Cancer Metabolism and the Evasion of Apoptotic Cell Death


Department of Hematology and Medical Oncology, Winship Cancer Institute, School of Medicine, Emory University, Atlanta, GA 30322, USA
*
Author to whom correspondence should be addressed.
Cancers 2019, 11(8), 1144; https://doi.org/10.3390/cancers11081144
Submission received: 1 July 2019
/
Revised: 29 July 2019
/
Accepted: 8 August 2019
/
Published: 9 August 2019
(This article belongs to the Special Issue Metabolic Reprogramming and Vulnerabilities in Cancer)
{kind=link}
{kind=link}
Abstract
:Cellular growth and proliferation depend upon the acquisition and synthesis of specific metabolites. These metabolites fuel the bioenergy, biosynthesis, and redox potential required for duplication of cellular biomass. Multicellular organisms maintain tissue homeostasis by balancing signals promoting proliferation and removal of cells via apoptosis. While apoptosis is in itself an energy dependent process activated by intrinsic and extrinsic signals, whether specific nutrient acquisition (elevated or suppressed) and their metabolism regulates apoptosis is less well investigated. Normal cellular metabolism is regulated by lineage specific intrinsic features and microenvironment driven extrinsic features. In the context of cancer, genetic abnormalities, unconventional microenvironments and/or therapy engage constitutive pro-survival signaling to re-program and rewire metabolism to maintain survival, growth, and proliferation. It thus becomes particularly relevant to understand whether altered nutrient acquisition and metabolism in cancer can also contribute to the evasion of apoptosis and consequently therapy resistance. Our review attempts to dissect a causal relationship between two cancer hallmarks, i.e., deregulated cellular energetics and the evasion of programmed cell death with primary focus on the intrinsic pathway of apoptosis.
1. Introduction
Cancer cells engage various mechanisms to evade apoptosis. The intrinsic pathway of apoptosis has been shown to be frequently disrupted in cancer cells and is closely regulated by cellular metabolism. While oncogenes and tumor suppressors reprogram tumor metabolism, microenvironment or therapy-imposed stresses can additionally rewire metabolism, creating new metabolic dependencies. Direct evidence that metabolic changes promote resistance to therapy via alterations of BCL-2 family expression is sparse. Metabolites like glucose and glutamine however have been shown to regulate the expression and binding properties of specific BCL-2 proteins. The strongest evidence that cancer metabolism regulates programmed cell death to promote resistance comes indirectly from numerous studies showing that targeting metabolism alters the apoptotic threshold consequently sensitizing resistant cancer to various therapies. There is opportunity for developing therapies that co-target metabolic vulnerabilities and the BCL-2 family of proteins. We thus review the intrinsic pathway of apoptosis and its close relationship with metabolism.
2. Apoptosis: Extrinsic and Intrinsic Programmed Cell Death
Apoptosis is a mode of programmed cell death essential for maintaining tissue homeostasis by elimination of unwanted, superfluous, and damaged cells [1]. Apoptosis occurs discretely in individual cells of our body and is a highly regulated energy dependent process. Deregulation of apoptosis is involved in the pathogenesis of several diseases like neurodegenerative conditions, which involve excessive apoptosis, as well as cancer, which, in contrast, is characterized by accumulation of cells exhibiting insufficient engagement of the apoptotic machinery and evasion of apoptosis [2,3,4]. There are two primary pathways that lead to apoptosis; the intrinsic pathway of apoptosis and the extrinsic pathway of apoptosis. Both pathways result in the activation of cysteine aspartyl-specific proteases or ‘caspases’, which are the final effectors of apoptosis and cleave several proteins leading to cell death [5,6,7]. The extrinsic pathway can be engaged by activation of death receptors of the tumor necrosis factor (TNF) superfamily such as Fas (Apo/CD95), TNF Receptor 1 (TNFR1), TNF-related apoptosis-inducing ligand (TRAIL) receptors, etc., located on the cell surface, by binding with their specific ligands [8,9]. On the other hand, induction of the intrinsic (mitochondrial) pathway is primarily regulated by the B cell lymphoma (BCL-2) family of proteins [10] and is activated by internal stress sensors in response to cellular stresses like nutrient deprivation, DNA damage, hypoxia, etc. Detachment of cells from the extracellular matrix can also induce a form of apoptotic cell death called ‘anoikis’, which acts to control the growth and re-attachment of detached cells to a different matrix [11]. Resistance to anoikis is an attribute of cancer cells with metastatic potential [12,13]. Anoikis can engage both extrinsic as well as the intrinsic pathway of apoptosis [13,14].
Apoptosis is biochemically and morphologically distinct from other forms of programmed cell death implicated in cancer such as ferroptosis [15,16]. Ferroptosis was originally discovered as an iron-dependent form of oxidative cell death induced by inhibition of cystine uptake by the cystine/glutamate antiporter (xCT) using small molecule inhibitors, such as Erastin or excess glutamate [15]. Ferroptosis is initiated by a decrease or inhibition of glutathione peroxidase 4 (GPX4) activity. GPX4 catalyzes the reduction of lipid peroxides while oxidizing glutathione (GSH), a key player in the cellular antioxidant defense. Depletion of GSH by inhibition of xCT, for instance, or direct inhibition of GPX4 results in accumulation of lipid peroxides, which are converted to lipid ROS by the action of reactive iron cations such as Fe2+ resulting in ferroptotic cell death. In another study, glutaminolysis, fueled by glutamine along with iron carrier, transferrin, were found to be critical regulators of ferroptosis [17]. Hence, ferroptosis is linked to dysregulation of iron and ROS metabolism and lipid peroxidation pathways and is closely linked to glutamine metabolism. While, ferroptosis warrants a mention here, this review will primarily focus on the intrinsic apoptotic pathway of cell death and its relationship with cancer metabolism.
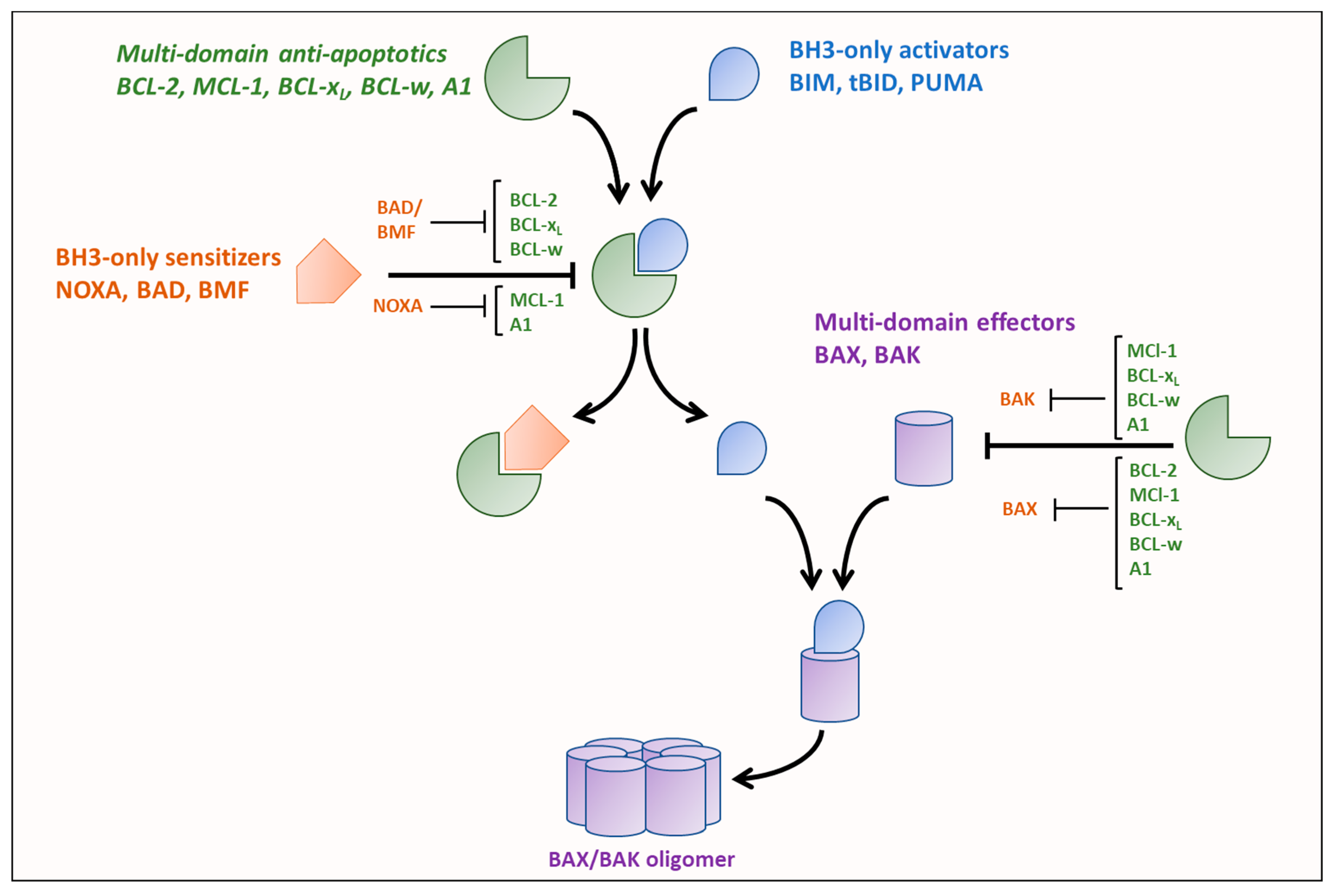
The key regulators of the intrinsic pathway of apoptosis, are the BCL-2 family of proteins that are broadly classified as either anti-apoptotic or pro-apoptotic. BCL-2 family members are characterized by containing at least one of four BCL-2 homology (BH1-4) domains. Induction of the intrinsic pathway of apoptosis is initiated by the release of pro-apoptotic BH3-only ‘activators’ (BCL-2 interacting mediator of cell death (BIM), P53-upregulated modulator of apoptosis (PUMA), or truncated BH3 interacting domain death agonist (tBID)) from anti-apoptotic multi-domain BCL-2 family members (BCL-2, BCL-xL, myeloid cell leukemia 1 (MCL-1), BCL-w and A1) to activate multi-domain ‘effectors’, BCL-2 associated X protein (BAX) and BCL-2 antagonist/killer (BAK) [18,19].
The activation of the intrinsic pathway of apoptosis is regulated by a delicate balance of pro- and anti-apoptotic BCL-2 proteins. BH3 domain-only proteins have different binding affinities for the multi-domain pro- and anti-apoptotic proteins dictated by differences in the amino acid sequences of their BH3 domains [20,21]. Additionally, the BH3 domain-only activator proteins bind to all multi-domain anti-apoptotics with high affinities while the BH-3 domain-only sensitizers have selective binding affinities for the anti-apoptotics, as shown in Figure 1. Furthermore, activators also have variable binding affinities for the effectors, for example, BIM has been shown to preferentially activate BAX while BID preferentially activates BAK [22]. BH3-only activators are released either by reduction in expression of an anti-apoptotic to which they are bound or displacement from the anti-apoptotic by a BH3-only sensitizer (such as NOXA, BCL-xL/BCL-2 associated death promoter (BAD), and BCL-2 modifying factor (BMF)). The released BH3-only activators can then activate BAX and BAK, promoting their oligomerization and result in subsequent mitochondrial outer membrane permeabilization (MOMP), thus releasing proteins of the intermembrane space such as cytochrome c, second mitochondria-derived activator of caspase (SMAC)/DIABLO, endonuclease G, and OMI into the cytosol [23] to activate distinct steps of the apoptotic cascade. Besides differences in binding tendencies, expression levels and post-translational modifications of the different BCL-2 family proteins also play a major role in determining if apoptosis is initiated [21,24,25,26].
The extrinsic pathway can interact with the intrinsic pathway through BID which is cleaved by caspase-8 to its truncated active form, tBID [27]. This cross-talk between the extrinsic and intrinsic pathways of apoptosis is crucial in cells that are unable to initiate apoptosis only by activation of death receptors and require additional engagement of the intrinsic pathway. This has important implications for approaches which use TRAIL and TRAIL receptor-based therapies to engage the extrinsic pathway as cancer cells which require tBID activation of intrinsic pathway may be rendered resistant by changes in the levels of BCL-2 proteins (such as overexpression of BCL-2) upon death receptor activation. [28]. Further, studies linking metabolism to activation of the extrinsic pathway of apoptosis by death receptor-ligand interactions are scarce and often require involvement of the intrinsic pathway [10,29,30,31,32]. Therefore, we have limited the scope of this review to the intrinsic pathway and its regulation by cancer metabolism.
3. Cancer and Deregulation of BCL-2 Family Proteins
Evasion of cell death is one of the hallmarks of cancer [33]. Cancer cells employ various means of preventing cell death including deregulation of the intrinsic apoptosis pathway. Upregulation of the anti-apoptotic BCL-2 proteins and loss of pro-apoptotic BH3 proteins has been observed in many cancers [34,35]. In fact, BCL-2 was identified in the t(14,18) chromosomal translocation in follicular lymphoma, which results in fusion of the BCL-2 gene locus with in the immunoglobulin heavy chain locus on chromosome 14 with reciprocal translocation of the variable heavy chain genes to chromosome 18 [36,37,38]. Upregulation of BCL-2 has also been observed in a number of other hematological malignancies as well as solid tumors [39]. Similarly, increased levels of BCL-xL and MCL-1 have been detected via different mechanisms and shown to promote tumorigenesis [39,40,41]. Many cancers engage mechanisms for stabilization of MCL-1, which is a highly unstable protein that exhibits rapid turnover due to proteasomal degradation [42,43,44]. On the other, hand downregulation of pro-apoptotic proteins is another means of avoiding apoptosis initiation. Loss of p53 tumor suppressor, which upregulates pro-apoptotic BH3-only proteins such as PUMA, BID, and NOXA, has been observed in various cancers [45,46,47,48]. Pro-apoptotic effectors—BAX and BAK—have also been found to be downregulated in multiple cancers [49,50,51].
However, despite of successful evasion of apoptosis, cancer cells are generally more susceptible to apoptosis than normal cells. The genetic complexity, microenvironment (hypoxia, nutrient deprivation/competition, and pH), and therapy-related stress bring cells more proximal to the apoptotic threshold of cancer cells. Cancer cells thus often express elevated levels of pro-apoptotic BH3-only proteins. Concordantly, to sequester unbound pro-apoptotics and thereby prevent MOMP, cancer cells generally upregulate anti-apoptotic BCL-2 family proteins and, as a result, contain higher levels of pro-apoptotic activators bound to anti-apoptotic proteins. Such cells exhibiting elevated levels of anti-apoptotics bound to pro-apoptotics and proximal to the apoptotic threshold are considered to be ‘primed’ for apoptosis [52].
4. Cancer Metabolism
Cancer cells exhibit altered nutrient acquisition and metabolism to sustain biosynthetic, bioenergetic and redox homeostasis demands. Alterations in specific metabolites also have implications on gene expression, protein expression and/function by, for example, epigenetic and posttranslational modifications. In particular, cancer cells exhibit elevated uptake of glucose and glutamine. Numerous cancer cell types exhibit elevated expression of the glucose transporter GLUT1 allowing for facilitative glucose uptake [53,54]. GLUT1 is a high affinity glucose transporter and proximal rate-limiting step in glucose metabolism [54,55,56]. It is the most frequently implicated GLUT family member in cancer and linked to poor survival and prognosis [57,58]. Glycolysis is the first major pathway involved in glucose catabolism enabling the retention of glucose within the cell when hexokinase phosphorylates glucose to glucose-6-phosphate. The ensuing steps of glycolysis then allow for the anaerobic synthesis of NADH, ATP, and pyruvate. Glucose-6-phosphate is also channeled into the pentose phosphate pathway (PPP) for synthesis of nucleotide precursors and production of nicotinamide adenine dinucleotide phosphate (NADPH), which is required for maintaining redox homeostasis as well as for reductive biosynthesis of fatty acids [59,60,61]. The irreversible oxidative arm of the PPP contributes to cellular NADPH pools while the reversible non-oxidative arm produces nucleotide precursor ribose-5-phosphate and glycolysis intermediates such as fructose-6-phosphate and glyceraldehyde-3-phosphate. Cancer cells utilize PPP to support cell growth and proliferation and for antioxidant defense via NADPH synthesis. Depending upon cellular requirements for NAPDH synthesis and/or ribose-5-phosphate, the oxidative and/or the nonoxidative arms of the PPP may be activated. The PPP was also identified as a prosurvival pathway in AML cells with high mammalian target of rapamycin complex 1 (mTORC1) activity and inhibition of glucose-6-phosphate dehydrogenase (G6PD), the first rate-limiting enzyme of the oxidative arm of the PPP, selectively targeted AML cells [62]. Further, mTORC1 has been shown to circumvent glycolysis inhibition via the oxidative PPP in a number of different cancers [63]. Pyruvate is further transported into the mitochondria and converted to acetyl-CoA, which feeds into the tricarboxylic acid (TCA) cycle producing various TCA cycle intermediates and NADH (reduced nicotinamide adenine dinucleotide) and FADH2 (flavin adenine dinucleotide). NADH and FADH2 fuel oxidative phosphorylation (OXPHOS) to generate adenosine triphosphate (ATP). Even though generation of lactate from glucose is less energy efficient cancer cells primarily convert glucose to lactate regardless of the oxygen concentration. This phenomenon of aerobic glycolysis is termed the ‘Warburg effect’ [64,65,66]. The inherent reliance of cancer cells on aerobic glycolysis despite the presence of functional mitochondria and clear requirement for mitochondrial metabolism [67,68,69] in tumor development suggests the unique biology sustained by glucose metabolized in the glycolysis/PPP vs. mitochondrial pathways.
Several studies have also shown that cancer cells are also highly dependent on glutamine as a source of carbon and nitrogen for the synthesis of nucleotides, amino acids including glutamate and aspartate, and hexosamines, as well as for anaplerotic replenishment of TCA cycle intermediates and OXPHOS [70,71,72,73]. Importantly, glutamine is metabolized to glutamate—an amino acid that is critical in the synthesis of GSH—and thus, glutamine is also important for redox homeostasis in cancer cells [74,75,76]. While low levels of ROS have been shown to upregulate pathways promoting proliferation and as well as adaptive stress responses promoting tumorigenesis, higher ROS can upregulate cell death pathways [77,78,79]. Due to their high metabolic activity, cancer cells are faced with higher levels of endogenous reactive oxygen species (ROS) [79,80,81,82]. This necessitates synthesis of antioxidants such as GSH in order to maintain redox homeostasis and cancer cells may, thus, upregulate the import of nutrients and metabolic enzymes involved in the antioxidant defense.
Activation of oncogenes such as RAS, AKT, and MYC as well as loss of tumor suppressor genes such as p53 drive aerobic glycolysis in cancer cells [83]. RAS promotes glycolysis through activation of mTOR and upregulation of hypoxia inducible factors (HIFs) [84,85,86]. HIF1α in turn upregulates several genes promoting glycolysis as well as glucose transporters (GLUT1 and GLUT3) [87,88]. Elstrom et al., 2004, demonstrated that activation of AKT and the ensuing increase in aerobic glycolysis was sufficient to maintain survival and leukemogenesis of growth factor-deprived FL5.12 cells [89]. Under normal conditions p53 inhibits glycolysis through downregulation of glucose transporters (GLUT1 and GLUT4) and hexokinase 2 (HK2) enzyme and inhibition of HIF1α [90,91,92]. p53 also upregulates TP53-induced glycolysis and apoptosis regulator (TIGAR), which indirectly inhibits phosphofructokinase 1 (PFK1), another important glycolysis enzyme, thereby channeling glucose-6-phosphate into the PPP [93]. Therefore, loss of p53 promotes a glycolytic phenotype. Yun et al., 2009, showed that colorectal cell lines with mutant KRAS or BRAF upregulated GLUT1 and exhibited higher glucose uptake and glycolysis, which could be targeted with an inhibitor of hexokinase, 3-bromopyruvate (3-BrPA) [94]. They further demonstrated that the cell lines with the mutant alleles had a growth advantage in low glucose conditions and low glucose conditions selected for clones with KRAS or BRAF mutations. Moreover, the upregulation of GLUT1 was found to be independent of HIF1α suggesting that low glucose conditions may select for KRAS or BRAF mutations while hypoxia may favor PI3K, c-MYC, or TP53 mutations [94]. MYC, on the other hand, upregulates glucose and glutamine metabolism independent of the PI3K/AKT pathway [95,96]. Cells with oncogenic KRAS mutations or MYC upregulation have been shown to be dependent on glutamine for synthesis of amino acids, nucleotides and GSH [97,98,99]. MYC upregulation was associated with induction of glutaminase (GLS1), glutamate dehydrogenase (GDH) and the glutamine transporter, ASCT2/SLC1A5 [96,100,101,102,103]. Thus, oncogenes and tumor suppressors reprogram tumor metabolism and microenvironment-driven nutrient deprivation/hypoxia can additionally rewire metabolism creating new metabolic dependencies.
5. Intersection of Cancer Metabolism and BCL-2 Proteins
Many members of the BCL-2 family have been shown to be under metabolic control and are regulated by nutrient deprivation stresses. While an elevation in β-oxidation, ATP synthesis, and redox balance have implications in anoikis resistance [104] and glutamine metabolism in ferroptosis [17], we will focus on studies highlighting metabolic regulation of the BCL-2 protein family in this section. The highly labile BCL-2 protein MCL-1 has been reported to be stabilized by high glucose metabolism through different mechanisms. Zhao et al., 2007, showed that MCL-1 levels are regulated by Glycogen Synthase Kinase 3α and 3β (GSK-3α/3β), which phosphorylates MCL-1 at serine-159 and targets it for ubiquitination and degradation. Increased glucose metabolism increases phosphorylation of GSK-3α/3β at serine 21 and 9 by protein kinase C, negatively regulating its activity, thereby, preventing phosphorylation of MCL-1 and its subsequent degradation [105]. Pradelli et al., 2009, showed that reduction in MCL-1 levels upon glucose deprivation is regulated by inhibition of its translation through AMP-activated protein Kinase (AMPK) activation and mTOR inhibition [106]. Further, NOXA has been shown to induce apoptosis under glucose deprivation or by glycolysis inhibition by glucose analog 2-deoxy glucose (2-DG) with concordant decline in MCL-1 levels in different cancers [107,108,109,110]. Additionally, glucose limitation induces apoptosis through the ERK2/eIF-2α/ATF4-dependent pathway and upregulation of BID which could be reversed by supplementation with glutamate and α-ketoglutarate [111].
Glucose deprivation has also been shown to induce BH3-only sensitizer PUMA via induction of p53 which regulates the transcription of PUMA [112]. Moreover, Coloff et al., 2010, showed that PUMA and BIM are both induced upon glucose deprivation but only PUMA expression level is suppressed again upon restoring mitochondrial metabolism by supplementation with methyl pyruvate in the absence of glucose [113]. Further, they also showed that AKT promotes glycolysis and is required to reduce PUMA levels and apoptosis upon interleukin-3 (IL-3) withdrawal. Another mechanism through which AKT may prevent apoptosis is by increasing the binding of hexokinase to the mitochondrial membrane, thereby promoting its interaction with the outer membrane voltage-dependent anion channel (VDAC), and, in turn, preventing VDAC closure and mitochondrial membrane hyperpolarization that precedes cytochrome c release [114]. Further investigation of the mechanism by which AKT inhibited apoptosis led to the finding that AKT inhibited tBID mediated BAX and BAK oligomerization on growth factor withdrawal by promoting association of hexokinase with the mitochondrial membrane. Glucose deprivation was found to attenuate AKT induced inhibition of tBID mediated apoptosis by promoting dissociation of hexokinase from the mitochondrial membrane. Supporting these observations, overexpression of tBID also promoted dissociation of hexokinase from the mitochondrial membrane which was attenuated by activated AKT. Moreover, tBID mediated apoptosis was also inhibited by ectopic expression of the catalytically active amino-terminal region of hexokinase II containing its mitochondria-binding domain, further confirming the direct involvement of hexokinase in apoptosis [115].
Interestingly, glucose deprivation and treatment with 2-DG were found to result in different cell fates in mantle cell lymphoma. 2-DG resulted in cell death by downregulation of MCL-1 via the AMPK/mTOR pathway, while on the other hand, glucose deprivation inhibited cell death and maintained expression levels of BCL-2, BCL-xL, as well as MCL-1 by activation of the AKT and the subsequent inactivating phosphorylation of GSK-3β [116]. 2-DG has been shown to enhance ABT-737-induced apoptosis in lymphoma cells via upregulation of BIM that was reversed by supplementation with mannose [117]. Further, CHOP was found to be induced suggesting that apoptosis was initiated via the UPR-CHOP-BIM axis. Oxidative stress by co-treatment with 2-DG and 6-aminonicotinamide, which inhibits the PPP was also found to result in mitochondrial dysfunction and stimulate the intrinsic pathway of apoptosis in malignant cells [118].
Besides glucose deprivation other nutrient stresses such as amino acid deprivation can affect apoptosis. For example, both amino acid and glucose deprivation can activate the GCN2 (general control non-derepressible 2) kinase that phosphorylates eIF2α which in turn upregulates ATF4 translation. Glutamine depletion was found to induce cell death via upregulation of PUMA and NOXA in MYCN-amplified neuroblastoma [111]. This increase in PUMA and NOXA was mediated by ATF4 induction via the GCN2-eIF2a pathway. Further, nutrient deprivation can also result in the shift in the binding of pro-apoptotics to anti-apoptotics along with inducing changes in the expression levels. We have shown that glutamine deprivation results in upregulation of BIM and increased binding of BIM to BCL-2 in multiple myeloma (MM) [119]. Treatment of glutamine-deprived MM or 6-diazo-5-oxo-L-norleucine (DON)-treated MM patient samples and MM cell lines with the BCL-2 antagonist venetoclax (ABT-199) resulted in increased apoptosis. The glutamine-deprivation induced sensitization to venetoclax was partially reversed with α-ketoglutarate supplementation supporting the role of glutamine metabolism in regulating BIM induction and elevation of BCL-2 dependence in MM cells.
Recently a link between the electron transport chain (ETC) activity and the BCL-2 proteins has emerged. Chan et al., 2015, identified BCL-2 in a large-scale RNA interference (RNAi) screen to be synthetically lethal to Isocitrate Dehydrogenase 1 (IDH1) mutant expressing Acute Myeloid Leukemia (AML) and found that venetoclax selectively targeted mutant IDH1/2 AML [120]. This dependence of mutant AML on BCL-2 was found to be mediated by accumulation of oncometabolite (R)-2-hydroxyglutarate, which in turn inhibited Complex IV of the electron transport chain. Moreover, treatment of mutant IDH1/2 AML cell lines with cell permeable precursor of (R)-2-hydroxyglutarate, octyl-(R)-2-hydroxyglutrate sensitized the cells to venetoclax (ABT-199) [120]. In another study, a loss-of-function CRISPR/Cas9 knockout screen in AML identified heme biosynthesis in regulating apoptosis and sensitivity to venetoclax. Disruption of heme biosynthesis was found to deplete the ETC Complexes II-IV with a relatively higher effect on Complex IV than Complexes II and III. Treatment with succinyl acetone (SA) was found to attenuate the activity of Complexes II-IV as well as synergize with venetoclax induced death [121]. The authors further proposed that depolarization of the mitochondrial membrane due to heme depletion affected the integrity of the mitochondrial membrane and potentiated MOMP and apoptosis. Recent work by Chen et al., 2019, further demonstrated that disruption of mitochondrial function and dynamics sensitizes AML to venetoclax [122]. In this work, a CRISPR/Cas9 knockout screen was used to identify genes whose deletion synergized with venetoclax treatment. Several genes involved in mitochondrial structure and function were identified. One of the top-scoring candidates CLPB, which encodes a mitochondrial AAA+ ATPase chaperonin was also found to be upregulated in AML patients. Depletion of CLPB was shown to impact mitochondrial structure, induce mitochondrial stress response via upregulation ATF4 and its downstream targets as well as sensitize cells to venetoclax [122]. Our recent observations (under review) demonstrate that the succinate ubiquinone reductase activity of Complex II of the ETC is a predictor and target for venetoclax sensitization in MM [123].
Several studies have also demonstrated cross-talk between apoptosis and autophagy. Cancer cells frequently encounter nutrient deprivation stress and autophagy can promote their survival by recycling biomolecules via degradation of endogenous cellular components. Autophagy has also been shown to prevent cell death by apoptosis by degrading apoptosis regulators (e.g., degradation of caspase-8 in some TRAIL-induced apoptotic cells) [124,125]. On the other hand, autophagy can promote cell death through the engagement of cell death pathways [126,127]. Additionally, anti-apoptotic BCL-2 family proteins (BCL-2, MCL-1, and BCL-xL) can to bind BH3 domain only autophagy regulator Beclin-1 [128], which plays a key role in the initiation of autophagy [129]. Binding of these BCL-2 proteins to Beclin-1 inhibits the formation of pre-autophagosomal structure and prevents autophagy from occurring. Downregulation of MCL-1 due to nutrient deprivation as well as deletion of MCL-1 in cortical neurons can attenuate its inhibitory effect on autophagy or apoptosis and either pathway can ensue depending upon expression of Beclin-1 or BAX [130]. Further, Beclin-1 can be displaced from the anti-apoptotic BCL-2 proteins by BH3 only proteins such as BID, PUMA, BAD, and NOXA [131,132]. Therefore, changes in the metabolic state can impact both apoptosis and pro-survival/pro-apoptotic autophagy and their cross-talk.
BCL-2 family members also play a key role in regulating glucose and lipid metabolism. Phosphorylation of BH3-only BAD at Ser-155 was found to upregulate glucose oxidation by the mitochondria through interaction with glucokinase enzyme, which catalyzes the conversion of glucose to glucose-6-phosphate in hepatocytes [133]. While glucokinase has limited expression in other cells of the body, this observation has important implications as its isoform hexokinase II, is overexpressed in various cancers [134,135]. Further, glucose deprivation was found to reduce the phosphorylation on BAD and result in subsequent BAD-dependent apoptosis. The phosphorylated form of BAD has also been shown to play a role in glucose-dependent secretion of insulin by β cells [136]. Phosphorylation of BH3-only NOXA by cyclin-dependent kinase 5 (CDK5) has been shown to increase glucose flux into the pentose phosphate pathway [137]. Additionally, glucose deprivation and inhibition of CDK5 were shown to result in apoptosis through dephosphorylation of NOXA and restoration of its pro-apoptotic function. On the other hand, high glucose concentration can promote phosphorylation of NOXA at Ser-13 position. This phosphorylated form of NOXA binds MCL-1 but is unable to initiate MOMP and apoptosis. In addition, tBID has been reported to inhibit carnitine palmitoyl-transferase-1 (CPT1), thereby, preventing lipid transport and fatty acid (FA) oxidation in the mitochondria [138]. This tBID induced inhibition of FA oxidation was found to result in accumulation of FA metabolites such as palmitoyl-CoA. Palmitoyl-CoA can affect mitochondrial integrity and function and is also a precursor for synthesis of ceramide, which is an important mediator of programmed cell death [139,140,141]. Thus, this work highlights a close relation between the lipid metabolism and the apoptotic machinery implicating a direct involvement of lipid metabolites in the process of apoptosis.
BCL-2 proteins have been shown to regulate mitochondrial morphology and dynamics apart from their role in MOMP [142]. BAX and BAK were found to promote mitochondrial fusion in healthy cells [143,144]. Further, BAX and BAK are involved in the process of mitochondrial permeability transition pore (MPTP) formation in necrosis [145,146]. BCL-xL has been shown to increase the rate of mitochondrial fission, fusion, and biomass in cultured neurons, as well as plays a neuroprotective role by promoting synapse formation [147,148]. In addition, BCL-2 proteins also play a role in regulation of metabolism at the inner mitochondrial membrane where they localize. BCL-2 was found to bind to cytochrome c oxidase and cyclophilin D and regulate mitochondrial respiration [149,150]. BCL-xL has been shown to maintain mitochondrial membrane potential by directly modulating F1F0-ATP synthase and preventing proton leak into the mitochondrial matrix [151]. Further, MCL-1 deletion has been shown to result in a number of mitochondrial defects, suggesting an alternating role of MCL-1 in maintaining mitochondrial integrity and function [152].
In summary, there is considerable evidence that BCL-2 proteins are under metabolic regulation and in some instances can also modulate metabolism. Alterations in tumor metabolism can potentiate the ability of the cell to evade apoptosis. Therefore, the metabolic state has important implications for therapy as cancer cells can indirectly become resistant to chemotherapy by rewiring/reprogramming metabolism and, thus, changing their BCL-2 protein landscape.
6. Implications for Therapy
Most chemoresistance involves reduced drug sensitivity and impaired ability to execute apoptosis. The inability to effectively target BCL-2 proteins accounts for resistance to bortezomib [153,154,155], rapamycin [156], cyclin-dependent kinase inhibitors [157], ABT 737 [158], and death receptor (Fas/TRAIL)-induced apoptosis [106] in various cancers including MM. MM, acute myelogenous and lymphocytic leukemia, and various solid tumor cells and refractory cancers subject to prior chemotherapy, exhibit reduced “priming”, i.e., exhibit suboptimal quantities of BH3 activators bound to anti-apoptotics, thereby increasing the threshold level required to induce apoptosis [159,160,161,162]. Thus, developing strategies to increase the primed state could potentially circumvent resistance. Such approaches can potentially sensitize to existing therapy and also enhance sensitivity to BH3 mimetics/BCL-2 protein antagonists providing alternative routes to apoptosis induction.
The BH3 activator BIM is highly expressed in cells of hematopoietic origin [163], and evaluation of BH3 activators bound to the anti-apoptotic BCl-2 proteins in MM demonstrated that BIM is the most relevant BH3 activator dictating BCL-2 dependence [164]. Despite heavy reliance of MM on MCL-1 [165,166] and correlation of MCL-1 levels with poor prognoses [40], specifically targeting BIM-BCL-2 interactions induces apoptosis even in MCL-1 dependent MM [164]. While MCL-1 binds and sequesters BIM, the presence of sufficient BIM bound to other anti-apoptotics such as BCL-2 when targeted can also induce apoptosis, the effectiveness of which is importantly dictated by the quantity and distribution of BIM among the anti-apoptotics [164].
BH3 mimetics are potent small molecules used to release BH3 activators bound to anti-apoptotics. Currently, venetoclax (ABT-199) is the most promising small molecule BCL-2 antagonist. MCL-1-selective antagonists are in clinical trial [167]. The requirement for MCL-1 in myocardial homeostasis may preclude targeting MCL-1 [168]. Also, targeting BCL-xL by ABT-737 is limited by its deleterious effects on normal platelet viability [169]. Clinically, venetoclax as a monotherapy is highly efficacious in Chronic Lymphocytic Leukemia (CLL) (that is BCL-2 dependent) [170]; however, effective only in a minority contingent of MM [171], with a recent phase 1 study suggesting only selective efficacy within the 11;14 myeloma patients [172].
Several studies have shown that co-targeting mitochondrial metabolism and the ETC can synergize with BH3 mimetics. We have shown that inhibition of glutamine metabolic pathways by glutamine deprivation or glutamine antagonist 6-diazo-5-oxo-L-norleucine (DON), modulates BCL-2 dependence, reducing the apoptotic threshold by increasing BIM-BCL-2 binding and the primed state, consequently increasing sensitivity to BH3 mimetics [119]. We have identified that targeting glutamine metabolism increases the primed state of the cell by increasing BIM binding to BCL-2 even across MM cells that are MCL-1 dependent i.e. having BIM bound primarily to MCL-1 [119]. In another study, DON was also shown to enhance apoptosis in combination with BCL-2 family antagonist Navitoclax (ABT-263) in glutamine addicted neuroblastoma and Ewing’s sarcoma cells overexpressing Myc [173]. Inhibition of GLS1 by CB-839 was found to reduce OXPHOS and selectively reduce proliferation and induce apoptosis in AML cells [174]. Supplementation with α-ketoglutarate and introduction of a hyperactive glutaminase C mutant rescued the cells from apoptosis. This study further showed that glutaminolysis inhibition by CB-839 synergized with venetoclax in AML. A recent study investigating the mechanism of action of combination treatment with venetoclax and hypomethylating agent azacitidine in older de novo AML patients revealed disruption of the TCA cycle, inhibition of ETC Complex II culminating in attenuation of OXPHOS leading to sensitization to venetoclax [175]. The inhibition of Complex II upon treatment with azacitidine was mediated by reduction in glutathione levels resulting in reduction of activating glutathionylation of Succinate Dehydrogenase A (SDHA) subunit of Complex II and selectively targeted the leukemic stem cell (LSC) population [175]. Further, in another study, investigation of the metabolism of LSCs from de novo AML patients revealed that they are dependent upon amino acid metabolism for the TCA cycle, and treatment with venetoclax and azacitidine depleted their amino acid pools as well as downregulated amino acid transporters [176]. Interestingly, AML blasts and LSCs from relapsed patients were found to be less reliant on amino acid metabolism and were resistant to venetoclax and azacitidine by utilizing fatty acid metabolism to support their TCA cycle. This suggests that relapse patients could potentially benefit from therapies combining venetoclax and azacitidine with inhibitors of fatty acid metabolism. This work illustrates how cancer cells can rewire their metabolism and develop resistance to therapy. Understanding the metabolism of relapsed/refractory cancer can help design combination therapies to overcome the resistance. Previously, Samudia et al., 2010, had demonstrated that de novo fatty acid synthesis supported fatty acid oxidation in leukemic cell lines OCI-AML3 and MOLM13. Inhibition of fatty acid oxidation or fatty acid synthesis by etomoxir (carnitine palmitoyltransferase-1 inhibitor) or orlistat (fatty acid synthase/lipolysis inhibitor) respectively, had anti-proliferative effects [177]. Moreover, treatment with etomoxir or orlistat sensitized these cell lines to apoptosis induction with ABT-737 (Bcl-xL, Bcl-2, and Bcl-w antagonist) and etomoxir also improved the efficacy of ABT-737 in a human AML murine model. While the precise mechanism of sensitization was not elucidated, this work indicated that fatty acid metabolism can be targeted to sensitize AML cells to BH3 mimetics. Additionally, recent work from our group has unveiled the importance of ETC function in modulating sensitivity of MM to venetoclax. We have demonstrated that inhibition of succinate ubiquinone reductase (SQR) by small molecule inhibitor thenoyltrifluoroacetone (TTFA) sensitizes venetoclax-resistant MM cells (under review, Nature Communications) [123]. This sensitization is mediated by upregulation of ATF4 upon TTFA treatment and subsequent increase in BIM and NOXA.
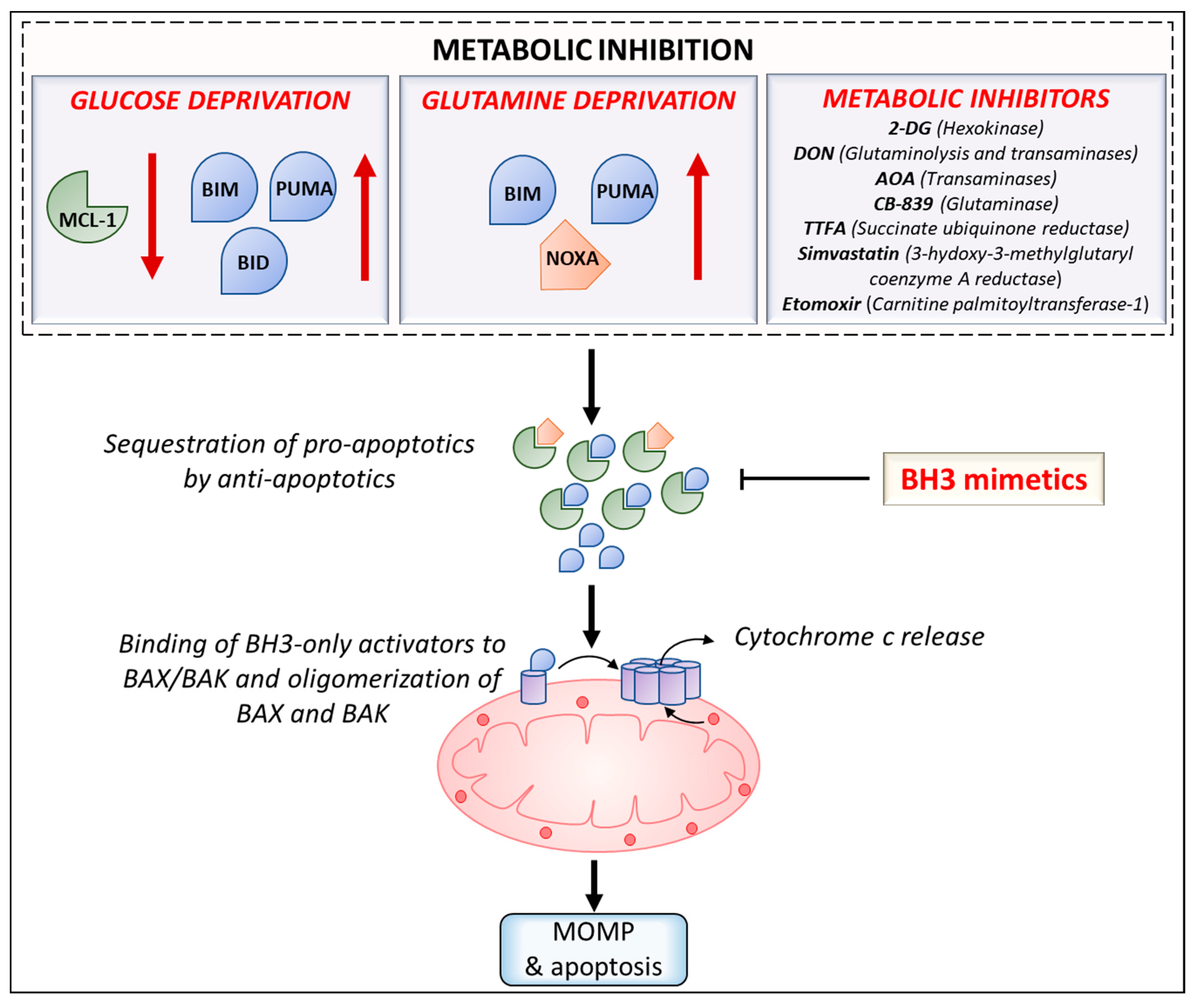
Lastly, efforts to repurpose FDA approved drugs for cancer treatment have led to investigation of efficacy of combining statins with BH3 mimetics. Several preclinical studies have shown that statins can act as anticancer agents as well as synergize with chemotherapeutics by enhancing cytotoxicity or by increasing cellular concentration of chemotherapeutics [178]. Inhibition of a key enzyme in the cholesterol synthesis pathway—3-hydoxy-3-methylglutaryl coenzyme A reductase (HMGCR)—by simvastatin was found to synergize with venetoclax in inducing apoptosis in diffuse large B cell lymphoma and AML [179]. HMGCR inhibition was shown to result in reduction in protein geranylgeranylation, subsequently leading to upregulation of PUMA and thus, priming the cancer cells for apoptosis. Furthermore, analysis of CLL clinical trials with venetoclax monotherapy revealed that statin users resulted in a higher rate of complete remission than patients without any background statin use. The mevalonate pathway has been shown to be upregulated in several cancers and its inhibition by statins and other inhibitors in combination with BH3 mimetics such as venetoclax can be an attractive therapeutic strategy for these malignancies [180]. Antibiotics constitute another promising group of compounds that can be repurposed for cancer therapy due to their inhibitory effects on mitochondrial function/metabolism. Ravà et al., 2018, reported that inhibition of mitochondrial translation by tigecycline sensitized MYC/BCL-2 double-hit lymphomas to venetoclax [181]. Related antibiotics doxycycline and tetracycline were also found to synergize with venetoclax. Further, tigecycline also synergized with venetoclax in treatment of mice engrafted with DHL cell lines or patient derived xenografts [181]. Recent work by Al-Zebeeby et al., 2019, on identifying metabolic inhibitors that can target resistance mechanisms in CLL has shown that inhibition of glutamine uptake and metabolism by various inhibitors (transaminase inhibitor amino-oxyacetic acid (AOA), ASCT2 inhibitor GPNA, glutaminase inhibitor CB-839, and glutamine: fructose-6-phosphate-amidotransferase (GFAT) inhibitor Azaserine) was found to sensitize BCL-xL antagonist (A-1331852) resistant K562 cells to A-1331852 induced apoptosis [182]. Additionally, inhibition of downstream metabolic pathways such as reductive carboxylation, fatty acid synthesis and cholesterol synthesis by ATP-citrate lyase (ACLY) inhibitor SB20499, fatty acid synthase (FASN) inhibitor GSK2194069 and HMGCR inhibition by statins, respectively, sensitized resistant cells to A-1331852. Unpublished work from our group has also shown that AOA treatment sensitizes multiple myeloma cell lines to venetoclax. In sum, these studies demonstrate the benefit of developing co-targeting approaches using metabolic inhibitors and BH3 mimetics for cancer therapy. Various strategies to induce sensitization to BH3 mimetics by increasing the ‘primed’ state of cancer cells using metabolic inhibition are outlined in Figure 2.
7. Conclusions
Altered metabolism and the evasion of apoptosis are hallmarks of cancer cells. Targeting tumor metabolic dependencies is an attractive strategy for therapy development. However, the efficacy of monotherapy using metabolic inhibitors is limited by emergence of adaptive metabolic changes. Cancers cells are largely dependent upon pro-survival proteins of the BCL-2 family for evasion of apoptosis. Recent advances in understanding regulation of apoptosis have shown that BCL-2 proteins are under tight metabolic control. Several BH3 mimetics have been developed to target the primed state of cancer cells. Co-targeting approaches combining metabolic inhibitors, that increase the ‘primed’ state, and BH3 mimetics can be used to achieve potent drug synergies and further, expand the application of these individual therapies. Therefore, concomitant inhibition of the two hallmarks of cancer i.e. altered metabolism and the evasion of apoptosis is a promising strategy to effectively target various cancers.
Funding
This work was supported by a National Institutes of Health: R01 CA208328 and Leukemia Lymphoma Society TRP Award #6573-19 to Mala Shanmugam and R01 CA192844 to Lawrence H Boise.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wideranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 2000, 1, 1201–1230. [Google Scholar] [CrossRef] [PubMed]
- Favaloro, B.; Allocati, N.; Graziano, V.; Di Ilio, C.; De Laurenzi, V. Role of apoptosis in disease. Aging 2012, 4, 330. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C. Dysregulation of apoptosis in cancer. J. Clin. Oncol. 1999, 17, 2941. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, D.W.; Thornberry, N.A. Caspases: Killer proteases. Trends Biochem. Sci. 1997, 22, 299–306. [Google Scholar] [CrossRef]
- Budihardjo, I.; Oliver, H.; Lutter, M.; Luo, X.; Wang, X. Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 1999, 15, 269–290. [Google Scholar] [CrossRef] [PubMed]
- Riedl, S.J.; Shi, Y. Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell Biol. 2004, 5, 897. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; El-Deiry, W.S. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene 2003, 22, 8628–8633. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef]
- Carr, R.M.; Qiao, G.; Qin, J.; Jayaraman, S.; Prabhakar, B.S.; Maker, A.V. Targeting the metabolic pathway of human colon cancer overcomes resistance to TRAIL-induced apoptosis. Cell Death Discov. 2016, 2, 16067. [Google Scholar] [CrossRef] [Green Version]
- Frisch, S.M.; Francis, H. Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 1994, 124, 619–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta 2013, 1833, 3481–3498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilmore, A. Anoikis. Cell Death Differ. 2005, 12, 1473. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, J.J.A. Molecular mechanisms of “detachment-induced apoptosis—Anoikis”. Apoptosis 2002, 7, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.J.C. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danial, N.N.; Korsmeyer, S.J. Cell death: Critical control points. Cell 2004, 116, 205–219. [Google Scholar] [CrossRef]
- Cheng, E.H.; Wei, M.C.; Weiler, S.; Flavell, R.A.; Mak, T.W.; Lindsten, T.; Korsmeyer, S.J. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol. Cell 2001, 8, 705–711. [Google Scholar] [CrossRef]
- Day, C.L.; Chen, L.; Richardson, S.J.; Harrison, P.J.; Huang, D.C.; Hinds, M.G. Solution structure of prosurvival Mcl-1 and characterization of its binding by proapoptotic BH3-only ligands. J. Biol. Chem. 2005, 280, 4738–4744. [Google Scholar] [CrossRef]
- Chen, L.; Willis, S.N.; Wei, A.; Smith, B.J.; Fletcher, J.I.; Hinds, M.G.; Colman, P.M.; Day, C.L.; Adams, J.M.; Huang, D.C. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell 2005, 17, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Sarosiek, K.A.; Chi, X.; Bachman, J.A.; Sims, J.J.; Montero, J.; Patel, L.; Flanagan, A.; Andrews, D.W.; Sorger, P.; Letai, A. BID preferentially activates BAK while BIM preferentially activates BAX, affecting chemotherapy response. Mol. Cell 2013, 51, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Ni Chonghaile, T.; Letai, A. Mimicking the BH3 domain to kill cancer cells. Oncogene 2008, 27 (Suppl. 1), S149–S157. [Google Scholar] [CrossRef] [Green Version]
- Ruvolo, P.P.; Deng, X.; May, W.S. Phosphorylation of Bcl2 and regulation of apoptosis. Leukemia 2001, 15, 515–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chipuk, J.E.; Moldoveanu, T.; Llambi, F.; Parsons, M.J.; Green, D.R. The BCL-2 family reunion. Mol. Cell 2010, 37, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Kutuk, O.; Letai, A. Regulation of Bcl-2 family proteins by posttranslational modifications. Curr. Mol. Med. 2008, 8, 102–118. [Google Scholar] [PubMed]
- Li, H.; Zhu, H.; Xu, C.-j.; Yuan, J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef]
- Yuan, X.; Gajan, A.; Chu, Q.; Xiong, H.; Wu, K.; Wu, G.S. Developing TRAIL/TRAIL death receptor-based cancer therapies. Cancer Metastasis Rev. 2018, 37, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Pinedo, C.; Ruiz-Ruiz, C.; Ruiz de Almodovar, C.; Palacios, C.; Lopez-Rivas, A. Inhibition of glucose metabolism sensitizes tumor cells to death receptor-triggered apoptosis through enhancement of death-inducing signaling complex formation and apical procaspase-8 processing. J. Biol. Chem. 2003, 278, 12759–12768. [Google Scholar] [CrossRef]
- Iurlaro, R.; Püschel, F.; León-Annicchiarico, C.L.; O’Connor, H.; Martin, S.J.; Palou-Gramón, D.; Lucendo, E.; Muñoz-Pinedo, C. Glucose deprivation induces ATF4-mediated apoptosis through TRAIL death receptors. Mol. Cell. Biol. 2017, 37, e00479-16. [Google Scholar] [CrossRef]
- Nam, S.Y.; Amoscato, A.A.; Lee, Y.J. Low glucose-enhanced TRAIL cytotoxicity is mediated through the ceramide-Akt-FLIP pathway. Oncogene 2002, 21, 337–346. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Haapa-Paananen, S.; Kaminskyy, V.O.; Kohonen, P.; Fey, V.; Zhivotovsky, B.; Kallioniemi, O.; Perala, M. Inhibition of the mitochondrial pyrimidine biosynthesis enzyme dihydroorotate dehydrogenase by doxorubicin and brequinar sensitizes cancer cells to TRAIL-induced apoptosis. Oncogene 2014, 33, 3538–3549. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Yip, K.W.; Reed, J.C. Bcl-2 family proteins and cancer. Oncogene 2008, 27, 6398. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Cory, S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 2007, 26, 1324. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y.; Finger, L.R.; Yunis, J.; Nowell, P.C.; Croce, C.M. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science 1984, 226, 1097–1099. [Google Scholar] [CrossRef] [PubMed]
- Graninger, W.B.; Seto, M.; Boutain, B.; Goldman, P.; Korsmeyer, S.J. Expression of Bcl-2 and Bcl-2-Ig fusion transcripts in normal and neoplastic cells. J. Clin. Investig. 1987, 80, 1512–1515. [Google Scholar] [CrossRef] [PubMed]
- Seto, M.; Jaeger, U.; Hockett, R.D.; Graninger, W.; Bennett, S.; Goldman, P.; Korsmeyer, S.J. Alternative promoters and exons, somatic mutation and deregulation of the Bcl-2-Ig fusion gene in lymphoma. EMBO J. 1988, 7, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Soini, Y.; Pääkkö, P.; Lehto, V. Histopathological evaluation of apoptosis in cancer. Am. J. Pathol. 1998, 153, 1041–1053. [Google Scholar] [CrossRef]
- Wuilleme-Toumi, S.; Robillard, N.; Gomez, P.; Moreau, P.; Le Gouill, S.; Avet-Loiseau, H.; Harousseau, J.L.; Amiot, M.; Bataille, R. Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia 2005, 19, 1248–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Guttikonda, S.; Roberts, L.; Uziel, T.; Semizarov, D.; Elmore, S.W.; Leverson, J.D.; Lam, L.T. Mcl-1 is critical for survival in a subgroup of non-small-cell lung cancer cell lines. Oncogene 2011, 30, 1963–1968. [Google Scholar] [CrossRef] [PubMed]
- Townsend, K.J.; Trusty, J.L.; Traupman, M.A.; Eastman, A.; Craig, R.W. Expression of the antiapoptotic MCL1 gene product is regulated by a mitogen activated protein kinase-mediated pathway triggered through microtubule disruption and protein kinase C. Oncogene 1998, 17, 1223. [Google Scholar] [CrossRef] [PubMed]
- Croxton, R.; Ma, Y.; Song, L.; Haura, E.B.; Cress, W.D. Direct repression of the Mcl-1 promoter by E2F1. Oncogene 2002, 21, 1359. [Google Scholar] [CrossRef] [PubMed]
- Derouet, M.; Thomas, L.; Cross, A.; Moots, R.J.; Edwards, S.W. Granulocyte macrophage colony-stimulating factor signaling and proteasome inhibition delay neutrophil apoptosis by increasing the stability of Mcl-1. J. Biol. Chem. 2004, 279, 26915–26921. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhang, L.; Hwang, P.M.; Kinzler, K.W.; Vogelstein, B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol. Cell 2001, 7, 673–682. [Google Scholar] [CrossRef]
- Sax, J.K.; Fei, P.; Murphy, M.E.; Bernhard, E.; Korsmeyer, S.J.; El-Deiry, W.S. BID regulation by p53 contributes to chemosensitivity. Nat. Cell Biol. 2002, 4, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Oda, E.; Ohki, R.; Murasawa, H.; Nemoto, J.; Shibue, T.; Yamashita, T.; Tokino, T.; Taniguchi, T.; Tanaka, N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000, 288, 1053–1058. [Google Scholar] [CrossRef]
- Villunger, A.; Michalak, E.M.; Coultas, L.; Müllauer, F.; Böck, G.; Ausserlechner, M.J.; Adams, J.M.; Strasser, A. p53-and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 2003, 302, 1036–1038. [Google Scholar] [CrossRef]
- Rosen, K.; Rak, J.; Jin, J.; Kerbel, R.S.; Newman, M.J.; Filmus, J. Downregulation of the pro-apoptotic protein Bak is required for the ras-induced transformation of intestinal epithelial cells. Curr. Biol. 1998, 8, 1331–1334. [Google Scholar] [CrossRef]
- Toshiyuki, M.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 2932–2999. [Google Scholar] [CrossRef]
- McCurrach, M.E.; Connor, T.M.; Knudson, C.M.; Korsmeyer, S.J.; Lowe, S.W. bax-deficiency promotes drug resistance and oncogenic transformation by attenuating p53-dependent apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 2345–2349. [Google Scholar] [CrossRef]
- Certo, M.; Del Gaizo Moore, V.; Nishino, M.; Wei, G.; Korsmeyer, S.; Armstrong, S.A.; Letai, A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 2006, 9, 351–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, T.; Seino, Y.; Fukumoto, H.; Koh, G.; Yano, H.; Inagaki, N.; Yamada, Y.; Inoue, K.; Manabe, T.; Imura, H. Over-expression of facilitative glucose transporter genes in human cancer. Biochem. Biophys. Res. Commun. 1990, 170, 223–230. [Google Scholar] [CrossRef]
- Medina, R.A.; Owen, G.I. Glucose transporters: Expression, regulation and cancer. Biol. Res. 2002, 35, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Enriquez, S.; Marin-Hernandez, A.; Gallardo-Perez, J.C.; Moreno-Sanchez, R. Kinetics of transport and phosphorylation of glucose in cancer cells. J. Cell. Physiol. 2009, 221, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Manolescu, A.R.; Witkowska, K.; Kinnaird, A.; Cessford, T.; Cheeseman, C. Facilitated hexose transporters: New perspectives on form and function. Physiology (Bethesda) 2007, 22, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Yongzhi, H.; Chen, S.; Luo, X.; Lin, Y.; Zhou, Y.; Jin, H.; Hou, B.; Deng, Y.; Tu, L.; et al. The prognostic value of GLUT1 in cancers: A systematic review and meta-analysis. Oncotarget 2017, 8, 43356–43367. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Yang, C.; Zhang, X.; Zheng, P.; Shen, W. Expression of glucose transporter 1 and prognosis in non-small cell lung cancer: A pooled analysis of 1665 patients. Oncotarget 2017, 8, 60954–60961. [Google Scholar] [CrossRef]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Gruning, N.M.; Kruger, A.; Tauqeer Alam, M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 2015, 90, 927–963. [Google Scholar] [CrossRef]
- Jiang, P.; Du, W.; Wu, M. Regulation of the pentose phosphate pathway in cancer. Protein Cell 2014, 5, 592–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulain, L.; Sujobert, P.; Zylbersztejn, F.; Barreau, S.; Stuani, L.; Lambert, M.; Palama, T.L.; Chesnais, V.; Birsen, R.; Vergez, F.; et al. High mTORC1 activity drives glycolysis addiction and sensitivity to G6PD inhibition in acute myeloid leukemia cells. Leukemia 2017, 31, 2326–2335. [Google Scholar] [CrossRef] [PubMed]
- Pusapati, R.V.; Daemen, A.; Wilson, C.; Sandoval, W.; Gao, M.; Haley, B.; Baudy, A.R.; Hatzivassiliou, G.; Evangelista, M.; Settleman, J. mTORC1-Dependent Metabolic Reprogramming Underlies Escape from Glycolysis Addiction in Cancer Cells. Cancer Cell 2016, 29, 548–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Pavlova, N.N.; Thompson, C.B. Cancer cell metabolism: The essential role of the nonessential amino acid, glutamine. EMBO J. 2017, 36, 1302–1315. [Google Scholar] [CrossRef]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, M.E. Glutathione: An overview of biosynthesis and modulation. Chem. Biol. Interact. 1998, 111, 1–14. [Google Scholar] [CrossRef]
- Amelio, I.; Cutruzzolá, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and glycine metabolism in cancer. J. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Amores-Sánchez, M.I.; Medina, M.Á. Glutamine, as a precursor of glutathione, and oxidative stress. J. Mol. Genet. Metab. 1999, 67, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef] [Green Version]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Drug Discov. 2009, 8, 579. [Google Scholar] [CrossRef]
- Wang, J.; Yi, J. Cancer cell killing via ROS: To increase or decrease, that is the question. Cancer Biol. Ther. 2008, 7, 1875–1884. [Google Scholar] [CrossRef]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar]
- Toyokuni, S.; Okamoto, K.; Yodoi, J.; Hiai, H. Persistent oxidative stress in cancer. FEBS Lett. 1995, 358, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Valko, M.; Rhodes, C.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, C.M.; Reczek, E.E.; James, M.F.; Brems, H.; Legius, E.; Cichowski, K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc. Natl. Acad. Sci. USA 2005, 102, 8573–8578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, K.G.; Fingar, D.C. Mammalian target of rapamycin (mTOR): Conducting the cellular signaling symphony. J. Biol. Chem. 2010, 285, 14071–14077. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Pore, N.; Behrooz, A.; Ismail-Beigi, F.; Maity, A. Regulation of glut1 mRNA by hypoxia-inducible factor-1. Interaction between H-ras and hypoxia. J. Biol. Chem. 2001, 276, 9519–9525. [Google Scholar] [CrossRef]
- Flier, J.S.; Mueckler, M.M.; Usher, P.; Lodish, H.F. Elevated levels of glucose transport and transporter messenger RNA are induced by ras or src oncogenes. Science 1987, 235, 1492–1495. [Google Scholar] [CrossRef]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef]
- Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Karnieli, E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004, 64, 2627–2633. [Google Scholar] [CrossRef]
- Mathupala, S.P.; Heese, C.; Pedersen, P.L. Glucose catabolism in cancer cells. The type II hexokinase promoter contains functionally active response elements for the tumor suppressor p53. J. Biol. Chem. 1997, 272, 22776–22780. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V.; An, W.G.; Romanova, L.Y.; Trepel, J.; Fojo, T.; Neckers, L. p53 inhibits hypoxia-inducible factor-stimulated transcription. J. Biol. Chem. 1998, 273, 11995–11998. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.; Markowitz, S.; Zhou, S.; et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef] [PubMed]
- Osthus, R.C.; Shim, H.; Kim, S.; Li, Q.; Reddy, R.; Mukherjee, M.; Xu, Y.; Wonsey, D.; Lee, L.A.; Dang, C.V. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J. Biol. Chem. 2000, 275, 21797–21800. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [Green Version]
- Gaglio, D.; Metallo, C.M.; Gameiro, P.A.; Hiller, K.; Danna, L.S.; Balestrieri, C.; Alberghina, L.; Stephanopoulos, G.; Chiaradonna, F. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol. Syst. Biol. 2011, 7, 523. [Google Scholar] [CrossRef] [PubMed]
- Brunelli, L.; Caiola, E.; Marabese, M.; Broggini, M.; Pastorelli, R. Capturing the metabolomic diversity of KRAS mutants in non-small-cell lung cancer cells. Oncotarget 2014, 5, 4722. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Yuneva, M.; Zamboni, N.; Oefner, P.; Sachidanandam, R.; Lazebnik, Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J. Cell Biol. 2007, 178, 93–105. [Google Scholar] [CrossRef] [Green Version]
- Yuneva, M.O.; Fan, T.W.; Allen, T.D.; Higashi, R.M.; Ferraris, D.V.; Tsukamoto, T.; Mates, J.M.; Alonso, F.J.; Wang, C.; Seo, Y.; et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012, 15, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Le, A.; Hancock, C.; Lane, A.N.; Dang, C.V.; Fan, T.W.; Phang, J.M. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc. Natl. Acad. Sci. USA 2012, 109, 8983–8988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, Z.T.; Grassian, A.R.; Song, L.; Jiang, Z.; Gerhart-Hines, Z.; Irie, H.Y.; Gao, S.; Puigserver, P.; Brugge, J.S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009, 461, 109–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Altman, B.J.; Coloff, J.L.; Herman, C.E.; Jacobs, S.R.; Wieman, H.L.; Wofford, J.A.; Dimascio, L.N.; Ilkayeva, O.; Kelekar, A. Glycogen synthase kinase 3α and 3β mediate a glucose-sensitive antiapoptotic signaling pathway to stabilize Mcl-1. Mol. Cell. Biol. 2007, 27, 4328–4339. [Google Scholar] [CrossRef] [PubMed]
- Pradelli, L.A.; Beneteau, M.; Chauvin, C.; Jacquin, M.A.; Marchetti, S.; Munoz-Pinedo, C.; Auberger, P.; Pende, M.; Ricci, J. Glycolysis inhibition sensitizes tumor cells to death receptors-induced apoptosis by AMP kinase activation leading to Mcl-1 block in translation. Oncogene 2010, 29, 1641. [Google Scholar] [CrossRef] [PubMed]
- Alves, N.L.; Derks, I.A.; Berk, E.; Spijker, R.; van Lier, R.A.; Eldering, E. The Noxa/Mcl-1 axis regulates susceptibility to apoptosis under glucose limitation in dividing T cells. Immunity 2006, 24, 703–716. [Google Scholar] [CrossRef]
- Wensveen, F.M.; Alves, N.L.; Derks, I.A.; Reedquist, K.A.; Eldering, E. Apoptosis induced by overall metabolic stress converges on the Bcl-2 family proteins Noxa and Mcl-1. Apoptosis 2011, 16, 708. [Google Scholar] [CrossRef]
- Leon-Annicchiarico, C.L.; Ramirez-Peinado, S.; Dominguez-Villanueva, D.; Gonsberg, A.; Lampidis, T.J.; Munoz-Pinedo, C. ATF4 mediates necrosis induced by glucose deprivation and apoptosis induced by 2-deoxyglucose in the same cells. FEBS J. 2015, 282, 3647–3658. [Google Scholar] [CrossRef] [Green Version]
- Ramirez-Peinado, S.; Alcazar-Limones, F.; Lagares-Tena, L.; El Mjiyad, N.; Caro-Maldonado, A.; Tirado, O.M.; Munoz-Pinedo, C. 2-deoxyglucose induces Noxa-dependent apoptosis in alveolar rhabdomyosarcoma. Cancer Res. 2011, 71, 6796–6806. [Google Scholar] [CrossRef]
- Shin, S.; Buel, G.R.; Wolgamott, L.; Plas, D.R.; Asara, J.M.; Blenis, J.; Yoon, S.O. ERK2 Mediates Metabolic Stress Response to Regulate Cell Fate. Mol. Cell 2015, 59, 382–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Coloff, J.L.; Ferguson, E.C.; Jacobs, S.R.; Cui, K.; Rathmell, J.C. Glucose metabolism attenuates p53 and Puma-dependent cell death upon growth factor deprivation. J. Biol. Chem. 2008, 283, 36344–36353. [Google Scholar] [CrossRef] [PubMed]
- Coloff, J.L.; Mason, E.F.; Altman, B.J.; Gerriets, V.A.; Liu, T.; Nichols, A.N.; Zhao, Y.; Wofford, J.A.; Jacobs, S.R.; Ilkayeva, O. Akt requires glucose metabolism to suppress puma expression and prevent apoptosis of leukemic T cells. J. Biol. Chem. 2011, 286, 5921–5933. [Google Scholar] [CrossRef] [PubMed]
- Gottlob, K.; Majewski, N.; Kennedy, S.; Kandel, E.; Robey, R.B.; Hay, N.J.G. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001, 15, 1406–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majewski, N.; Nogueira, V.; Robey, R.B.; Hay, N. Akt inhibits apoptosis downstream of BID cleavage via a glucose-dependent mechanism involving mitochondrial hexokinases. Mol. Cell. Biol. 2004, 24, 730–740. [Google Scholar] [CrossRef] [PubMed]
- MacFarlane, M.; Robinson, G.L.; Cain, K. Glucose—A sweet way to die. Cell Cycle 2012, 11, 3919–3925. [Google Scholar] [CrossRef] [PubMed]
- Zagorodna, O.; Martin, S.M.; Rutkowski, D.T.; Kuwana, T.; Spitz, D.R.; Knudson, C.M. 2-deoxyglucose-induced toxicity is regulated by Bcl-2 family members and is enhanced by antagonizing Bcl-2 in lymphoma cell lines. Oncogene 2012, 31, 2738–2749. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, R.; Sharma, P.K.; Jadon, S.P.; Varshney, R. A combination of 2-deoxy-D-glucose and 6-aminonicotinamide induces cell cycle arrest and apoptosis selectively in irradiated human malignant cells. Tumour Biol. 2012, 33, 1021–1030. [Google Scholar] [CrossRef]
- Bajpai, R.; Matulis, S.M.; Wei, C.; Nooka, A.K.; Von Hollen, H.E.; Lonial, S.; Boise, L.H.; Shanmugam, M. Targeting glutamine metabolism in multiple myeloma enhances BIM binding to BCL-2 eliciting synthetic lethality to venetoclax. Oncogene 2016, 35, 3955–3964. [Google Scholar] [CrossRef]
- Chan, S.M.; Thomas, D.; Corces-Zimmerman, M.R.; Xavy, S.; Rastogi, S.; Hong, W.J.; Zhao, F.; Medeiros, B.C.; Tyvoll, D.A.; Majeti, R. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat. Med. 2015, 21, 178–184. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.H.; Xie, A.; Rutter, J.C.; Ahn, Y.R.; Lloyd-Cowden, J.M.; Nichols, A.G.; Soderquist, R.S.; Koves, T.R.; Muoio, D.M.; MacIver, N.J.; et al. Systematic Dissection of the Metabolic-Apoptotic Interface in AML Reveals Heme Biosynthesis to Be a Regulator of Drug Sensitivity. Cell Metab. 2019, 29, 1217–1231. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Glytsou, C.; Zhou, H.; Narang, S.; Reyna, D.E.; Lopez, A.; Sakellaropoulos, T.; Gong, Y.; Kloetgen, A.; Yap, Y.S. Targeting mitochondrial structure sensitizes acute myeloid leukemia to Venetoclax treatment. Cancer Discov. 2019. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, R.; Achreja, A.; Sharma, A.; Wei, C.; Edgar, C.L.; Siddiqa, A.; Gupta, V.A.; Matulis, S.M.; McBrayer, S.K.; Mittal, A.; et al. Succinate ubiquinone reductase predicts and regulates venetoclax sensitivity in multiple myeloma. Nature Commun. 2019. Revised and Resubmitted. [Google Scholar]
- Han, J.; Hou, W.; Goldstein, L.A.; Lu, C.; Stolz, D.B.; Yin, X.-M.; Rabinowich, H. Involvement of protective autophagy in TRAIL resistance of apoptosis-defective tumor cells. J. Biol. Chem. 2008, 283, 19665–19677. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhou, X.; Qiao, J.; Bao, A. Autophagy is a regulator of TRAIL-induced apoptosis in NSCLC A549 cells. J. Cell Commun. Signal. 2017, 11, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Denton, D.; Nicolson, S.; Kumar, S. Cell death by autophagy: Facts and apparent artefacts. Cell Death Differ. 2012, 19, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Yuan, J. Autophagy in cell death: An innocent convict? J. Clin. Investig. 2005, 115, 2679–2688. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Kleeman, L.K.; Jiang, H.H.; Gordon, G.; Goldman, J.E.; Berry, G.; Herman, B.; Levine, B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J. Virol. 1998, 72, 8586–8596. [Google Scholar]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Germain, M.; Nguyen, A.P.; Le Grand, J.N.; Arbour, N.; Vanderluit, J.L.; Park, D.S.; Opferman, J.T.; Slack, R.S. MCL-1 is a stress sensor that regulates autophagy in a developmentally regulated manner. EMBO J. 2011, 30, 395–407. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Criollo, A.; Tasdemir, E.; Vicencio, J.M.; Tajeddine, N.; Hickman, J.A.; Geneste, O.; Kroemer, G. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-XL. Autophagy 2007, 3, 374–376. [Google Scholar] [CrossRef] [PubMed]
- Erlich, S.; Mizrachy, L.; Segev, O.; Lindenboim, L.; Zmira, O.; Adi-Harel, S.; Hirsch, J.A.; Stein, R.; Pinkas-Kramarski, R. Differential interactions between Beclin 1 and Bcl-2 family members. Autophagy 2007, 3, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Danial, N.N.; Gramm, C.F.; Scorrano, L.; Zhang, C.Y.; Krauss, S.; Ranger, A.M.; Datta, S.R.; Greenberg, M.E.; Licklider, L.J.; Lowell, B.B.; et al. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature 2003, 424, 952–956. [Google Scholar] [CrossRef] [PubMed]
- Mathupala, S.P.; Rempel, A.; Pedersen, P.L. Aberrant glycolytic metabolism of cancer cells: A remarkable coordination of genetic, transcriptional, post-translational, and mutational events that lead to a critical role for type II hexokinase. J. Bioenerg. Biomembr. 1997, 29, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.A. Mammalian hexokinases and their abnormal expression in cancer. Br. J. Biomed. Sci. 2000, 57, 170–178. [Google Scholar] [PubMed]
- Danial, N.N.; Walensky, L.D.; Zhang, C.-Y.; Choi, C.S.; Fisher, J.K.; Molina, A.J.; Datta, S.R.; Pitter, K.L.; Bird, G.H.; Wikstrom, J.D. Dual role of proapoptotic BAD in insulin secretion and beta cell survival. Nat. Med. 2008, 14, 144. [Google Scholar] [CrossRef]
- Lowman, X.H.; McDonnell, M.A.; Kosloske, A.; Odumade, O.A.; Jenness, C.; Karim, C.B.; Jemmerson, R.; Kelekar, A. The proapoptotic function of Noxa in human leukemia cells is regulated by the kinase Cdk5 and by glucose. Mol. Cell 2010, 40, 823–833. [Google Scholar] [CrossRef]
- Giordano, A.; Calvani, M.; Petillo, O.; Grippo, P.; Tuccillo, F.; Melone, M.A.B.; Bonelli, P.; Calarco, A.; Peluso, G. tBid induces alterations of mitochondrial fatty acid oxidation flux by malonyl-CoA-independent inhibition of carnitine palmitoyltransferase-1. Cell Death Differ. 2005, 12, 603. [Google Scholar] [CrossRef]
- De Pablo, M.A.; Susin, S.A.; Jacotot, E.; Larochette, N.; Costantini, P.; Ravagnan, L.; Zamzami, N.; Kroemer, G. Palmitate induces apoptosis via a direct effect on mitochondria. Apoptosis 1999, 4, 81–87. [Google Scholar] [CrossRef]
- Listenberger, L.L.; Ory, D.S.; Schaffer, J.E. Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J. Biol. Chem. 2001, 276, 14890–14895. [Google Scholar] [CrossRef]
- Pettus, B.J.; Chalfant, C.E.; Hannun, Y.A. Ceramide in apoptosis: An overview and current perspectives. Biochim. Biophys. Acta 2002, 1585, 114–125. [Google Scholar] [CrossRef]
- Autret, A.; Martin, S.J. Emerging role for members of the Bcl-2 family in mitochondrial morphogenesis. Mol. Cell 2009, 36, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.; Wei, Q.; Feng, L.; Dong, G.; Tao, Y.; Mei, L.; Xie, Z.-J.; Dong, Z. Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proc. Natl. Acad. Sci. USA 2007, 104, 11649–11654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karbowski, M.; Norris, K.L.; Cleland, M.M.; Jeong, S.-Y.; Youle, R.J. Role of Bax and Bak in mitochondrial morphogenesis. Nature 2006, 443, 658. [Google Scholar] [CrossRef] [PubMed]
- Whelan, R.S.; Konstantinidis, K.; Wei, A.-C.; Chen, Y.; Reyna, D.E.; Jha, S.; Yang, Y.; Calvert, J.W.; Lindsten, T.; Thompson, C.B. Bax regulates primary necrosis through mitochondrial dynamics. Proc. Natl. Acad. Sci. USA 2012, 109, 6566–6571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karch, J.; Kwong, J.Q.; Burr, A.R.; Sargent, M.A.; Elrod, J.W.; Peixoto, P.M.; Martinez-Caballero, S.; Osinska, H.; Cheng, E.H.; Robbins, J. Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. eLife 2013, 2, e00772. [Google Scholar] [CrossRef] [PubMed]
- Berman, S.B.; Chen, Y.-b.; Qi, B.; McCaffery, J.M.; Rucker, E.B.; Goebbels, S.; Nave, K.-A.; Arnold, B.A.; Jonas, E.A.; Pineda, F.J. Bcl-xL increases mitochondrial fission, fusion, and biomass in neurons. J. Cell Biol. 2009, 184, 707–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonas, E.A.; Porter, G.A.; Alavian, K.N. Bcl-xL in neuroprotection and plasticity. Front. Physiol. 2014, 5, 355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.X.; Pervaiz, S. Involvement of cytochrome c oxidase subunits Va and Vb in the regulation of cancer cell metabolism by Bcl-2. Cell Death Differ. 2010, 17, 408–420. [Google Scholar] [CrossRef]
- Eliseev, R.A.; Malecki, J.; Lester, T.; Zhang, Y.; Humphrey, J.; Gunter, T.E. Cyclophilin D interacts with Bcl2 and exerts an anti-apoptotic effect. J. Biol. Chem. 2009, 284, 9692–9699. [Google Scholar] [CrossRef]
- Alavian, K.N.; Li, H.; Collis, L.; Bonanni, L.; Zeng, L.; Sacchetti, S.; Lazrove, E.; Nabili, P.; Flaherty, B.; Graham, M.; et al. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat. Cell Biol. 2011, 13, 1224–1233. [Google Scholar] [CrossRef] [PubMed]
- Perciavalle, R.M.; Stewart, D.P.; Koss, B.; Lynch, J.; Milasta, S.; Bathina, M.; Temirov, J.; Cleland, M.M.; Pelletier, S.; Schuetz, J.D.; et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat. Cell Biol. 2012, 14, 575–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podar, K.; Gouill, S.L.; Zhang, J.; Opferman, J.T.; Zorn, E.; Tai, Y.T.; Hideshima, T.; Amiot, M.; Chauhan, D.; Harousseau, J.L.; et al. A pivotal role for Mcl-1 in Bortezomib-induced apoptosis. Oncogene 2008, 27, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xu, J.; He, J.; Zheng, Y.; Li, H.; Lu, Y.; Qian, J.; Lin, P.; Weber, D.M.; Yang, J.; et al. A critical role of autocrine sonic hedgehog signaling in human CD138+ myeloma cell survival and drug resistance. Blood 2014, 124, 2061–2071. [Google Scholar] [CrossRef] [PubMed]
- Pepper, C.; Hoy, T.; Bentley, D.P. Bcl-2/Bax ratios in chronic lymphocytic leukaemia and their correlation with in vitro apoptosis and clinical resistance. Br. J. Cancer 1997, 76, 935–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, J.R.; Hippo, Y.; Robert, F.; Chen, S.M.; Malina, A.; Lin, C.J.; Trojahn, U.; Wendel, H.G.; Charest, A.; Bronson, R.T.; et al. mTORC1 promotes survival through translational control of Mcl-1. Proc. Natl. Acad. Sci. USA 2008, 105, 10853–10858. [Google Scholar] [CrossRef] [Green Version]
- Eguchi, T.; Itadani, H.; Shimomura, T.; Kawanishi, N.; Hirai, H.; Kotani, H. Expression levels of p18INK4C modify the cellular efficacy of cyclin-dependent kinase inhibitors via regulation of Mcl-1 expression in tumor cell lines. Mol. Cancer Ther. 2009, 8, 1460–1472. [Google Scholar] [CrossRef]
- Nguyen, M.; Marcellus, R.C.; Roulston, A.; Watson, M.; Serfass, L.; Murthy Madiraju, S.R.; Goulet, D.; Viallet, J.; Belec, L.; Billot, X.; et al. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc. Natl. Acad. Sci. USA 2007, 104, 19512–19517. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Carlson, N.; Takeyama, K.; Dal Cin, P.; Shipp, M.; Letai, A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell 2007, 12, 171–185. [Google Scholar] [CrossRef]
- Ni Chonghaile, T.; Sarosiek, K.A.; Vo, T.T.; Ryan, J.A.; Tammareddi, A.; Moore Vdel, G.; Deng, J.; Anderson, K.C.; Richardson, P.; Tai, Y.T.; et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science 2011, 334, 1129–1133. [Google Scholar] [CrossRef]
- Montero, J.; Sarosiek, K.A.; DeAngelo, J.D.; Maertens, O.; Ryan, J.; Ercan, D.; Piao, H.; Horowitz, N.S.; Berkowitz, R.S.; Matulonis, U.; et al. Drug-induced death signaling strategy rapidly predicts cancer response to chemotherapy. Cell 2015, 160, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Hogdal, L.J.; Benito, J.M.; Bucci, D.; Han, L.; Borthakur, G.; Cortes, J.; DeAngelo, D.J.; Debose, L.; Mu, H.; et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014, 4, 362–375. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, L.A.; Cullen, L.; Visvader, J.; Lindeman, G.J.; Print, C.; Bath, M.L.; Huang, D.C.; Strasser, A. The proapoptotic BH3-only protein bim is expressed in hematopoietic, epithelial, neuronal, and germ cells. Am. J. Pathol. 2000, 157, 449–461. [Google Scholar] [CrossRef]
- Morales, A.A.; Kurtoglu, M.; Matulis, S.M.; Liu, J.; Siefker, D.; Gutman, D.M.; Kaufman, J.L.; Lee, K.P.; Lonial, S.; Boise, L.H. Distribution of Bim determines Mcl-1 dependence or codependence with Bcl-xL/Bcl-2 in Mcl-1-expressing myeloma cells. Blood 2011, 118, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Gojo, I.; Fenton, R.G. Myeloid cell factor-1 is a critical survival factor for multiple myeloma. Blood 2002, 99, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Derenne, S.; Monia, B.; Dean, N.M.; Taylor, J.K.; Rapp, M.J.; Harousseau, J.L.; Bataille, R.; Amiot, M. Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-x(L) is an essential survival protein of human myeloma cells. Blood 2002, 100, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Xiang, W.; Yang, C.-Y.; Bai, L. MCL-1 inhibition in cancer treatment. Onco Targets Ther. 2018, 11, 7301–7314. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.L.; Roberts, D.J.; Kubli, D.A.; Lee, Y.; Quinsay, M.N.; Owens, J.B.; Fischer, K.M.; Sussman, M.A.; Miyamoto, S.; Gustafsson, A.B. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev. 2013, 27, 1365–1377. [Google Scholar] [CrossRef] [Green Version]
- Schoenwaelder, S.M.; Jarman, K.E.; Gardiner, E.E.; Hua, M.; Qiao, J.; White, M.J.; Josefsson, E.C.; Alwis, I.; Ono, A.; Willcox, A.; et al. Bcl-xL-inhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood 2011, 118, 1663–1674. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Touzeau, C.; Dousset, C.; Le Gouill, S.; Sampath, D.; Leverson, J.D.; Souers, A.J.; Maiga, S.; Bene, M.C.; Moreau, P.; Pellat-Deceunynck, C.; et al. The Bcl-2 specific BH3 mimetic ABT-199: A promising targeted therapy for t(11;14) multiple myeloma. Leukemia 2014, 28, 210–212. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Vij, R.; Kaufman, J.L.; Mikhael, J.R.; Facon, T.; Pegourie, B.; Benboubker, L.; Moreau, P.; Amiot, M.; Alzate, S.; et al. Safety and Efficacy of Venetoclax (ABT-199/GDC-0199) Monotherapy for Relapsed/Refractory Multiple Myeloma: Phase 1 Preliminary Results. Blood 2015, 126, 4219. [Google Scholar]
- Olsen, R.R.; Mary-Sinclair, M.N.; Yin, Z.; Freeman, K.W. Antagonizing Bcl-2 family members sensitizes neuroblastoma and Ewing’s sarcoma to an inhibitor of glutamine metabolism. PLoS ONE 2015, 10, e0116998. [Google Scholar] [CrossRef] [PubMed]
- Jacque, N.; Ronchetti, A.M.; Larrue, C.; Meunier, G.; Birsen, R.; Willems, L.; Saland, E.; Decroocq, J.; Maciel, T.T.; Lambert, M.; et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 2015, 126, 1346–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D’Alessandro, A.; Culp-Hill, R.; Riemondy, K.A.; Gillen, A.E. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 2018, 24, 1859. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740. [Google Scholar] [CrossRef] [PubMed]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Investig. 2010, 120, 142–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadi, Y.; Karimian, R.; Panahi, Y. Effects of statins on the chemoresistance-The antagonistic drug-drug interactions versus the anti-cancer effects. Biomed. Pharmacother. 2018, 108, 1856–1865. [Google Scholar] [CrossRef]
- Lee, J.S.; Roberts, A.; Juarez, D.; Vo, T.T.; Bhatt, S.; Herzog, L.O.; Mallya, S.; Bellin, R.J.; Agarwal, S.K.; Salem, A.H.; et al. Statins enhance efficacy of venetoclax in blood cancers. Sci. Transl. Med. 2018, 10, eaaq1240. [Google Scholar] [CrossRef]
- Clendening, J.W.; Pandyra, A.; Boutros, P.C.; El Ghamrasni, S.; Khosravi, F.; Trentin, G.A.; Martirosyan, A.; Hakem, A.; Hakem, R.; Jurisica, I.; et al. Dysregulation of the mevalonate pathway promotes transformation. Proc. Natl. Acad. Sci. USA 2010, 107, 15051–15056. [Google Scholar] [CrossRef] [Green Version]
- Rava, M.; D’Andrea, A.; Nicoli, P.; Gritti, I.; Donati, G.; Doni, M.; Giorgio, M.; Olivero, D.; Amati, B. Therapeutic synergy between tigecycline and venetoclax in a preclinical model of MYC/BCL2 double-hit B cell lymphoma. Sci. Transl. Med. 2018, 10, eaan8723. [Google Scholar] [CrossRef] [PubMed]
- Al-Zebeeby, A.; Vogler, M.; Milani, M.; Richards, C.; Alotibi, A.; Greaves, G.; Dyer, M.J.S.; Cohen, G.M.; Varadarajan, S. Targeting intermediary metabolism enhances the efficacy of BH3 mimetic therapy in hematologic malignancies. Haematologica 2019, 104, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Unified model of BCL-2 protein family interactions. BH3-only activators (BIM, tBID, and PUMA) can bind to the multi-domain anti-apoptotics (BCL-2, MCL-2, BCL-xL, BCL-w, and A1) as well as the multi-domain pro-apoptotic effectors (BAX and BAK), whereas each of the BH3-only sensitizers (NOXA, BAD, and BMF) can only bind to a subset of the multi-domain anti-apoptotics. Binding of BH3-only activator to BAX/BAK promotes the oligomerization of BAX/BAK. BAX and BAK can be inhibited by a subset of the anti-apoptotics.
Figure 1.
Unified model of BCL-2 protein family interactions. BH3-only activators (BIM, tBID, and PUMA) can bind to the multi-domain anti-apoptotics (BCL-2, MCL-2, BCL-xL, BCL-w, and A1) as well as the multi-domain pro-apoptotic effectors (BAX and BAK), whereas each of the BH3-only sensitizers (NOXA, BAD, and BMF) can only bind to a subset of the multi-domain anti-apoptotics. Binding of BH3-only activator to BAX/BAK promotes the oligomerization of BAX/BAK. BAX and BAK can be inhibited by a subset of the anti-apoptotics.

Figure 2.
Metabolic regulation of BCL-2 proteins inducing sensitization to BCL-2 antagonists. Glucose/glutamine deprivation can modulate the levels of BCL-2 proteins resulting in changes in binding of BH3-only proteins with the anti-apoptotic proteins. Treatment with BH3-mimetics can release activators from anti-apoptotics. Unbound/released activators then activate BAX/BAK and initiate MOMP. A subset of metabolic inhibitors (pre-clinical leads and small molecules in clinical trials) that elevate the primed state and increase sensitivity to BCL-2-targeted BH3 mimetics have been shown along with their targets. Abbreviations: 2-DG: 2-deoxyglucose; DON: 6-diazo-5-oxo-L-norleucine; AOA: aminooxyacetic acid; TTFA: thenoyl trifluoracetone.
Figure 2.
Metabolic regulation of BCL-2 proteins inducing sensitization to BCL-2 antagonists. Glucose/glutamine deprivation can modulate the levels of BCL-2 proteins resulting in changes in binding of BH3-only proteins with the anti-apoptotic proteins. Treatment with BH3-mimetics can release activators from anti-apoptotics. Unbound/released activators then activate BAX/BAK and initiate MOMP. A subset of metabolic inhibitors (pre-clinical leads and small molecules in clinical trials) that elevate the primed state and increase sensitivity to BCL-2-targeted BH3 mimetics have been shown along with their targets. Abbreviations: 2-DG: 2-deoxyglucose; DON: 6-diazo-5-oxo-L-norleucine; AOA: aminooxyacetic acid; TTFA: thenoyl trifluoracetone.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sharma, A.; Boise, L.H.; Shanmugam, M. Cancer Metabolism and the Evasion of Apoptotic Cell Death. Cancers 2019, 11, 1144. https://doi.org/10.3390/cancers11081144
AMA Style
Sharma A, Boise LH, Shanmugam M. Cancer Metabolism and the Evasion of Apoptotic Cell Death. Cancers. 2019; 11(8):1144. https://doi.org/10.3390/cancers11081144
Chicago/Turabian StyleSharma, Aditi, Lawrence H. Boise, and Mala Shanmugam. 2019. "Cancer Metabolism and the Evasion of Apoptotic Cell Death" Cancers 11, no. 8: 1144. https://doi.org/10.3390/cancers11081144
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.