Reactive Oxygen Species in the Tumor Microenvironment: An Overview


{kind=link}
{kind=link}
Abstract
1. Introduction
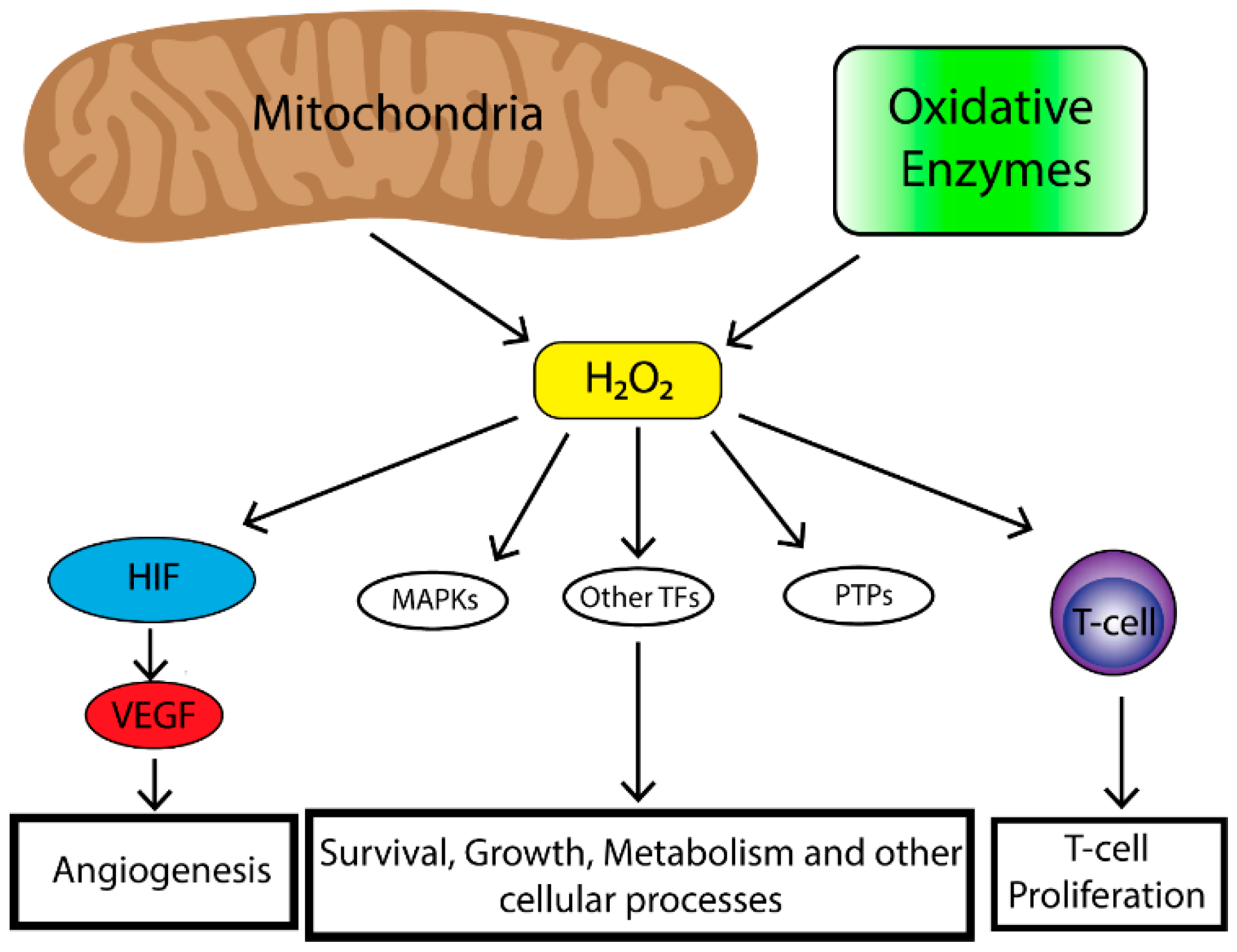
2. Role of Reactive Oxygen Species (ROS) in Tumorigenesis
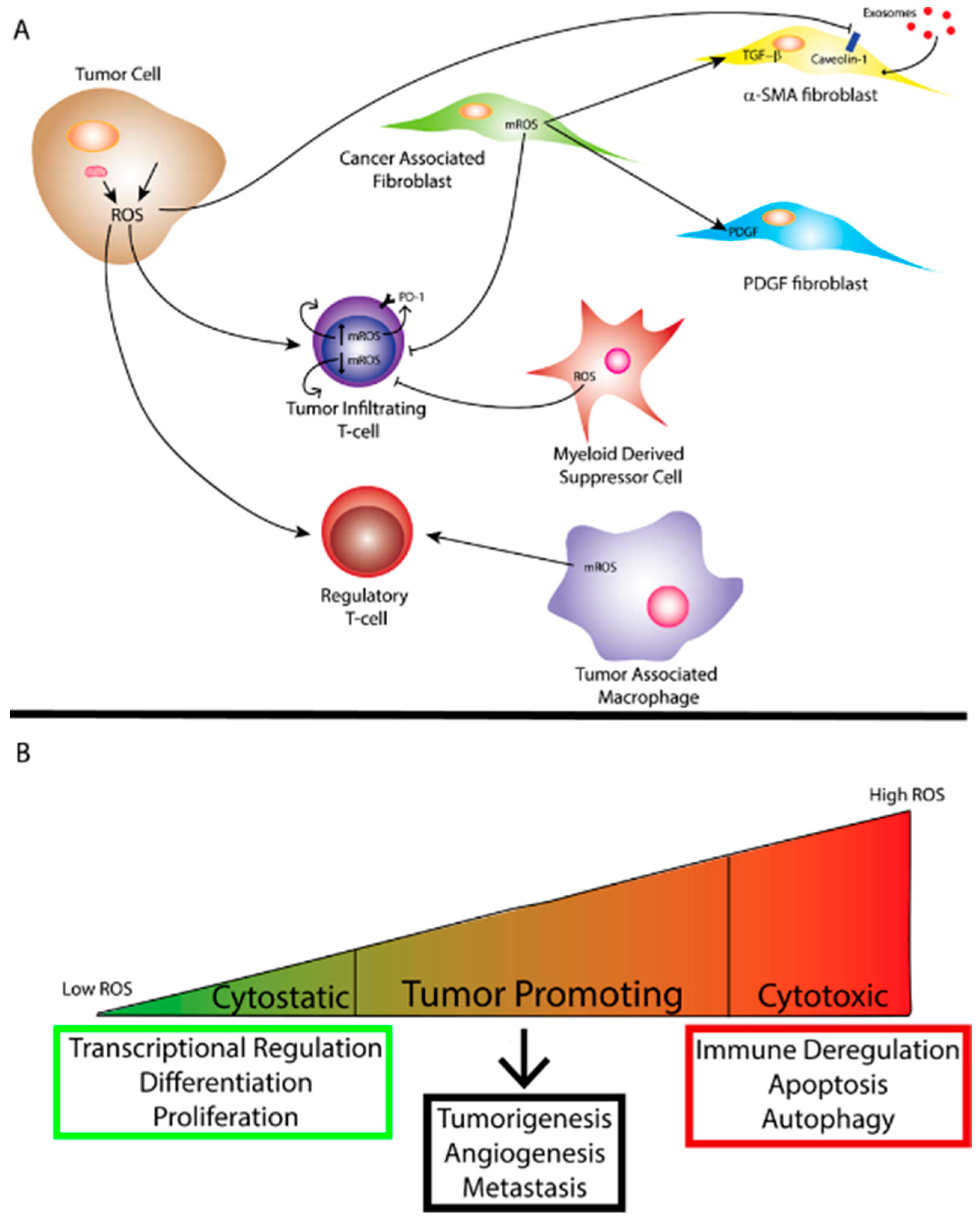
3. Cancer-Associated Fibroblasts, ROS and the Tumor Microenvironment (TME)
4. Tumor Immunity and ROS
5. The Microbiome and ROS
6. Therapeutic Implications of ROS Targeting
7. Conclusions
Funding
Conflicts of Interest
References
- Rhee, S.G. Cell signaling. H2O2 a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef] [PubMed]
- Diebold, L.; Chandel, N.S. Mitochondrial ROS regulation of proliferating cells. Free Radic. Biol. Med. 2016, 100, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Chandel, N.S. Reactive oxygen species-dependent signaling regulates cancer. Cell Mol. Life Sci. 2009, 66, 3663–3673. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Chandel, N.S. Mitochondrial metabolism and cancer. Ann. N. Y. Acad. Sci. 2009, 1177, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.; Chandel, N.S. Regulation of redox balance in cancer and T cells. J. Biol. Chem. 2018, 293, 7499–7507. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Balliet, R.M.; Rivadeneira, D.B.; Chiavarina, B.; Pavlides, S.; Wang, C.; Whitaker-Menezes, D.; Daumer, K.M.; Lin, Z.; Witkiewicz, A.K.; et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle 2010, 9, 3256–3276. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Scholer-Dahirel, A.; Mechta-Grigoriou, F. The role of reactive oxygen species and metabolism on cancer cells and their microenvironment. Semin. Cancer Biol. 2014, 25, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Storz, P. Reactive oxygen species in tumor progression. Front. Biosci. 2005, 10, 1881–1896. [Google Scholar] [CrossRef] [PubMed]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Nickel, A.; Kohlhaas, M.; Maack, C. Mitochondrial reactive oxygen species production and elimination. J. Mol. Cell. Cardiol. 2014, 73, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef] [PubMed]
- Fridovich, I. Superoxide anion radical (O2-), superoxide dismutases, and related matters. J. Biol. Chem. 1997, 272, 18515–18517. [Google Scholar] [CrossRef]
- Han, D.; Antunes, F.; Canali, R.; Rettori, D.; Cadenas, E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J. Biol Chem. 2003, 278, 5557–5563. [Google Scholar] [CrossRef]
- Orr, A.L.; Vargas, L.; Turk, C.N.; Baaten, J.E.; Matzen, J.T.; Dardov, V.J.; Attle, S.J.; Li, J.; Quackenbush, D.C.; Goncalves, R.L.; et al. Suppressors of superoxide production from mitochondrial complex III. Nat. Chem. Biol. 2015, 11, 834–836. [Google Scholar] [CrossRef]
- Winterbourn, C.C. The biological chemistry of hydrogen peroxide. Methods Enzymol. 2013, 528, 3–25. [Google Scholar]
- Echtay, K.S.; Murphy, M.P.; Smith, R.A.; Talbot, D.A.; Brand, M.D. Superoxide activates mitochondrial uncoupling protein 2 from the matrix side. Studies using targeted antioxidants. J. Biol. Chem. 2002, 277, 47129–47135. [Google Scholar] [CrossRef] [PubMed]
- Frederick, R.L.; Shaw, J.M. Moving mitochondria: Establishing distribution of an essential organelle. Traffic 2007, 8, 1668–1675. [Google Scholar] [CrossRef] [PubMed]
- Al-Mehdi, A.B.; Pastukh, V.M.; Swiger, B.M.; Reed, D.J.; Patel, M.R.; Bardwell, G.C.; Pastukh, V.V.; Alexeyev, M.F.; Gillespie, M.N. Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci. Signal 2012, 5, ra47. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Reyes, I.; Diebold, L.P.; Kong, H.; Schieber, M.; Huang, H.; Hensley, C.T.; Mehta, M.M.; Wang, T.; Santos, J.H.; Woychik, R.; et al. TCA Cycle and Mitochondrial Membrane Potential Are Necessary for Diverse Biological Functions. Mol. Cell 2016, 61, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [PubMed]
- Pugh, C.W.; Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: Role of the HIF system. Nat. Med. 2003, 9, 677–684. [Google Scholar] [CrossRef] [PubMed]
- De Smet, F.; Segura, I.; De Bock, K.; Hohensinner, P.J.; Carmeliet, P. Mechanisms of vessel branching: Filopodia on endothelial tip cells lead the way. Arter. Thromb. Vasc. Biol. 2009, 29, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. VEGF-A: A critical regulator of blood vessel growth. Eur. Cytokine Netw. 2009, 20, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Hoeben, A.; Landuyt, B.; Highley, M.S.; Wildiers, H.; Van Oosterom, A.T.; De Bruijn, E.A. Vascular endothelial growth factor and angiogenesis. Pharmacol. Rev. 2004, 56, 549–580. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Gill, T.; Levine, A.D. Mitochondria-derived hydrogen peroxide selectively enhances T cell receptor-initiated signal transduction. J. Biol. Chem. 2013, 288, 26246–26255. [Google Scholar] [CrossRef] [PubMed]
- Ames, B.N.; Shigenaga, M.K.; Hagen, T.M. Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. USA 1993, 90, 7915–7922. [Google Scholar] [CrossRef] [PubMed]
- Schafer, Z.T.; Grassian, A.R.; Song, L.; Jiang, Z.; Gerhart-Hines, Z.; Irie, H.Y.; Gao, S.; Puigserver, P.; Brugge, J.S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009, 461, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Irani, K.; Xia, Y.; Zweier, J.L.; Sollott, S.J.; Der, C.J.; Fearon, E.R.; Sundaresan, M.; Finkel, T.; Goldschmidt-Clermont, P.J. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 1997, 275, 1649–1652. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Mambo, E.; Sidransky, D. Mitochondrial DNA mutations in human cancer. Oncogene 2006, 25, 4663–4674. [Google Scholar] [CrossRef]
- Park, J.S.; Sharma, L.K.; Li, H.; Xiang, R.; Holstein, D.; Wu, J.; Lechleiter, J.; Naylor, S.L.; Deng, J.J.; Lu, J.; et al. A heteroplasmic, not homoplasmic, mitochondrial DNA mutation promotes tumorigenesis via alteration in reactive oxygen species generation and apoptosis. Hum. Mol. Genet. 2009, 18, 1578–1589. [Google Scholar] [CrossRef] [PubMed]
- Sharma, L.K.; Fang, H.; Liu, J.; Vartak, R.; Deng, J.; Bai, Y. Mitochondrial respiratory complex I dysfunction promotes tumorigenesis through ROS alteration and AKT activation. Hum. Mol. Genet. 2011, 20, 4605–4616. [Google Scholar] [CrossRef]
- Woo, D.K.; Green, P.D.; Santos, J.H.; D’Souza, A.D.; Walther, Z.; Martin, W.D.; Christian, B.E.; Chandel, N.S.; Shadel, G.S. Mitochondrial genome instability and ROS enhance intestinal tumorigenesis in APC(Min/+) mice. Am. J. Pathol. 2012, 180, 24–31. [Google Scholar] [CrossRef]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Ostman, A.; Augsten, M. Cancer-associated fibroblasts and tumor growth--bystanders turning into key players. Curr. Opin. Genet. Dev. 2009, 19, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Serini, G.; Gabbiani, G. Mechanisms of myofibroblast activity and phenotypic modulation. Exp. Cell Res. 1999, 250, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Madar, S.; Goldstein, I.; Rotter, V. ‘Cancer associated fibroblasts’—More than meets the eye. Trends Mol. Med. 2013, 19, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Madar, S.; Brosh, R.; Buganim, Y.; Ezra, O.; Goldstein, I.; Solomon, H.; Kogan, I.; Goldfinger, N.; Klocker, H.; Rotter, V. Modulated expression of WFDC1 during carcinogenesis and cellular senescence. Carcinogenesis 2009, 30, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Buganim, Y.; Madar, S.; Rais, Y.; Pomeraniec, L.; Harel, E.; Solomon, H.; Kalo, E.; Goldstein, I.; Brosh, R.; Haimov, O.; et al. Transcriptional activity of ATF3 in the stromal compartment of tumors promotes cancer progression. Carcinogenesis 2011, 32, 1749–1757. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, A.A.; Cullen, K.J. Paracrine/autocrine regulation of breast cancer by the insulin-like growth factors. Breast Cancer Res. Treat. 1998, 47, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Noel, A.; De Pauw-Gillet, M.C.; Purnell, G.; Nusgens, B.; Lapiere, C.M.; Foidart, J.M. Enhancement of tumorigenicity of human breast adenocarcinoma cells in nude mice by matrigel and fibroblasts. Br. J. Cancer 1993, 68, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Kuperwasser, C.; Chavarria, T.; Wu, M.; Magrane, G.; Gray, J.W.; Carey, L.; Richardson, A.; Weinberg, R.A. Reconstruction of functionally normal and malignant human breast tissues in mice. Proc. Natl. Acad. Sci. USA 2004, 101, 4966–4971. [Google Scholar] [CrossRef]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef]
- Chauhan, V.P.; Boucher, Y.; Ferrone, C.R.; Roberge, S.; Martin, J.D.; Stylianopoulos, T.; Bardeesy, N.; DePinho, R.A.; Padera, T.P.; Munn, L.L.; et al. Compression of pancreatic tumor blood vessels by hyaluronan is caused by solid stress and not interstitial fluid pressure. Cancer Cell 2014, 26, 14–15. [Google Scholar] [CrossRef] [PubMed]
- Jezierska-Drutel, A.; Rosenzweig, S.A.; Neumann, C.A. Role of oxidative stress and the microenvironment in breast cancer development and progression. Adv. Cancer Res. 2013, 119, 107–125. [Google Scholar] [PubMed]
- Radisky, E.S.; Radisky, D.C. Stromal induction of breast cancer: Inflammation and invasion. Rev. Endocr. Metab. Disord. 2007, 8, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Zavadil, J.; Haley, J.; Kalluri, R.; Muthuswamy, S.K.; Thompson, E. Epithelial-mesenchymal transition. Cancer Res. 2008, 68, 9574–9577. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef] [PubMed]
- McDonald, L.T.; Russell, D.L.; Kelly, R.R.; Xiong, Y.; Motamarry, A.; Patel, R.K.; Jones, J.A.; Watson, P.M.; Turner, D.P.; Watson, D.K.; et al. Hematopoietic stem cell-derived cancer-associated fibroblasts are novel contributors to the pro-tumorigenic microenvironment. Neoplasia 2015, 17, 434–448. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Acar, A.; Eaton, E.N.; Mellody, K.T.; Scheel, C.; Ben-Porath, I.; Onder, T.T.; Wang, Z.C.; Richardson, A.L.; Weinberg, R.A.; et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc. Natl. Acad. Sci. USA 2010, 107, 20009–20014. [Google Scholar] [CrossRef] [PubMed]
- Mueller, L.; Goumas, F.A.; Affeldt, M.; Sandtner, S.; Gehling, U.M.; Brilloff, S.; Walter, J.; Karnatz, N.; Lamszus, K.; Rogiers, X.; et al. Stromal fibroblasts in colorectal liver metastases originate from resident fibroblasts and generate an inflammatory microenvironment. Am. J. Pathol. 2007, 171, 1608–1618. [Google Scholar] [CrossRef]
- Toullec, A.; Gerald, D.; Despouy, G.; Bourachot, B.; Cardon, M.; Lefort, S.; Richardson, M.; Rigaill, G.; Parrini, M.C.; Lucchesi, C.; et al. Oxidative stress promotes myofibroblast differentiation and tumour spreading. EMBO Mol. Med. 2010, 2, 211–230. [Google Scholar] [CrossRef]
- Jain, M.; Rivera, S.; Monclus, E.A.; Synenki, L.; Zirk, A.; Eisenbart, J.; Feghali-Bostwick, C.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial reactive oxygen species regulate transforming growth factor-beta signaling. J. Biol. Chem. 2013, 288, 770–777. [Google Scholar] [CrossRef]
- Artaud-Macari, E.; Goven, D.; Brayer, S.; Hamimi, A.; Besnard, V.; Marchal-Somme, J.; Ali, Z.E.; Crestani, B.; Kerdine-Romer, S.; Boutten, A.; et al. Nuclear factor erythroid 2-related factor 2 nuclear translocation induces myofibroblastic dedifferentiation in idiopathic pulmonary fibrosis. Antioxid. Redox Signal. 2013, 18, 66–79. [Google Scholar] [CrossRef]
- Sampson, N.; Koziel, R.; Zenzmaier, C.; Bubendorf, L.; Plas, E.; Jansen-Durr, P.; Berger, P. ROS signaling by NOX4 drives fibroblast-to-myofibroblast differentiation in the diseased prostatic stroma. Mol. Endocrinol. 2011, 25, 503–515. [Google Scholar] [CrossRef]
- Frijhoff, J.; Dagnell, M.; Augsten, M.; Beltrami, E.; Giorgio, M.; Ostman, A. The mitochondrial reactive oxygen species regulator p66Shc controls PDGF-induced signaling and migration through protein tyrosine phosphatase oxidation. Free Radic. Biol. Med. 2014, 68, 268–277. [Google Scholar] [CrossRef]
- Dagnell, M.; Frijhoff, J.; Pader, I.; Augsten, M.; Boivin, B.; Xu, J.; Mandal, P.K.; Tonks, N.K.; Hellberg, C.; Conrad, M.; et al. Selective activation of oxidized PTP1B by the thioredoxin system modulates PDGF-beta receptor tyrosine kinase signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 13398–13403. [Google Scholar] [CrossRef]
- Meng, T.C.; Fukada, T.; Tonks, N.K. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol. Cell. 2002, 9, 387–399. [Google Scholar] [CrossRef]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 1995, 270, 296–299. [Google Scholar] [CrossRef]
- Salmeen, A.; Park, B.O.; Meyer, T. The NADPH oxidases NOX4 and DUOX2 regulate cell cycle entry via a p53-dependent pathway. Oncogene 2010, 29, 4473–4484. [Google Scholar] [CrossRef]
- Sotgia, F.; Martinez-Outschoorn, U.E.; Howell, A.; Pestell, R.G.; Pavlides, S.; Lisanti, M.P. Caveolin-1 and cancer metabolism in the tumor microenvironment: Markers, models, and mechanisms. Annu. Rev. Pathol. 2012, 7, 423–467. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Pavlides, S.; Whitaker-Menezes, D.; Daumer, K.M.; Milliman, J.N.; Chiavarina, B.; Migneco, G.; Witkiewicz, A.K.; Martinez-Cantarin, M.P.; Flomenberg, N.; et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: Implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle 2010, 9, 2423–2433. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Lin, Z.; Flomenberg, N.; Howell, A.; Pestell, R.G.; Lisanti, M.P.; Sotgia, F. Cytokine production and inflammation drive autophagy in the tumor microenvironment: Role of stromal caveolin-1 as a key regulator. Cell Cycle 2011, 10, 1784–1793. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Lin, Z.; Trimmer, C.; Flomenberg, N.; Wang, C.; Pavlides, S.; Pestell, R.G.; Howell, A.; Sotgia, F.; Lisanti, M.P. Cancer cells metabolically “fertilize” the tumor microenvironment with hydrogen peroxide, driving the Warburg effect: Implications for PET imaging of human tumors. Cell Cycle 2011, 10, 2504–2520. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Caveolae and signalling in cancer. Nat. Rev. Cancer 2015, 15, 225–237. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Trimmer, C.; Lin, Z.; Whitaker-Menezes, D.; Chiavarina, B.; Zhou, J.; Wang, C.; Pavlides, S.; Martinez-Cantarin, M.P.; Capozza, F.; et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFkappaB activation in the tumor stromal microenvironment. Cell Cycle 2010, 9, 3515–3533. [Google Scholar] [CrossRef]
- Asterholm, I.W.; Mundy, D.I.; Weng, J.; Anderson, R.G.; Scherer, P.E. Altered mitochondrial function and metabolic inflexibility associated with loss of caveolin-1. Cell Metab. 2012, 15, 171–185. [Google Scholar]
- Svensson, K.J.; Christianson, H.C.; Wittrup, A.; Bourseau-Guilmain, E.; Lindqvist, E.; Svensson, L.M.; Morgelin, M.; Belting, M. Exosome uptake depends on ERK1/2-heat shock protein 27 signaling and lipid Raft-mediated endocytosis negatively regulated by caveolin-1. J. Biol. Chem. 2013, 288, 17713–17724. [Google Scholar] [CrossRef]
- Zhao, H.; Achreja, A.; Iessi, E.; Logozzi, M.; Mizzoni, D.; Di Raimo, R.; Nagrath, D.; Fais, S. The key role of extracellular vesicles in the metastatic process. Biochim. Biophys. Acta 2017, 1869, 64–77. [Google Scholar] [CrossRef]
- Cho, J.A.; Park, H.; Lim, E.H.; Lee, K.W. Exosomes from breast cancer cells can convert adipose tissue-derived mesenchymal stem cells into myofibroblast-like cells. Int. J. Oncol. 2012, 40, 130–138. [Google Scholar]
- Paggetti, J.; Haderk, F.; Seiffert, M.; Janji, B.; Distler, U.; Ammerlaan, W.; Kim, Y.J.; Adam, J.; Lichter, P.; Solary, E.; et al. Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood 2015, 126, 1106–1117. [Google Scholar] [CrossRef]
- Gangoda, L.; Boukouris, S.; Liem, M.; Kalra, H.; Mathivanan, S. Extracellular vesicles including exosomes are mediators of signal transduction: Are they protective or pathogenic? Proteomics 2015, 15, 260–271. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 2016, 5, e10250. [Google Scholar] [CrossRef]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell. 2010, 40, 280–293. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef]
- Carroll, B.; Otten, E.G.; Manni, D.; Stefanatos, R.; Menzies, F.M.; Smith, G.R.; Jurk, D.; Kenneth, N.; Wilkinson, S.; Passos, J.F.; et al. Oxidation of SQSTM1/p62 mediates the link between redox state and protein homeostasis. Nat. Commun. 2018, 9, 256. [Google Scholar] [CrossRef]
- Li, L.; Chen, Y.; Gibson, S.B. Starvation-induced autophagy is regulated by mitochondrial reactive oxygen species leading to AMPK activation. Cell. Signal. 2013, 25, 50–65. [Google Scholar] [CrossRef]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef]
- Guo, J.Y.; Karsli-Uzunbas, G.; Mathew, R.; Aisner, S.C.; Kamphorst, J.J.; Strohecker, A.M.; Chen, G.; Price, S.; Lu, W.; Teng, X.; et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013, 27, 1447–1461. [Google Scholar] [CrossRef]
- Lan, S.H.; Wu, S.Y.; Zuchini, R.; Lin, X.Z.; Su, I.J.; Tsai, T.F.; Lin, Y.J.; Wu, C.T.; Liu, H.S. Autophagy suppresses tumorigenesis of hepatitis B virus-associated hepatocellular carcinoma through degradation of microRNA-224. Hepatology 2014, 59, 505–517. [Google Scholar] [CrossRef]
- Liu, X.D.; Yao, J.; Tripathi, D.N.; Ding, Z.; Xu, Y.; Sun, M.; Zhang, J.; Bai, S.; German, P.; Hoang, A.; et al. Autophagy mediates HIF2alpha degradation and suppresses renal tumorigenesis. Oncogene 2015, 34, 2450–2460. [Google Scholar] [CrossRef]
- Cianfanelli, V.; Fuoco, C.; Lorente, M.; Salazar, M.; Quondamatteo, F.; Gherardini, P.F.; De Zio, D.; Nazio, F.; Antonioli, M.; D’Orazio, M.; et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat. Cell Biol. 2015, 17, 706. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gelinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Capparelli, C.; Whitaker-Menezes, D.; Guido, C.; Balliet, R.; Pestell, T.G.; Howell, A.; Sneddon, S.; Pestell, R.G.; Martinez-Outschoorn, U.; Lisanti, M.P.; et al. CTGF drives autophagy, glycolysis and senescence in cancer-associated fibroblasts via HIF1 activation, metabolically promoting tumor growth. Cell Cycle 2012, 11, 2272–2284. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xue, L.; Zhang, X.; Bu, S.; Zhu, X.; Lai, D. Autophagy protects ovarian cancer-associated fibroblasts against oxidative stress. Cell Cycle 2016, 15, 1376–1385. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, S.; Vera, I.; Gandara, R.; Sneddon, S.; Pestell, R.G.; Mercier, I.; Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; et al. Warburg meets autophagy: Cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid. Redox Signal. 2012, 16, 1264–1284. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Pavlides, S.; Howell, A.; Pestell, R.G.; Tanowitz, H.B.; Sotgia, F.; Lisanti, M.P. Stromal-epithelial metabolic coupling in cancer: Integrating autophagy and metabolism in the tumor microenvironment. Int. J. Biochem. Cell Biol. 2011, 43, 1045–1051. [Google Scholar] [CrossRef] [PubMed]
- Vasievich, E.A.; Huang, L. The suppressive tumor microenvironment: A challenge in cancer immunotherapy. Mol. Pharm. 2011, 8, 635–641. [Google Scholar] [CrossRef]
- Cemerski, S.; Cantagrel, A.; Van Meerwijk, J.P.; Romagnoli, P. Reactive oxygen species differentially affect T cell receptor-signaling pathways. J. Biol. Chem. 2002, 277, 19585–19593. [Google Scholar] [CrossRef]
- Devadas, S.; Zaritskaya, L.; Rhee, S.G.; Oberley, L.; Williams, M.S. Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation: Selective regulation of mitogen-activated protein kinase activation and fas ligand expression. J. Exp. Med. 2002, 195, 59–70. [Google Scholar] [CrossRef]
- Jackson, S.H.; Devadas, S.; Kwon, J.; Pinto, L.A.; Williams, M.S. T cells express a phagocyte-type NADPH oxidase that is activated after T cell receptor stimulation. Nat. Immunol. 2004, 5, 818–827. [Google Scholar] [CrossRef]
- Kaminski, M.M.; Sauer, S.W.; Kaminski, M.; Opp, S.; Ruppert, T.; Grigaravicius, P.; Grudnik, P.; Grone, H.J.; Krammer, P.H.; Gulow, K. T cell activation is driven by an ADP-dependent glucokinase linking enhanced glycolysis with mitochondrial reactive oxygen species generation. Cell Rep. 2012, 2, 1300–1315. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, M.; Kiessling, M.; Suss, D.; Krammer, P.H.; Gulow, K. Novel role for mitochondria: Protein kinase Ctheta-dependent oxidative signaling organelles in activation-induced T-cell death. Mol. Cell. Biol. 2007, 27, 3625–3639. [Google Scholar] [CrossRef]
- Scharping, N.E.; Menk, A.V.; Moreci, R.S.; Whetstone, R.D.; Dadey, R.E.; Watkins, S.C.; Ferris, R.L.; Delgoffe, G.M. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 2016, 45, 701–703. [Google Scholar] [CrossRef]
- Siska, P.J.; Beckermann, K.E.; Mason, F.M.; Andrejeva, G.; Greenplate, A.R.; Sendor, A.B.; Chiang, Y.J.; Corona, A.L.; Gemta, L.F.; Vincent, B.G.; et al. Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight 2017, 2, 93411. [Google Scholar] [CrossRef] [PubMed]
- Ligtenberg, M.A.; Mougiakakos, D.; Mukhopadhyay, M.; Witt, K.; Lladser, A.; Chmielewski, M.; Riet, T.; Abken, H.; Kiessling, R. Coexpressed Catalase Protects Chimeric Antigen Receptor-Redirected T Cells as well as Bystander Cells from Oxidative Stress-Induced Loss of Antitumor Activity. J. Immunol. 2016, 196, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Maeda, A.; Nishimura, H.; Kurosaki, T.; Honjo, T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc. Natl. Acad. Sci. USA 2001, 98, 13866–13871. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jie, H.B.; Lei, Y.; Gildener-Leapman, N.; Trivedi, S.; Green, T.; Kane, L.P.; Ferris, R.L. PD-1/SHP-2 inhibits Tc1/Th1 phenotypic responses and the activation of T cells in the tumor microenvironment. Cancer Res. 2015, 75, 508–518. [Google Scholar] [CrossRef]
- Chamoto, K.; Chowdhury, P.S.; Kumar, A.; Sonomura, K.; Matsuda, F.; Fagarasan, S.; Honjo, T. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc. Natl. Acad. Sci. USA 2017, 114, E761–E770. [Google Scholar] [CrossRef]
- Scharping, N.E.; Menk, A.V.; Whetstone, R.D.; Zeng, X.; Delgoffe, G.M. Efficacy of PD-1 Blockade is Potentiated by Metformin-Induced Reduction of Tumor Hypoxia. Cancer Immunol. Res. 2017, 5, 9–16. [Google Scholar] [CrossRef]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv4. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef]
- Reck, M.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fulop, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef]
- Delgoffe, G.M.; Kole, T.P.; Zheng, Y.; Zarek, P.E.; Matthews, K.L.; Xiao, B.; Worley, P.F.; Kozma, S.C.; Powell, J.D. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 2009, 30, 832–844. [Google Scholar] [CrossRef]
- Zeng, H.; Yang, K.; Cloer, C.; Neale, G.; Vogel, P.; Chi, H. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature 2013, 499, 485–490. [Google Scholar] [CrossRef]
- Shi, L.Z.; Wang, R.; Huang, G.; Vogel, P.; Neale, G.; Green, D.R.; Chi, H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 2011, 208, 1367–1376. [Google Scholar] [CrossRef]
- Dang, E.V.; Barbi, J.; Yang, H.Y.; Jinasena, D.; Yu, H.; Zheng, Y.; Bordman, Z.; Fu, J.; Kim, Y.; Yen, H.R.; et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 2011, 146, 772–784. [Google Scholar] [CrossRef]
- Maj, T.; Wang, W.; Crespo, J.; Zhang, H.; Wang, W.; Wei, S.; Zhao, L.; Vatan, L.; Shao, I.; Szeliga, W.; et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol. 2017, 18, 1332–1341. [Google Scholar] [CrossRef]
- Kunisada, Y.; Eikawa, S.; Tomonobu, N.; Domae, S.; Uehara, T.; Hori, S.; Furusawa, Y.; Hase, K.; Sasaki, A.; Udono, H. Attenuation of CD4(+) CD25(+) Regulatory T Cells in the Tumor Microenvironment by Metformin, a Type 2 Diabetes Drug. EBioMedicine 2017, 25, 154–164. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Singer, B.D.; Steinert, E.M.; Martinez, C.A.; Mehta, M.M.; Martinez-Reyes, I.; Gao, P.; Helmin, K.A.; Abdala-Valencia, H.; Sena, L.A.; et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature 2019, 565, 495–499. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef]
- Qu, P.; Boelte, K.C.; Lin, P.C. Negative regulation of myeloid-derived suppressor cells in cancer. Immunol. Investig. 2012, 41, 562–580. [Google Scholar] [CrossRef]
- OuYang, L.Y.; Wu, X.J.; Ye, S.B.; Zhang, R.X.; Li, Z.L.; Liao, W.; Pan, Z.Z.; Zheng, L.M.; Zhang, X.S.; Wang, Z.; et al. Tumor-induced myeloid-derived suppressor cells promote tumor progression through oxidative metabolism in human colorectal cancer. J. Transl. Med. 2015, 13, 47. [Google Scholar] [CrossRef]
- Wei, J.; Zhang, M.; Zhou, J. Myeloid-derived suppressor cells in major depression patients suppress T-cell responses through the production of reactive oxygen species. Psychiatry Res. 2015, 228, 695–701. [Google Scholar] [CrossRef]
- Liu, Y.; Wei, J.; Guo, G.; Zhou, J. Norepinephrine-induced myeloid-derived suppressor cells block T-cell responses via generation of reactive oxygen species. Immunopharmacol. Immunotoxicol. 2015, 37, 359–365. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Sena, L.A.; Chandel, N.S. Mitochondria in the regulation of innate and adaptive immunity. Immunity 2015, 42, 406–417. [Google Scholar] [CrossRef]
- Kraaij, M.D.; Savage, N.D.; van der Kooij, S.W.; Koekkoek, K.; Wang, J.; van den Berg, J.M.; Ottenhoff, T.H.; Kuijpers, T.W.; Holmdahl, R.; van Kooten, C.; et al. Induction of regulatory T cells by macrophages is dependent on production of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 17686–17691. [Google Scholar] [CrossRef]
- Lin, X.; Zheng, W.; Liu, J.; Zhang, Y.; Qin, H.; Wu, H.; Xue, B.; Lu, Y.; Shen, P. Oxidative stress in malignant melanoma enhances tumor necrosis factor-alpha secretion of tumor-associated macrophages that promote cancer cell invasion. Antioxid. Redox Signal. 2013, 19, 1337–1355. [Google Scholar] [CrossRef]
- Purcell, R.V.; Pearson, J.; Aitchison, A.; Dixon, L.; Frizelle, F.A.; Keenan, J.I. Colonization with enterotoxigenic Bacteroides fragilis is associated with early-stage colorectal neoplasia. PLoS ONE 2017, 12, e0171602. [Google Scholar] [CrossRef]
- Wu, S.; Rhee, K.J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F.; et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef]
- Gur, C.; Ibrahim, Y.; Isaacson, B.; Yamin, R.; Abed, J.; Gamliel, M.; Enk, J.; Bar-On, Y.; Stanietsky-Kaynan, N.; Coppenhagen-Glazer, S.; et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity 2015, 42, 344–355. [Google Scholar] [CrossRef]
- Mangerich, A.; Knutson, C.G.; Parry, N.M.; Muthupalani, S.; Ye, W.; Prestwich, E.; Cui, L.; McFaline, J.L.; Mobley, M.; Ge, Z.; et al. Infection-induced colitis in mice causes dynamic and tissue-specific changes in stress response and DNA damage leading to colon cancer. Proc. Natl. Acad. Sci. USA 2012, 109, E1820–E1829. [Google Scholar] [CrossRef]
- Huycke, M.M.; Abrams, V.; Moore, D.R. Enterococcus faecalis produces extracellular superoxide and hydrogen peroxide that damages colonic epithelial cell DNA. Carcinogenesis 2002, 23, 529–536. [Google Scholar] [CrossRef]
- Goodwin, A.C.; Destefano Shields, C.E.; Wu, S.; Huso, D.L.; Wu, X.; Murray-Stewart, T.R.; Hacker-Prietz, A.; Rabizadeh, S.; Woster, P.M.; Sears, C.L.; et al. Polyamine catabolism contributes to enterotoxigenic Bacteroides fragilis-induced colon tumorigenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 15354–15359. [Google Scholar] [CrossRef]
- Bernstein, H.; Bernstein, C.; Payne, C.M.; Dvorak, K. Bile acids as endogenous etiologic agents in gastrointestinal cancer. World J. Gastroenterol. 2009, 15, 3329–3340. [Google Scholar] [CrossRef]
- Devkota, S.; Wang, Y.; Musch, M.W.; Leone, V.; Fehlner-Peach, H.; Nadimpalli, A.; Antonopoulos, D.A.; Jabri, B.; Chang, E.B. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature 2012, 487, 104–108. [Google Scholar] [CrossRef]
- Jones, R.M.; Mercante, J.W.; Neish, A.S. Reactive oxygen production induced by the gut microbiota: Pharmacotherapeutic implications. Curr. Med. Chem. 2012, 19, 1519–1529. [Google Scholar] [CrossRef]
- Clark, A.; Mach, N. The Crosstalk between the Gut Microbiota and Mitochondria during Exercise. Front. Physiol. 2017, 8, 319. [Google Scholar] [CrossRef]
- Yardeni, T.; Tanes, C.E.; Bittinger, K.; Mattei, L.M.; Schaefer, P.M.; Singh, L.N.; Wu, G.D.; Murdock, D.G.; Wallace, D.C. Host mitochondria influence gut microbiome diversity: A role for ROS. Sci. Signal. 2019, 12, eaaw3159. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef]
- Ota, S.; Horigome, K.; Ishii, T.; Nakai, M.; Hayashi, K.; Kawamura, T.; Kishino, A.; Taiji, M.; Kimura, T. Metformin suppresses glucose-6-phosphatase expression by a complex I inhibition and AMPK activation-independent mechanism. Biochem. Biophys. Res. Commun. 2009, 388, 311–316. [Google Scholar] [CrossRef]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Averet, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607–614. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Kurelac, I.; Umesh Ganesh, N.; Iorio, M.; Porcelli, A.M.; Gasparre, G. The multifaceted effects of metformin on tumor microenvironment. Semin. Cell Dev. Biol. 2019. [Google Scholar] [CrossRef]
- Stuart, S.D.; Schauble, A.; Gupta, S.; Kennedy, A.D.; Keppler, B.R.; Bingham, P.M.; Zachar, Z. A strategically designed small molecule attacks alpha-ketoglutarate dehydrogenase in tumor cells through a redox process. Cancer Metab. 2014, 2, 4. [Google Scholar] [CrossRef]
- Alistar, A.; Morris, B.B.; Desnoyer, R.; Klepin, H.D.; Hosseinzadeh, K.; Clark, C.; Cameron, A.; Leyendecker, J.; D’Agostino, R., Jr.; Topaloglu, U.; et al. Safety and tolerability of the first-in-class agent CPI-613 in combination with modified FOLFIRINOX in patients with metastatic pancreatic cancer: A single-centre, open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2017, 18, 770–778. [Google Scholar] [CrossRef]
- Reisman, S.A.; Lee, C.Y.; Meyer, C.J.; Proksch, J.W.; Sonis, S.T.; Ward, K.W. Topical application of the synthetic triterpenoid RTA 408 protects mice from radiation-induced dermatitis. Radiat. Res. 2014, 181, 512–520. [Google Scholar] [CrossRef]
- Reisman, S.A.; Lee, C.Y.; Meyer, C.J.; Proksch, J.W.; Ward, K.W. Topical application of the synthetic triterpenoid RTA 408 activates Nrf2 and induces cytoprotective genes in rat skin. Arch. Derm. Res. 2014, 306, 447–454. [Google Scholar] [CrossRef]
- Reisman, S.A.; Goldsberry, A.R.; Lee, C.Y.; O’Grady, M.L.; Proksch, J.W.; Ward, K.W.; Meyer, C.J. Topical application of RTA 408 lotion activates Nrf2 in human skin and is well-tolerated by healthy human volunteers. BMC Dermatol. 2015, 15, 10. [Google Scholar] [CrossRef]
- Probst, B.L.; Trevino, I.; McCauley, L.; Bumeister, R.; Dulubova, I.; Wigley, W.C.; Ferguson, D.A. RTA 408, A Novel Synthetic Triterpenoid with Broad Anticancer and Anti-Inflammatory Activity. PLoS ONE 2015, 10, e0122942. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weinberg, F.; Ramnath, N.; Nagrath, D. Reactive Oxygen Species in the Tumor Microenvironment: An Overview. Cancers 2019, 11, 1191. https://doi.org/10.3390/cancers11081191
Weinberg F, Ramnath N, Nagrath D. Reactive Oxygen Species in the Tumor Microenvironment: An Overview. Cancers. 2019; 11(8):1191. https://doi.org/10.3390/cancers11081191
Chicago/Turabian StyleWeinberg, Frank, Nithya Ramnath, and Deepak Nagrath. 2019. "Reactive Oxygen Species in the Tumor Microenvironment: An Overview" Cancers 11, no. 8: 1191. https://doi.org/10.3390/cancers11081191
APA StyleWeinberg, F., Ramnath, N., & Nagrath, D. (2019). Reactive Oxygen Species in the Tumor Microenvironment: An Overview. Cancers, 11(8), 1191. https://doi.org/10.3390/cancers11081191