Emerging Role of USP8, HMGA, and Non-Coding RNAs in Pituitary Tumorigenesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods and Objectives
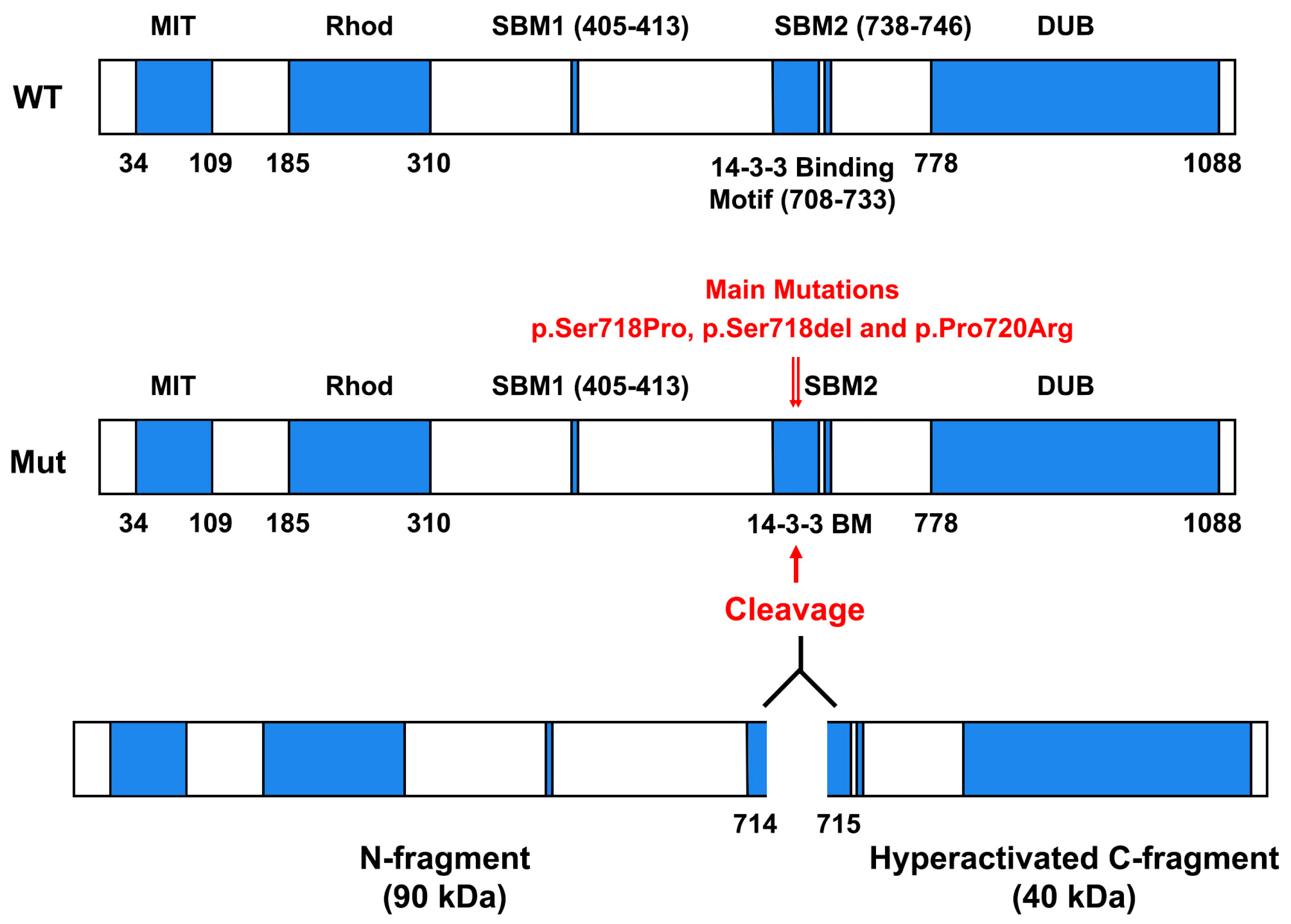
3. Somatic Mutations in Ubiquitin-Specific Protease 8 (USP8) Gene in ACTH-Secreting PitNETs
- (1)
- Microtubule-interacting and trafficking domain (MIT) (aa 34 to aa 109) that is required for efficient abscission at the end of cytokinesis, together with components of the ESCRT-III complex.
- (2)
- Rhodanese-like domain (Rhod) (aa 185 to aa 310).
- (3)
- SH3-binding motif (SBM) (aa. 405 to aa 413).
- (4)
- 14-3-3-binding motif (14-3-3 BM) (aa 708 to aa 733). Vertebrate USP8 protein sequences have a well-conserved 14-3-3 BM, which consists in RSYSSP sequence. In particular, 14-3-3 proteins, a protein family composed of seven isoforms in humans, are important regulatory proteins that can bind to a consensus sequence, RSXpSXP (where X represents any amino acid and pS represents phosphorylated Serine), controlling the functions and the cellular compartmentalization of several 14-3-3 BM-carrying proteins [23]. Intriguingly, it has been found that the binding of 14-3-3 protein to 14-3-3 BM of murine USP8 strongly decreased its deubiquitinase activity on ubiquitinated Epidermal Growth Factor Receptor [24].
- (5)
- Deubiquitinase catalytic domain (DUB) (aa 778 to aa 1088) that removes the conjugated ubiquitin molecules from the target proteins.
3.1. Clinicopathological Features of ACTH-PitNET Patients Carrying USP8 Mutations
3.2. USP8 Involvement on other Human Neoplasias: High Expression of USP8 in Lung and Cervical Carcinomas
3.3. USP48, BRAF, and RASD1 Mutations on ACTH-PitNETs
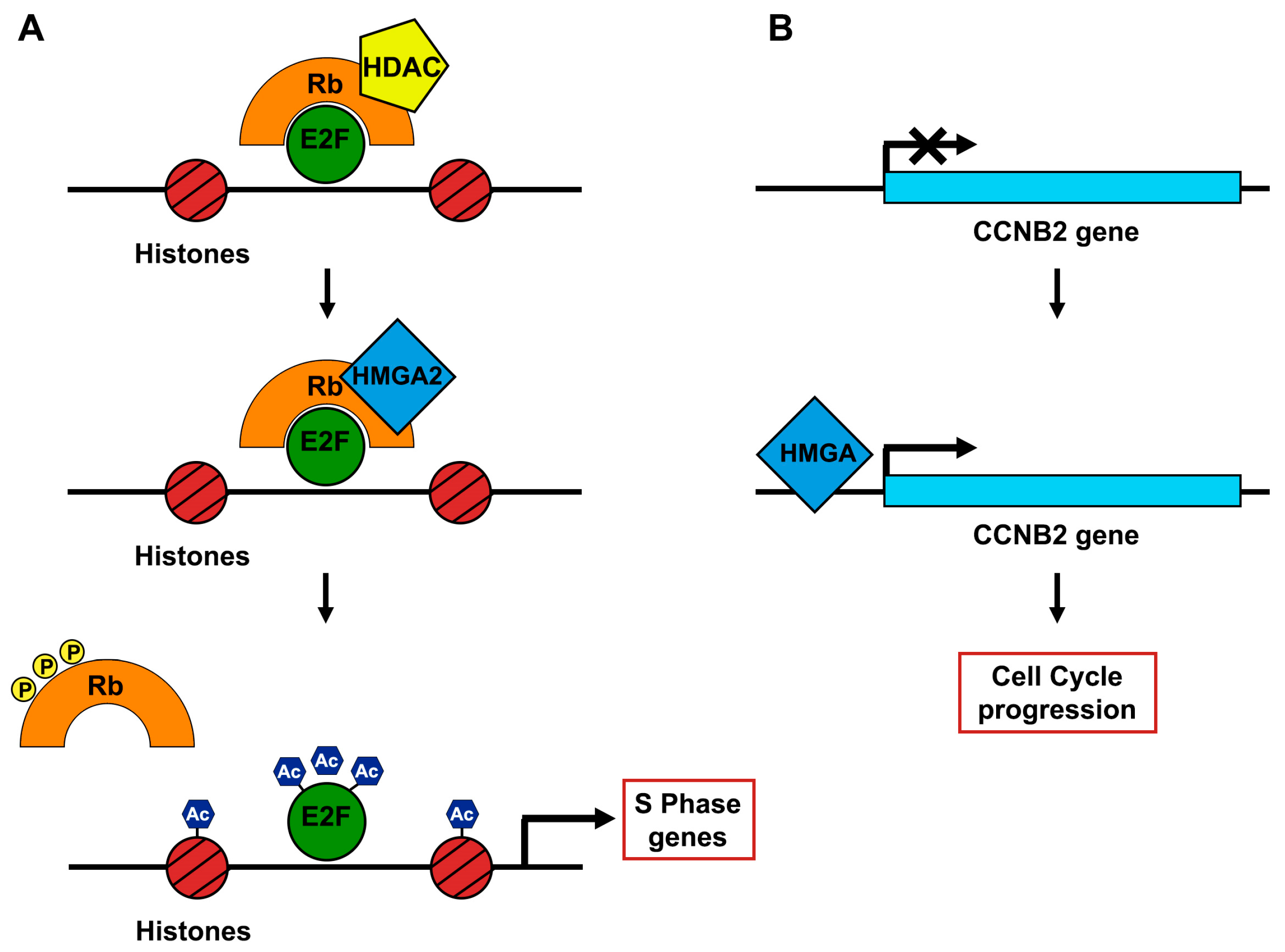
4. HMGA Proteins as Drivers of Pituitary Tumorigenesis
- (1)
- In steady state, pRB and Histone deacetylase 1 (HDAC1) are complexed with E2F1, strongly inhibiting its activity.
- (2)
- HMGA2 displaces HDAC1 interacting with pRB.
- (3)
- The shift of HDAC1 enrolls several enzymes that promote the acetylation of both E2F1 and histones. Thus, E2F1 is activated in its “free” form (Figure 3).
4.1. HMGA Overexpression Correlates with a More Aggressive Phenotype of PitNETs
4.2. Regulation of HMGA Expression by Non-Coding RNAs in Pituitary Tumors
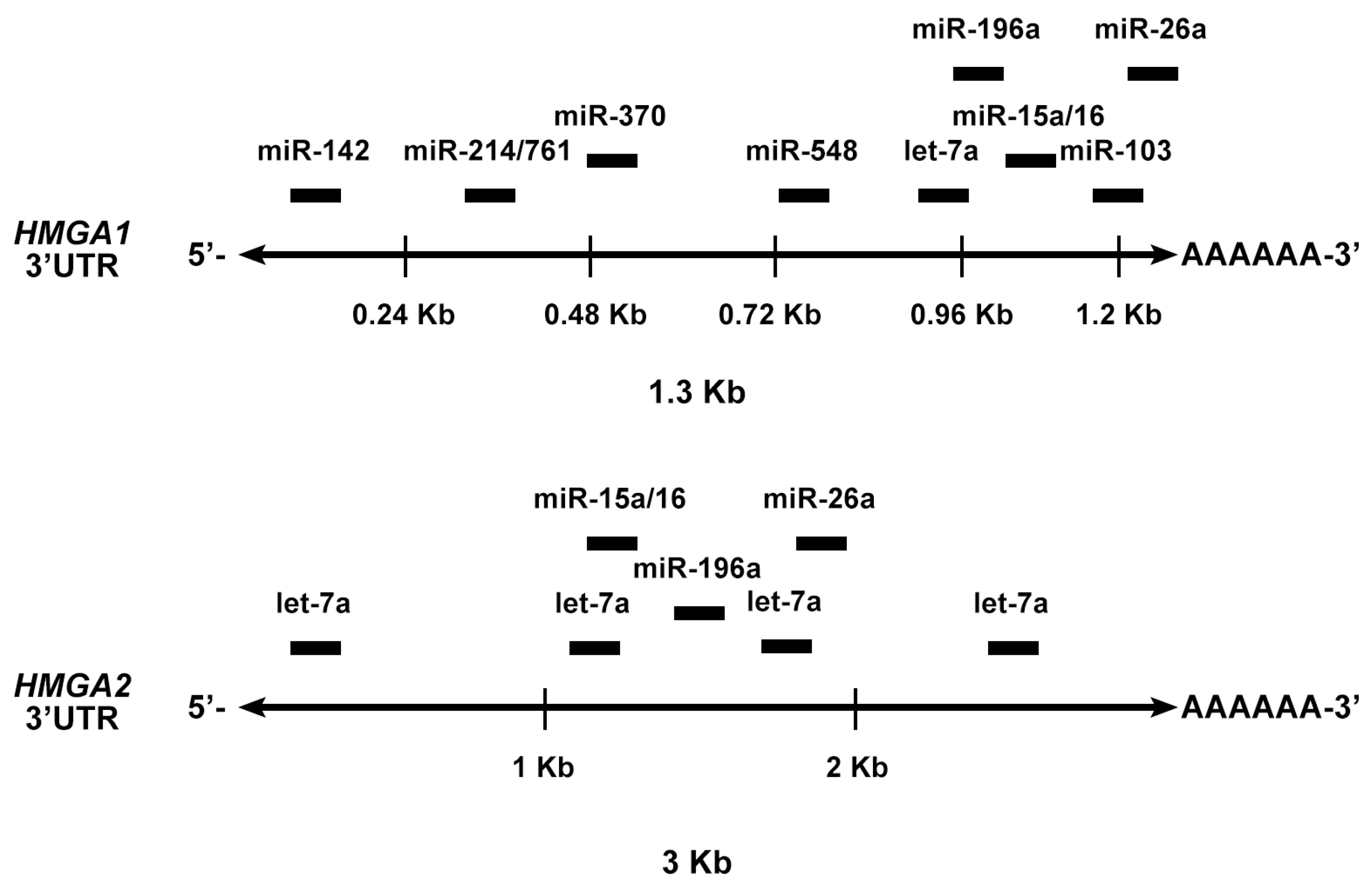
4.3. MiRNAs Targeting the HMGA Genes
4.4. Deregulated Expression of lncRNAs Controlling HMGA Expression Levels in PitNETs
5. Deregulated Expression of other miRNAs in Pituitary Tumorigenesis
6. Circular RNAs Associated with miRNAs in NFPA
7. Other lncRNAs Involved in Pituitary Tumorigenesis
8. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Ezzat, S.; Asa, S.L.; Couldwell, W.T.; Barr, C.E.; Dodge, W.E.; Vance, M.L.; McCutcheon, I.E. The prevalence of pituitary adenomas: A systematic review. Cancer 2004, 101, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Kopczak, A.; Renner, U.; Karl Stalla, G. Advances in understanding pituitary tumors. F1000Prime Rep. 2014, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Gejman, R.; Swearingen, B.; Hedley-Whyte, E.T. Role of Ki-67 proliferation index and p53 expression in predicting progression of pituitary adenomas. Hum. Pathol. 2008, 39, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Mete, O.; Lopes, M.B. Overview of the 2017 WHO Classification of Pituitary Tumors. Endocr. Pathol. 2017, 28, 228–243. [Google Scholar] [CrossRef] [PubMed]
- Asa, S.L.; Casar-Borota, O.; Chanson, P.; Delgrange, E.; Earls, P.; Ezzat, S.; Grossman, A.; Ikeda, H.; Inoshita, N.; Karavitaki, N.; et al. From pituitary adenoma to pituitary neuroendocrine tumor (PitNET): An International Pituitary Pathology Club proposal. Endocr. Relat. Cancer 2017, 24, C5–C8. [Google Scholar] [CrossRef] [PubMed]
- Trouillas, J. In search of a prognostic classification of endocrine pituitary tumors. Endocr. Pathol. 2014, 25, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Melmed, S. Pathogenesis of pituitary tumors. Nat. Rev. Endocrinol. 2011, 7, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Vierimaa, O.; Georgitsi, M.; Lehtonen, R.; Vahteristo, P.; Kokko, A.; Raitila, A.; Tuppurainen, K.; Ebeling, T.M.; Salmela, P.I.; Paschke, R.; et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science 2006, 312, 1228–1230. [Google Scholar] [CrossRef]
- Trivellin, G.; Daly, A.F.; Faucz, F.R.; Yuan, B.; Rostomyan, L.; Larco, D.O.; Schernthaner-Reiter, M.H.; Szarek, E.; Leal, L.F.; Caberg, J.H.; et al. Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N. Engl. J. Med. 2014, 371, 2363–2374. [Google Scholar] [CrossRef]
- Formosa, R.; Xuereb-Anastasi, A.; Vassallo, J. Aip regulates cAMP signalling and GH secretion in GH3 cells. Endocr. Relat. Cancer 2013, 20, 495–505. [Google Scholar] [CrossRef]
- Kasuki Jomori de Pinho, L.; Vieira Neto, L.; Armondi Wildemberg, L.E.; Gasparetto, E.L.; Marcondes, J.; de Almeida Nunes, B.; Takiya, C.M.; Gadelha, M.R. Low aryl hydrocarbon receptor-interacting protein expression is a better marker of invasiveness in somatotropinomas than Ki-67 and p53. Neuroendocrinology 2011, 94, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Kasuki, L.; Vieira Neto, L.; Wildemberg, L.E.; Colli, L.M.; de Castro, M.; Takiya, C.M.; Gadelha, M.R. AIP expression in sporadic somatotropinomas is a predictor of the response to octreotide LAR therapy independent of SSTR2 expression. Endocr. Relat. Cancer 2012, 19, L25–L29. [Google Scholar] [CrossRef] [PubMed]
- Bates, B.; Zhang, L.; Nawoschik, S.; Kodangattil, S.; Tseng, E.; Kopsco, D.; Kramer, A.; Shan, Q.; Taylor, N.; Johnson, J.; et al. Characterization of Gpr101 expression and G-protein coupling selectivity. Brain Res. 2006, 1087, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kober, P.; Bujko, M.; Oledzki, J.; Tysarowski, A.; Siedlecki, J.A. Methyl-CpG binding column-based identification of nine genes hypermethylated in colorectal cancer. Mol. Carcinog. 2011, 50, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Pellegata, N.S.; Quintanilla-Martinez, L.; Siggelkow, H.; Samson, E.; Bink, K.; Hofler, H.; Fend, F.; Graw, J.; Atkinson, M.J. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc. Natl. Acad Sci. USA 2006, 103, 15558–15563. [Google Scholar] [CrossRef] [PubMed]
- de Kock, L.; Sabbaghian, N.; Plourde, F.; Srivastava, A.; Weber, E.; Bouron-Dal Soglio, D.; Hamel, N.; Choi, J.H.; Park, S.H.; Deal, C.L.; et al. Pituitary blastoma: A pathognomonic feature of germ-line DICER1 mutations. Acta Neuropathol. 2014, 128, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Freda, P.U.; Beckers, A.M.; Katznelson, L.; Molitch, M.E.; Montori, V.M.; Post, K.D.; Vance, M.L.; Endocrine, S. Pituitary incidentaloma: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2011, 96, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Vlotides, G.; Cruz-Soto, M.; Rubinek, T.; Eigler, T.; Auernhammer, C.J.; Melmed, S. Mechanisms for growth factor-induced pituitary tumor transforming gene-1 expression in pituitary folliculostellate TtT/GF cells. Mol. Endocrinol. 2006, 20, 3321–3335. [Google Scholar] [CrossRef] [PubMed]
- Lines, K.E.; Stevenson, M.; Thakker, R.V. Animal models of pituitary neoplasia. Mol. Cell Endocrinol. 2016, 421, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Nieman, L.K. Recent Updates on the Diagnosis and Management of Cushing’s Syndrome. Endocrinol. Metab. (Seoul) 2018, 33, 139–146. [Google Scholar] [CrossRef]
- Ballmann, C.; Thiel, A.; Korah, H.E.; Reis, A.C.; Saeger, W.; Stepanow, S.; Kohrer, K.; Reifenberger, G.; Knobbe-Thomsen, C.B.; Knappe, U.J.; et al. USP8 Mutations in Pituitary Cushing Adenomas-Targeted Analysis by Next-Generation Sequencing. J. Endocr. Soc. 2018, 2, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.Y.; Song, Z.J.; Chen, J.H.; Wang, Y.F.; Li, S.Q.; Zhou, L.F.; Mao, Y.; Li, Y.M.; Hu, R.G.; Zhang, Z.Y.; et al. Recurrent gain-of-function USP8 mutations in Cushing’s disease. Cell Res. 2015, 25, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.K. The 14-3-3 proteins: Integrators of diverse signaling cues that impact cell fate and cancer development. Trends Cell Biol. 2009, 19, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, E.; Kitamura, N.; Komada, M. 14-3-3-dependent inhibition of the deubiquitinating activity of UBPY and its cancellation in the M phase. Exp. Cell Res. 2007, 313, 3624–3634. [Google Scholar] [CrossRef] [PubMed]
- Reincke, M.; Sbiera, S.; Hayakawa, A.; Theodoropoulou, M.; Osswald, A.; Beuschlein, F.; Meitinger, T.; Mizuno-Yamasaki, E.; Kawaguchi, K.; Saeki, Y.; et al. Mutations in the deubiquitinase gene USP8 cause Cushing’s disease. Nat. Genet. 2015, 47, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Perez-Rivas, L.G.; Theodoropoulou, M.; Ferrau, F.; Nusser, C.; Kawaguchi, K.; Stratakis, C.A.; Faucz, F.R.; Wildemberg, L.E.; Assie, G.; Beschorner, R.; et al. The Gene of the Ubiquitin-Specific Protease 8 Is Frequently Mutated in Adenomas Causing Cushing’s Disease. J. Clin. Endocrinol. Metab. 2015, 100, E997–E1004. [Google Scholar] [CrossRef] [PubMed]
- Kontogeorgos, G.; Stefaneanu, L.; Kovacs, K.; Cheng, Z. Localization of Epidermal Growth Factor (EGF) and Epidermal Growth Factor Receptor (EGFr) in Human Pituitary Adenomas and Nontumorous Pituitaries: An Immunocytochemical Study. Endocr. Pathol. 1996, 7, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Theodoropoulou, M.; Arzberger, T.; Gruebler, Y.; Jaffrain-Rea, M.L.; Schlegel, J.; Schaaf, L.; Petrangeli, E.; Losa, M.; Stalla, G.K.; Pagotto, U. Expression of epidermal growth factor receptor in neoplastic pituitary cells: Evidence for a role in corticotropinoma cells. J. Endocrinol. 2004, 183, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Onguru, O.; Scheithauer, B.W.; Kovacs, K.; Vidal, S.; Jin, L.; Zhang, S.; Ruebel, K.H.; Lloyd, R.V. Analysis of epidermal growth factor receptor and activated epidermal growth factor receptor expression in pituitary adenomas and carcinomas. Mod. Pathol. 2004, 17, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Honda, J.; Oomizu, S.; Kiuchi, Y.; Komatsu, N.; Takeuchi, S.; Takahashi, S. Identification of epidermal growth factor mRNA-expressing cells in the mouse anterior pituitary. Neuroendocrinology 2000, 71, 155–162. [Google Scholar] [CrossRef]
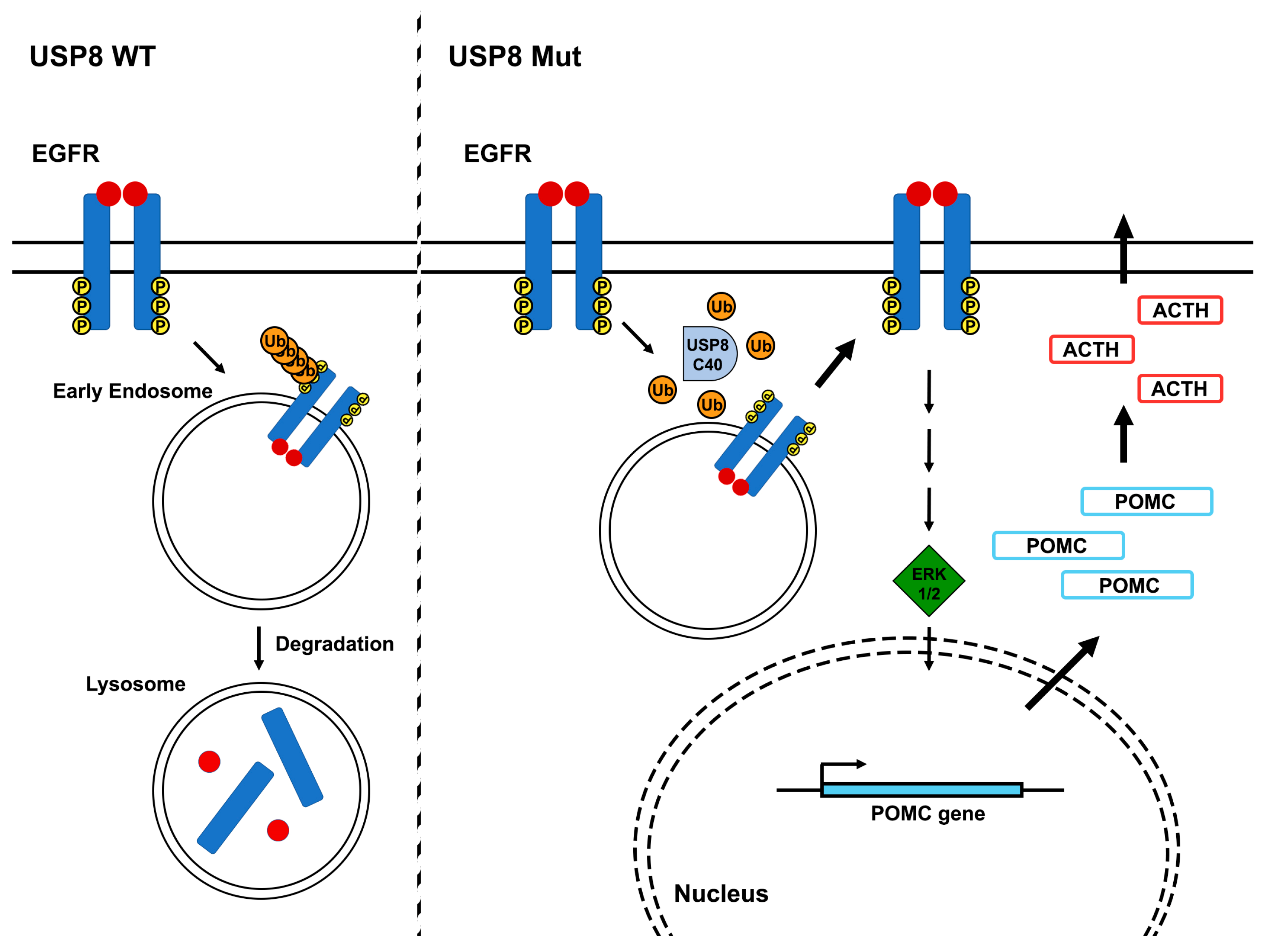
- Fukuoka, H.; Cooper, O.; Ben-Shlomo, A.; Mamelak, A.; Ren, S.G.; Bruyette, D.; Melmed, S. EGFR as a therapeutic target for human, canine, and mouse ACTH-secreting pituitary adenomas. J. Clin. Investig. 2011, 121, 4712–4721. [Google Scholar] [CrossRef] [PubMed]
- Araki, T.; Liu, X.; Kameda, H.; Tone, Y.; Fukuoka, H.; Tone, M.; Melmed, S. EGFR Induces E2F1-Mediated Corticotroph Tumorigenesis. J. Endocr. Soc. 2017, 1, 127–143. [Google Scholar] [CrossRef] [PubMed]
- Araki, T.; Liu, N.A.; Tone, Y.; Cuevas-Ramos, D.; Heltsley, R.; Tone, M.; Melmed, S. E2F1-mediated human POMC expression in ectopic Cushing’s syndrome. Endocr. Relat. Cancer 2016, 23, 857–870. [Google Scholar] [CrossRef] [PubMed]
- Fusco, A.; Fedele, M. Roles of HMGA proteins in cancer. Nat. Rev. Cancer 2007, 7, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Albani, A.; Perez-Rivas, L.G.; Dimopoulou, C.; Zopp, S.; Colon-Bolea, P.; Roeber, S.; Honegger, J.; Flitsch, J.; Rachinger, W.; Buchfelder, M.; et al. The USP8 mutational status may predict long-term remission in patients with Cushing’s disease. Clin. Endocrinol. (Oxf.) 2018, 89, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Losa, M.; Mortini, P.; Pagnano, A.; Detomas, M.; Cassarino, M.F.; Pecori Giraldi, F. Clinical characteristics and surgical outcome in USP8-mutated human adrenocorticotropic hormone-secreting pituitary adenomas. Endocrine 2019, 63, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Perez-Rivas, L.G.; Theodoropoulou, M.; Puar, T.H.; Fazel, J.; Stieg, M.R.; Ferrau, F.; Assie, G.; Gadelha, M.R.; Deutschbein, T.; Fragoso, M.C.; et al. Somatic USP8 mutations are frequent events in corticotroph tumor progression causing Nelson’s tumor. Eur. J. Endocrinol. 2018, 178, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Zhao, C.; Wei, N.; Wu, X.; Cui, J.; Xing, Y. High Expression of Ubiquitin-Specific Protease 8 (USP8) Is Associated with Poor Prognosis in Patients with Cervical Squamous Cell Carcinoma. Med. Sci. Monit. 2018, 24, 4934–4943. [Google Scholar] [CrossRef] [PubMed]
- Byun, S.; Lee, S.Y.; Lee, J.; Jeong, C.H.; Farrand, L.; Lim, S.; Reddy, K.; Kim, J.Y.; Lee, M.H.; Lee, H.J.; et al. USP8 is a novel target for overcoming gefitinib resistance in lung cancer. Clin. Cancer Res. 2013, 19, 3894–3904. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Shiba-Ishii, A.; Nakagawa, T.; Husni, R.E.; Sakashita, S.; Takeuchi, T.; Noguchi, M. Ubiquitin-specific protease 8 is a novel prognostic marker in early-stage lung adenocarcinoma. Pathol. Int. 2017, 67, 292–301. [Google Scholar] [CrossRef]
- Kim, Y.; Shiba-Ishii, A.; Nakagawa, T.; Iemura, S.I.; Natsume, T.; Nakano, N.; Matsuoka, R.; Sakashita, S.; Lee, S.; Kawaguchi, A.; et al. Stratifin regulates stabilization of receptor tyrosine kinases via interaction with ubiquitin-specific protease 8 in lung adenocarcinoma. Oncogene 2018, 37, 5387–5402. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jian, X.; Deng, S.; Ma, Z.; Shou, X.; Shen, Y.; Zhang, Q.; Song, Z.; Li, Z.; Peng, H.; et al. Identification of recurrent USP48 and BRAF mutations in Cushing’s disease. Nat. Commun. 2018, 9, 3171. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Shi, Y.; Zhao, Y. USP8 mutation in Cushing’s disease. Oncotarget 2015, 6, 18240–18241. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Mitsui, T.; Ishida, M.; Izawa, M.; Arita, J. Rasd1 is an estrogen-responsive immediate early gene and modulates expression of late genes in rat anterior pituitary cells. Endocr. J. 2017, 64, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Uzilov, A.V.; Cheesman, K.C.; Fink, M.Y.; Newman, L.C.; Pandya, C.; Lalazar, Y.; Hefti, M.; Fowkes, M.; Deikus, G.; Lau, C.Y.; et al. Identification of a novel RASD1 somatic mutation in a USP8-mutated corticotroph adenoma. Cold Spring Harb. Mol. Case Stud. 2017, 3, a001602. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.R.; Lehn, D.A.; Reeves, R. Alternative processing of mRNAs encoding mammalian chromosomal high-mobility-group proteins HMG-I and HMG-Y. Mol. Cell Biol. 1989, 9, 2114–2123. [Google Scholar] [CrossRef] [PubMed]
- Thanos, D.; Maniatis, T. The high mobility group protein HMG I(Y) is required for NF-kappa B-dependent virus induction of the human IFN-beta gene. Cell 1992, 71, 777–789. [Google Scholar] [CrossRef]
- Foti, D.; Chiefari, E.; Fedele, M.; Iuliano, R.; Brunetti, L.; Paonessa, F.; Manfioletti, G.; Barbetti, F.; Brunetti, A.; Croce, C.M.; et al. Lack of the architectural factor HMGA1 causes insulin resistance and diabetes in humans and mice. Nat. Med. 2005, 11, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Benson, K.F.; Ashar, H.R.; Chada, K. Mutation responsible for the mouse pygmy phenotype in the developmentally regulated factor HMGI-C. Nature 1995, 376, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Anand, A.; Chada, K. In vivo modulation of Hmgic reduces obesity. Nat. Genet. 2000, 24, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Forzati, F.; Esposito, F.; Arra, C.; Palma, G.; Barbieri, A.; Palmieri, D.; Fedele, M.; Pierantoni, G.M.; De Martino, I.; et al. Hmga1/Hmga2 double knock-out mice display a “superpygmy” phenotype. Biol. Open 2014, 3, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Pallante, P.; Sepe, R.; Puca, F.; Fusco, A. High mobility group a proteins as tumor markers. Front. Med. (Lausanne) 2015, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- Berlingieri, M.T.; Pierantoni, G.M.; Giancotti, V.; Santoro, M.; Fusco, A. Thyroid cell transformation requires the expression of the HMGA1 proteins. Oncogene 2002, 21, 2971–2980. [Google Scholar] [CrossRef] [PubMed]
- Battista, S.; Fidanza, V.; Fedele, M.; Klein-Szanto, A.J.; Outwater, E.; Brunner, H.; Santoro, M.; Croce, C.M.; Fusco, A. The expression of a truncated HMGI-C gene induces gigantism associated with lipomatosis. Cancer Res. 1999, 59, 4793–4797. [Google Scholar] [PubMed]
- Arlotta, P.; Tai, A.K.; Manfioletti, G.; Clifford, C.; Jay, G.; Ono, S.J. Transgenic mice expressing a truncated form of the high mobility group I-C protein develop adiposity and an abnormally high prevalence of lipomas. J. Biol. Chem. 2000, 275, 14394–14400. [Google Scholar] [CrossRef] [PubMed]
- Fedele, M.; Battista, S.; Kenyon, L.; Baldassarre, G.; Fidanza, V.; Klein-Szanto, A.J.; Parlow, A.F.; Visone, R.; Pierantoni, G.M.; Outwater, E.; et al. Overexpression of the HMGA2 gene in transgenic mice leads to the onset of pituitary adenomas. Oncogene 2002, 21, 3190–3198. [Google Scholar] [CrossRef] [PubMed]
- Fedele, M.; Pentimalli, F.; Baldassarre, G.; Battista, S.; Klein-Szanto, A.J.; Kenyon, L.; Visone, R.; De Martino, I.; Ciarmiello, A.; Arra, C.; et al. Transgenic mice overexpressing the wild-type form of the HMGA1 gene develop mixed growth hormone/prolactin cell pituitary adenomas and natural killer cell lymphomas. Oncogene 2005, 24, 3427–3435. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, D.; Valentino, T.; De Martino, I.; Esposito, F.; Cappabianca, P.; Wierinckx, A.; Vitiello, M.; Lombardi, G.; Colao, A.; Trouillas, J.; et al. PIT1 upregulation by HMGA proteins has a role in pituitary tumorigenesis. Endocr. Relat. Cancer 2012, 19, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Finelli, P.; Pierantoni, G.M.; Giardino, D.; Losa, M.; Rodeschini, O.; Fedele, M.; Valtorta, E.; Mortini, P.; Croce, C.M.; Larizza, L.; et al. The High Mobility Group A2 gene is amplified and overexpressed in human prolactinomas. Cancer Res. 2002, 62, 2398–2405. [Google Scholar] [PubMed]
- Fedele, M.; Visone, R.; De Martino, I.; Troncone, G.; Palmieri, D.; Battista, S.; Ciarmiello, A.; Pallante, P.; Arra, C.; Melillo, R.M.; et al. HMGA2 induces pituitary tumorigenesis by enhancing E2F1 activity. Cancer Cell 2006, 9, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Fedele, M.; Fusco, A. Role of the high mobility group A proteins in the regulation of pituitary cell cycle. J. Mol. Endocrinol. 2010, 44, 309–318. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Steno, A.; Bocko, J.; Rychly, B.; Chorvath, M.; Celec, P.; Fabian, M.; Belan, V.; Steno, J. Nonfunctioning pituitary adenomas: Association of Ki-67 and HMGA-1 labeling indices with residual tumor growth. Acta Neurochir. (Wien.) 2014, 156, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.L.; Qian, Z.R.; Rahman, M.M.; Yoshimoto, K.; Yamada, S.; Kudo, E.; Sano, T. Increased expression of HMGA1 correlates with tumour invasiveness and proliferation in human pituitary adenomas. Histopathology 2010, 56, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Fedele, M.; Paciello, O.; De Biase, D.; Monaco, M.; Chiappetta, G.; Vitiello, M.; Barbieri, A.; Rea, D.; Luciano, A.; Papparella, S.; et al. HMGA2 cooperates with either p27(kip1) deficiency or Cdk4(R24C) mutation in pituitary tumorigenesis. Cell Cycle 2018, 17, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Deveson, I.W.; Hardwick, S.A.; Mercer, T.R.; Mattick, J.S. The Dimensions, Dynamics, and Relevance of the Mammalian Noncoding Transcriptome. Trends Genet. 2017, 33, 464–478. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
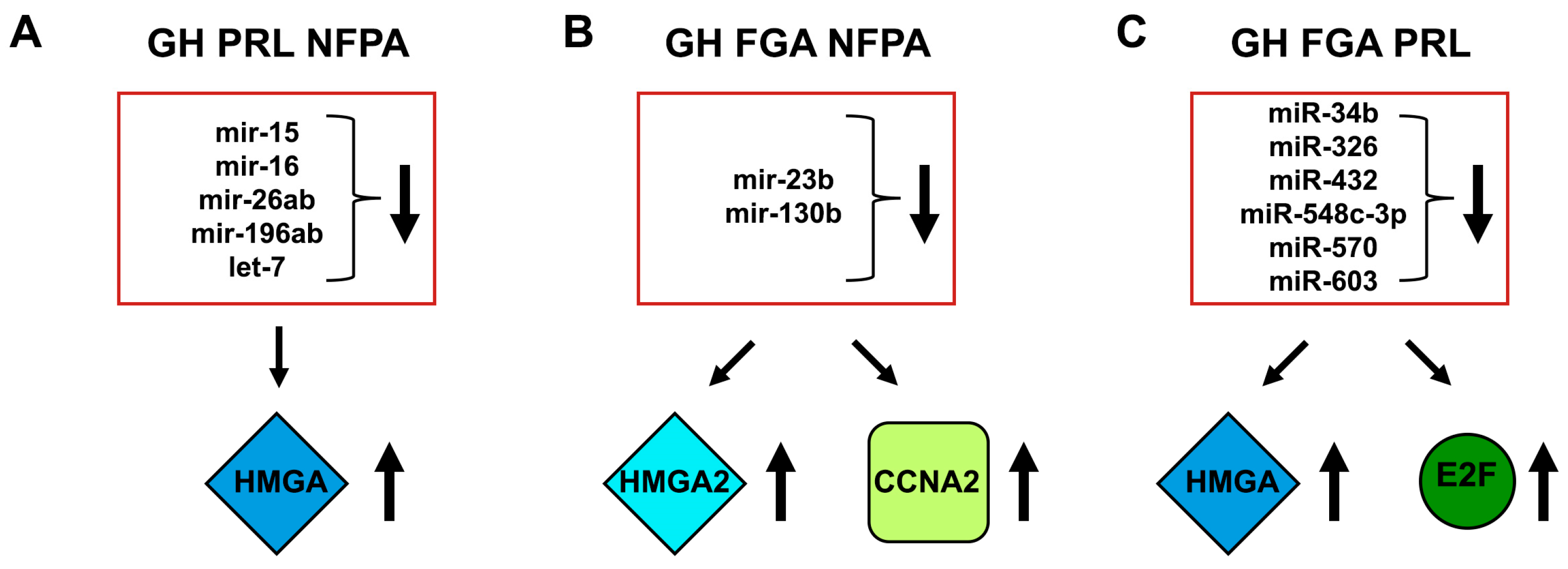
- Palmieri, D.; D’Angelo, D.; Valentino, T.; De Martino, I.; Ferraro, A.; Wierinckx, A.; Fedele, M.; Trouillas, J.; Fusco, A. Downregulation of HMGA-targeting microRNAs has a critical role in human pituitary tumorigenesis. Oncogene 2012, 31, 3857–3865. [Google Scholar] [CrossRef]
- Leone, V.; Langella, C.; D’Angelo, D.; Mussnich, P.; Wierinckx, A.; Terracciano, L.; Raverot, G.; Lachuer, J.; Rotondi, S.; Jaffrain-Rea, M.L.; et al. Mir-23b and miR-130b expression is downregulated in pituitary adenomas. Mol. Cell Endocrinol. 2014, 390, 1–7. [Google Scholar] [CrossRef]
- D’Angelo, D.; Palmieri, D.; Mussnich, P.; Roche, M.; Wierinckx, A.; Raverot, G.; Fedele, M.; Croce, C.M.; Trouillas, J.; Fusco, A. Altered microRNA expression profile in human pituitary GH adenomas: Down-regulation of miRNA targeting HMGA1, HMGA2, and E2F1. J. Clin. Endocrinol. Metab. 2012, 97, E1128–E1138. [Google Scholar] [CrossRef] [PubMed]
- Balakirev, E.S.; Ayala, F.J. Pseudogenes: Are they “junk” or functional DNA? Annu. Rev. Genet. 2003, 37, 123–151. [Google Scholar] [CrossRef] [PubMed]
- Mighell, A.J.; Smith, N.R.; Robinson, P.A.; Markham, A.F. Vertebrate pseudogenes. FEBS Lett. 2000, 468, 109–114. [Google Scholar] [CrossRef]
- Esposito, F.; De Martino, M.; Petti, M.G.; Forzati, F.; Tornincasa, M.; Federico, A.; Arra, C.; Pierantoni, G.M.; Fusco, A. HMGA1 pseudogenes as candidate proto-oncogenic competitive endogenous RNAs. Oncotarget 2014, 5, 8341–8354. [Google Scholar] [CrossRef] [PubMed]
- Esposito, F.; De Martino, M.; Forzati, F.; Fusco, A. HMGA1-pseudogene overexpression contributes to cancer progression. Cell Cycle 2014, 13, 3636–3639. [Google Scholar] [CrossRef] [PubMed]
- De Martino, M.; Palma, G.; Azzariti, A.; Arra, C.; Fusco, A.; Esposito, F. The HMGA1 Pseudogene 7 Induces miR-483 and miR-675 Upregulation by Activating Egr1 through a ceRNA Mechanism. Genes 2017, 8, 330. [Google Scholar] [CrossRef] [PubMed]
- De Martino, M.; Forzati, F.; Marfella, M.; Pellecchia, S.; Arra, C.; Terracciano, L.; Fusco, A.; Esposito, F. HMGA1P7-pseudogene regulates H19 and Igf2 expression by a competitive endogenous RNA mechanism. Sci. Rep. 2016, 6, 37622. [Google Scholar] [CrossRef] [PubMed]
- De Martino, M.; Forzati, F.; Arra, C.; Fusco, A.; Esposito, F. HMGA1-pseudogenes and cancer. Oncotarget 2016, 7, 28724–28735. [Google Scholar] [CrossRef] [PubMed]
- Esposito, F.; De Martino, M.; D’Angelo, D.; Mussnich, P.; Raverot, G.; Jaffrain-Rea, M.L.; Fraggetta, F.; Trouillas, J.; Fusco, A. HMGA1-pseudogene expression is induced in human pituitary tumors. Cell Cycle 2015, 14, 1471–1475. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, S.; Wu, H.; Yue, L.; Lei, B.; Xiong, Y.; Wei, Z.; Zheng, N. The long noncoding RNA CASC2 inhibits pituitary adenoma progressionby inhibiting HMGA2 expression. Int. J. Clin. Exp. Med. 2017, 10, 14458–14467. [Google Scholar]
- D’Angelo, D.; Mussnich, P.; Sepe, R.; Raia, M.; Del Vecchio, L.; Cappabianca, P.; Pellecchia, S.; Petrosino, S.; Saggio, S.; Solari, D.; et al. RPSAP52 lncRNA is overexpressed in pituitary tumors and promotes cell proliferation by acting as miRNA sponge for HMGA proteins. J. Mol. Med. (Berl.) 2019, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.G.; He, D.S.; Zhou, J.; Yao, B.; Xiao, W.W.; Chen, C.H.; Zhu, Y.H.; Wang, H.J. Differential expression of microRNAs in GH-secreting pituitary adenomas. Diagn. Pathol. 2010, 5, 79. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.Q.; Wang, R.J.; Diao, C.F.; Li, J.W.; Su, J.L.; Zhang, S. The PTTG1-targeting miRNAs miR-329, miR-300, miR-381, and miR-655 inhibit pituitary tumor cell tumorigenesis and are involved in a p53/PTTG1 regulation feedback loop. Oncotarget 2015, 6, 29413–29427. [Google Scholar] [CrossRef] [PubMed]
- Butz, H.; Liko, I.; Czirjak, S.; Igaz, P.; Khan, M.M.; Zivkovic, V.; Balint, K.; Korbonits, M.; Racz, K.; Patocs, A. Down-regulation of Wee1 kinase by a specific subset of microRNA in human sporadic pituitary adenomas. J. Clin. Endocrinol. Metab. 2010, 95, E181–E191. [Google Scholar] [CrossRef] [PubMed]
- Trivellin, G.; Butz, H.; Delhove, J.; Igreja, S.; Chahal, H.S.; Zivkovic, V.; McKay, T.; Patocs, A.; Grossman, A.B.; Korbonits, M. MicroRNA miR-107 is overexpressed in pituitary adenomas and inhibits the expression of aryl hydrocarbon receptor-interacting protein in vitro. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E708–E719. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.B.; Li, W.Q.; Lin, S.J.; Wang, C.D.; Cai, L.; Lu, J.L.; Chen, Y.X.; Su, Z.P.; Shang, H.B.; Yang, W.L.; et al. MicroRNA expression profile of bromocriptine-resistant prolactinomas. Mol. Cell Endocrinol. 2014, 395, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Du, Y.; Yang, X.; Mo, Y.; Fan, C.; Xiong, F.; Ren, D.; Ye, X.; Li, C.; Wang, Y.; et al. Circular RNAs function as ceRNAs to regulate and control human cancer progression. Mol. Cancer 2018, 17, 79. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Hu, B.; Feng, Y.; Wang, Z.; Wang, X.; Zhu, D.; Zhu, Y.; Jiang, X.; Wang, H. CircOMA1 mediated miR-145-5p suppresses tumor growth of nonfunctioning pituitary adenomas by targeting TPT1. J. Clin. Endocrinol. Metab. 2019, 104, 2419–2434. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, Y.; Mehta, K.R.; Danila, D.C.; Scolavino, S.; Johnson, S.R.; Klibanski, A. A pituitary-derived MEG3 isoform functions as a growth suppressor in tumor cells. J. Clin. Endocrinol. Metab. 2003, 88, 5119–5126. [Google Scholar] [CrossRef]
- Miyoshi, N.; Wagatsuma, H.; Wakana, S.; Shiroishi, T.; Nomura, M.; Aisaka, K.; Kohda, T.; Surani, M.A.; Kaneko-Ishino, T.; Ishino, F. Identification of an imprinted gene, Meg3/Gtl2 and its human homologue MEG3, first mapped on mouse distal chromosome 12 and human chromosome 14q. Genes Cells 2000, 5, 211–220. [Google Scholar] [CrossRef]
- Zhao, J.; Dahle, D.; Zhou, Y.; Zhang, X.; Klibanski, A. Hypermethylation of the promoter region is associated with the loss of MEG3 gene expression in human pituitary tumors. J. Clin. Endocrinol. Metab. 2005, 90, 2179–2186. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Hou, B.; Ye, Z.; Ling, C.; Guo, Y. Knockdown of long non-coding RNA AFAP1-AS1 inhibits growth and promotes apoptosis in pituitary adenomas. Int. J. Clin. Exp. Pathol. 2018, 11, 1238–1246. [Google Scholar]
- Li, Z.; Li, C.; Liu, C.; Yu, S.; Zhang, Y. Expression of the long non-coding RNAs MEG3, HOTAIR, and MALAT-1 in non-functioning pituitary adenomas and their relationship to tumor behavior. Pituitary 2015, 18, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Li, C.; Xie, W.; Wang, Z.; Gao, H.; Cao, L.; Hao, L.; Zhang, Y. Long non-coding RNA C5orf66-AS1 is downregulated in pituitary null cell adenomas and is associated with their invasiveness. Oncol. Rep. 2017, 38, 1140–1148. [Google Scholar] [CrossRef]
- Raveh, E.; Matouk, I.J.; Gilon, M.; Hochberg, A. The H19 Long non-coding RNA in cancer initiation, progression and metastasis—A proposed unifying theory. Mol. Cancer 2015, 14, 184. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Yu, C.; Ni, H.; Liang, W.; Yan, H.; Jin, W. Expression of the long non-coding RNA H19 and MALAT-1 in growth hormone-secreting pituitary adenomas and its relationship to tumor behavior. Int. J. Dev. Neurosci. 2018, 67, 46–50. [Google Scholar] [CrossRef]
- D’Angelo, D.; Borbone, E.; Palmieri, D.; Uboldi, S.; Esposito, F.; Frapolli, R.; Pacelli, R.; D’Incalci, M.; Fusco, A. The impairment of the High Mobility Group A (HMGA) protein function contributes to the anticancer activity of trabectedin. Eur. J. Cancer 2013, 49, 1142–1151. [Google Scholar] [CrossRef]
- Fedele, M.; De Martino, I.; Pivonello, R.; Ciarmiello, A.; Del Basso De Caro, M.L.; Visone, R.; Palmieri, D.; Pierantoni, G.M.; Arra, C.; Schmid, H.A.; et al. SOM230, a new somatostatin analogue, is highly effective in the therapy of growth hormone/prolactin-secreting pituitary adenomas. Clin. Cancer Res. 2007, 13, 2738–2744. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Angelo, D.; De Martino, M.; Arra, C.; Fusco, A. Emerging Role of USP8, HMGA, and Non-Coding RNAs in Pituitary Tumorigenesis. Cancers 2019, 11, 1302. https://doi.org/10.3390/cancers11091302
D’Angelo D, De Martino M, Arra C, Fusco A. Emerging Role of USP8, HMGA, and Non-Coding RNAs in Pituitary Tumorigenesis. Cancers. 2019; 11(9):1302. https://doi.org/10.3390/cancers11091302
Chicago/Turabian StyleD’Angelo, Daniela, Marco De Martino, Claudio Arra, and Alfredo Fusco. 2019. "Emerging Role of USP8, HMGA, and Non-Coding RNAs in Pituitary Tumorigenesis" Cancers 11, no. 9: 1302. https://doi.org/10.3390/cancers11091302
APA StyleD’Angelo, D., De Martino, M., Arra, C., & Fusco, A. (2019). Emerging Role of USP8, HMGA, and Non-Coding RNAs in Pituitary Tumorigenesis. Cancers, 11(9), 1302. https://doi.org/10.3390/cancers11091302