Mutations in the ND2 Subunit of Mitochondrial Complex I Are Sufficient to Confer Increased Tumorigenic and Metastatic Potential to Cancer Cells



 ,
, 
Abstract
1. Introduction
2. Results
2.1. Mitochondrial Supercomplex Assembly in L929 and L929dt Cells
2.2. Activity of Respiratory Complexes in L929 and L929dt Cells
2.3. Metabolic Features of L929 and L929dt Cells
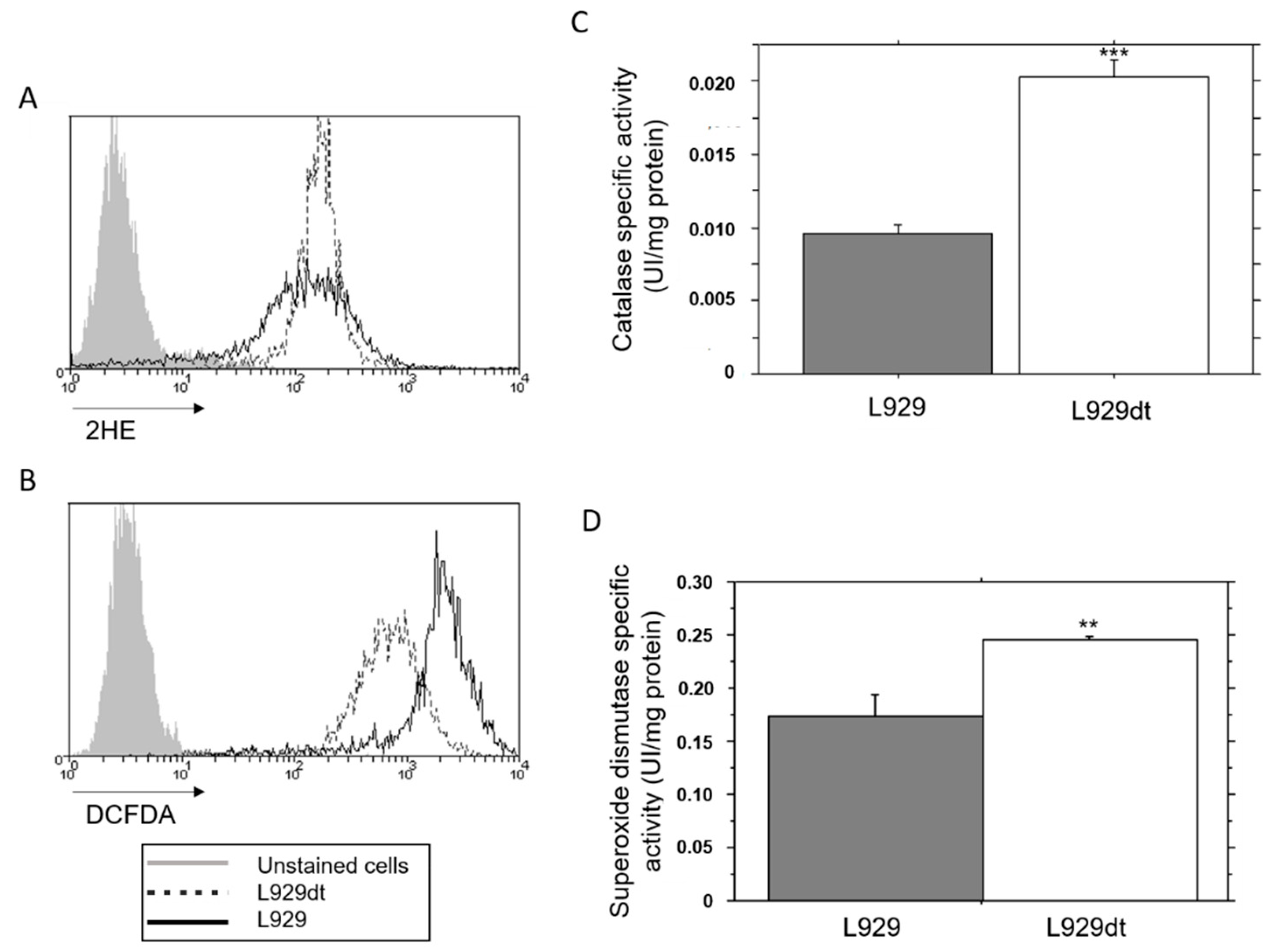
2.4. Endogenous ROS Generation and Catalase Activity in L929 and L929dt Cells
2.5. In Vivo Tumor Development of L929 and L929dt Cells
2.6. Determination of Mutations in Complex I Mitochondrial DNA in L929dt Cells Compared with Parental L929 Cells
2.7. Experiments with Cybrid Cells
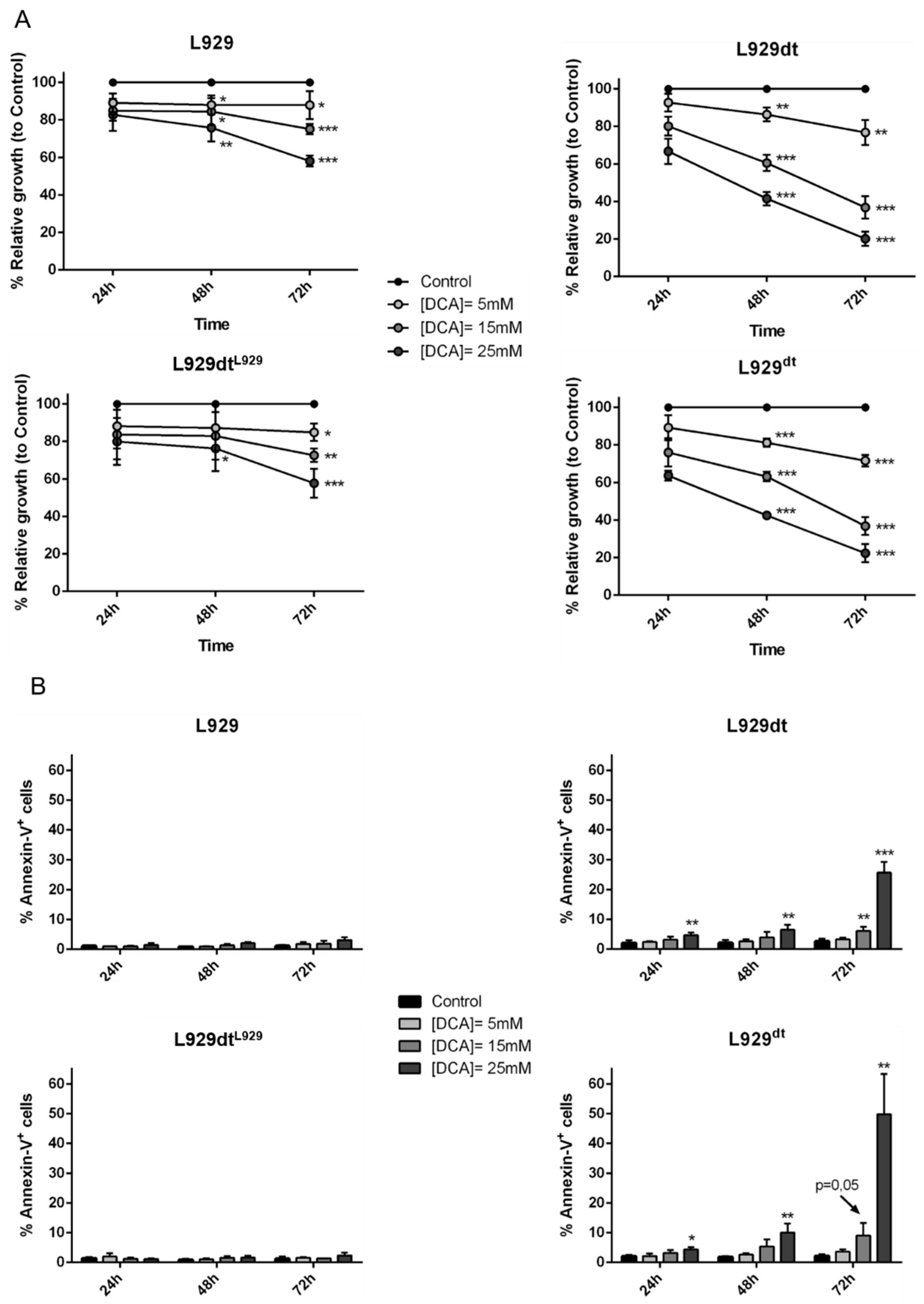
2.8. DCA Effect on L929 and L929dt and Cybrid Cells Proliferation, Cell Death and MHC-I Expression
3. Discussion
4. Materials and Methods
4.1. Cells and Mice
4.2. Flow Cytometry Determination of MHC-I Surface Expression, Apoptosis Induction and ROS Production
4.3. Mitochondrial Superoxide Production Analysis
4.4. Cell Viability Assays
4.5. Supplementation with DCA or Galactose
4.6. Blue Native Polyacrylamide Electrophoresis
4.7. Analysis of Mitochondrial Supercomplex Assembly by Immunoblot
4.8. In Gel Complex I Activity
4.9. OXPHOS Performance
4.10. Analysis of Mitochondrial DNA Mutations
4.11. Generation of Cybrid Cells
4.12. Determination of Catalase and Superoxide Dismutase Specific Activity
4.13. In Vivo Tumor Development Experiments
4.14. Histological Studies
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Weinhouse, S.; Warburg, O.; Burk, D.; Schade, A.L. On Respiratory Impairment in Cancer Cells. Science 1956, 124, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Acín-Pérez, R.; Fernández-Silva, P.; Peleato, M.L.; Pérez-Martos, A.; Enriquez, J.A. Respiratory Active Mitochondrial Supercomplexes. Mol. Cell 2008, 32, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acín-Pérez, R.; Latorre-Pellicer, A.; Colás, C.; Balsa, E.; Perales-Clemente, E.; Quirós, P.; Calvo, E.; Rodríguez-Hernández, M.; et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 2013, 340, 1567–1570. [Google Scholar] [CrossRef] [PubMed]
- Greggio, C.; Jha, P.; Kulkarni, S.S.; Lagarrigue, S.; Broskey, N.T.; Boutant, M.; Wang, X.; Alonso, S.C.; Ofori, E.; Auwerx, J.; et al. Enhanced Respiratory Chain Supercomplex Formation in Response to Exercise in Human Skeletal Muscle. Cell Metab. 2017, 25, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lostao, L.; Anel, A.; Pardo, J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef] [PubMed]
- Villalba, M.; Rathore, M.G.; López-Royuela, N.; Krzywinska, E.; Garaude, J.; Allende-Vega, N. From tumor cell metabolism to tumor immune escape. Int. J. Biochem. Cell Biol. 2013, 45, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Romero, I.; Aptsiauri, N.; García-Lora, A. Generation of MHC class I diversity in primary tumors and selection of the malignant phenotype. Int. J. Cancer 2016, 138, 271–280. [Google Scholar] [CrossRef]
- Jeon, S.M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumor cell survival during enery stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef]
- Cruz-Bermúdez, A.; Vallejo, C.G.; Vicente-Blanco, R.J.; Gallardo, M.E.; Fernández-Moreno, M.A.; Quintanilla, M.; Garesse, R. Enhanced tumorigenicity by mitochondrial DNA mild mutations. Oncotarget 2015, 6, 13628–13643. [Google Scholar] [CrossRef]
- Gasparre, G.; Porcelli, A.M.; Lenaz, G.; Romeo, G. Relevance of Mitochondrial Genetics and Metabolism in Cancer Development. Cold Spring Harb. Perspect. Biol. 2013, 5, a011411. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J.I. ROS-Generating Mitochondrial DNA Mutations Can Regulate Tumor Cell Metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Santidrian, A.; Matsuno-Yagi, A.; Ritland, M.; Seo, B.; LeBoeuf, S.; Gay, L.; Yagi, T.; Felding-Habermann, B. Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J. Clin. Investig. 2013, 123, 1068–1081. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Guha, M.; Dong, D.; Whelan, K.; Ruthel, G.; Uchikado, Y.; Natsugoe, S.; Nakagawa, H.; Avadhani, N. Disruption of cytochrome c oxidase function induces the Warburg effect and metabolic reprogramming. Oncogene 2016, 35, 1585–1595. [Google Scholar] [CrossRef] [PubMed]
- Catalan, E.; Charni, S.; Jaime, P.; Aguilò, J.I.; Enriquez, J.A.; Naval, J.; Pardo, J.; Villalba, M.; Anel, A. MHC-I modulation due to changes in tumor cell metabolism regulates tumor sensitivity to CTL and NK cells. OncoImmunology 2015, 4, e985924. [Google Scholar] [CrossRef] [PubMed]
- Acín-PérezR; Bayona-Bafaluy, M.P.; Fernández-Silva, P.; Moreno-Loshuertos, R.; Pérez-Martos, A.; Bruno, C.; Moraes, C.T.; Enríquez, J.A. Respiratory Complex III Is Required to Maintain Complex I in Mammalian Mitochondria. Mol. Cell 2004, 13, 805–815. [Google Scholar]
- Selinsky, C.L.; Howell, M.D. Soluble Tumor Necrosis Factor Receptor Type I Enhances Tumor Development and Persistence in Vivo. Cell. Immunol. 2000, 200, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, R.V.; Galoforo, S.S.; Berns, C.M.; Armour, E.P.; McEachern, D.; Corry, P.M.; Lee, Y.J. Comparison of tumor growth betweenHSP25- andHSP27-transfected murine L929 cells in nude mice. Int. J. Cancer 1997, 72, 871–877. [Google Scholar] [CrossRef]
- Agnoletto, C.; Brunelli, L.; Melloni, E.; Pastorelli, R.; Casciano, F.; Rimondi, E.; Rigolin, G.M.; Cuneo, A.; Secchiero, P.; Zauli, G. The anti-leukemic activity of sodium dichloroacetate in p53mutated/null cells is mediated by a p53-independent ILF3/p21 pathway. Oncotarget 2014, 6, 2385–2396. [Google Scholar]
- Agnoletto, C.; Melloni, E.; Casciano, F.; Rigolin, G.M.; Rimondi, E.; Celeghini, C.; Brunelli, L.; Cuneo, A.; Secchiero, P.; Zauli, G. Sodium dichloroacetate exhibits anti-leukemic activity in B-chronic lymphocytic leukemia (B-CLL) and synergizes with the p53 activator Nutlin-3. Oncotarget 2014, 5, 4347–4360. [Google Scholar] [CrossRef][Green Version]
- Allende-Vega, N.; Krzywinska, E.; Orecchioni, S.; López-Royuela, N.; Reggiani, F.; Talarico, G.; Rossi, J.F.; Rossignol, R.; Hicheri, Y.; Cartron, G.; et al. The presence of wild type p53 in hematological cancers improves the efficacy of combinational therapy targeting metabolism. Oncotarget 2015, 6, 19228–19245. [Google Scholar] [CrossRef]
- Garaude, J.; Acín-Pérez, R.; Martínez-Cano, S.; Enamorado, M.; Ugolini, M.; Nistal-Villán, E.; Hervás-Stubbs, S.; Pelegrín, P.; Sander, L.E.; Enríquez, J.; et al. Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nat. Immunol. 2016, 17, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Kang, T.; Chen, L.; Zhang, W.; Liao, Y.; Chen, J.; Shi, Y. Detection of mitochondrial DNA mutations by high-throughput sequencing in the blood of breast cancer patients. Int. J. Mol. Med. 2014, 33, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.J.; Bai, R.K.; Wong, L.J. Comprehensive scanning of somatic mitochondrial DNA mutations in breast cancer. Cancer Res. 2002, 62, 972–976. [Google Scholar] [PubMed]
- Lam, E.T.; Bracci, P.M.; Holly, E.A.; Chu, C.; Poon, A.; Wan, E.; White, K.; Kwok, P.Y.; Pawlikowska, L.; Tranah, G.J. Mitochondrial DNA sequence variation and risk of pancreatic cancer. Cancer Res. 2011, 72, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Prior, S.; Griffiths, A.; Baxter, J.; Hodder, S.; Silvester, K.; Lewis, P. Mitochondrial DNA mutations in oral squamous cell carcinoma. Carcinogenesis 2006, 27, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Zhou, S.; Chang, S.; McFate, T.; Verma, A.; Califano, J. Mitochondrial Mutations Contribute to HIF1a Accumulation via Increased Reactive Oxygen Species and Up-regulated Pyruvate Dehydrogenease Kinase 2 in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2009, 15, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Kachhap, S.; Sun, W.; Wu, G.; Chuang, A.; Poeta, L.; Grumbine, L.; Mithani, S.K.; Chatterjee, A.; Koch, W.; et al. Frequency and phenotypic implications of mitochondrial DNA mutations in human squamous cell cancers of the head and neck. Proc. Natl. Acad. Sci. USA 2007, 104, 7540–7545. [Google Scholar] [CrossRef] [PubMed]
- Qiao, C.; Zhou, C.; Zhang, S.; Guo, R.; Zhang, F.; Qian, S.; Huan, Y.; Song, Y.; Liao, H.; Li, C.; et al. Analysis of ND4 gene mutations in acute myelogenous leukemia. Chin. J. Hematol. 2014, 35, 708–712. [Google Scholar]
- Yeung, K.Y.; Dickinson, A.; Donoghue, J.F.; Polekhina, G.; White, S.J.; Grammatopoulos, D.K.; McKenzie, M.; Johns, T.G.; John, J.C. The identification of mitochondrial DNA variants in glioblastoma multiforme. Acta Neuropathol. Commun. 2014, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Loshuertos, R.; Acín-Pérez, R.; Fernández-Silva, P.; Movilla, N.; Pérez-Martos, A.; Rodriguez de Cordoba, S.; Gallardo, M.; Enríquez, J. Differences in reactive oxygen species production explain the phenotypes associated with common mouse mitochondrial DNA variants. Nat. Genet. 2006, 38, 1261–1268. [Google Scholar] [CrossRef]
- Koshikawa, N.; Akimoto, M.; Hayashi, J.-I.; Nagase, H.; Takenaga, K. Association of predicted pathogenic mutations in mitochondrial ND genes with distant metastasis in NSCLC and colon cancer. Sci. Rep. 2017, 7, 15535. [Google Scholar] [CrossRef] [PubMed]
- Zamzami, N. Mitochondrial control of nuclear apoptosis. J. Exp. Med. 1996, 183, 1533–1544. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.; Filigheddu, N.; Bravo-San Pedro, J.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef] [PubMed]
- Molon, B.; Calì, B.; Viola, A. T Cells and Cancer: How Metabolism Shapes Immunity. Front. Immunol. 2016, 7, 435. [Google Scholar] [CrossRef] [PubMed]
- Charni, S.; Aguilo, J.I.; Garaude, J.; De Bettignies, G.; Jacquet, C.; Hipskind, R.A.; Singer, D.; Anel, A.; Villalba, M. ERK5 Knockdown Generates Mouse Leukemia Cells with Low MHC Class I Levels That Activate NK Cells and Block Tumorigenesis. J. Immunol. 2009, 182, 3398–3405. [Google Scholar] [CrossRef]
- Charni, S.; De Bettignies, G.; Rathore, M.G.; Aguiló, J.I.; Elsen, P.J.V.D.; Haouzi, D.; Hipskind, R.A.; Enriquez, J.A.; Sanchez-Beato, M.; Pardo, J.; et al. Oxidative Phosphorylation Induces De Novo Expression of the MHC Class I in Tumor Cells through the ERK5 Pathway. J. Immunol. 2010, 185, 3498–3503. [Google Scholar] [CrossRef] [PubMed]
- Sawhney, R.S.; Liu, W.; Brattain, M.G. A novel role of ERK5 in integrin-mediated cell adhesion and motility in cancer cells via Fak signaling. J. Cell. Physiol. 2009, 219, 152–161. [Google Scholar] [CrossRef]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef]
- Bosque, A.; Alava, M.A.; Paz-Artal, E.; Naval, J.; Allende, L.M.; Anel, A.; Aguilò, J.I. The induction of Bim expression in human T-cell blasts is dependent on nonapoptotic Fas/CD95 signaling. Blood 2007, 109, 1627–1635. [Google Scholar] [CrossRef]
- Devadas, S.; Zaritskaya, L.; Rhee, S.G.; Oberley, L.; Williams, M.S. Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation: Selective regulation of mitogen-activated protein kinase activation and fas ligand expression. J. Exp. Med. 2002, 195, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Alley, M.C.; Scudiero, D.A.; Monks, A.; Hursey, M.L.; Czerwinski, M.J.; Fine, D.; Abbot, B.J.; Mayo, J.G.; Shoemaker, R.H.; Boyd, M.R. Sensibility of drug screening with panels of human tumor cell lines using microculture tetrazolium assay. Cancer Res. 1988, 48, 589–601. [Google Scholar] [PubMed]
- Schägger, H.; Pfeiffer, K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 2000, 19, 1777–1783. [Google Scholar] [CrossRef] [PubMed]
- Hofhaus, G.; Shakeley, R.M.; Attardi, G. Use of polarography to detect respiration defects in cell cultures. Methods Enzymol. 1996, 264, 476–483. [Google Scholar] [PubMed]
- Chen, E.I. Mitochondrial dysfunction and cancer metastasis. J. Bioenerg. Biomembr. 2012, 44, 619–622. [Google Scholar] [CrossRef]
- Bayona-Bafaluy, M.P.; Acin-Perez, R.; Mullikin, J.C.; Park, J.S.; Moreno-Loshuertos, R.; Hu, P.; Perez-Martos, A.; Fernandez-Silva, P.; Bai, Y.; Enriquez, J.A. Revisiting the mouse mitochondrial DNA sequence. Nucleic Acids Res. 2003, 31, 5349–5355. [Google Scholar] [CrossRef]
- Chomyn, A. Platelet-mediated transformation of human mitochondrial DNA-less cells. Methods Enzymol. 1996, 264, 334–339. [Google Scholar]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metastasis Detection | ||||
|---|---|---|---|---|
| L929 | L929dt | dtL929 | L929dt | |
| Mouse 1 | --- | 1 (adjacent, 2 gr) | --- | 2 (adjacent, 3 gr; Stomach, 0.4 gr) |
| Mouse 2 | --- | --- | --- | --- |
| Mouse 3 | --- | --- | --- | 2 (adjacent, 1.5 gr; Seminal, 1.2 gr) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marco-Brualla, J.; Al-Wasaby, S.; Soler, R.; Romanos, E.; Conde, B.; Justo-Méndez, R.; Enríquez, J.A.; Fernández-Silva, P.; Martínez-Lostao, L.; Villalba, M.; et al. Mutations in the ND2 Subunit of Mitochondrial Complex I Are Sufficient to Confer Increased Tumorigenic and Metastatic Potential to Cancer Cells. Cancers 2019, 11, 1027. https://doi.org/10.3390/cancers11071027
Marco-Brualla J, Al-Wasaby S, Soler R, Romanos E, Conde B, Justo-Méndez R, Enríquez JA, Fernández-Silva P, Martínez-Lostao L, Villalba M, et al. Mutations in the ND2 Subunit of Mitochondrial Complex I Are Sufficient to Confer Increased Tumorigenic and Metastatic Potential to Cancer Cells. Cancers. 2019; 11(7):1027. https://doi.org/10.3390/cancers11071027
Chicago/Turabian StyleMarco-Brualla, Joaquín, Sameer Al-Wasaby, Ruth Soler, Eduardo Romanos, Blanca Conde, Raquel Justo-Méndez, José A. Enríquez, Patricio Fernández-Silva, Luis Martínez-Lostao, Martín Villalba, and et al. 2019. "Mutations in the ND2 Subunit of Mitochondrial Complex I Are Sufficient to Confer Increased Tumorigenic and Metastatic Potential to Cancer Cells" Cancers 11, no. 7: 1027. https://doi.org/10.3390/cancers11071027
APA StyleMarco-Brualla, J., Al-Wasaby, S., Soler, R., Romanos, E., Conde, B., Justo-Méndez, R., Enríquez, J. A., Fernández-Silva, P., Martínez-Lostao, L., Villalba, M., Moreno-Loshuertos, R., & Anel, A. (2019). Mutations in the ND2 Subunit of Mitochondrial Complex I Are Sufficient to Confer Increased Tumorigenic and Metastatic Potential to Cancer Cells. Cancers, 11(7), 1027. https://doi.org/10.3390/cancers11071027