Determination of the Potential Tumor-Suppressive Effects of Gsdme in a Chemically Induced and in a Genetically Modified Intestinal Cancer Mouse Model

 ,
,  and
and
Abstract
1. Introduction
2. Results
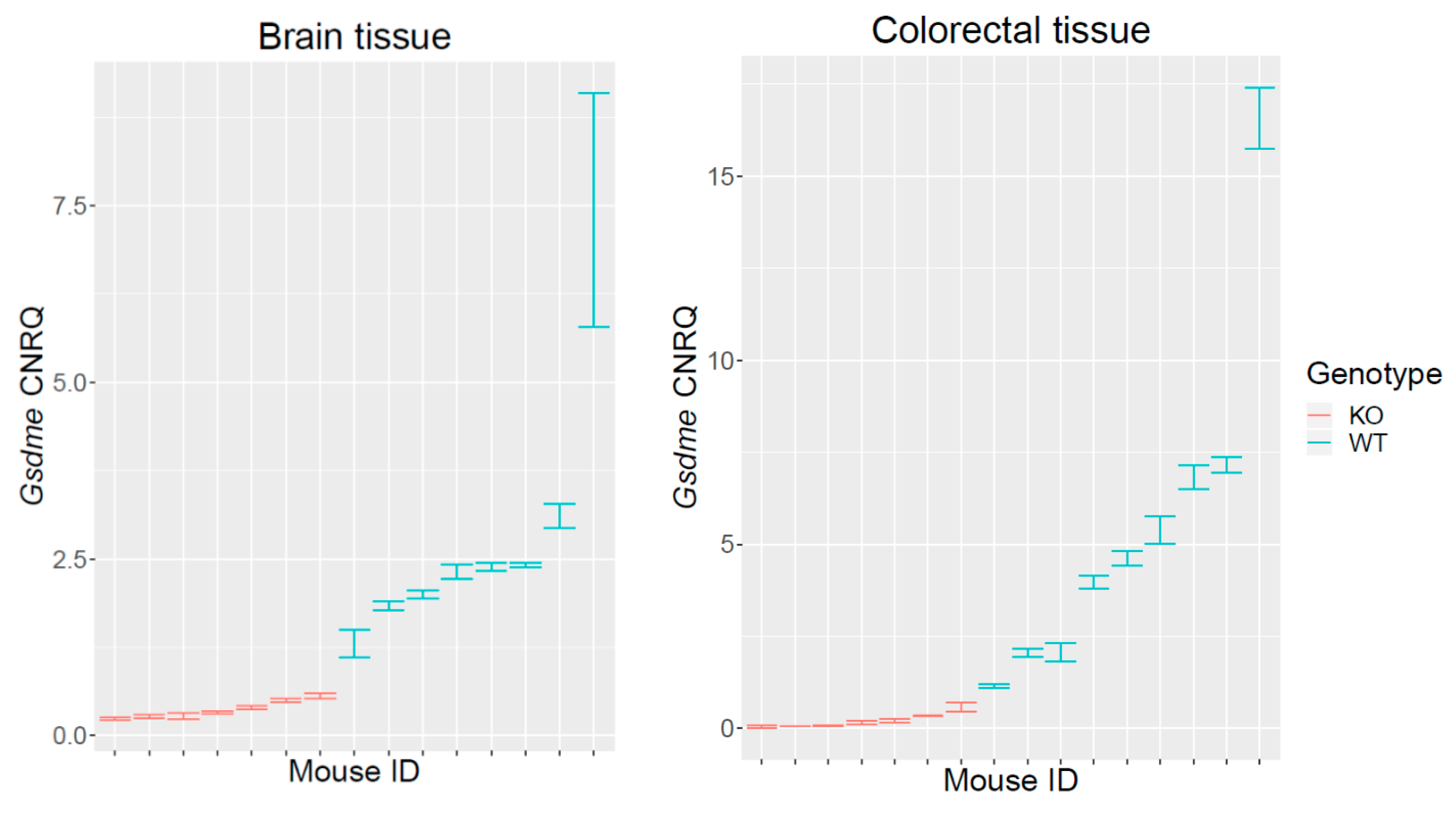
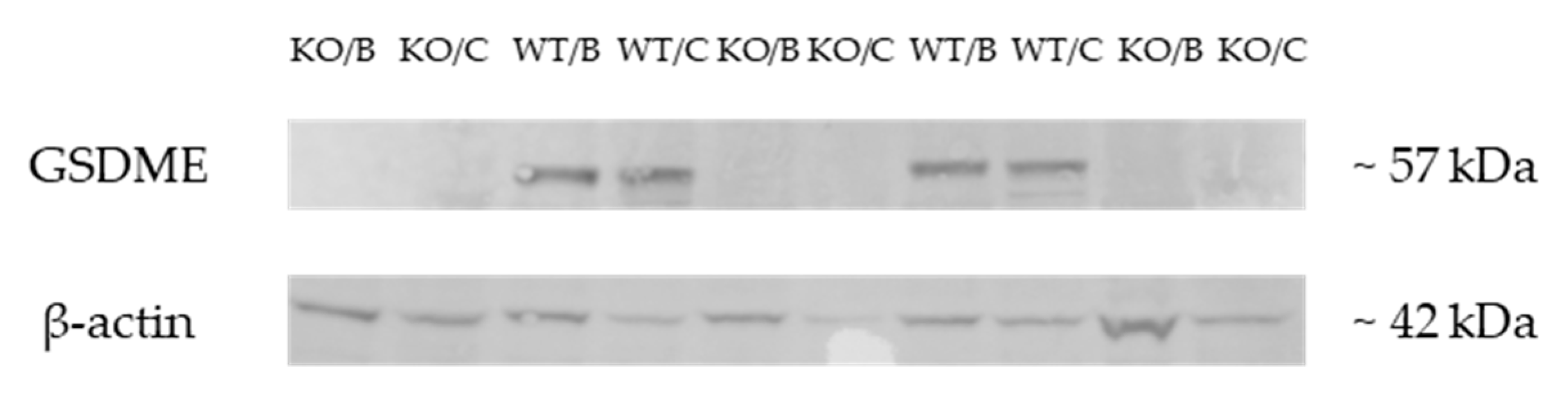
2.1. Validation of the Gsdme KO Mouse Model
2.2. Chemically Induced Intestinal Cancer Mouse Model
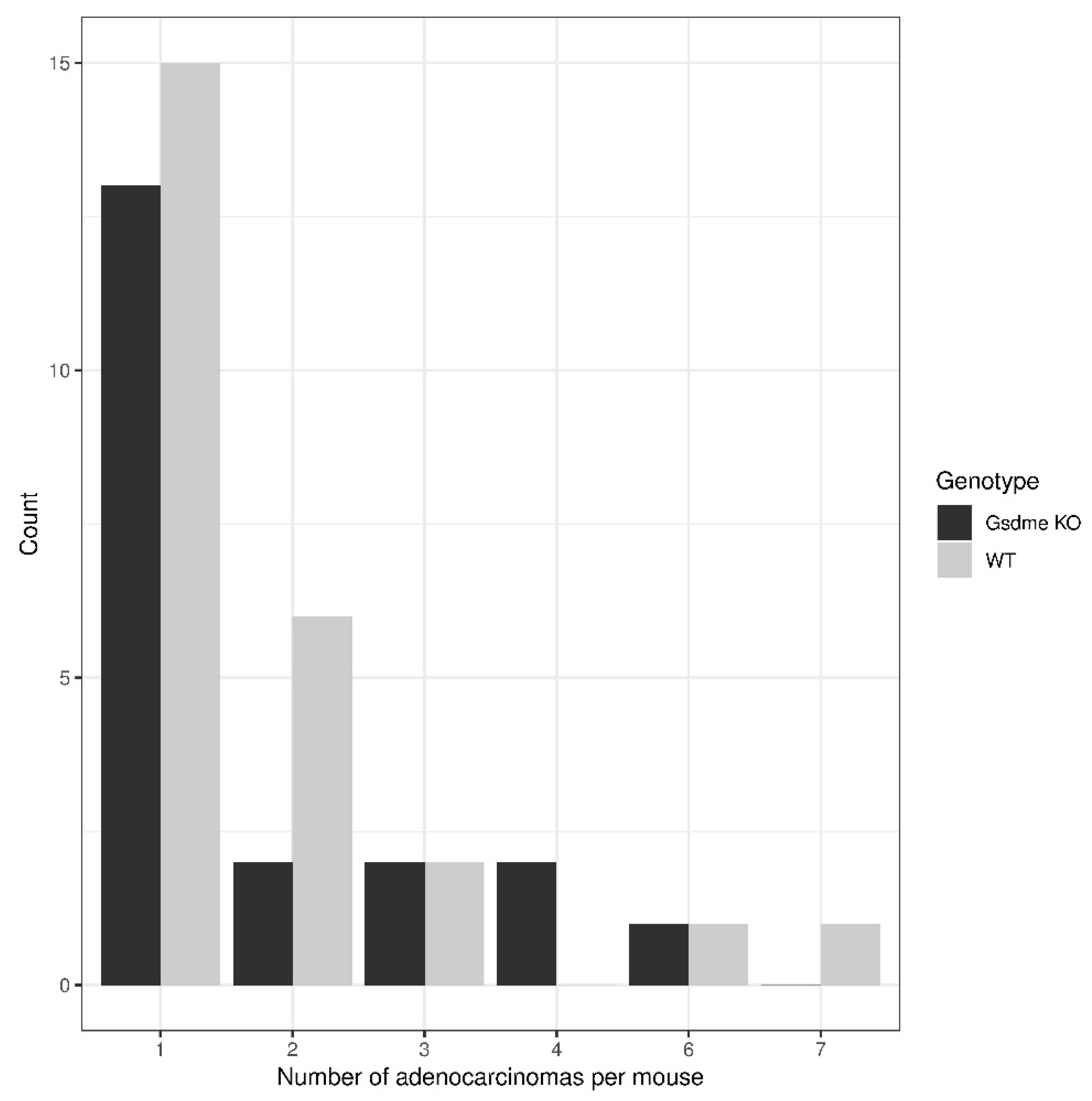
2.2.1. Intestinal Pathology in the Chemically Induced Model
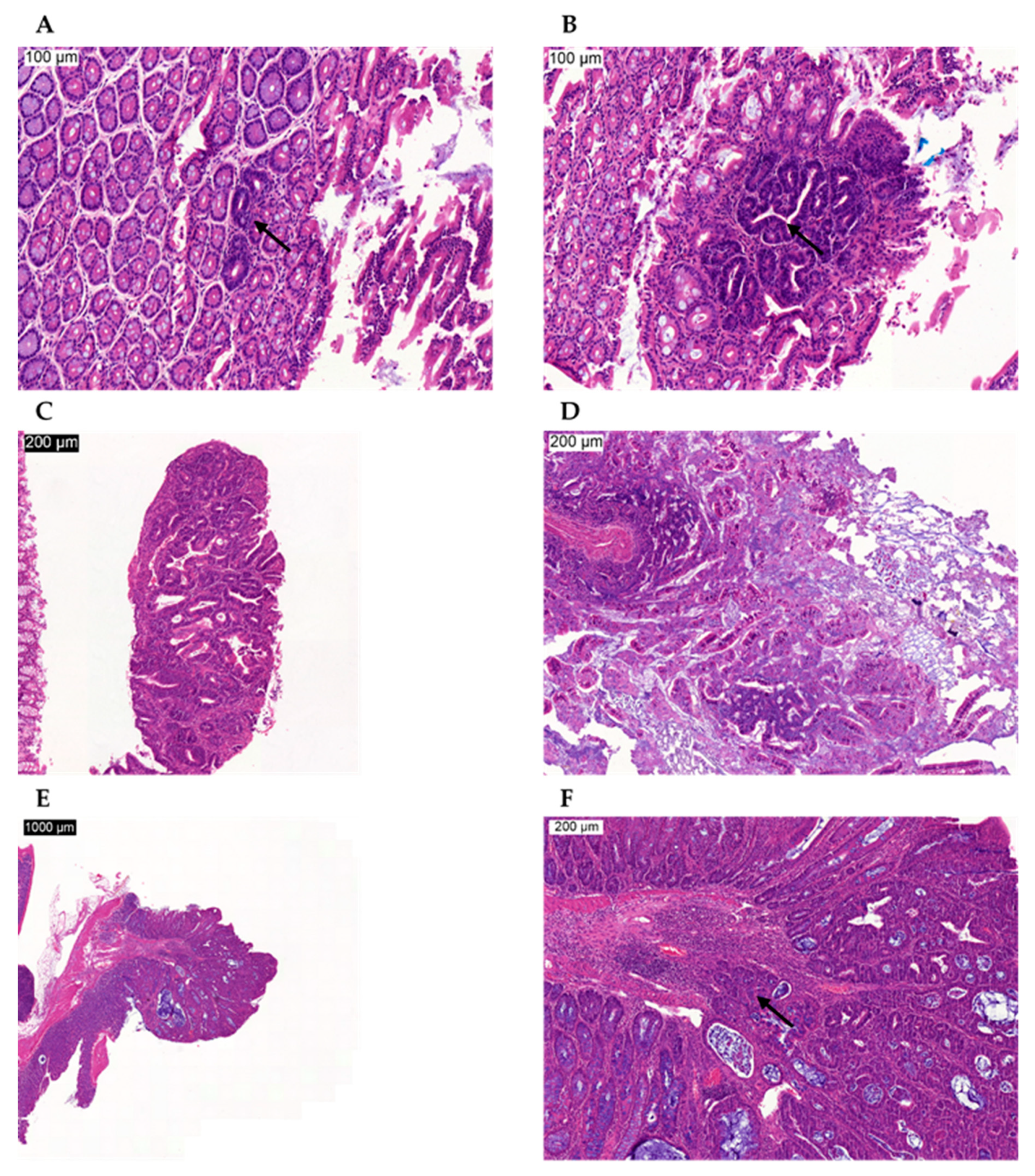
Proliferative Lesions in the Chemically Induced Model
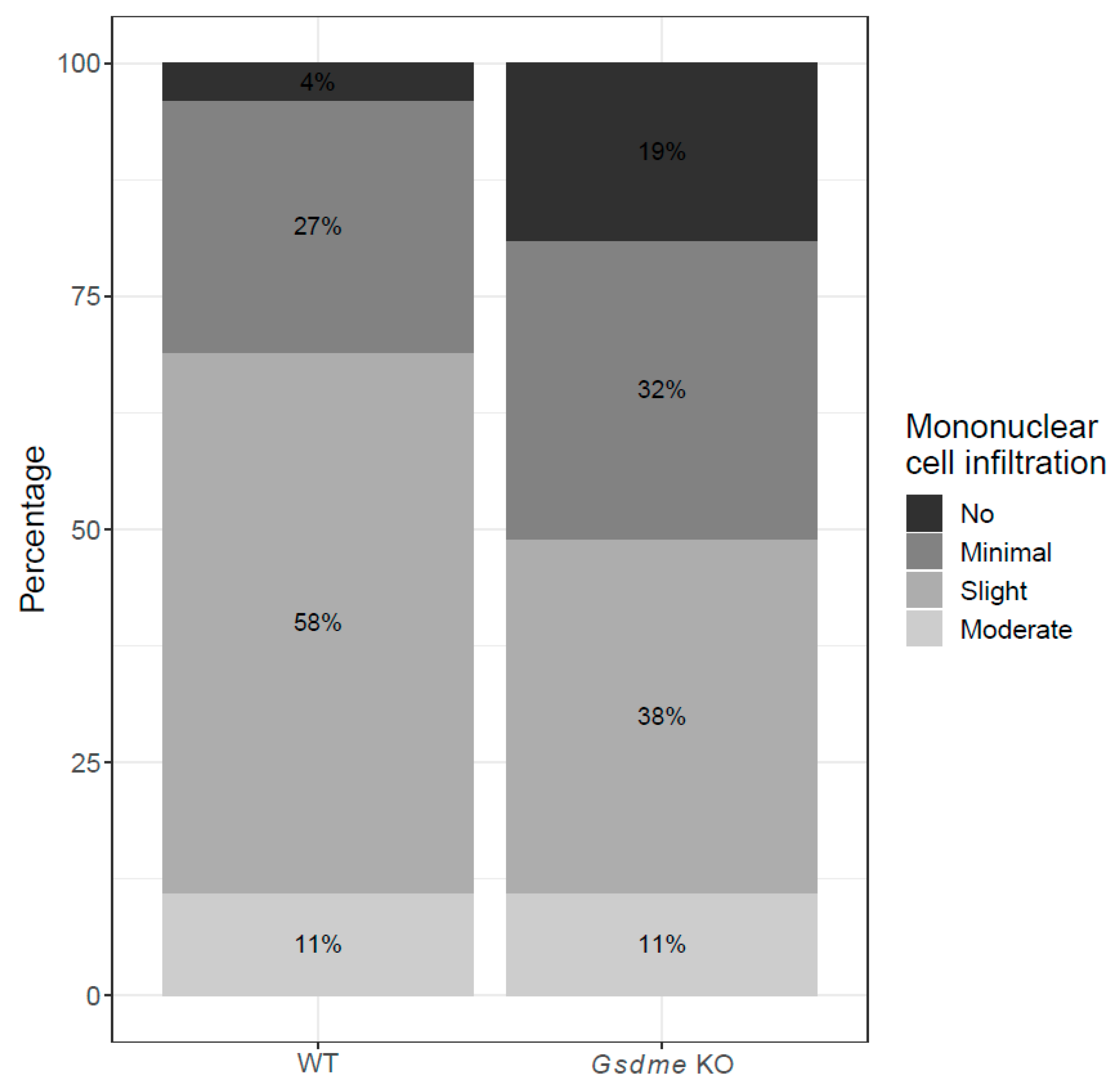
2.2.2. Other Organs Analyzed in the Chemically Induced Model
2.3. Genetically Modified Intestinal Cancer Mouse Model
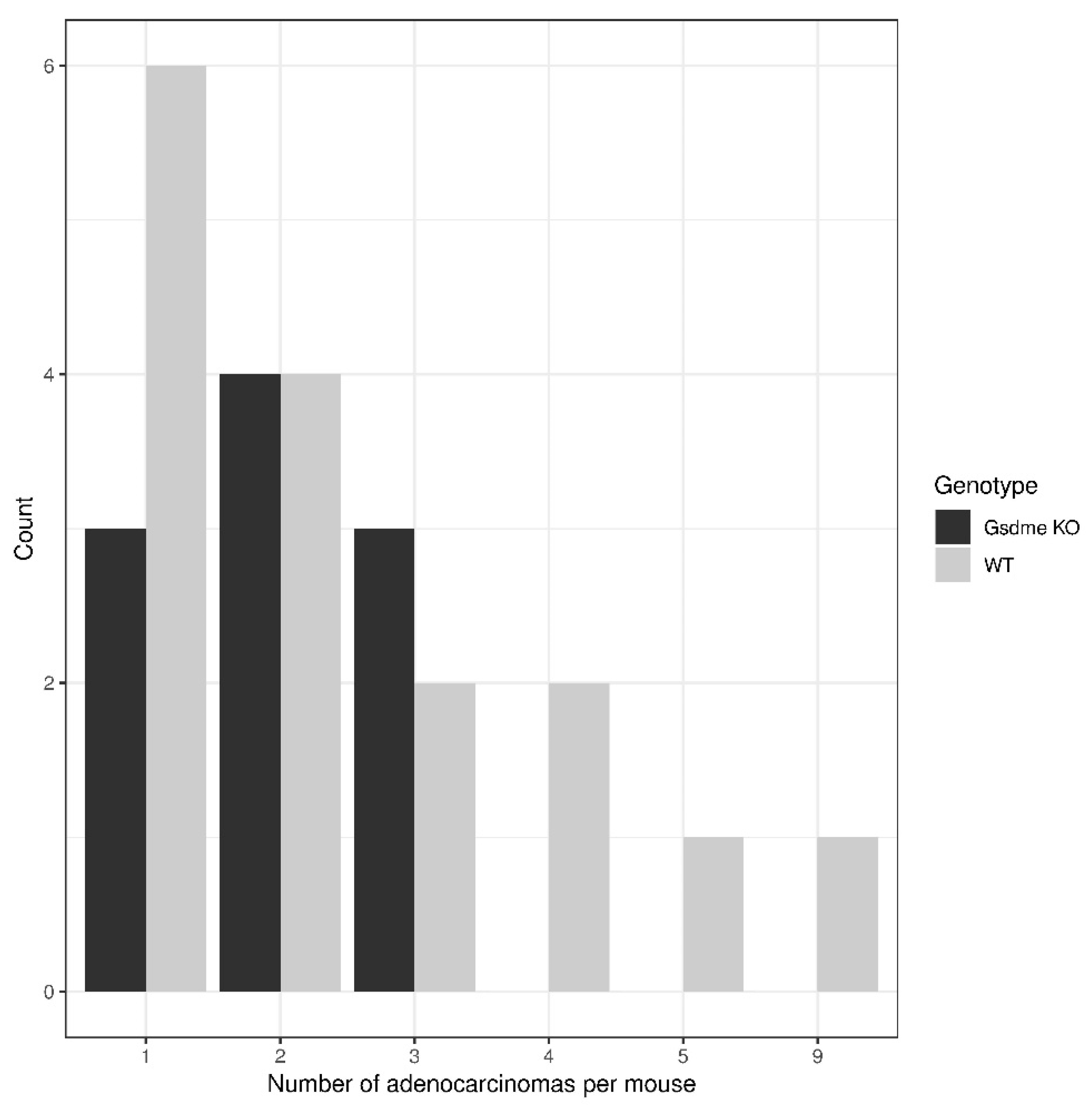
2.3.1. Intestinal Pathology in the Genetically Modified Model
Proliferative Lesions in the Genetically Modified Model
2.3.2. Other Organs Analyzed in the Genetically Modified Model
3. Discussion
4. Materials and Methods
4.1. Gsdme Knockout Mouse
4.1.1. Chemically Induced Intestinal Cancer Mouse Model
4.1.2. Genetically Modified Intestinal Cancer Mouse Model
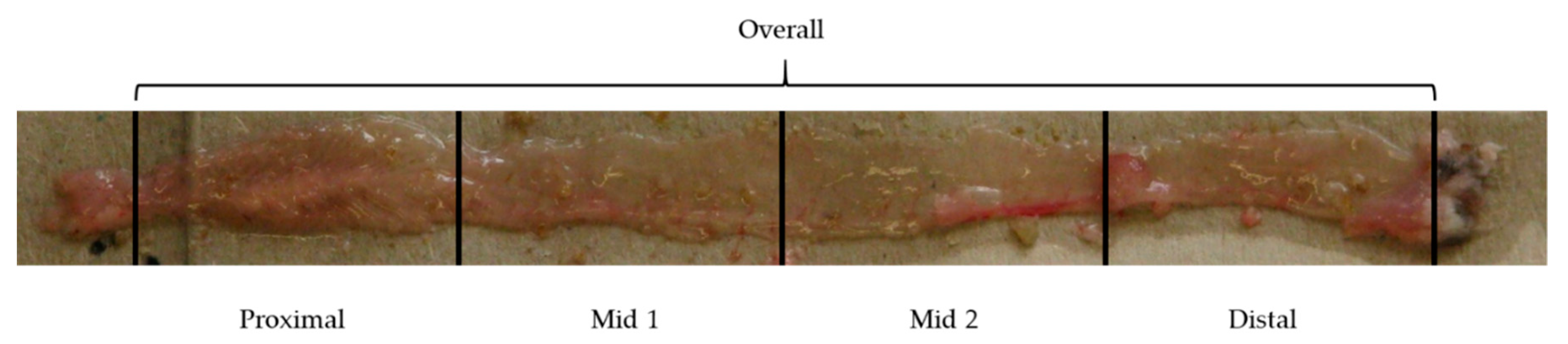
4.2. Dissection and Histology
4.3. qRT-PCR
4.4. Western Blot
4.5. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van Laer, L.; Huizing, E.H.; Verstreken, M.; van Zuijlen, D.; Wauters, J.G.; Bossuyt, P.J.; Van de Heyning, P.; McGuirt, W.T.; Smith, R.J.; Willems, P.J.; et al. Nonsyndromic hearing impairment is associated with a mutation in DFNA5. Nat. Genet. 1998, 20, 194–197. [Google Scholar] [CrossRef] [PubMed]
- de Beeck, K.O.; Van Camp, G.; Thys, S.; Cools, N.; Callebaut, I.; Vrijens, K.; Van Nassauw, L.; Van Tendeloo, V.F.I.; Timmermans, J.P.; Van Laer, L. The DFNA5 gene, responsible for hearing loss and involved in cancer, encodes a novel apoptosis-inducing protein. Eur. J. Hum. Genet. 2011, 19, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Van Rossom, S.; de Beeck, K.O.; Franssens, V.; Swinnen, E.; Schepers, A.; Ghillebert, R.; Caldara, M.; Van Camp, G.; Winderickx, J. The splicing mutant of the human tumor suppressor protein DFNA5 induces programmed cell death when expressed in the yeast Saccharomyces cerevisiae. Front. Oncol. 2012, 2, 77. [Google Scholar] [CrossRef] [PubMed]
- Van Rossom, S.; de Beeck, K.O.; Hristovska, V.; Winderickx, J.; Van Camp, G. The deafness gene DFNA5 induces programmed cell death through mitochondria and MAPK-related pathways. Front. Cell. Neurosci. 2015, 9, 231. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, J.; de Beeck, K.O.; Fransen, E.; Croes, L.; Beyens, M.; Suls, A.; Berghe, W.V.; Peeters, M.; Van Camp, G. Methylation analysis of Gasdermin E shows great promise as a biomarker for colorectal cancer. Cancer Med. 2019, 8, 2133–2145. [Google Scholar] [CrossRef] [PubMed]
- Croes, L.; Beyens, M.; Fransen, E.; Ibrahim, J.; Berghe, W.V.; Suls, A.; Peeters, M.; Pauwels, P.; Van Camp, G.; de Beeck, K.O. Large-scale analysis of DFNA5 methylation reveals its potential as biomarker for breast cancer. Clin. Epigenetics 2018, 10, 51. [Google Scholar] [CrossRef] [PubMed]
- Croes, L.; de Beeck, K.O.; Pauwels, P.; Berghe, W.V.; Peeters, M.; Fransen, E.; Van Camp, G. DFNA5 promoter methylation a marker for breast tumorigenesis. Oncotarget 2017, 8, 31948–31958. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-J.; Tang, L.; Shen, D.-W.; Wang, C.; Yuan, Q.-Y.; Gao, W.; Wang, Y.-K.; Xu, R.-H.; Zhang, H. The expression and regulation of DFNA5 in human hepatocellular carcinoma DFNA5 in hepatocellular carcinoma. Mol. Biol. Rep. 2013, 40, 6525–6531. [Google Scholar] [CrossRef]
- Yokomizo, K.; Harada, Y.; Kijima, K.; Shinmura, K.; Sakata, M.; Sakuraba, K.; Kitamura, Y.; Shirahata, A.; Goto, T.; Mizukami, H.; et al. Methylation of the DFNA5 gene is frequently detected in colorectal cancer. Anticancer Res. 2012, 32, 1319–1322. [Google Scholar]
- de Beeck, K.O.; Van Laer, L.; Van Camp, G. DFNA5, a gene involved in hearing loss and cancer: A review. Ann. Otol. Rhinol. Laryngol. 2012, 121, 197–207. [Google Scholar] [CrossRef]
- Fujikane, T.; Nishikawa, N.; Toyota, M.; Suzuki, H.; Nojima, M.; Maruyama, R.; Ashida, M.; Ohe-Toyota, M.; Kai, M.; Nishidate, T.; et al. Genomic screening for genes upregulated by demethylation revealed novel targets of epigenetic silencing in breast cancer. Breast Cancer Res. Treat. 2009, 122, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Lebron, C.; Nagpal, J.K.; Chae, Y.K.; Chang, X.; Huang, Y.; Chuang, T.; Yamashita, K.; Trink, B.; Ratovitski, E.A.; et al. Methylation of the DFNA5 increases risk of lymph node metastasis in human breast cancer. Biochem. Biophys. Res. Commun. 2008, 370, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Chang, X.; Yamashita, K.; Nagpal, J.K.; Baek, J.H.; Wu, G.; Trink, B.; Ratovitski, E.A.; Mori, M.; Sidransky, D. Aberrant promoter methylation and tumor suppressive activity of the DFNA5 gene in colorectal carcinoma. Oncogene 2008, 27, 3624–3634. [Google Scholar] [CrossRef] [PubMed]
- Akino, K.; Toyota, M.; Suzuki, H.; Imai, T.; Maruyama, R.; Kusano, M.; Nishikawa, N.; Watanabe, Y.; Sasaki, Y.; Abe, T.; et al. Identification of DFNA5 as a target of epigenetic inactivation in gastric cancer. Cancer Sci. 2006, 98, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Masuda, Y.; Futamura, M.; Kamino, H.; Nakamura, Y.; Kitamura, N.; Ohnishi, S.; Miyamoto, Y.; Ichikawa, H.; Ohta, T.; Ohki, M.; et al. The potential role of DFNA5, a hearing impairment gene, in p53-mediated cellular response to DNA damage. J. Hum. Genet. 2006, 51, 652–664. [Google Scholar] [CrossRef] [PubMed]
- Lage, H.; Helmbach, H.; Grottke, C.; Dietel, M.; Schadendorf, D. DFNA5 (ICERE-1) contributes to acquired etoposide resistance in melanoma cells. FEBS Lett. 2001, 494, 54–59. [Google Scholar] [CrossRef]
- Thompson, D.A.; Weigel, R.J. Characterization of a gene that is inversely correlated with estrogen receptor expression (ICERE-1) in breast carcinomas. Eur. J. Biochem. 1998, 252, 169–177. [Google Scholar] [CrossRef] [PubMed]
- De Robertis, M.; Massi, E.; Poeta, M.L.; Carotti, S.; Morini, S.; Cecchetelli, L.; Signori, E.; Fazio, V.M. The AOM/DSS murine model for the study of colon carcinogenesis: From pathways to diagnosis and therapy studies. J. Carcinog. 2011, 10, 9. [Google Scholar]
- Neufert, C.; Becker, C.; Neurath, M.F. An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat. Protoc. 2007, 2, 1998–2004. [Google Scholar] [CrossRef]
- Smits, R.; van der Houven van Oordt, W.; Luz, A.; Zurcher, C.; Jagmohan-Changur, S.; Breukel, C.; Khan, P.M.; Fodde, R. Apc1638N: A mouse model for familial adenomatous polyposis-associated desmoid tumors and cutaneous cysts. Gastroenterology 1998, 114, 275–283. [Google Scholar] [CrossRef]
- Fodde, R.; Edelmann, W.; Yang, K.; van Leeuwen, C.; Carlson, C.; Renault, B.; Breukel, C.; Alt, E.; Lipkin, M.; Khan, P.M. A targeted chain-termination mutation in the mouse Apc gene results in multiple intestinal tumors. Proc. Natl. Acad. Sci. USA 1994, 91, 8969–8973. [Google Scholar] [CrossRef] [PubMed]
- Thaker, A.I.; Shaker, A.; Rao, M.S.; Ciorba, M.A. Modeling colitis-associated cancer with azoxymethane (AOM) and dextran sulfate sodium (DSS). J. Vis. Exp. 2012, 10, e4100. [Google Scholar] [CrossRef] [PubMed]
- Nandan, M.O.; Yang, V.W. Genetic and Chemical Models of Colorectal Cancer in Mice. Curr. Colorectal Cancer Rep. 2010, 6, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, D.W.; Giardina, C.; Tanaka, T. Mouse models for the study of colon carcinogenesis. Carcinogenesis 2008, 30, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Boivin, G.P.; Washington, K.; Yang, K.; Ward, J.M.; Pretlow, T.P.; Russell, R.; Besselsen, D.G.; Godfrey, V.L.; Doetschman, T.; Dove, W.F.; et al. Pathology of mouse models of intestinal cancer: Consensus report and recommendations. Gastroenterology 2003, 124, 762–777. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.M.; Treuting, P.M. Rodent intestinal epithelial carcinogenesis: Pathology and preclinical models. Toxicol. Pathol. 2013, 42, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Guda, K.; Upender, M.B.; Belinsky, G.; Flynn, C.; Nakanishi, M.; Marino, J.N.; Ried, T.; Rosenberg, D.W. Carcinogen-induced colon tumors in mice are chromosomally stable and are characterized by low-level microsatellite instability. Oncogene 2004, 23, 3813–3821. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.-W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell Death Dis. 2004, 118, 285–296. [Google Scholar]
- Becker, C.; Fantini, M.C.; Schramm, C.; Lehr, H.A.; Wirtz, S.; Nikolaev, A.; Burg, J.; Strand, S.; Kiesslich, R.; Huber, S.; et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Cell Death Dis. 2004, 21, 491–501. [Google Scholar] [CrossRef]
- Baek, S.J.; Okazaki, R.; Lee, S.-H.; Martinez, J.; Kim, J.-S.; Yamaguchi, K.; Mishina, Y.; Martin, D.W.; Shoieb, A.; McEntee, M.F.; et al. Nonsteroidal anti-inflammatory drug-activated gene-1 over expression in transgenic mice suppresses intestinal neoplasia. Gastroenterology 2006, 131, 1553–1560. [Google Scholar] [CrossRef]
- Pretlow, T.P.; Edelmann, W.; Kucherlapati, R.; Pretlow, T.G.; Augenlicht, L.H. Spontaneous aberrant crypt foci in Apc1638N mice with a mutant Apc allele. Am. J. Pathol. 2003, 163, 1757–1763. [Google Scholar] [CrossRef]
- Betton, G.R.; Whiteley, L.O.; Anver, M.R.; Brown, R. Gastrointestinal Tract; Springer: Berlin/Heidelberg, Germany, 2001. [Google Scholar]
- Greaves, P. Liver and pancreas. In Histopathology of Preclinical Toxicity Studies; Elsevier: Amsterdam, The Netherlands, 2007; ISBN 9780444538611. [Google Scholar]
- Ward, J.M. Dose response to a single injection of azoxymethane in rats. Induction of tumors in the gastrointestinal tract, auditory sebaceous glands, kidney, liver and preputial gland. Vet. Pathol. 1975, 12, 165–177. [Google Scholar] [CrossRef]
- Rowlatt, C.; Franks, L.M.; Sheriff, M.U.; Chesterman, F.C. Naturally occurring tumors and other lesions of the digestive tract in untreated C57BL mice. J. Natl. Cancer Inst. 1969, 43, 1353–1364. [Google Scholar]
- Suzuki, R.; Kohno, H.; Sugie, S.; Nakagama, H. Strain differences in the susceptibility to azoxymethane and dextran sodium sulfate-induced colon carcinogenesis in mice. Carcinogenesis 2005, 27, 162–169. [Google Scholar] [CrossRef]
- Lee, S.M.; Kim, N.; Son, H.J.; Park, J.H.; Nam, R.H.; Ham, M.H.; Choi, D.; Sohn, S.H.; Shin, E.; Hwang, Y.-J.; et al. The Effect of Sex on the Azoxymethane/Dextran Sulfate Sodium-treated Mice Model of Colon Cancer. J. Cancer Prev. 2016, 21, 271–278. [Google Scholar] [CrossRef]
- Amos-Landgraf, J.M.; Heijmans, J.; Wielenga, M.C.B.; Dunkin, E.; Krentz, K.J.; Clipson, L.; Ederveen, A.G.; Groothuis, P.G.; Mosselman, S.; Muncan, V.; et al. Sex disparity in colonic adenomagenesis involves promotion by male hormones, not protection by female hormones. Proc. Natl. Acad. Sci. USA 2014, 111, 16514–16519. [Google Scholar] [CrossRef]
- Diwan, B.A.; Blackman, K.E. Differential susceptibility of 3 sublines of C57BL/6 mice to the induction of colorectal tumors by 1,2-dimethylhydrazine. Cancer Lett. 1980, 9, 111–115. [Google Scholar] [CrossRef]
- Nambiar, P.R.; Girnun, G.; Lillo, N.A.; Guda, K.; Whiteley, H.E.; Rosenberg, D.W. Preliminary Analysis of Azoxymethane Induced Colon Tumors in Inbred Mice Commonly Used as Transgenic/Knockout Progenitors. Int. J. Oncol. 2003, 22, 145–150. [Google Scholar] [CrossRef]
- Tanaka, T.; Kohno, H.; Suzuki, R.; Yamada, Y.; Sugie, S.; Mori, H. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci. 2003, 94, 965–973. [Google Scholar] [CrossRef]
- Rogers, C.; Fernandes-Alnemri, T.; Mayes, L.; Alnemri, D.; Cingolani, G.; Alnemri, E.S. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 2017, 8, 14128. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Yu, J.; Li, S.; Qi, J.; Chen, Z.; Wu, Y.; Guo, J.; Wang, K.; Sun, X.; Zheng, J. Cleavage of GSDME by caspase-3 determines lobaplatin-induced pyroptosis in colon cancer cells. Cell Death Dis. 2019, 10, 193. [Google Scholar] [CrossRef]
- Zhang, C.-C.; Li, C.-G.; Wang, Y.-F.; Xu, L.-H.; He, X.-H.; Zeng, Q.-Z.; Zeng, C.-Y.; Mai, F.-Y.; Hu, B.; Ouyang, D.-Y. Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in A549 lung cancer cells via caspase-3/GSDME activation. Apoptosis 2019, 24, 312–325. [Google Scholar] [CrossRef]
- Lu, H.; Zhang, S.; Wu, J.; Chen, M.; Cai, M.-C.; Fu, Y.; Li, W.; Wang, J.; Zhao, X.; Yu, Z.; et al. Molecular Targeted Therapies Elicit Concurrent Apoptotic and GSDME-Dependent Pyroptotic Tumor Cell Death. Clin. Cancer Res. 2018, 24, 6066–6077. [Google Scholar] [CrossRef]
- Wang, Y.; Yin, B.; Li, D.; Wang, G.; Han, X.; Sun, X. GSDME mediates caspase-3-dependent pyroptosis in gastric cancer. Biochem. Biophys. Res. Commun. 2017, 495, 1418–1425. [Google Scholar] [CrossRef]
- Strzyz, P. Cell death: Pulling the apoptotic trigger for necrosis. Nat. Rev. Mol. Cell Biol. 2017, 18, 72. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kroemer, G. Secondary Necrosis: Accidental No More. Trends Cancer 2017, 3, 1–2. [Google Scholar] [CrossRef]
- Stoll, G.; Ma, Y.; Yang, H.; Kepp, O.; Zitvogel, L.; Kroemer, G. Pro-necrotic molecules impact local immunosurveillance in human breast cancer. Oncoimmunology 2017, 6, e1299302. [Google Scholar] [CrossRef]
- Nolte, T.; Brander-Weber, P.; Dangler, C.; Deschl, U.; Elwell, M.R.; Greaves, P.; Hailey, R.; Leach, M.W.; Pandiri, A.R.; Rogers, A.; et al. Nonproliferative and Proliferative Lesions of the Gastrointestinal Tract, Pancreas and Salivary Glands of the Rat and Mouse. J. Toxicol. Pathol. 2016, 29, 1S–125S. [Google Scholar] [CrossRef]
- R Core Team. R. A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria, 2019. Available online: https://www.R-project.org/ (accessed on 5 December 2018).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lesion | Chemical Experiment—Large Intestine | Genetic Experiment—Small Intestine | ||||||
|---|---|---|---|---|---|---|---|---|
| Gsdme KO (n = 46) | WT (n = 54) | Δ (%) | p-Value | Apc1638N/+ Gsdme KO (n = 13) | Apc1638N/+ Gsdme WT (n = 24) | Δ (%) | p-Value | |
| OVERALL | ||||||||
| moderate mucosal inflammation | 19 | 20 | 4.3 | 0.73 | 3 | 16 | −43.6 | 0.010 |
| marked mucosal inflammation | 4 | 1 | 6.8 | 0.014 | 0 | 0 | 0.0 | - |
| glandular cyst | 46 | 54 | 0.0 | - | 0 | 0 | 0.0 | - |
| typical hyperplasia | 21 | 26 | −2.4 | 0.70 | 1 | 9 | −29.8 | 0.034 |
| atypical hyperplasia | 18 | 13 | 15.0 | 0.081 | 5 | 9 | 1.0 | 0.90 |
| adenoma | 3 | 1 | 4.6 | 0.57 | 0 | 0 | 0.0 | - |
| adenocarcinoma | 20 | 25 | −2.8 | 0.67 | 10 | 16 | 10.2 | 0.55 |
| proliferative change | 34 | 36 | 7.2 | 0.22 | 11 | 23 | −11.2 | 0.21 |
| PROXIMAL | ||||||||
| moderate mucosal inflammation | 6 | 4 | 5.6 | 0.31 | 2 | 9 | −22.1 | 0.13 |
| marked mucosal inflammation | 0 | 0 | 0.0 | - | 0 | 0 | 0.0 | - |
| glandular cyst | 25 | 43 | −25.3 | 0.043 | 0 | 0 | 0.0 | - |
| typical hyperplasia | 0 | 0 | 0.0 | - | 0 | 7 | −29.2 | 0.0077 |
| atypical hyperplasia | 0 | 0 | 0.0 | - | 3 | 6 | −1.9 | 0.97 |
| adenoma | 0 | 0 | 0.0 | - | 0 | 0 | 0.00 | - |
| adenocarcinoma | 0 | 0 | 0.0 | - | 8 | 13 | 7.3 | 0.74 |
| proliferative change | 0 | 0 | 0.0 | - | 10 | 22 | −14.8 | 0.17 |
| MID 1 | ||||||||
| moderate mucosal inflammation | 9 | 7 | 6.6 | 0.37 | 1 | 5 | −13.1 | 0.30 |
| marked mucosal inflammation | 1 | 0 | 2.2 | 0.053 | 0 | 0 | 0.0 | - |
| glandular cyst | 21 | 47 | −41.3 | 0.00038 | 0 | 0 | 0.0 | - |
| typical hyperplasia | 2 | 4 | −3.1 | 0.64 | 0 | 2 | −8.3 | 0.19 |
| atypical hyperplasia | 5 | 0 | 10.9 | 0.023 | 1 | 5 | −13.1 | 0.29 |
| adenoma | 0 | 0 | 0.0 | - | 0 | 0 | 0.0 | - |
| adenocarcinoma | 1 | 2 | −1.5 | 0.69 | 5 | 8 | 5.2 | 0.77 |
| proliferative change | 8 | 6 | 6.3 | 0.53 | 6 | 12 | −3.8 | 0.83 |
| MID 2 | ||||||||
| moderate mucosal inflammation | 12 | 12 | 3.9 | 0.57 | 0 | 7 | −29.2 | 0.0088 |
| marked mucosal inflammation | 3 | 0 | 6.5 | 0.019 | 0 | 0 | 0.0 | - |
| glandular cyst | 35 | 44 | −5.4 | 0.79 | 0 | 0 | 0.0 | - |
| typical hyperplasia | 8 | 12 | −4.8 | 0.25 | 0 | 2 | −8.3 | 0.19 |
| atypical hyperplasia | 9 | 6 | 8.5 | 0.27 | 2 | 1 | 11.2 | 0.24 |
| adenoma | 1 | 0 | 2.2 | 0.50 | 0 | 0 | 0.0 | - |
| adenocarcinoma | 12 | 13 | 2.0 | 0.68 | 0 | 4 | −16.7 | 0.049 |
| proliferative change | 24 | 21 | 13.3 | 0.20 | 2 | 6 | −9.6 | 0.48 |
| DISTAL | ||||||||
| moderate mucosal inflammation | 7 | 11 | −5.2 | 0.51 | 0 | 3 | −12.5 | 0.098 |
| marked mucosal inflammation | 0 | 1 | −1.9 | 0.49 | 0 | 0 | 0.0 | - |
| glandular cyst | 43 | 53 | −4.7 | 0.57 | 0 | 0 | 0.0 | - |
| typical hyperplasia | 18 | 19 | 3.9 | 0.19 | 1 | 0 | 7.7 | 0.15 |
| atypical hyperplasia | 7 | 10 | −3.3 | 0.95 | 0 | 0 | 0.0 | - |
| adenoma | 2 | 1 | 2.5 | 0.72 | 0 | 0 | 0.0 | - |
| adenocarcinoma | 13 | 18 | −5.1 | 0.46 | 3 | 3 | 10.6 | 0.43 |
| proliferative change | 28 | 32 | 1.6 | 0.63 | 4 | 3 | 18.3 | 0.20 |
| Overall: 100 Mice | |||
| 46 Gsdme KO | 54 WT | ||
31  | 15  | 23 | 31 |
| 20 Weeks: 37 Mice | |||
| 13 Gsdme KO | 24 WT | ||
| 9 | 4 | 10 | 14 |
| 22 Weeks: 37 Mice | |||
| 24 Gsdme KO | 13 WT | ||
| 16 | 8 | 4 | 9 |
| 24 Weeks: 26 Mice | |||
| 9 Gsdme KO | 17 WT | ||
| 7 | 2 | 9 | 8 |
| Overall: 37 Mice | |||
|---|---|---|---|
| 13 Apc1638N/+ Gsdme KO | 24 Apc1638N/+ Gsdme WT | ||
| 8 | 5 | 16 | 8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Croes, L.; Fransen, E.; Hylebos, M.; Buys, K.; Hermans, C.; Broeckx, G.; Peeters, M.; Pauwels, P.; Op de Beeck, K.; Van Camp, G. Determination of the Potential Tumor-Suppressive Effects of Gsdme in a Chemically Induced and in a Genetically Modified Intestinal Cancer Mouse Model. Cancers 2019, 11, 1214. https://doi.org/10.3390/cancers11081214
Croes L, Fransen E, Hylebos M, Buys K, Hermans C, Broeckx G, Peeters M, Pauwels P, Op de Beeck K, Van Camp G. Determination of the Potential Tumor-Suppressive Effects of Gsdme in a Chemically Induced and in a Genetically Modified Intestinal Cancer Mouse Model. Cancers. 2019; 11(8):1214. https://doi.org/10.3390/cancers11081214
Chicago/Turabian StyleCroes, Lieselot, Erik Fransen, Marieke Hylebos, Kimberly Buys, Christophe Hermans, Glenn Broeckx, Marc Peeters, Patrick Pauwels, Ken Op de Beeck, and Guy Van Camp. 2019. "Determination of the Potential Tumor-Suppressive Effects of Gsdme in a Chemically Induced and in a Genetically Modified Intestinal Cancer Mouse Model" Cancers 11, no. 8: 1214. https://doi.org/10.3390/cancers11081214
APA StyleCroes, L., Fransen, E., Hylebos, M., Buys, K., Hermans, C., Broeckx, G., Peeters, M., Pauwels, P., Op de Beeck, K., & Van Camp, G. (2019). Determination of the Potential Tumor-Suppressive Effects of Gsdme in a Chemically Induced and in a Genetically Modified Intestinal Cancer Mouse Model. Cancers, 11(8), 1214. https://doi.org/10.3390/cancers11081214