Increased Non-Homologous End Joining Makes DNA-PK a Promising Target for Therapeutic Intervention in Uveal Melanoma


Abstract
1. Introduction
2. Results
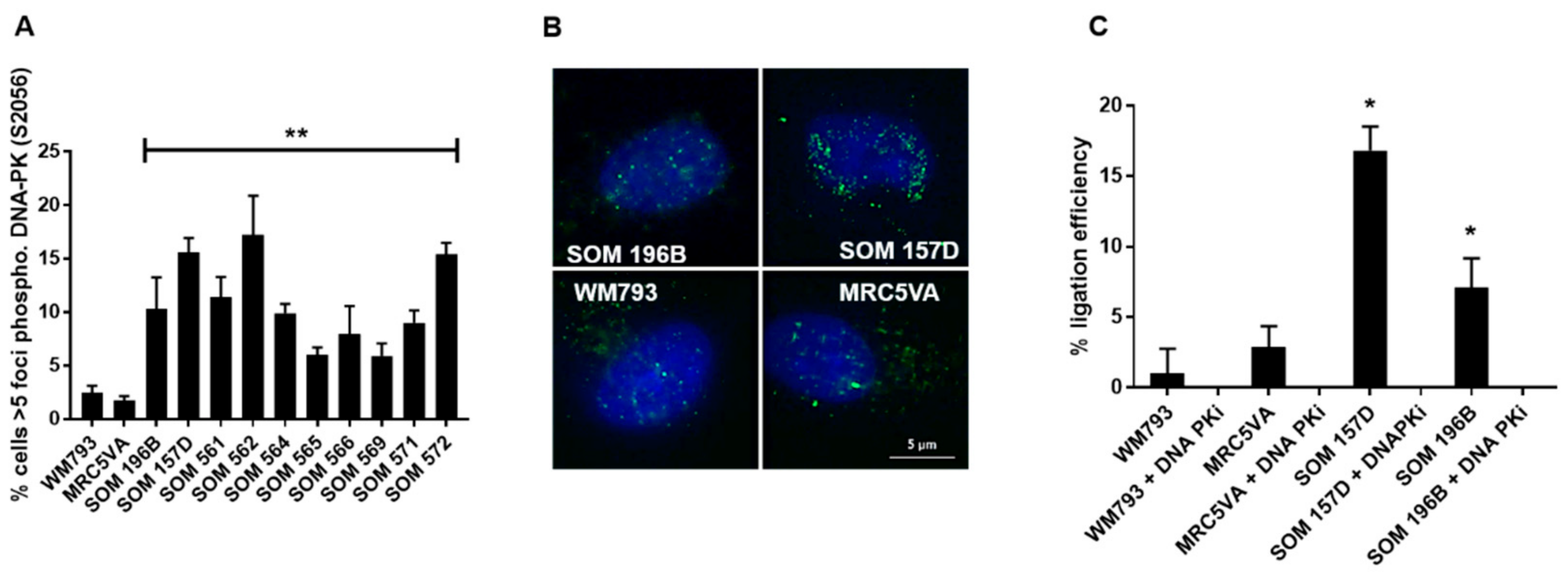
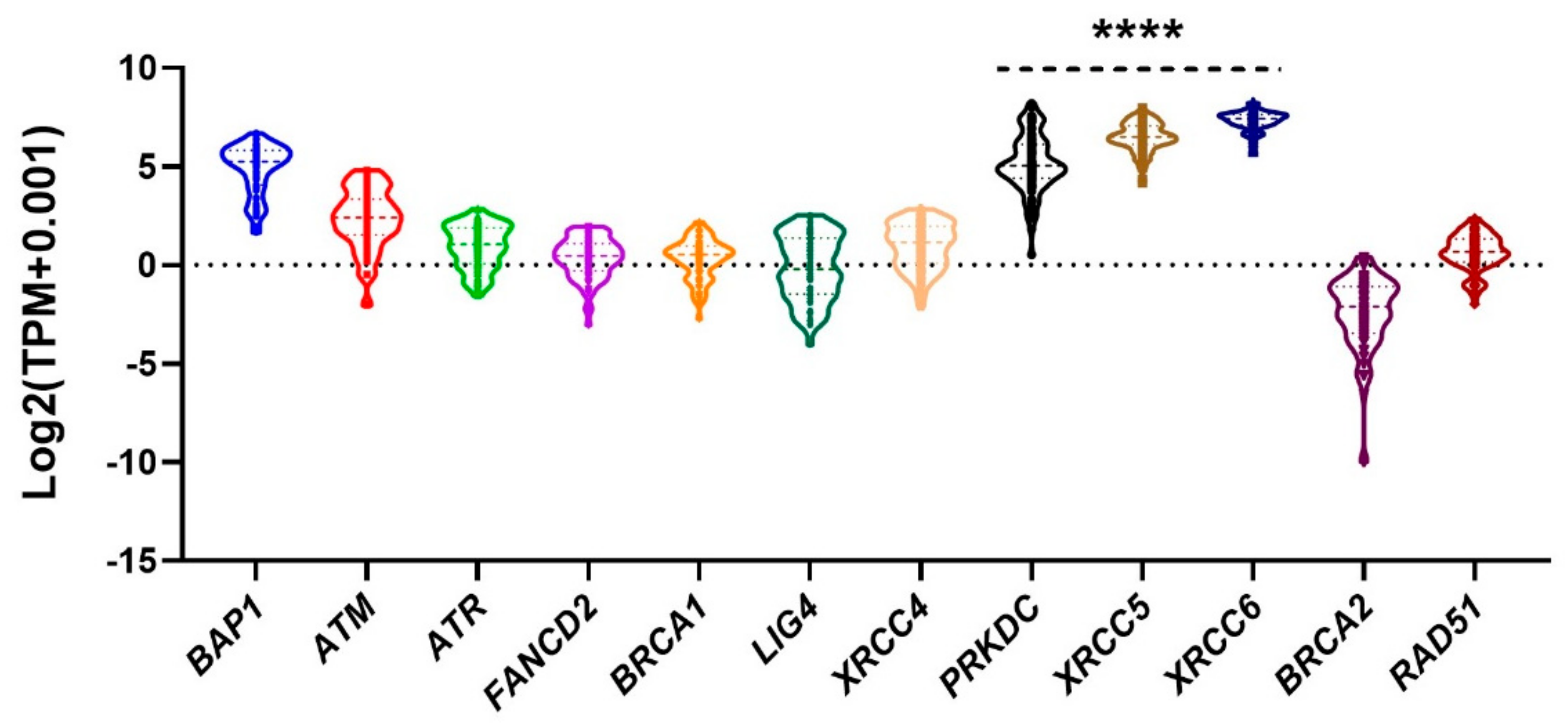
2.1. NHEJ is Elevated in UM
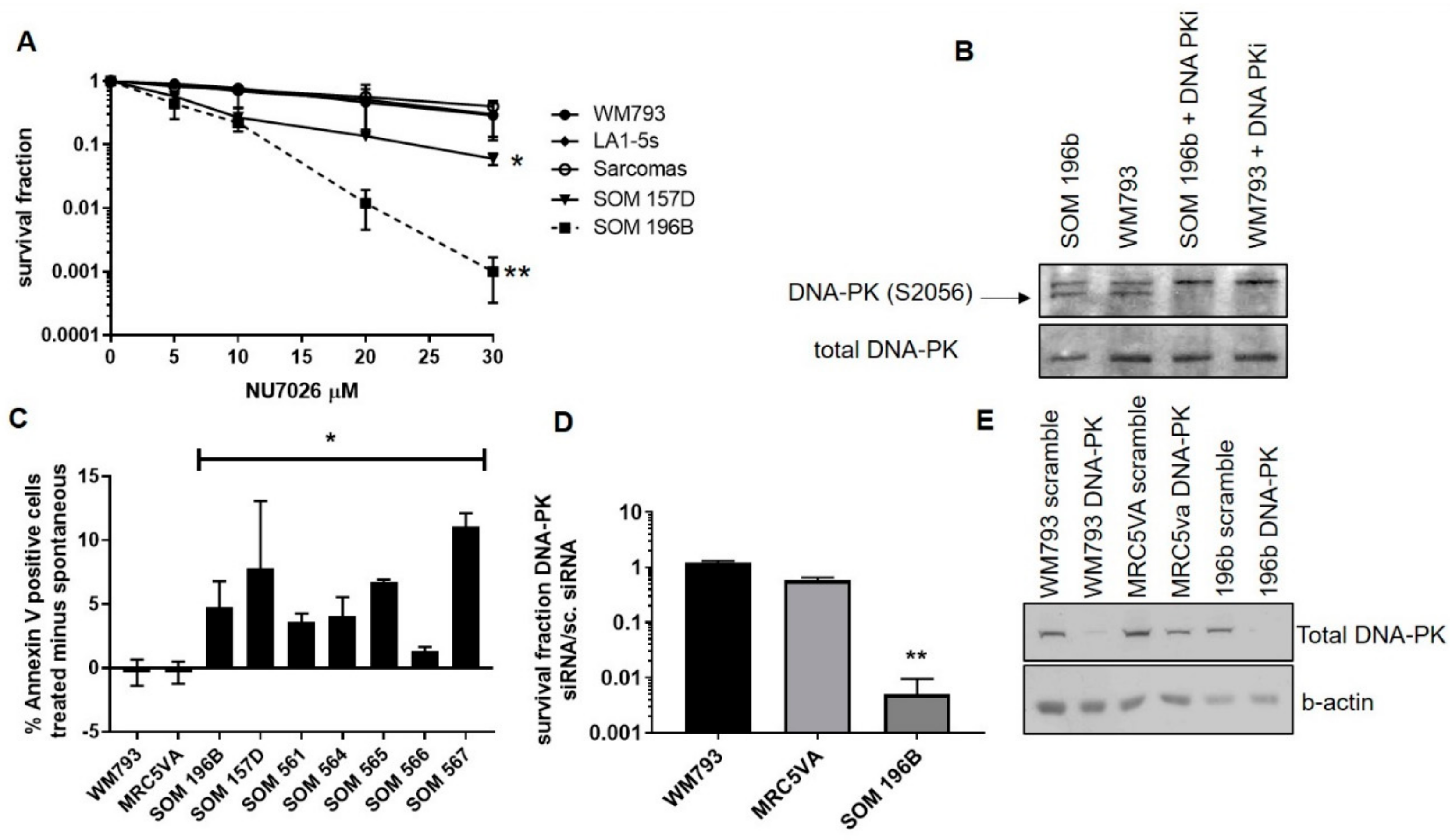
2.2. Inhibition of DNA-PK Mediated NHEJ is Toxic to UM and Induces Apoptosis
2.3. siRNA-Mediated Depletion of DNA-PKcs is Toxic to UM
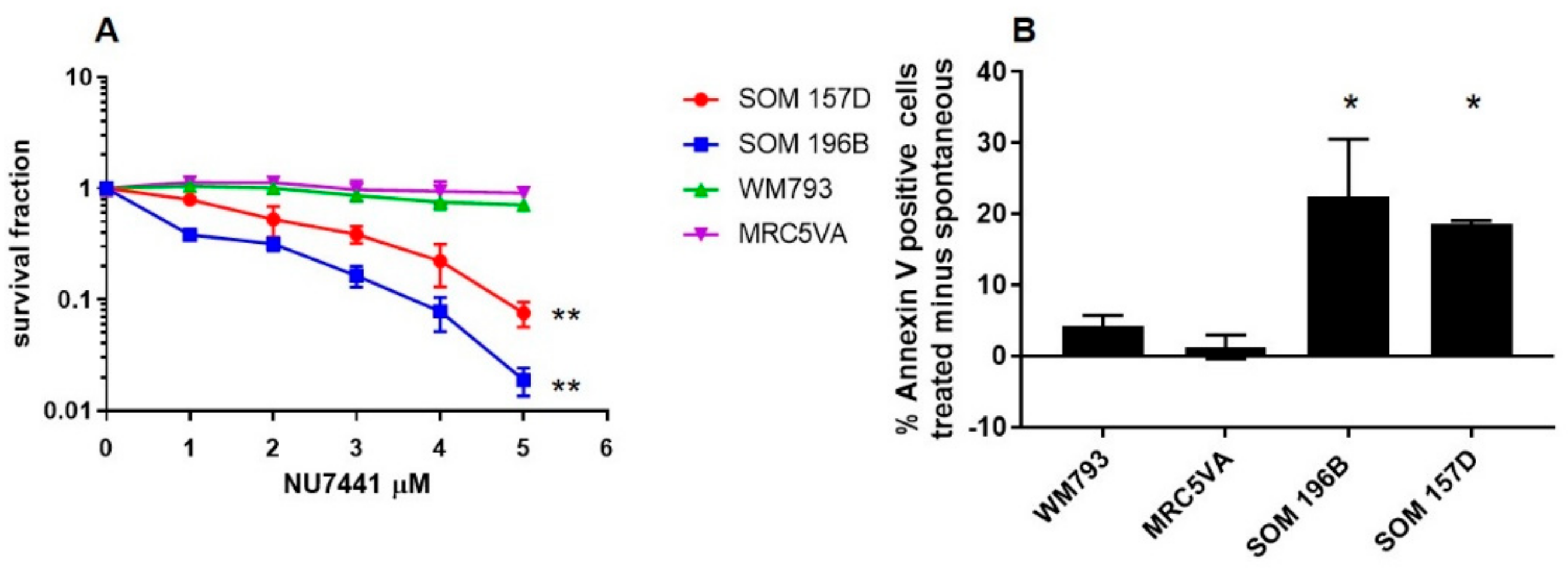
2.4. Confirmation of UM Sensitivity to DNA-PK Inhibition with the Alternative Inhibitor NU7441
2.5. DNA-PK Inhibition Potentiates the Cytotoxic Effects of IR in UM Cells and Sensitizes UM to ICL Agents
2.6. HR Functionality in UM Following Inhibition of NHEJ
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. DNA Repair Protein Foci Formation Analysis
4.3. End-Joining Efficiency Assay
4.4. The Cancer Genome Atlas (TCGA) UM Omics Data Analysis for Upregulation of NHEJ
4.5. Toxicity Assays
4.6. Protein Expression by Western Blotting
4.7. Annexin V Apoptosis Assay
4.8. siRNA Transfection
4.9. Sister Chromatid Exchange Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Singh, A.D.; Turell, M.E.; Topham, A.K. Uveal melanoma: Trends in incidence, treatment, and survival. Ophthalmology 2011, 118, 1881–1885. [Google Scholar] [CrossRef] [PubMed]
- Aronow, M.E.; Topham, A.K.; Singh, A.D. Uveal melanoma: 5-Year update on incidence, treatment, and survival (SEER 1973–2013). Ocular Oncol. Pathol. 2018, 4, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Papakostas, T.D.; Lane, A.M.; Morrison, M.; Gragoudas, E.S.; Kim, I.K. Long-term outcomes after proton beam irradiation in patients with large choroidal melanomas. JAMA Ophthalmol. 2017, 135, 1191–1196. [Google Scholar] [CrossRef] [PubMed]
- Karydis, I.; Gangi, A.; Wheater, M.J.; Choi, J.; Wilson, I.; Thomas, K.; Pearce, N.; Takhar, A.; Gupta, S.; Hardman, D.; et al. Percutaneous hepatic perfusion with melphalan in uveal melanoma: A safe and effective treatment modality in an orphan disease. J. Surg. Oncol. 2018, 117, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Kujala, E.; Makitie, T.; Kivela, T. Very long-term prognosis of patients with malignant uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2003, 44, 4651–4659. [Google Scholar] [CrossRef] [PubMed]
- Nichols, E.E.; Richmond, A.; Daniels, A.B. Tumor characteristics, genetics, management, and the risk of metastasis in uveal melanoma. Semin. Ophthalmol. 2016, 31, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Nichols, E.E.; Richmond, A.; Daniels, A.B. Micrometastatic dormancy in uveal melanoma: a comprehensive review of the evidence, mechanisms, and implications for future adjuvant therapies. Int. Ophthalmol. Clin. 2017, 57, 1–10. [Google Scholar] [CrossRef]
- Prescher, G.; Bornfeld, N.; Hirche, H.; Horsthemke, B.; Jockel, K.H.; Becher, R. Prognostic implications of monosomy 3 in uveal melanoma. Lancet 1996, 347, 1222–1225. [Google Scholar] [CrossRef]
- Sisley, K.; Rennie, I.G.; Parsons, M.A.; Jacques, R.; Hammond, D.W.; Bell, S.M.; Potter, A.M.; Rees, R.C. Abnormalities of chromosomes 3 and 8 in posterior uveal melanoma correlate with prognosis. Genes Chromosomes Cancer 1997, 19, 22–28. [Google Scholar] [CrossRef]
- White, V.A.; Chambers, J.D.; Courtright, P.D.; Chang, W.Y.; Horsman, D.E. Correlation of cytogenetic abnormalities with the outcome of patients with uveal melanoma. Cancer 1998, 83, 354–359. [Google Scholar] [CrossRef]
- Sisley, K.; Parsons, M.A.; Garnham, J.; Potter, A.M.; Curtis, D.; Rees, R.C.; Rennie, I.G. Association of specific chromosome alterations with tumour phenotype in posterior uveal melanoma. Br. J. Cancer 2000, 82, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Landreville, S.; Agapova, O.A.; Harbour, J.W. Emerging insights into the molecular pathogenesis of uveal melanoma. Future Oncol. 2008, 4, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O’Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 2009, 457, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in GNA11 in uveal melanoma. N. Eng. J. Med. 2010, 363, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Onken, M.D.; Worley, L.A.; Long, M.D.; Duan, S.; Council, M.L.; Bowcock, A.M.; Harbour, J.W. Oncogenic mutations in GNAQ occur early in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2008, 49, 5230–5234. [Google Scholar] [CrossRef]
- Sisley, K.; Doherty, R.; Cross, N.A. What hope for the future? GNAQ and uveal melanoma. Br. J. Ophthalmol. 2011, 95, 620–623. [Google Scholar] [CrossRef]
- Vader, M.J.C.; Madigan, M.C.; Versluis, M.; Suleiman, H.M.; Gezgin, G.; Gruis, N.A.; Out-Luiting, J.J.; Bergman, W.; Verdijk, R.M.; Jager, M.J.; et al. GNAQ and GNA11 mutations and downstream YAP activation in choroidal nevi. Br. J. Cancer 2017, 117, 884–887. [Google Scholar] [CrossRef]
- Steeb, T.; Wessely, A.; Ruzicka, T.; Heppt, M.V.; Berking, C. How to MEK the best of uveal melanoma: A systematic review on the efficacy and safety of MEK inhibitors in metastatic or unresectable uveal melanoma. Eur. J. Cancer 2018, 103, 41–51. [Google Scholar] [CrossRef]
- Chalasani, R.; Giblin, M.; Conway, R.M. Role of topical chemotherapy for primary acquired melanosis and malignant melanoma of the conjunctiva and cornea: Review of the evidence and recommendations for treatment. Clin. Exp. Ophthalmol. 2006, 34, 708–714. [Google Scholar] [CrossRef]
- Vogl, T.; Eichler, K.; Zangos, S.; Herzog, C.; Hammerstingl, R.; Balzer, J.; Gholami, A. Preliminary experience with transarterial chemoembolization (TACE) in liver metastases of uveal malignant melanoma: Local tumor control and survival. J. Cancer Res. Clin. Oncol. 2007, 133, 177–184. [Google Scholar] [CrossRef]
- Gravells, P.; Hoh, L.; Canovas, D.; Rennie, I.G.; Sisley, K.; Bryant, H.E. Resistance of uveal melanoma to the interstrand cross-linking agent mitomycin C is associated with reduced expression of CYP450R. Br. J. Cancer 2011, 104, 1098–1105. [Google Scholar] [CrossRef] [PubMed]
- Her, J.; Bunting, S.F. How cells ensure correct repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10502–10511. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Halbrook, J.; Nickoloff, J.A. Interactive competition between homologous recombination and non-homologous end joining. Mol. Cancer Res. MCR 2003, 1, 913–920. [Google Scholar] [PubMed]
- Hoh, L.; Gravells, P.; Canovas, D.; Ul-Hassan, A.; Rennie, I.G.; Bryant, H.; Sisley, K. Atypically low spontaneous sister chromatid exchange formation in uveal melanoma. Genes Chromosomes Cancer 2011, 50, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Gravells, P.; Hoh, L.; Solovieva, S.; Patil, A.; Dudziec, E.; Rennie, I.G.; Sisley, K.; Bryant, H.E. Reduced FANCD2 influences spontaneous SCE and RAD51 foci formation in uveal melanoma and Fanconi anaemia. Oncogene 2013, 32, 5338–5346. [Google Scholar] [CrossRef] [PubMed]
- Adamo, A.; Collis, S.J.; Adelman, C.A.; Silva, N.; Horejsi, Z.; Ward, J.D.; Martinez-Perez, E.; Boulton, S.J.; La Volpe, A. Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol. Cell 2010, 39, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.P.; Chan, D.W.; Kobayashi, J.; Burma, S.; Asaithamby, A.; Morotomi-Yano, K.; Botvinick, E.; Qin, J.; Chen, D.J. Cell cycle dependence of DNA-dependent protein kinase phosphorylation in response to DNA double strand breaks. J. Biol. Chem. 2005, 280, 14709–14715. [Google Scholar] [CrossRef] [PubMed]
- Dogrusoz, M.; Ruschel, T.A.; Cao, J.; Colak, S.; van Pelt, S.I.; Kroes, W.G.M.; Teunisse, A.; Alsafadi, S.; van Duinen, S.G.; Luyten, G.P.M.; et al. Differential expression of DNA repair genes in prognostically-favorable versus unfavorable uveal melanoma. Cancers 2019, 11, 1104. [Google Scholar] [CrossRef]
- Davidson, D.; Coulombe, Y.; Martinez-Marignac, V.L.; Amrein, L.; Grenier, J.; Hodkinson, K.; Masson, J.Y.; Aloyz, R.; Panasci, L. Irinotecan and DNA-PKcs inhibitors synergize in killing of colon cancer cells. Investig. N. Drugs 2012, 30, 1248–1256. [Google Scholar] [CrossRef]
- Veuger, S.J.; Curtin, N.J.; Richardson, C.J.; Smith, G.C.; Durkacz, B.W. Radiosensitization and DNA repair inhibition by the combined use of novel inhibitors of DNA-dependent protein kinase and poly (ADP-ribose) polymerase-1. Cancer Res. 2003, 63, 6008–6015. [Google Scholar]
- Sonoda, E.; Sasaki, M.S.; Morrison, C.; Yamaguchi-Iwai, Y.; Takata, M.; Takeda, S. Sister chromatid exchanges are mediated by homologous recombination in vertebrate cells. Mol. Cell. Biol. 1999, 19, 5166–5169. [Google Scholar] [CrossRef] [PubMed]
- Talens, F.; Jalving, M.; Gietema, J.A.; Van Vugt, M.A. Therapeutic targeting and patient selection for cancers with homologous recombination defects. Expert Opin. Drug Discov. 2017, 12, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Latt, S.A.; Stetten, G.; Juergens, L.A.; Buchanan, G.R.; Gerald, P.S. Induction by alkylating agents of sister chromatid exchanges and chromatid breaks in Fanconi’s anemia. Proc. Natl. Acad. Sci. USA 1975, 72, 4066–4070. [Google Scholar] [CrossRef] [PubMed]
- Pace, P.; Mosedale, G.; Hodskinson, M.R.; Rosado, I.V.; Sivasubramaniam, M.; Patel, K.J. Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science 2010, 329, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Zitzmann, S.; Westermann, F.; Arnold, K.; Brouwers, S.; Schwab, M.; Savelyeva, L. Increased rates of spontaneous sister chromatid exchange in lymphocytes of BRCA2+/- carriers of familial breast cancer clusters. Cancer Lett. 2004, 210, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, S.K.; Jette, N.; Lees-Miller, S.P. Non-homologous end joining: Emerging themes and unanswered questions. DNA Repair 2014, 17, 2–8. [Google Scholar] [CrossRef]
- Shen, H.; Schultz, M.; Kruh, G.D.; Tew, K.D. Increased expression of DNA-dependent protein kinase confers resistance to adriamycin. Biochimica et Biophysica Acta 1998, 1381, 131–138. [Google Scholar] [CrossRef]
- Eriksson, A.; Lewensoh, R.; Larsson, R.; Nilsson, A. DNA-dependent protein kinase in leukaemia cells and correlation with drug sensitivity. Anticancer Res. 2002, 22, 1787–1793. [Google Scholar]
- Kuo, C.Y.; Chou, W.C.; Wu, C.C.; Wong, T.S.; Kakadiya, R.; Lee, T.C.; Su, T.L.; Wang, H.C. Repairing of N-mustard derivative BO-1055 induced DNA damage requires NER, HR, and MGMT-dependent DNA repair mechanisms. Oncotarget 2015, 6, 25770–25783. [Google Scholar] [CrossRef]
- De Koning, L.; Decaudin, D.; El Botty, R.; Nicolas, A.; Carita, G.; Schuller, M.; Ouine, B.; Cartier, A.; Naguez, A.; Fleury, J.; et al. PARP inhibition increases the response to chemotherapy in uveal melanoma. Cancers 2019, 11, 751. [Google Scholar] [CrossRef]
- Woodward, J.K.; Nichols, C.E.; Rennie, I.G.; Parsons, M.A.; Murray, A.K.; Sisley, K. An in vitro assay to assess uveal melanoma invasion across endothelial and basement membrane barriers. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1708–1714. [Google Scholar]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 2018, 173, 400–416e411. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell 2017, 32, 204–220e215. [Google Scholar] [CrossRef] [PubMed]
- Vivian, J.; Rao, A.A.; Nothaft, F.A.; Ketchum, C.; Armstrong, J.; Novak, A.; Pfeil, J.; Narkizian, J.; Deran, A.D.; Musselman-Brown, A.; et al. Toil enables reproducible, open source, big biomedical data analyses. Nat. Biotech. 2017, 35, 314–316. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mean SCE/Cell | Median SCE/Cell | Range | |
|---|---|---|---|
| WM793 + DMSO | 13.69 | 13 | 9–19 |
| WM793 + DNAPKi | 29.27 | 24 | 14–54 |
| SOM 196B + DMSO | 4.25 | 4 | 1–9 |
| SOM 196 + DNAPKi | 6.78 | 6.25 | 3–18 |
| SOM 157d + DMSO | 5.97 | 6 | 2–13 |
| SOM 157d + DNAPKi | 9.97 | 8 | 4–26 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doherty, R.E.; Bryant, H.E.; Valluru, M.K.; Rennie, I.G.; Sisley, K. Increased Non-Homologous End Joining Makes DNA-PK a Promising Target for Therapeutic Intervention in Uveal Melanoma. Cancers 2019, 11, 1278. https://doi.org/10.3390/cancers11091278
Doherty RE, Bryant HE, Valluru MK, Rennie IG, Sisley K. Increased Non-Homologous End Joining Makes DNA-PK a Promising Target for Therapeutic Intervention in Uveal Melanoma. Cancers. 2019; 11(9):1278. https://doi.org/10.3390/cancers11091278
Chicago/Turabian StyleDoherty, Rachel E., Helen E. Bryant, Manoj K. Valluru, Ian G. Rennie, and Karen Sisley. 2019. "Increased Non-Homologous End Joining Makes DNA-PK a Promising Target for Therapeutic Intervention in Uveal Melanoma" Cancers 11, no. 9: 1278. https://doi.org/10.3390/cancers11091278
APA StyleDoherty, R. E., Bryant, H. E., Valluru, M. K., Rennie, I. G., & Sisley, K. (2019). Increased Non-Homologous End Joining Makes DNA-PK a Promising Target for Therapeutic Intervention in Uveal Melanoma. Cancers, 11(9), 1278. https://doi.org/10.3390/cancers11091278