1. Introduction
Multidrug resistance (MDR) is a severe and pervasive clinical problem which actually represents the leading cause of cancer treatment failure. MDR occurs when cancer cells develop resistance to structurally and mechanistically unrelated classes of anticancer drugs [
1] and involves several complex mechanisms [
2], among which the overexpression of ATP-binding cassette (ABC) transporters is the most prominent. ABC transporters regulate the ATP-dependent efflux of toxic endogenous molecules and chemotherapeutics out of the cell. Among the 49 ABC transporters known so far, P-glycoprotein (P-gp, MDR1, ABCB1), multi-drug resistance protein 1 (MRP1, ABCC1) and breast cancer resistance protein (BCRP, ABCG2) appear to play the most important roles in inducing resistance in several cancers, such as lung, breast, colon, ovarian, central nervous system (CNS) cancers, and melanomas [
3,
4,
5]. Moreover, ABC efflux transporters are also found in excretory epithelial cells of the gastrointestinal tract (GI), kidney, liver, and in endothelial cells of physiological barriers such as the blood-brain barrier (BBB) [
6,
7]. Thus, drug efflux pumps are key elements for drug delivery and efficacy in target tumor cells, as well as for CNS distribution. Therefore, their modulation offers a novel strategy to enhance the penetration and improve the efficacy of drugs into the brain, especially for drug-resistant CNS tumors [
8].
Glioblastoma Multiforme (GBM) is the most aggressive (World Health Organization grade IV) brain tumor in adults and the most frequent one. The majority of GBM patients do not live for longer than 1 year [
9] and the average 5-year survival rate for patients is less than 3% [
10]. After standard treatment, which requires surgical resection followed by concomitant radiotherapy and temozolomide therapy, fatal relapses commonly occur, and recurrent GBM tends to be even more aggressive and resistant to medical treatment than its primary counterpart [
11].
In the past two decades, molecular characterization of genomic alterations in GBM has offered an array of altered pathways that can be specifically targeted. Given their pivotal role in tumorigenesis, tyrosine kinases (TKs) have been indicated as an interesting target to treat several cancers [
12]. Abnormal or deregulated activity of at least one TK is responsible for oncogenic activity across a broad range of tumors, and in particular of 67.3% of GBM [
13]. Coordinately, small-molecule compounds that inhibit the kinase domain of specific TKs have been widely introduced into clinical practice for a number of advanced solid cancers in the last two decades [
14]. However, to date, kinase inhibitors have shown minimal efficacy in GBM clinical trials due to the unique characteristics of GBM: (i.) its infiltrative, invasive, and aggressive nature; (ii.) intrinsic and/or acquired drug resistance; (iii.) unsuccessful drug delivery across BBB [
15].
In the last years, we have focused on the development of novel TK inhibitors based on the pyrazolo[3,4-
d]pyrimidine scaffold. These compounds act as ATP-competitive tyrosine kinase inhibitors [
16] against the Src-Family Kinases (SFKs) [
17]. SFKs are involved in the regulation of cell proliferation, survival, invasion and angiogenesis, thus playing a crucial role in the development and progression of many cancer types, including GBM [
18]. In particular, elevated Src activity/phosphorylation are found in GBM samples and cancer cell lines [
19,
20].
The implication of SFKs in glioma biology has led to the clinical evaluation of the SFK inhibitor- dasatinib, as a novel therapeutic option in particular for recurrent GBM [
21,
22,
23]. However, its usefulness was limited by pharmacokinetics aspects, i.e., short half-life, sub-optimal CNS penetration, and inadequate delivery to the tumor. In vitro and in vivo studies have demonstrated that dasatinib is a substrate for the efflux transporters P-gp and BCRP, which are highly expressed in the BBB [
24,
25] and GBM cells [
26].
It was shown that TKIs interact with efflux transporters either as their substrates or inhibitors. In some cases, TKIs which are substrates for efflux transporters, when applied in high concentrations, can act as inhibitors of transporters [
27,
28,
29,
30]. Some of TKIs acting as ABC transporters inhibitors either entered clinical trials or were clinically approved [
31,
32,
33]. TKIs may also modulate MDR by regulating the expression levels of MDR proteins [
34]. Considering the dual inhibitory potential of TKIs, these anticancer agents are valuable for combining with other chemotherapeutics [
35].
The inhibition of drug efflux transporters often alters the pharmacokinetic properties of the co-administered substrate drug. The increase of oral bioavailability or decrease in biliary excretion and clearance usually result in higher systemic exposure and toxicity. Thus, lapatinib and gefitinib have been reported to strengthen the bioavailability of certain antitumor agents [
36,
37].
The anticancer activity of pyrazolo[3,4-
d]pyrimidines has been proved in vitro against several cancer cell lines, both from solid tumors (osteosarcoma [
38], prostate [
39], neuroblastoma [
40,
41], glioblastoma [
42,
43], rhabdomyosarcoma [
44], mesothelioma [
45], medulloblastoma [
46], medullary thyroid carcinoma [
47]) and hematological tumors (leukemia [
48] and Burkitt lymphomas [
49]), and further confirmed by in vivo studies in mouse models of neuroblastoma [
40], leukemia [
48], and glioblastoma [
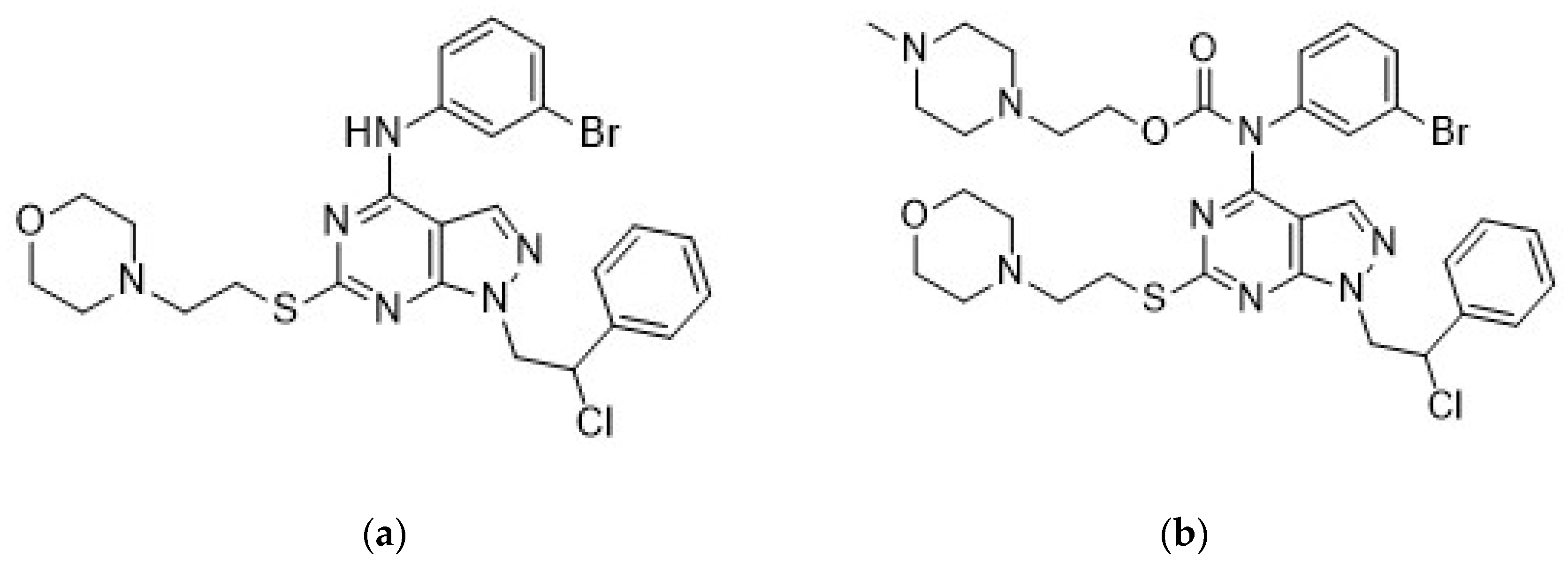
50]. In particular, the combination of compound Si306 (
Figure 1a), a pyrazolo[3,4-
d]pyrimidine derivative with radiotherapy strongly potentiated the suppression of U87 xenograft growth in nude mice with respect to control and single treatments [
50]. Moreover, following intraperitoneal (i.p.) injection, Si306 brain concentration progressively increased during the following 24 h, and the compound appeared to accumulate in the brain. Single oral treatment with Si306 prolonged by 30% the survival of mice orthotopically injected with U87 cells [
50]. Overall, this evidence confirmed the ability of Si306 to efficiently reach the brain. Analogous results were obtained with the prodrug of Si306 (pro-Si306,
Figure 1b), which showed an enhanced solubility and good efficacy in an orthotopic model of GBM with respect to the parental drug [
50].
In this study, we investigated the effect of Si306 and its prodrug on P-gp-mediated efflux, interaction with cytochrome P450 isoform 3A4 (CYP3A4), and suggested their potential use as sensitizers in combination with chemotherapeutics. Additionally, in order to widen the characterization of Si306, which may be useful for the design of preclinical studies, we explored its plasma and brain pharmacokinetics after both oral (p.o.) administration and intravenous (i.v.) injection, including plasma protein binding, and its acute toxicity in mice.
3. Discussion
In the last ten years, pharmaceutical research in oncology has produced many agents targeting important pathways that are considered as being crucial for the development and progression of different tumor types. Although these targeted agents have shown impressive preclinical results, the real clinical benefits have not been as satisfying as expected. It is clear that a great deal of effort was invested into understanding the molecular mechanisms involved in cancer development, while the evaluation of drug interactions with cellular components different from the target molecule was neglected.
New TKIs are being approved from the FDA each year on a regular basis [
53], either given alone or in combination with conventional chemotherapeutics. Although TKIs have revolutionized cancer therapy, increasing resistance to TKIs has been documented [
54]. Enhanced efflux of TKIs due to the over-expression of P-gp and BCRP in cancer cells is usually responsible for the poor chemotherapeutic response [
55,
56,
57]. Accumulating evidence continues to show that numerous TKIs are substrates of P-gp, so once they are entered into the cells, they are pumped out by ABC transporters [
58]. Therefore, the ability of a TKI drug to inhibit P-gp is considered as an important feature for successful anticancer treatment, especially in the case of GBM, whose therapy is additionally compromised by the presence of the BBB.
Recently, we reported an exploratory pharmacokinetic study after i.p. injection of Si306 and pro-Si306 [
50]. The data showed a prolonged overall exposure to the active Si306 after the injection of the prodrug, probably due to the slow hydrolysis into the blood of pro-Si306. The concentration of prodrug quantified into the brain was found higher than the quantity of Si306 measured in the brain of mice treated only with the free drug. Moreover, in comparison to the parental drug, after p.o. administration, pro-Si306 demonstrated a comparable efficacy with a slightly increasing median survival time of mice in an orthotopic GBM model [
50]. Finally, the PK profile of pro-Si306 administrated p.o. was evaluated. Differently from i.p. injection, pro-Si306 was detected neither into the plasma nor in the brain, probably because of a complete hydrolysis occurred into the GI tract [
50].
Considering the results obtained in our previous study, herein we decided to evaluate the effect of Si306 and pro-Si306 on P-gp efflux pump, which accounts for GI absorption and BBB penetration of different substrates, including anticancer drugs and TKIs. The effect of our compounds has been investigated compared with a reference compound, the well-known TKI dasatinib. The cell growth inhibition reported for dasatinib ranges between 7 and 50 μM in GBM cell lines [
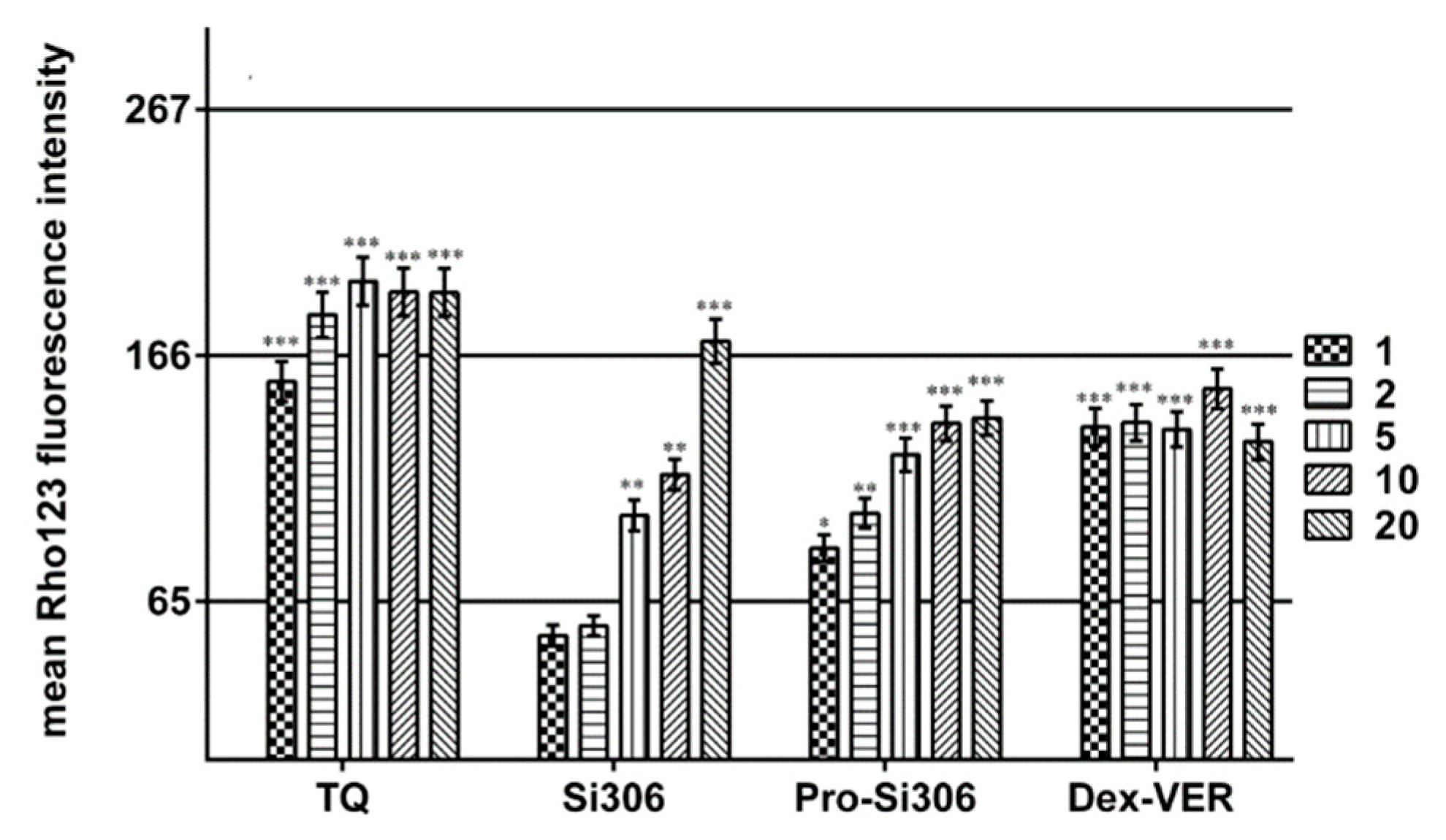
59]. These concentrations are above those clinically relevant. We showed that the novel c-Src inhibitor Si306 and its prodrug possess higher cell growth inhibitory potential with almost double efficacy in comparison with dasatinib. In addition, the effect of Si306 and pro-Si306 was not affected by the presence of MDR phenotype. Both compounds did not change the P-gp expression at mRNA and protein level, differently from dasatinib. Indeed, after 72 h treatment, dasatinib increased P-gp expression. This implies that the application of our novel c-Src inhibitors could not further increase P-gp expression in MDR GBM cells. Moreover, Si306 and pro-Si306 induced significant suppression of P-gp activity and their effect was dose-dependent. They directly interacted with P-gp, showing significant P-gp inhibition after 30 min of treatment.
Other authors reported transient decrease of P-gp expression achieved by 1 μM dasatinib after 24 h in MCF-7 (MCF-7/Adr) cell line, reversing its MDR to doxorubicin after 72 h. This effect on P-gp expression enabled reversal of doxorubicin resistance by 3.35-fold when 0.5 μM dasatinib was applied for 72 h. Dasatinib inhibited the cell growth of (MCF-7/Adr) with low efficacy, reaching IC
50 at 40 μM [
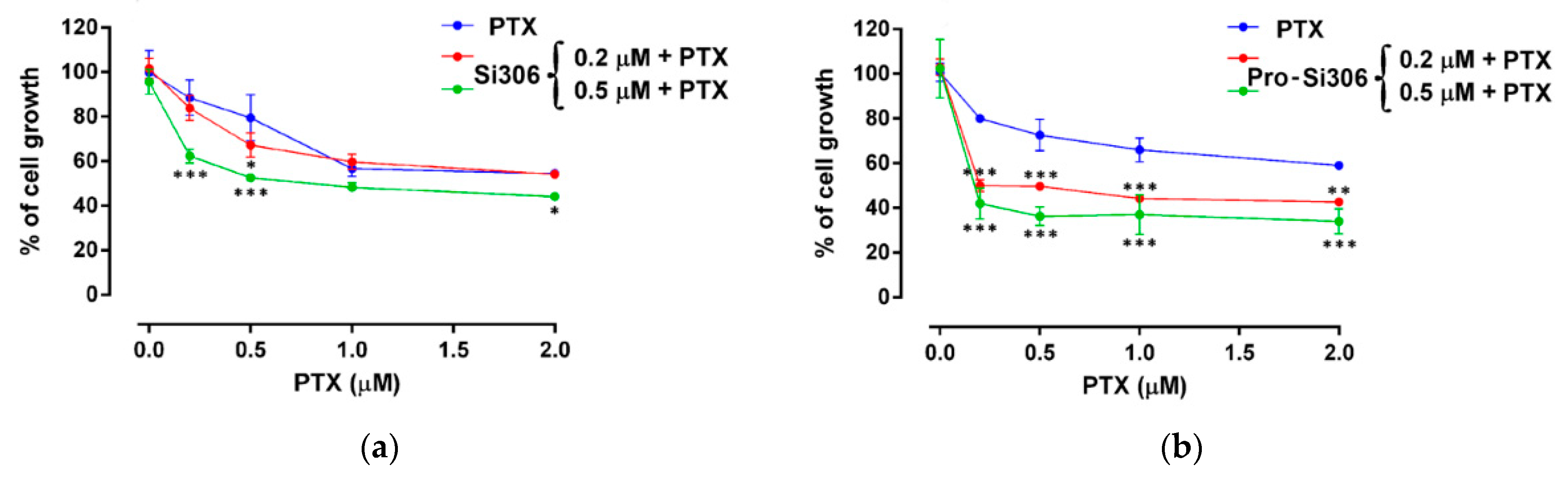
60]. In our experimental setting of 72 h simultaneous combined treatment, 0.5 μM of Si306 and pro-Si306 reverted PTX resistance in MDR GBM cells by 3.34 and 7.98-fold, respectively. The sensitization of U87-TxR cells was achieved by concentrations that were significantly lower than those necessary for the optimal cell growth inhibition or P-gp inhibition induced by Si306 and pro-Si306. Therefore, even sub-cytotoxic concentrations of new c-Src inhibitors were able to increase PTX intracellular accumulation by inhibiting P-gp. It is possible that the inhibitory effect of drug and prodrug on P-gp activity increases over time enabling the enhancement of PTX efficacy. Stronger effects regarding P-gp inhibition and reversal of PTX resistance were observed after pro-Si306 application, indicating a more potent activity of prodrug against P-gp.
Human cytochrome P450 is a family of enzymes responsible for the essential biotransformation of xenobiotics [
61]. Both P-gp and cytochrome P450 are involved in the first line of xenobiotic defense system, sometimes referred to as a cellular ADME system [
62]. In addition, drug metabolism via the cytochrome P450 system has emerged as an important determinant in the occurrence of several drug interactions that can result in drug toxicity, reduced pharmacological effect, and adverse drug reactions. Predominant isoforms of cytochrome P450 involved in drug metabolism are CYP2C9, CYP2D6 and CYP3A4 which metabolize up to 80–90% of all xenobiotics [
63,
64,
65,
66]. In particular, it has been estimated that CYP3A4 metabolizes up to 50% of all reported drugs and a few of them are also metabolized by other isoforms. In the liver and small intestine, p-glycoprotein and CYP3A4 are both expressed and create a functional drug efflux-metabolism “alliance”, which establishes an absorption barrier against xenobiotics [
67]. Moreover, it was shown that the second generation of P-gp inhibitors are metabolized by CYP3A4, thus competing with anticancer drugs for CYP-mediated clearing, finally leading to the adverse toxicity [
62,
68]. Coordinately, it is important to elucidate whether P-gp inhibitors (Si306 and its prodrug) may interact with cytochrome P450. Testosterone hydroxylation is in general used today as one of the characteristic assays for testing the inhibition of P450 isoform 3A4. The major product of testosterone metabolism is 6β-hydroxytestosterone but several other hydroxylations occur, including those at the 2β and 15β positions [
69,
70,
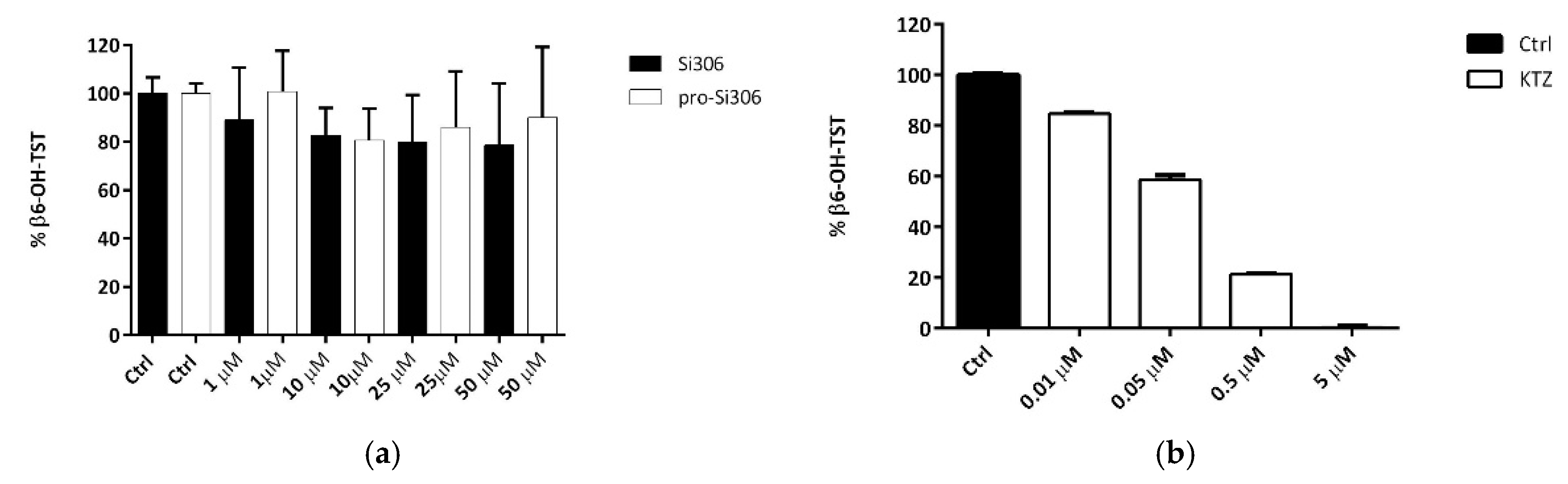
71]. The evidence that the testosterone 6β-hydroxylation is not also inhibited by the two compounds at the highest concentration used clearly indicates that both Si306 and its prodrug did not interact with the CYP3A4.
It is well-known that drug permeability through biological membranes depends on factors such as plasma protein-binding, hydrophobicity and molecular size of the drug. Among these factors, protein-binding of the drug, which is closely-related to hydrophobicity, plays an important role in drug pharmacokinetics and pharmacodynamics. HSA is the major protein component of blood plasma (about 60%) and binds a number of relatively insoluble endogenous compounds. AGP primarily binds basic drugs (e.g., amines) and it is also reported to bind hydrophobic compounds [
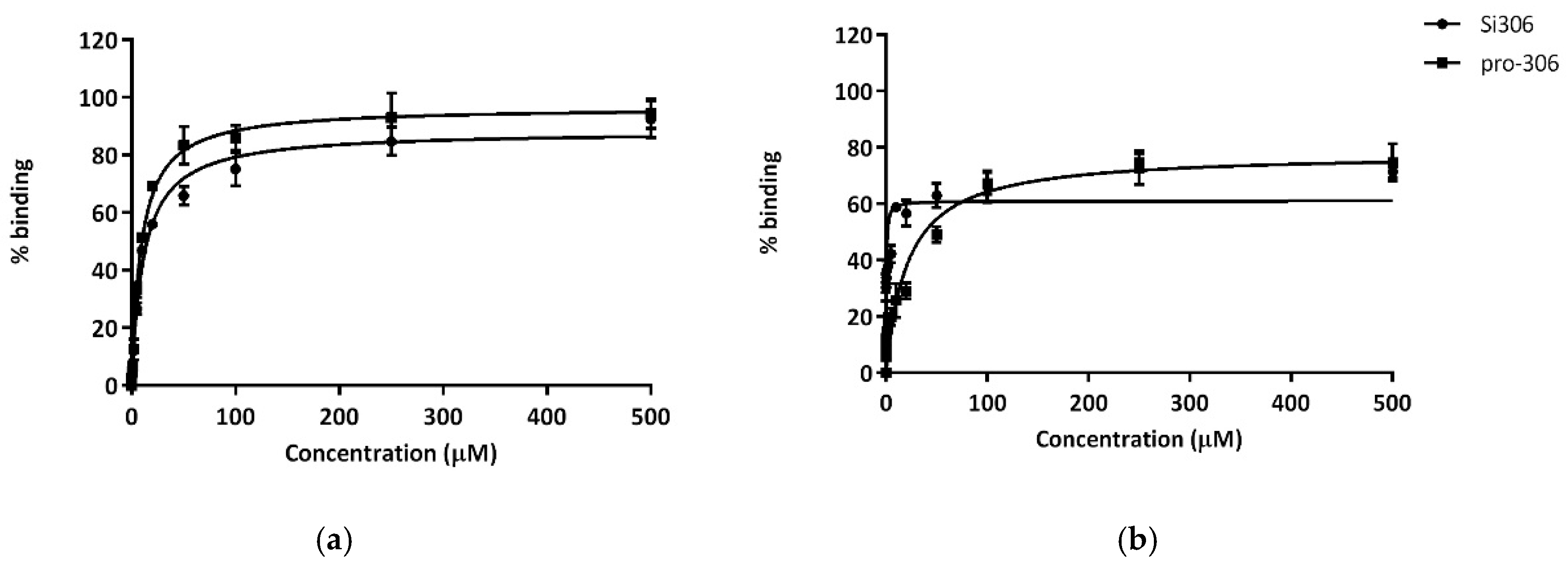
72]. For these reasons, the binding to albumin and alpha 1-acid glycoprotein of Si306 and pro-Si306 was evaluated. Both pro-Si306 and its parental drug Si306 showed high affinity for HSA and AGP plasmatic proteins (drugs that are highly bound are characterized by a K
D value less than 100 µM). For AGP, K
D values for Si306 and pro-Si306 were comparable, both being weak basic compounds. On the other hand, the very poorly water soluble [
50] Si306 displayed a very high affinity for albumin, 60-fold greater than pro-Si306. This feature may suggest a plasmatic large fraction of bound drug, which could be the reason for its slow plasma clearance kinetics.
From our previous study, pro-Si306 emerged as an effective potential drug against GBM, but unfortunately is not suitable for an oral administration due to the complete hydrolysis into the GI apparatus (mainly stomach) [
50]. On the other hand, Si306 can be administrated p.o. and this is one of the most important advantages in the use of TKIs, offering logistic flexibility and a good compliance in patients [
73].
To prove that Si306 could be considered a good drug candidate for the treatment of GBM that is worthy of further development, a PK study after p.o. administration and i.v. injection has been performed.
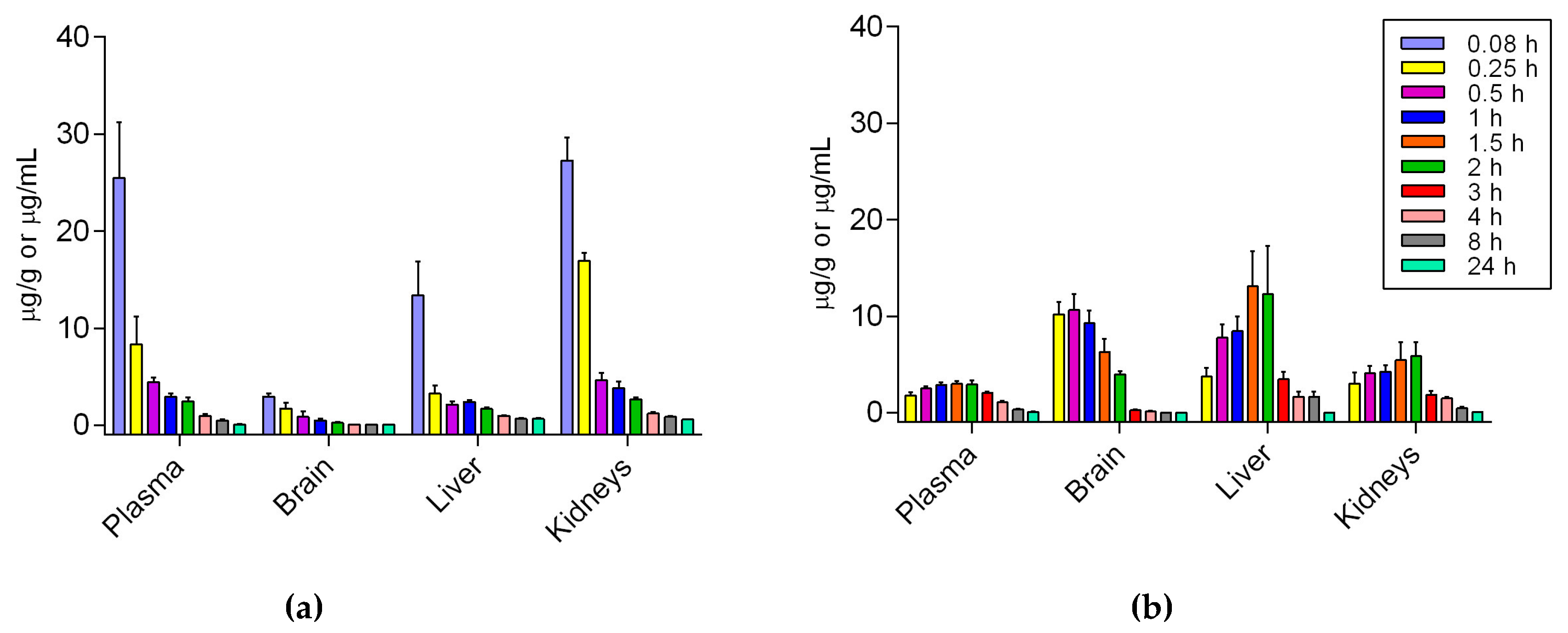
The peak plasma concentration of Si306 was observed at 5 min for i.v. and 1.5 h for p.o. The plasma half-life (t
1/2) resulted comparable between the two routes of administration and the oral bioavailability of Si306 has been estimated around 33.3% in mice, being higher than that reported for dasatinib (14% in mice) [
74]. After both i.v. and p.o. administration Si306 was able to reach the brain and pass the BBB. In particular, after i.v. injection the maximum concentration into the brain (2.94 μg/g,
Table 6) was observed after 5 min, while after oral administration a large amount of our drug has been detected (10.65 μg/g,
Table 6) into the brain after 30 min. In both cases, the concentration of the compound slowly decreased during the next 24 h. Overall, our new results demonstrated a good absorption of Si306 by GI and, most interestingly, an optimal brain penetration, with a 12-fold higher AUC brain/plasma when Si306 was orally administered with respect to the i.v. injection. The higher and very fast brain accumulation of oral Si306 could be related to the greater inhibition of p-gp, and possibly saturation of p-gp sites at the BBB, which could be simply a consequence of the orally administrated dose, which was twice that of the i.v. [
75].
In this context, being the Si306 considered able to block intestinal p-gp and since direct inhibition of p-gp in the small intestine was found to improve the oral bioavailability of p-gp substrates (dramatic increase in intestinal absorption of paclitaxel was observed when p-gp inhibitors were co-administrated [
75]), the idea of combination with other drugs substrates of p-gp could be proposed.
Finally, in order to verify the safety of Si306 in vivo, an intravenously single-dose toxicity study was performed. The compound was very well tolerated by mice, even at a high dose (100 mg/Kg). Moreover, no mortality or changes in animal behavior were observed following the administration. At fourth day from the injection of Si306, mice were sacrificed, and their organs were analyzed. Any microscopic difference in the architecture of the main metabolic organs was observed, indicating very good tolerability and a lack of acute toxicity.
4. Materials and Methods
4.1. Drugs
A pyrazolo[3,4-
d]pyrimidine derivative Si306 as well as its prodrug, pro-Si306, were previously synthesized and characterized [
50]. Dasatinib as well as PTX and Dex-VER were purchased from Sigma-Aldrich (Milan, Italy and Taufkirchen, Germany suppliers). Dr. Sven Rottenberg from The Netherlands Cancer Institute in Amsterdam kindly provided TQ for our research purposes. Aliquots of 20 mM of both Si306 and pro-Si306 were dissolved in dimethyl sulfoxide (DMSO) and kept at room temperature. TQ was also dissolved in DMSO (10 µM stocks) but kept at −20 °C. PTX aliquots were stored at −20 °C in absolute ethanol (1 mM), while sterile water dilution of Dex-VER was kept at room temperature. Immediately before treatments, the aforementioned drugs were diluted in sterile water.
4.2. Cells and Culture Conditions
Human GBM cell lines U87 and LN-229 were purchased from the American Type Culture Collection, Rockville, MD, USA. U87-TxR cells were selected from U87 cells by continuous exposure to increasing concentrations of PTX [
76]. LN-299, U87 and U87-TxR cells were grown in Minimum Essential Medium with 10% fetal bovine serum, 2 mM L-glutamine and 5000 U/mL penicillin, 5 mg/mL streptomycin solution. Cell lines were maintained at 37 °C in a humidified 5% CO
2 atmosphere. They were sub-cultured at 72 h intervals using 0.25% trypsin/EDTA and 16,000 cells/cm
2 were seeded into a fresh medium.
4.3. MTT Assay
MTT assay was used to assess the viability of U87 and U87-TxR cells (AppliChem GmbH, Taufkirchen, Germany). After reaching the confluence in 25 cm2 tissue flasks, U87 and U87-TxR cells were trypsinized. Cells were seeded at the optimal density determined in initial experiments: 4000 cell/well into flat-bottomed 96-well tissue culture plates. Following the overnight incubation, cells were treated with Si306, pro-Si306 and dasatinib (1–25 µM) for 72 h. In addition, the combined effects of Si306 and pro-Si306 with PTX were evaluated in MDR GBM cells. To that end, two concentrations of Si306 and pro-Si306 (0.2 μM and 0.5 μM) were combined with increasing concentrations of PTX (0.1–2 μM) in simultaneous treatment. The sensitization of U87-TxR cells to PTX was studied after 72 h. Briefly, MTT was added to each well in a final concentration of 0.2 mg/mL for 4 h to induce the formation of the formazan in cells with intact/viable mitochondria. Then, produced formazan was dissolved in DMSO, and the absorbance was measured at 540 nm on a microplate reader (LKB 5060-006 Micro Plate Reader, Vienna, Austria). IC50 values were calculated by nonlinear regression analysis using GraphPad Prism 6.0 for Windows (GraphPad Software, La Jolla, CA, USA).
4.4. RNA Extraction and Reverse Transcription (RT) Reaction
U87-TxR cells, untreated and treated with 5 μM of each compound Si306, pro-Si306 and dasatinib for 72 h were used to isolate total RNA and perform the RT reaction. Isolation was done by using Trizol® reagent (Invitrogen Life Technologies, Waltham, MA, USA). Quantification of RNA and its quality determination were performed by spectrophotometry and by agarose gel electrophoresis, respectively. For the RT reactions, 2 µg of total RNA were used, while a high-capacity cDNA reverse transcription kit (Applied Biosystems, Waltham, MA, USA) was employed.
4.5. Quantitative Real-Time PCR
To assess mRNA
mdr1 expression level in U87-TxR cells upon 5 μM Si306, pro-Si306 and dasatinib treatments, the quantitative real time PCR (qRT-PCR) was used. The PCR reactions according to the manufacturer’s recommendations were performed in the presence of 100 ng cDNA combined with primers specific for
MDR1 and
ACTB, as an internal control for normalization [
77,
78], and a Maxima SYBR Green/ROX qPCR Master Mix (Thermo Scientific, Waltham, MA, USA) on an ABI PRISM 7000 Sequence Detection System (Applied Biosystems, Waltham, MA, USA). Analyses of triplicate samples in terms of relative gene expression were completed using the 2
−ΔΔCt method [
79].
4.6. Flow-Cytometric Analysis of P-Glycoprotein Expression
To assess the P-glycoprotein expression level in MDR GBM cells, flow-cytometry was employed. U87-TxR cells seeded in adherent 6-well plates were treated with 5 µM Si306, pro-Si306 or dasatinib for 72 h. Then, cells were collected by trypsinization, washed in PBS, and then directly immuno-stained by FITC-conjugated anti-P-gp antibody (BD Biosciences, Winnersh, Berkshire, UK). To discriminate the level of background fluorescence, an isotype control IgG2bκ (Abcam, Cambridge, United Kingdom) was used. The fluorescence of FITC-conjugated anti-P-gp was analyzed on assessed on the fluorescence channel 1 (FL1) of the CyFlow Space Partec flow-cytometer (Sysmex Partec GmbH, Görlitz, Germany). After gating to exclude the cell debris and dead cells, a minimum of 10,000 cells per sample was assayed. Summit Dako Software was used for the analyses of results obtained by flow-cytometry.
4.7. Rho 123 Accumulation Assay
Rho 123 accumulation was analyzed by flow-cytometry utilizing the ability of Rho 123, which is a substrate for P-glycoprotein, to emit fluorescence. The intensity of the fluorescence is proportional to Rho 123 accumulation in the cell. Studies were carried out with Si306, pro-Si306, dasatinib, Dex-VER and TQ in U87-TxR cells. U87 cells were used as a positive control for Rho 123 accumulation. In simultaneous treatment that lasted 30 min, 5 μM Rho 123 was applied along with 5 μM of Si306, pro-Si306 and dasatinib. To obtain IC50 values for P-gp inhibition, 5 μM Rho 123 was simultaneously applied with the increasing concentrations of Si306, pro-Si306, Dex-VER (1, 2, 5, 10 and 20 μM) and TQ (1, 2, 5, 10 and 20 nM). Samples were incubated 30 min at 37 °C in 5% CO2. At the end of the accumulation period, the cells were pelleted by centrifugation, washed with PBS and placed in cold PBS. The samples were kept on ice in a dark setting until the analysis on CyFlow Space Partec flow-cytometer (Sysmex Partec GmbH, Germany). The orange fluorescence of Rho 123 was assessed on fluorescence channel 2 (FLH-2) at 585 nM. A minimum of 10,000 events was assayed for each sample and the obtained results were analyzed using Summit Dako Software (ver. 4.3, Fort Collins, CO, USA).
4.8. Data Collection and Statistical Methods
Statistical analyses for P-gp inhibition and combination treatments were performed by two-way ANOVA test (GraphPad Prism 6 software) using Dunnett’s multiple comparisons test. Statistical significance was accepted if p < 0.05.
4.9. Binding Fluorimetric Assay
In order to determine the binding affinity for Si306 and pro-Si306 to HSA (human serum albumin) and AGP (alpha-1-acid glycoprotein), the protein fluorescence has been monitored by spectroscopy. The quantitative analysis was performed using black 96 multiwell plates: in each well, a fixed concentration of HSA or AGP (10 µM in phosphate buffer 1 mM) was added with different amounts of the tested compound (0.1 µM to 500 µM). Plates were gently shacked and after 30 min at room temperature for equilibration, spectra were recorded from 300 to 400 nm, after excitation at 295 nm, by using a Perkin Elmer EnVision Multilabel Reader 2014 spectrofluorimeter. Finally, spectra were acquired with EnVision Manager ver.1.13 software (Walthman, MA, USA). The obtained non fluorescence quenching percentages were plotted against drug concentrations and the relative KD values were calculated using GraphPad software (version 6.0).
4.10. CYP450 Inhibition Study
The inhibition of cytochrome P450 3A4 isoform by Si306 and pro-Si306 was measured by determining its activity on the substrate testosterone.
The activity of CYP3A4 was evaluated by measuring the 6β-hydroxylated testosterone formation (CYP3A4-specific reaction). The amount of 6β-hydroxylated metabolite was assessed in the absence and presence of Si306, pro-Si306 and ketoconazole (testosterone concentration of 100 μM in phosphate buffer 0.025 M, pH 7.4). The compounds were solubilized in DMSO and added in a test tube to give final concentrations 1, 10, 25, 50 and 0.01, 0.05, 0.5, 5 μM, (DMSO did not exceed 2%). Ketoconazole was used as standard selective inhibitor of CYP3A4 isoenzyme [
55]. Incubations were carried out in a mix containing human liver microsomes (1 mg/mL of protein) in phosphate buffer 0.025 M, pH 7.4, NADPH (200 μM) previously solubilized in a MgCl
2 48 mM solution. The final incubation volume was 0.25 mL. After a 40 min-incubation period at 37 °C, the reaction was stopped by adding 1 mL of acetonitrile in the presence of corticosterone 2 μM as internal standard. The samples were centrifuged for 20 min at 5000 rpm. Concentrations of testosterone and its metabolite 6β-hydroxytestosterone were assessed by the HPLC method with UV detection. Calibration curve for quantitative analysis of 6β-hydroxytestosterone in the samples was obtained using commercial 6β-hydroxytestosterone purchased from Sigma Aldrich. For the statistical analysis, GraphPad InStat 3.0 (GraphPad Software, San Diego, CA, USA) was used.
4.11. Animals
4.11.1. For PK and BD Studies
Naive male BALB/C mice (weight 20 ± 2 g) were ordered from Charles River (Milan, Italy). All animals were pathogen free and approximately 4–6 weeks old when they arrived. The adaption period to the environment was not less than seven days. All the procedures used on animals in this study were approved by Institutional Animal Use and Care Committee at Università Cattolica del S. Cuore, Rome and Università degli Studi di Siena and authorized by the Italian Ministry of Health, according to Legislative Decree 116/92, which implemented the European Directive 86/609/EEC on laboratory animal protection in Italy. Methods for all the conducted experiments were performed in accordance with regulations, standards and guidelines of the Animal Use and Care Committee of Università Cattolica del S. Cuore, Rome and Università degli Studi di Siena.
4.11.2. For Acute Toxicity Studies
Male BALB/c mice (20 ± 2) g, supplied by Harlan Italy S.r.l., San Pietro al Natisone, Udine, Italy, were acclimated at 25 °C and 55% of humidity under natural light/dark conditions for 1 week before the proposed experiments. Animal welfare was routinely checked by veterinarians of the Service for Animal Welfare and all efforts were made to minimize suffering.
4.12. In Vivo Administration of Si306
Si306 was dissolved in a mixture of Tween80 (10% v/v), benzyl alcohol (1% v/v) and a 10 mM solution of citric acid. The compound was administered intravenously as a single dose of 25 mg/kg in 200 μL of volume. At several time points (0.08, 0.25, 0.5, 1, 2, 4, 8, 24 h), after drug administration, mice were treated i.p. with heparin (5000 U/kg) and sacrificed under CO2. Five animals were used for each time point. Blood, brain, liver and kidneys were collected for the following quantitative analysis. Approximately 500–600 μL of blood was collected from each animal and transferred to a tube containing 10 μL of heparin and mixed briefly.
For the oral PK studies, mice received a single dose of 50 mg/kg of Si306 (dissolved in the same vehicle used for i.v. studies) in 400 μL of final volume by gavage. At nine time-points (0.25, 0.5, 1, 1.5, 2, 3, 4, 8, 24 h) after administration, mice were sacrificed under CO
2. Blood and organs were collected and analyzed by LC-MS. The pharmacokinetic parameters, including area under concentration-time curve (AUC), maximum plasma concentration (C
max), half-time (t
1/2), apparent volume of distribution (V), plasma clearance (CL) and mean residence time (MRT), were calculated by non-compartmental analysis using PKSolver software (ver. 1.0, Nanjing, China) [
80].
4.12.1. Sample Preparation
Blood samples were centrifuged at 5000 rpm for 20 min to separate the plasma fraction, which was subsequently collected in a test tube. Acetonitrile (1 mL, with Si34 as internal standard at the concentration of 5 μg/mL) was added to each sample to denature proteins. Samples were centrifuged at 5000 rpm for 20 min, and the supernatant was recovered, dried under vacuum, solubilized again in methanol (100 μL), and analyzed. Brain and the other organs were homogenized using a T10 basic ULTRA-TURRAX
® homogenizer (Bioclass, Pistoia, Italy); in order to extract the compound from the tissue, acetonitrile internal standard solution was added, and the homogenate was centrifuged at 5000 rpm for 20 min. The supernatant was recovered, filtered and analyzed by LC-MS. The quantification of each compound was performed by reference to the appropriate calibration curve. Representative LC-MS chromatograms for Si306 and for Si34 are reported in
Figure S2 and Figure S3 respectively. Mass spectra are also shown in
Figure S4.
4.12.2. Recovery and Matrix Effect
The matrix effect was evaluated in samples by comparing the peak areas of the Si306 reference solutions reconstituted in blank organ extracts (for plasma, brain, liver and kidneys) against those of the analyte dissolved in mobile phase. The recovery of Si306 was evaluated by comparing the peak area of Si306 in spiked plasma and other organs samples with the peak areas of analyte spiked after extraction into plasma or organs’ extracts [
81]. The matrix effect of the IS was evaluated in a similar manner.
4.12.3. UV/HPLC-MS Instrumentation and Analysis Condition
LC analyses were performed by using Agilent 1100 LC/MSD VL system (G1946C) (Agilent Technologies, Palo Alto, CA, USA) constituted by a vacuum solvent degassing unit, a binary high-pressure gradient pump, a 1100 series UV detector, and a 1100 MSD model VL benchtop mass spectrometer. MSD single-quadrupole instrument was equipped with the orthogonal spray API-ES (Agilent Technologies, Palo Alto, CA, USA). The pressure of the nebulizing gas and the flow of the drying gas (nitrogen used for both) were set at 40 psi, 9 L/min, respectively. The capillary voltage, the fragmentor voltage, and the vaporization temperature were 3000 V, 70 V, and 350 °C, respectively. MSD was used in the positive ion mode. Spectra were acquired over the scan range m/z 50–1500 using a step size of 0.1. Chromatographic analysis for CYP3A4 interaction studies was performed using a Phenomenex Luna C18 100A column (250 × 4.6 mm, 5 μm particle size) at room temperature, at flow rate of 0.6 mL/min, and injection volume was 20 μL, operating with a gradient elution of acetonitrile (ACN) and water (H
2O): t = 0 min ACN 0%, t = 2 min ACN 0%, t = 40 min ACN 70%, t = 45 min ACN 70%, t = 50 min ACN 0%. UV detection was monitored at 254 nm. Chromatographic analysis for Si306 and its internal standard Si34 was performed using a Kinetex EVO C18 100A column (150 × 4.6 mm, 5 μm particle size) at room temperature, at flow rate of 0.6 mL/min, and injection volume was 20 μL, operating with a gradient elution of acetonitrile (ACN) and water (H
2O): t = 0 min ACN 0%, t = 3 min ACN 0%, t = 12 min ACN 98%, t = 18 min ACN 98%. Representative LC-MS chromatograms and mass spectra have been reported in
Supporting Information.
4.13. Evaluation of Acute Safety Profile in Mice
The study design includes three groups of five mice each. All mice were injected with compound through the tail vein in a single systemic dose (0.05 mL per dose). The test compound was solubilized in PBS/25% DMSO. The first group was treated with dose levels of 50 mg/kg; the second received a high-dose level of 100 mg/kg, whereas the third was inoculated with the only vehicle as the control. All mice were monitored for survival and the presence of any drug-related adverse effect (local signs of inflammation, weight loss, diarrhea, and behavioral alterations) throughout the experimental period. Mice were sacrificed 4 days after treatment, and liver, kidney, and brain tissue specimens were aseptically removed, fixed in 4% formaldehyde solution, and processed for paraffin embedding. Histopathological changes in tissue specimens were evaluated in 10 randomly selected tissue sections from each organ for all the samples. Sections cut at 2 μm were stained with hematoxylin and eosin (H&E) and analyzed by a pathologist with an optical microscope (Leica, Wetzlar, Germany).

 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}