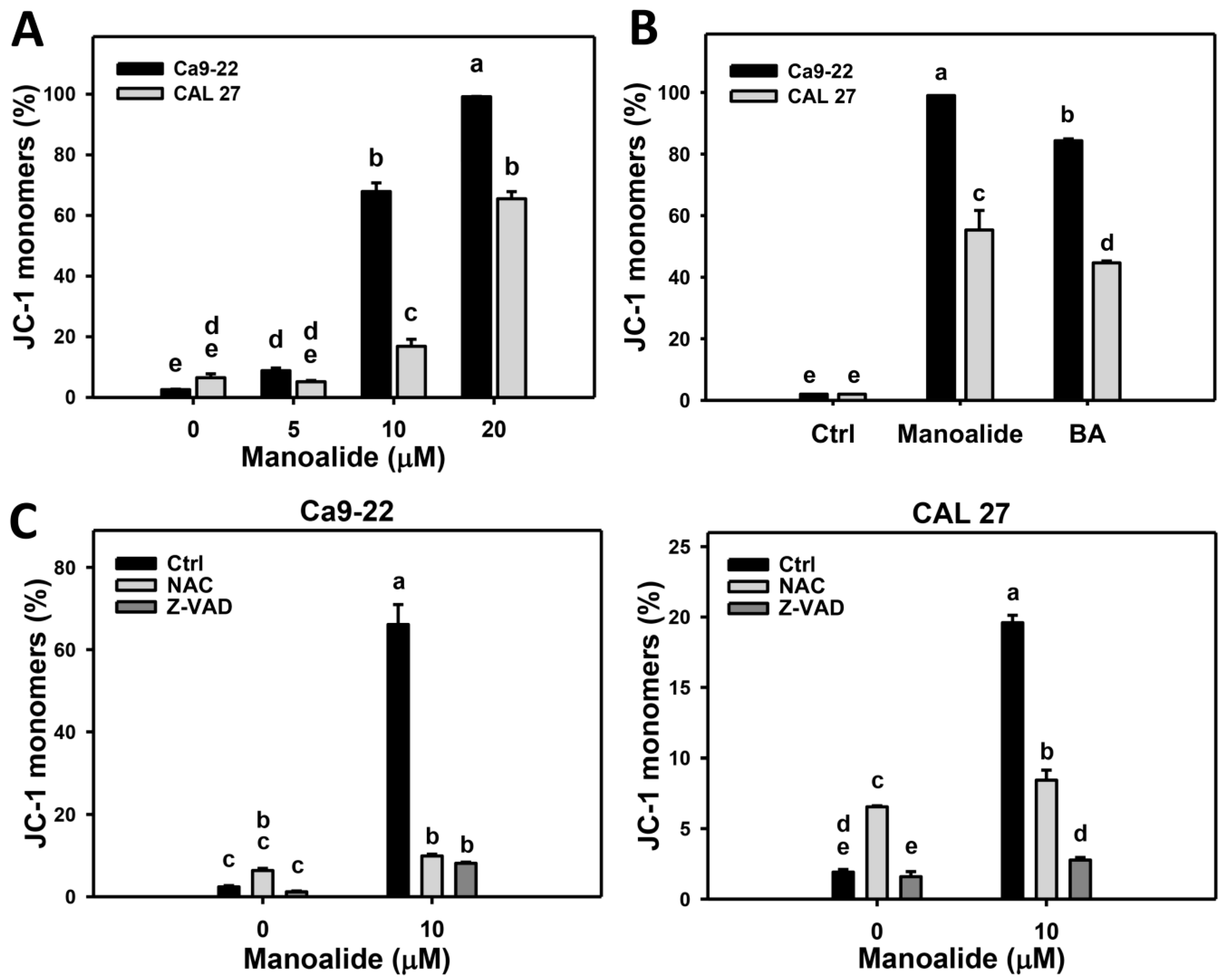
2.6. Membrane Potential (MitoMP) of Manoalide-Treated Oral Cancer Cells with or Without Pretreatments of NAC or Z-VAD
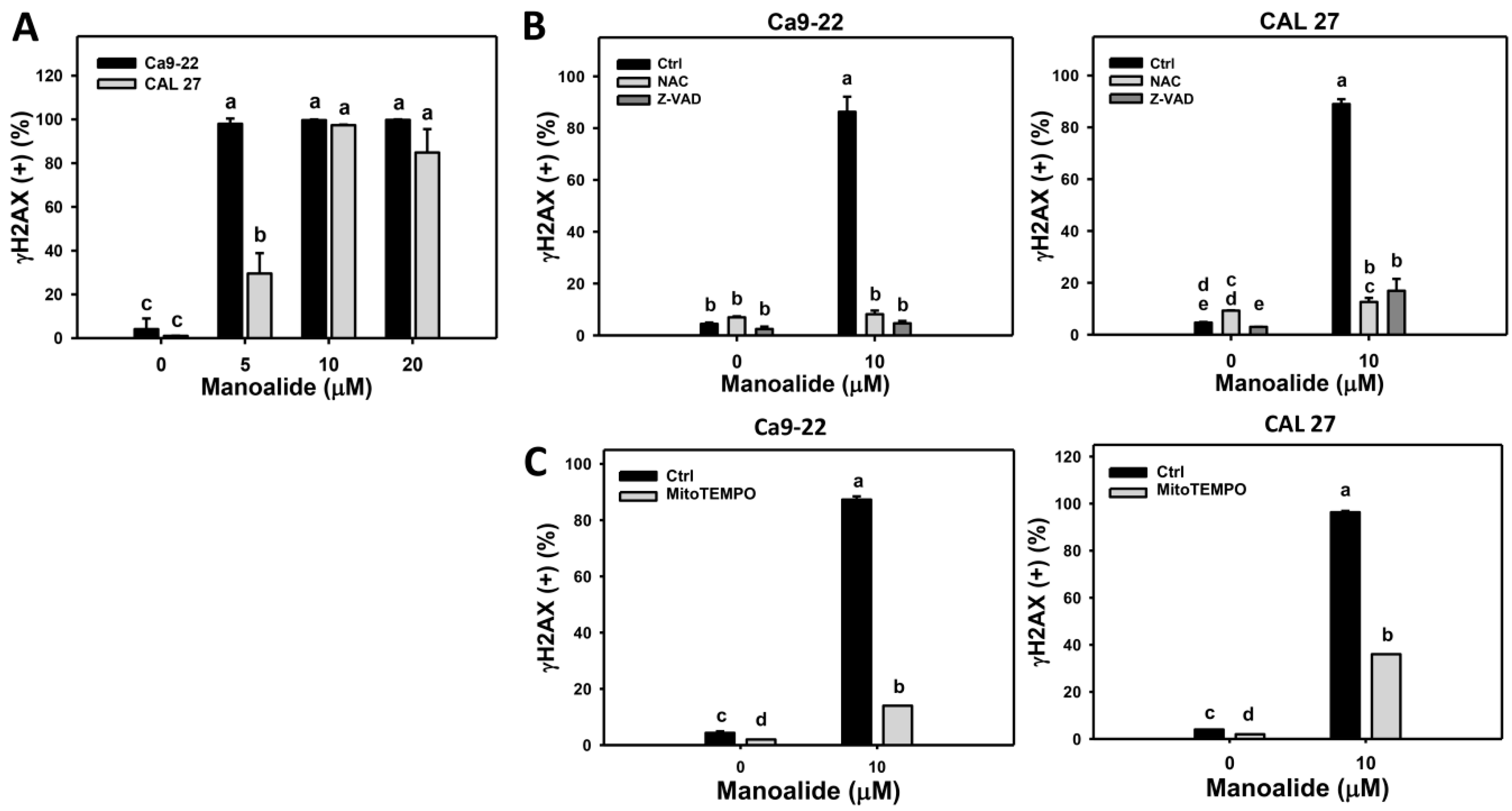
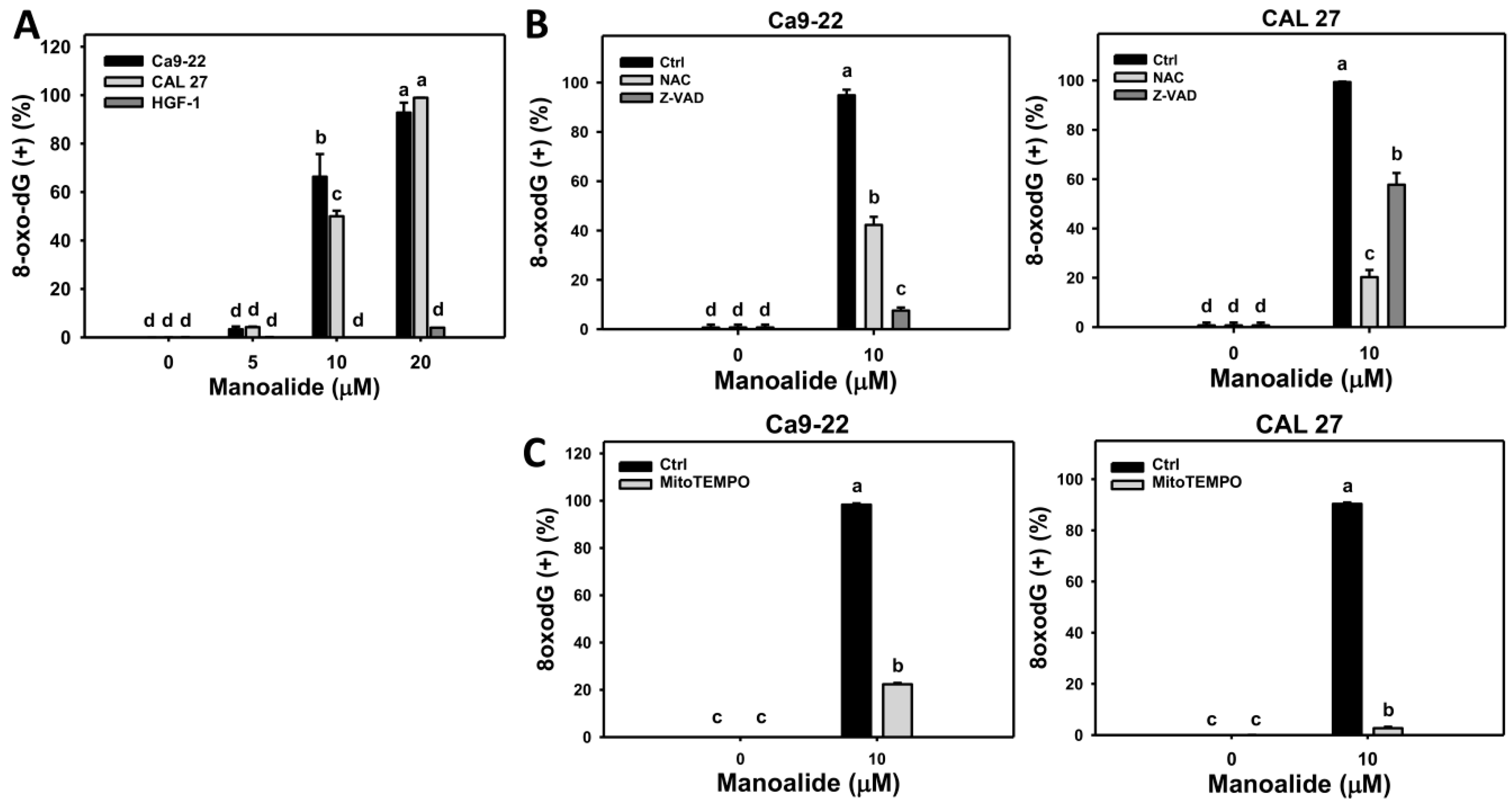
2.7. Flow Cytometry-Based DNA Damage Changes of Manoalide-Treated Oral Cancer Cells with or Without Pretreatments of NAC or Z-VAD
3. Discussion
3.1. Manoalide Sensitivity in Different Cancer Cells
3.2. Manoalide Induced Oxidative Stress Contributes to Preferential Killing Against Oral Cancer Cells
3.3. Manoalide Induced Oxidative stress Contributes to Apoptosis and DNA Damage Against Oral Cancer Cells
3.4. Potential Target Molecules of Manoalide
4. Materials and Methods
4.1. Cell and Drug Information
4.2. Cell Viability Assay
4.3. Cell Cycle Assay
4.4. Annexin V/7AAD Assay for Apoptosis
4.5. Western Blotting and c-Cas 3-Based Flow Cytometry for Apoptosis
4.6. ROS Assay
4.7. MitoSOX Assay
4.8. MitoMP Assay
4.9. γH2AX Assay
4.10. 8-oxodG Assay
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Montero, P.H.; Patel, S.G. Cancer of the oral cavity. Surg. Oncol. Clin. N. Am. 2015, 24, 491–508. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.C.; Huang, Y.L.; Lee, C.H.; Chen, M.J.; Lin, L.M.; Tsai, C.C. Betel quid chewing, cigarette smoking and alcohol consumption related to oral cancer in Taiwan. J. Oral Pathol. Med. 1995, 24, 450–453. [Google Scholar] [CrossRef] [PubMed]
- Petersen, P.E. Oral cancer prevention and control—The approach of the World Health Organization. Oral Oncol. 2009, 45, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Silverman, S., Jr. Oral cancer: Complications of therapy. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 1999, 88, 122–126. [Google Scholar] [CrossRef]
- Singh, R.; Sharma, M.; Joshi, P.; Rawat, D.S. Clinical status of anti-cancer agents derived from marine sources. Anticancer Agents Med. Chem. 2008, 8, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Sithranga Boopathy, N.; Kathiresan, K. Anticancer drugs from marine flora: An overview. J. Oncol. 2010, 2010, 214186. [Google Scholar] [CrossRef]
- Farooqi, A.A.; Fayyaz, S.; Hou, M.F.; Li, K.T.; Tang, J.Y.; Chang, H.W. Reactive oxygen species and autophagy modulation in non-marine drugs and marine drugs. Mar. Drugs 2014, 12, 5408–5424. [Google Scholar] [CrossRef]
- Lee, M.G.; Liu, Y.C.; Lee, Y.L.; El-Shazly, M.; Lai, K.H.; Shih, S.P.; Ke, S.C.; Hong, M.C.; Du, Y.C.; Yang, J.C.; et al. Heteronemin, a marine sesterterpenoid-type metabolite, induces apoptosis in prostate LNcap cells via oxidative and ER stress combined with the inhibition of topoisomerase II and Hsp90. Mar. Drugs 2018, 16, 204. [Google Scholar] [CrossRef]
- Mehbub, M.F.; Lei, J.; Franco, C.; Zhang, W. Marine sponge derived natural products between 2001 and 2010: Trends and opportunities for discovery of bioactives. Mar. Drugs 2014, 12, 4539–4577. [Google Scholar] [CrossRef]
- Mehbub, M.F.; Perkins, M.V.; Zhang, W.; Franco, C.M.M. New marine natural products from sponges (Porifera) of the order Dictyoceratida (2001 to 2012); A promising source for drug discovery, exploration and future prospects. Biotechnol. Adv. 2016, 34, 473–491. [Google Scholar] [CrossRef]
- Calcabrini, C.; Catanzaro, E.; Bishayee, A.; Turrini, E.; Fimognari, C. Marine sponge natural products with anticancer potential: An updated review. Mar. Drugs 2017, 15, 310. [Google Scholar] [CrossRef] [PubMed]
- Perdicaris, S.; Vlachogianni, T.; Valavanidis, A. Bioactive natural substances from marine sponges: New developments and prospects for future pharmaceuticals. Nat. Prod. Chem. Res. 2013, 1, 114. [Google Scholar] [CrossRef]
- de Silva, E.D.; Scheuer, P.J. Manoalide, an antibiotic sesterterpenoid from the marine sponge Luffariella variabilis (polejaeff). Tetrahedron Lett. 1980, 21, 1611–1614. [Google Scholar] [CrossRef]
- Soriente, A.; De Rosa, M.M.; Scettri, A.; Sodano, G.; Terencio, M.C.; Paya, M.; Alcaraz, M.J. Manoalide. Curr. Med. Chem. 1999, 6, 415–431. [Google Scholar] [PubMed]
- Muallem, S.; Loessberg, P.; Sachs, G.; Wheeler, L.A. Agonist-sensitive and -insensitive intracellular Ca2+ pools. Separate Ca2+-releasing mechanisms revealed by manoalide and benzohydroquinone. Biochem. J. 1991, 279, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; Mong, S.; Wu, H.L.; Clark, M.A.; Wheeler, L.; Crooke, S.T. Inhibition of phosphoinositide-specific phospholipase C by manoalide. Mol. Pharmacol. 1987, 32, 587–593. [Google Scholar] [PubMed]
- Wheeler, L.A.; Sachs, G.; De Vries, G.; Goodrum, D.; Woldemussie, E.; Muallem, S. Manoalide, a natural sesterterpenoid that inhibits calcium channels. J. Biol. Chem. 1987, 262, 6531–6538. [Google Scholar] [PubMed]
- Kijjoa, A.; Sawangwong, P. Drugs and cosmetics from the sea. Mar. Drugs 2004, 2, 73–82. [Google Scholar] [CrossRef]
- Kobayashi, J.; Zeng, C.M.; Ishibashi, M.; Sasaki, T. Luffariolides F and G, new manoalide derivatives from the Okinawan marine sponge Luffariella sp. J. Nat. Prod. 1993, 56, 436–439. [Google Scholar] [CrossRef]
- Thambidurai, Y.; D, S.; SKM, H.; T, A.K. Free radical scavenging activity of marine sponges collected from Kovalam, Chennai. Asian J. Pharm. Clin. Res. 2017, 10, 321. [Google Scholar] [CrossRef]
- Utkina, N.K. Antioxidant activity of zyzzyanones and makaluvamines from the marine sponge Zyzzya fuliginosa. Nat. Prod. Commun. 2013, 8, 1551–1552. [Google Scholar] [CrossRef] [PubMed]
- Chairman, K.; Singh, A.J.A.R.; Alagumuthu, G. Cytotoxic and antioxidant activity of selected marine sponges. Asian Pac. J. Trop. Dis. 2012, 2, 234–238. [Google Scholar] [CrossRef]
- Abdillah, S.; Nurhayati, A.P.D.; Nurhatika, S.; Setiawan, E.; Heffen, W.L. Cytotoxic and antioxidant activities of marine sponge diversity at Pecaron Bay Pasir Putih Situbondo East Java, Indonesia. J. Pharm. Res. 2013, 6, 685–689. [Google Scholar] [CrossRef]
- Rivera, A.P.; Uy, M.M. In vitro antioxidant and cytotoxic activities of some marine sponges collected off Misamis Oriental Coast, Philippines. E-J. Chem. 2012, 9, 354–358. [Google Scholar] [CrossRef]
- de Carvalho, D.D.; Sadok, A.; Bourgarel-Rey, V.; Gattacceca, F.; Penel, C.; Lehmann, M.; Kovacic, H. Nox1 downstream of 12-lipoxygenase controls cell proliferation but not cell spreading of colon cancer cells. Int. J. Cancer 2008, 122, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Bouayed, J.; Bohn, T. Exogenous antioxidants—Double-edged swords in cellular redox state: Health beneficial effects at physiologic doses versus deleterious effects at high doses. Oxid. Med. Cell Longev. 2010, 3, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Suzuki-Karasaki, Y.; Suzuki-Karasaki, M.; Uchida, M.; Ochiai, T. Depolarization controls TRAIL-sensitization and tumor-selective killing of cancer cells: Crosstalk with ROS. Front. Oncol. 2014, 4, 128. [Google Scholar] [CrossRef] [PubMed]
- Hseu, Y.C.; Lee, M.S.; Wu, C.R.; Cho, H.J.; Lin, K.Y.; Lai, G.H.; Wang, S.Y.; Kuo, Y.H.; Kumar, K.J.; Yang, H.L. The chalcone flavokawain B induces G2/M cell-cycle arrest and apoptosis in human oral carcinoma HSC-3 cells through the intracellular ROS generation and downregulation of the Akt/p38 MAPK signaling pathway. J. Agric. Food Chem. 2012, 60, 2385–2397. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.C.; El-Shazly, M.; Juan, Y.S.; Chang, C.Y.; Su, J.H.; Chen, Y.C.; Shih, S.P.; Chen, H.M.; Wu, Y.C.; Lu, M.C. Cracking the cytotoxicity code: Apoptotic induction of 10-acetylirciformonin B is mediated through ROS generation and mitochondrial dysfunction. Mar. Drugs 2014, 12, 3072–3090. [Google Scholar] [CrossRef]
- Tang, J.Y.; Wu, C.Y.; Shu, C.W.; Wang, S.C.; Chang, M.Y.; Chang, H.W. A novel sulfonyl chromen-4-ones (CHW09) preferentially kills oral cancer cells showing apoptosis, oxidative stress, and DNA damage. Environ. Toxicol. 2018, 33, 1195–1203. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Takahashi, A.; Kajihara, A.; Yamakawa, N.; Imai, Y.; Ota, I.; Okamoto, N.; Mori, E.; Noda, T.; Furusawa, Y.; et al. Depression of p53-independent Akt survival signals in human oral cancer cells bearing mutated p53 gene after exposure to high-LET radiation. Biochem. Biophys. Res. Commun. 2012, 423, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.C.; Yang, J.I.; Lee, J.C.; Tseng, C.N.; Chan, Y.C.; Hseu, Y.C.; Tang, J.Y.; Chuang, L.Y.; Huang, H.W.; Chang, F.R.; et al. Anti-proliferative effect of methanolic extract of Gracilaria tenuistipitata on oral cancer cells involves apoptosis, DNA damage, and oxidative stress. BMC Complement. Altern. Med. 2012, 12, 142. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Rajesh, M.; Yoshihiro, K.; Hasko, G.; Pacher, P. Simple quantitative detection of mitochondrial superoxide production in live cells. Biochem. Biophys. Res. Commun. 2007, 358, 203–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, H.; Liu, L.; Yang, Y.; Xun, W.; Wei, K.; Zeng, G. Betulinic acid inhibits cell proliferation in human oral squamous cell carcinoma via modulating ROS-regulated p53 signaling. Oncol. Res. 2017, 25, 1141–1152. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.Y.; Park, C.; Lee, J.H.; Chung, K.T.; Kwon, T.K.; Kim, G.Y.; Choi, B.T.; Choi, Y.H. Naringenin-induced apoptosis is attenuated by Bcl-2 but restored by the small molecule Bcl-2 inhibitor, HA 14-1, in human leukemia U937 cells. Toxicol. In Vitro 2009, 23, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.J.; Yang, L.X. Gamma-H2AX—A novel biomarker for DNA double-strand breaks. In Vivo 2008, 22, 305–309. [Google Scholar] [PubMed]
- Roszkowski, K.; Jozwicki, W.; Blaszczyk, P.; Mucha-Malecka, A.; Siomek, A. Oxidative damage DNA: 8-oxoGua and 8-oxodG as molecular markers of cancer. Med. Sci. Monit. 2011, 17, CR329–CR333. [Google Scholar] [CrossRef] [Green Version]
- Vaughan, L.; Glanzel, W.; Korch, C.; Capes-Davis, A. Widespread use of misidentified cell line KB (HeLa): Incorrect attribution and its impact revealed through mining the scientific literature. Cancer Res. 2017, 77, 2784–2788. [Google Scholar] [CrossRef]
- Yen, C.Y.; Hou, M.F.; Yang, Z.W.; Tang, J.Y.; Li, K.T.; Huang, H.W.; Huang, Y.H.; Lee, S.Y.; Fu, T.F.; Hsieh, C.Y.; et al. Concentration effects of grape seed extracts in anti-oral cancer cells involving differential apoptosis, oxidative stress, and DNA damage. BMC Complement. Altern. Med. 2015, 15, 94. [Google Scholar] [CrossRef]
- Chiu, C.C.; Haung, J.W.; Chang, F.R.; Huang, K.J.; Huang, H.M.; Huang, H.W.; Chou, C.K.; Wu, Y.C.; Chang, H.W. Golden berry-derived 4beta-hydroxywithanolide E for selectively killing oral cancer cells by generating ROS, DNA damage, and apoptotic pathways. PLoS ONE 2013, 8, e64739. [Google Scholar] [CrossRef]
- Chang, H.W.; Li, R.N.; Wang, H.R.; Liu, J.R.; Tang, J.Y.; Huang, H.W.; Chan, Y.H.; Yen, C.Y. Withaferin A induces oxidative stress-mediated apoptosis and DNA damage in oral cancer cells. Front. Physiol. 2017, 8, 634. [Google Scholar] [CrossRef] [PubMed]
- Zuco, V.; Supino, R.; Righetti, S.C.; Cleris, L.; Marchesi, E.; Gambacorti-Passerini, C.; Formelli, F. Selective cytotoxicity of betulinic acid on tumor cell lines, but not on normal cells. Cancer Lett. 2002, 175, 17–25. [Google Scholar] [CrossRef]
- Shankar, E.; Zhang, A.; Franco, D.; Gupta, S. Betulinic acid-mediated apoptosis in human prostate cancer cells involves p53 and nuclear factor-kappa B (NF-kappaB) pathways. Molecules 2017, 22, 264. [Google Scholar] [CrossRef]
- Lin, S.C.; Liu, C.J.; Chiu, C.P.; Chang, S.M.; Lu, S.Y.; Chen, Y.J. Establishment of OC3 oral carcinoma cell line and identification of NF-kappa B activation responses to areca nut extract. J. Oral Pathol. Med. 2004, 33, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Hamroun, D.; Kato, S.; Ishioka, C.; Claustres, M.; Beroud, C.; Soussi, T. The UMD TP53 database and website: Update and revisions. Hum. Mutat. 2006, 27, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.H.; Huang, Z.W.; Ho, F.M.; Chan, W.H. Berberine impairs embryonic development in vitro and in vivo through oxidative stress-mediated apoptotic processes. Environ. Toxicol. 2018, 33, 280–294. [Google Scholar] [CrossRef] [PubMed]
- Hung, J.H.; Chen, C.Y.; Omar, H.A.; Huang, K.Y.; Tsao, C.C.; Chiu, C.C.; Chen, Y.L.; Chen, P.H.; Teng, Y.N. Reactive oxygen species mediate Terbufos-induced apoptosis in mouse testicular cell lines via the modulation of cell cycle and pro-apoptotic proteins. Environ. Toxicol. 2016, 31, 1888–1898. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.T.; Huang, C.Y.; Tang, J.Y.; Liaw, C.C.; Li, R.N.; Liu, J.R.; Sheu, J.H.; Chang, H.W. Reactive oxygen species mediate soft corals-derived sinuleptolide-induced antiproliferation and DNA damage in oral cancer cells. Onco Targets Ther. 2017, 10, 3289–3297. [Google Scholar] [CrossRef]
- Chang, Y.T.; Huang, C.Y.; Li, K.T.; Li, R.N.; Liaw, C.C.; Wu, S.H.; Liu, J.R.; Sheu, J.H.; Chang, H.W. Sinuleptolide inhibits proliferation of oral cancer Ca9-22 cells involving apoptosis, oxidative stress, and DNA damage. Arch. Oral Biol. 2016, 66, 147–154. [Google Scholar] [CrossRef]
- Norbury, C.J.; Zhivotovsky, B. DNA damage-induced apoptosis. Oncogene 2004, 23, 2797–2808. [Google Scholar] [CrossRef] [Green Version]
- Inoue, M.; Sato, E.F.; Nishikawa, M.; Park, A.M.; Kira, Y.; Imada, I.; Utsumi, K. Mitochondrial generation of reactive oxygen species and its role in aerobic life. Curr. Med. Chem. 2003, 10, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.L.; Sedlic, F.; Bosnjak, Z.; Nilakantan, V. SOD1 and MitoTEMPO partially prevent mitochondrial permeability transition pore opening, necrosis, and mitochondrial apoptosis after ATP depletion recovery. Free Radic. Biol. Med. 2010, 49, 1550–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardo, D.; Dennis, E.A. Cobra venom phospholipase A2 inhibition by manoalide. A novel type of phospholipase inhibitor. J. Biol. Chem. 1985, 260, 7234–7240. [Google Scholar] [PubMed]
- Miao, J.Y.; Kaji, K.; Hayashi, H.; Araki, S. Inhibitors of phospholipase promote apoptosis of human endothelial cells. J. Biochem. 1997, 121, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Yarla, N.; Satyakumar, K.; Srinivasu, D.; DSVGK K, A.G.; Dharmapuri, G.; Putta, G.; Jagarlapoodi, S.; Bheeram, V.; Sadu, S.; Duddukuri, G. Phospholipase A2: A potential therapeutic target in inflammation and cancer (In silico, In vitro, In vivo and clinical approach). J. Cancer Sci. Ther. 2015, 7, 8. [Google Scholar]
- Mason, R.P. Calcium channel blockers, apoptosis and cancer: Is there a biologic relationship? J. Am. Coll. Cardiol. 1999, 34, 1857–1866. [Google Scholar] [CrossRef]
- Yang, C.Y.; Meng, C.L. Regulation of PG synthase by EGF and PDGF in human oral, breast, stomach, and fibrosarcoma cancer cell lines. J. Dent. Res. 1994, 73, 1407–1415. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.H.; Yeh, J.M.; Chan, W.H. Hazardous impacts of silver nanoparticles on mouse oocyte maturation and fertilization and fetal development through induction of apoptotic processes. Environ. Toxicol. 2018, 33, 1039–1049. [Google Scholar] [CrossRef]
- Wang, T.S.; Lin, C.P.; Chen, Y.P.; Chao, M.R.; Li, C.C.; Liu, K.L. CYP450-mediated mitochondrial ROS production involved in arecoline N-oxide-induced oxidative damage in liver cell lines. Environ. Toxicol. 2018, 33, 1029–1038. [Google Scholar] [CrossRef]
- Chen, C.Y.; Yen, C.Y.; Wang, H.R.; Yang, H.P.; Tang, J.Y.; Huang, H.W.; Hsu, S.H.; Chang, H.W. Tenuifolide B from Cinnamomum tenuifolium stem selectively inhibits proliferation of oral cancer cells via apoptosis, ROS generation, mitochondrial depolarization, and DNA damage. Toxins (Basel) 2016, 8, 319. [Google Scholar] [CrossRef]
- Yeh, C.C.; Tseng, C.N.; Yang, J.I.; Huang, H.W.; Fang, Y.; Tang, J.Y.; Chang, F.R.; Chang, H.W. Antiproliferation and induction of apoptosis in Ca9-22 oral cancer cells by ethanolic extract of Gracilaria tenuistipitata. Molecules 2012, 17, 10916–10927. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.F.; Chang, H.W.; Cheng, J.S.; Lee, H.P.; Yen, C.Y.; Tsai, W.L.; Cheng, J.T.; Li, Y.J.; Huang, W.C.; Lee, C.H.; et al. Map1lc3b and Sqstm1 modulated autophagy for tumorigenesis and prognosis in certain subsites of oral squamous cell carcinoma. J. Clin. Med. 2018, 7, 478. [Google Scholar] [CrossRef] [PubMed]
- Vignon, C.; Debeissat, C.; Georget, M.T.; Bouscary, D.; Gyan, E.; Rosset, P.; Herault, O. Flow cytometric quantification of all phases of the cell cycle and apoptosis in a two-color fluorescence plot. PLoS ONE 2013, 8, e68425. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.W.; Tang, J.Y.; Ou-Yang, F.; Wang, H.R.; Guan, P.Y.; Huang, C.Y.; Chen, C.Y.; Hou, M.F.; Sheu, J.H.; Chang, H.W. Sinularin selectively kills breast cancer cells showing G2/M arrest, apoptosis, and oxidative DNA damage. Molecules 2018, 23, 849. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Hamerlik, P.; Stockhausen, M.T.; Ehrmann, J.; Hlobilkova, A.; Laursen, H.; Kalita, O.; Kolar, Z.; Poulsen, H.S.; Broholm, H.; et al. Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene 2010, 29, 5095–5102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, S.; Kakasi, B.; Bercsenyi, M. Flow cytometric detection of oxidative DNA damage in fish spermatozoa exposed to cadmium—Short communication. Acta Vet. Hung. 2016, 64, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.R.; Gray, K.; Figg, N.; Kumar, S.; Bennett, M.R. The methyl xanthine caffeine inhibits DNA damage signaling and reactive species and reduces atherosclerosis in ApoE(-/-) mice. Arter. Thromb. Vasc. Biol. 2012, 32, 2461–2467. [Google Scholar] [CrossRef] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.-R.; Tang, J.-Y.; Wang, Y.-Y.; Farooqi, A.A.; Yen, C.-Y.; Yuan, S.-S.F.; Huang, H.-W.; Chang, H.-W. Manoalide Preferentially Provides Antiproliferation of Oral Cancer Cells by Oxidative Stress-Mediated Apoptosis and DNA Damage. Cancers 2019, 11, 1303. https://doi.org/10.3390/cancers11091303
Wang H-R, Tang J-Y, Wang Y-Y, Farooqi AA, Yen C-Y, Yuan S-SF, Huang H-W, Chang H-W. Manoalide Preferentially Provides Antiproliferation of Oral Cancer Cells by Oxidative Stress-Mediated Apoptosis and DNA Damage. Cancers. 2019; 11(9):1303. https://doi.org/10.3390/cancers11091303
Chicago/Turabian StyleWang, Hui-Ru, Jen-Yang Tang, Yen-Yun Wang, Ammad Ahmad Farooqi, Ching-Yu Yen, Shyng-Shiou F. Yuan, Hurng-Wern Huang, and Hsueh-Wei Chang. 2019. "Manoalide Preferentially Provides Antiproliferation of Oral Cancer Cells by Oxidative Stress-Mediated Apoptosis and DNA Damage" Cancers 11, no. 9: 1303. https://doi.org/10.3390/cancers11091303