Tumor Mesenchymal Stromal Cells Regulate Cell Migration of Atypical Teratoid Rhabdoid Tumor through Exosome-Mediated miR155/SMARCA4 Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Culture and Maintenance of ATRT Cells and tMSCs
2.2. Mir-155 Sponge Construction
2.3. Exosome Purification from Conditioned Medium and Identification by CD63 Surface Marker
2.4. Exosome Uptake Analysis with PKH67 Labeling
2.5. Heparin Inhibition of Exosome Uptake
2.6. GW4869 Treatment
2.7. Wound Healing Assay
2.8. Transmission Electron Microscopy
2.9. Extraction of RNA in Exosomes
2.10. Microarray Analysis for MicroRNA Expression in Exosome
2.11. TaqMan® MicroRNA Assays
2.12. Western Blotting
2.13. Xenotransplantation of ATRT Cells and 3T-MRI
2.14. Statistical Analysis
3. Results
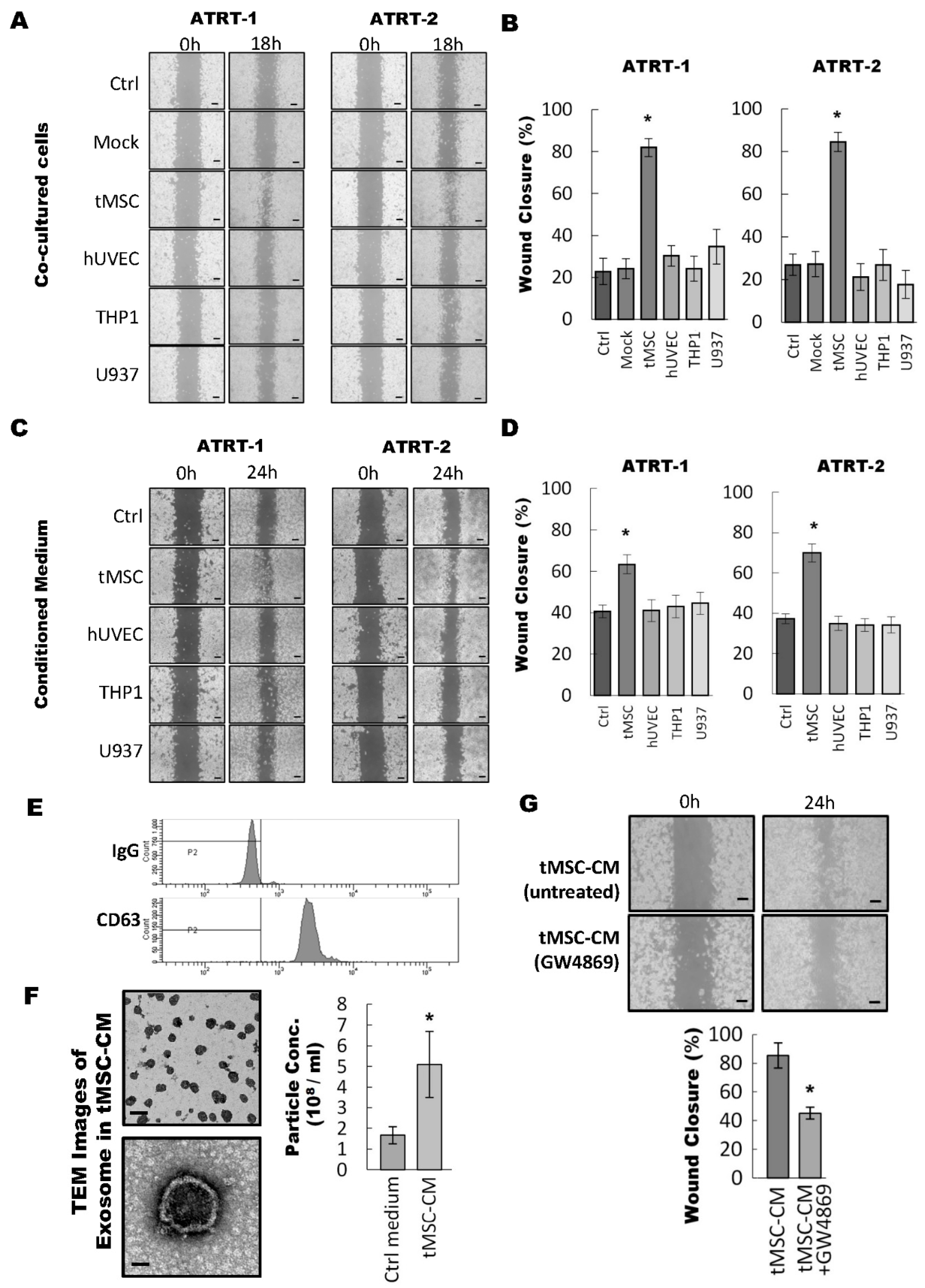
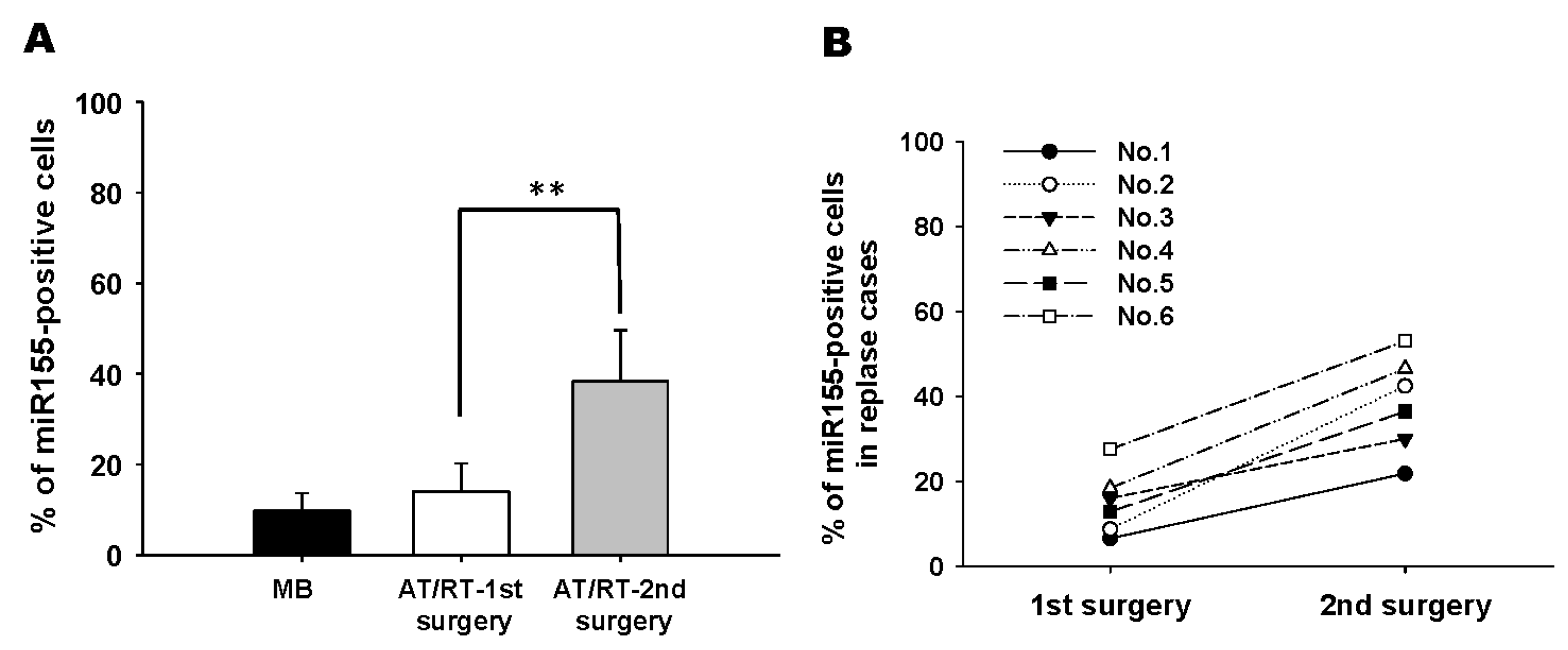
3.1. Tumor-Associated Mesenchymal Stromal Cells Enhances Migratory Ability of ATRT Cell Lines Through an Exosome-Dependent Mechanism
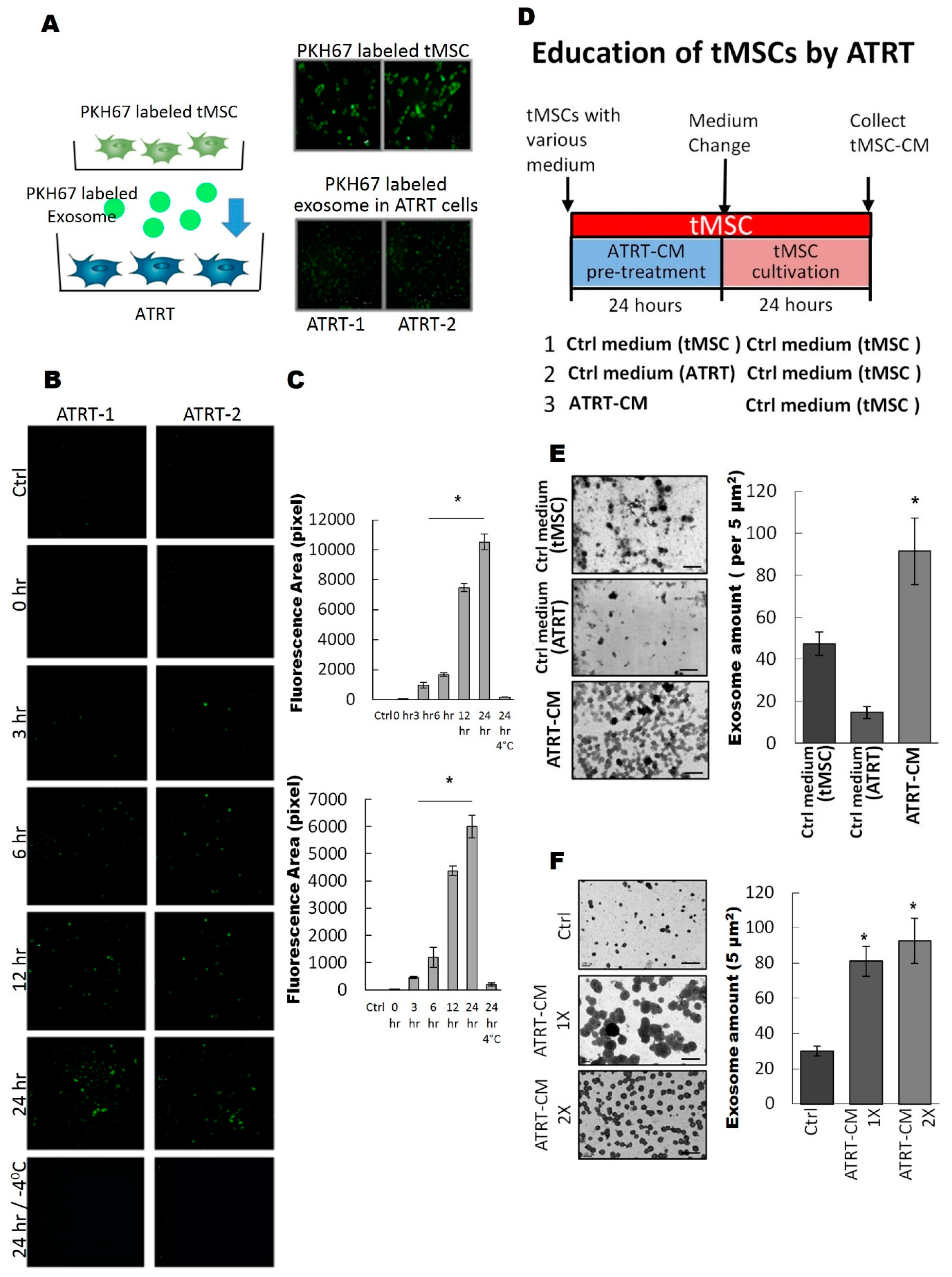
3.2. ATRT Cells Promote Exosome Released from tMSCs via a Paracrine Mechanism
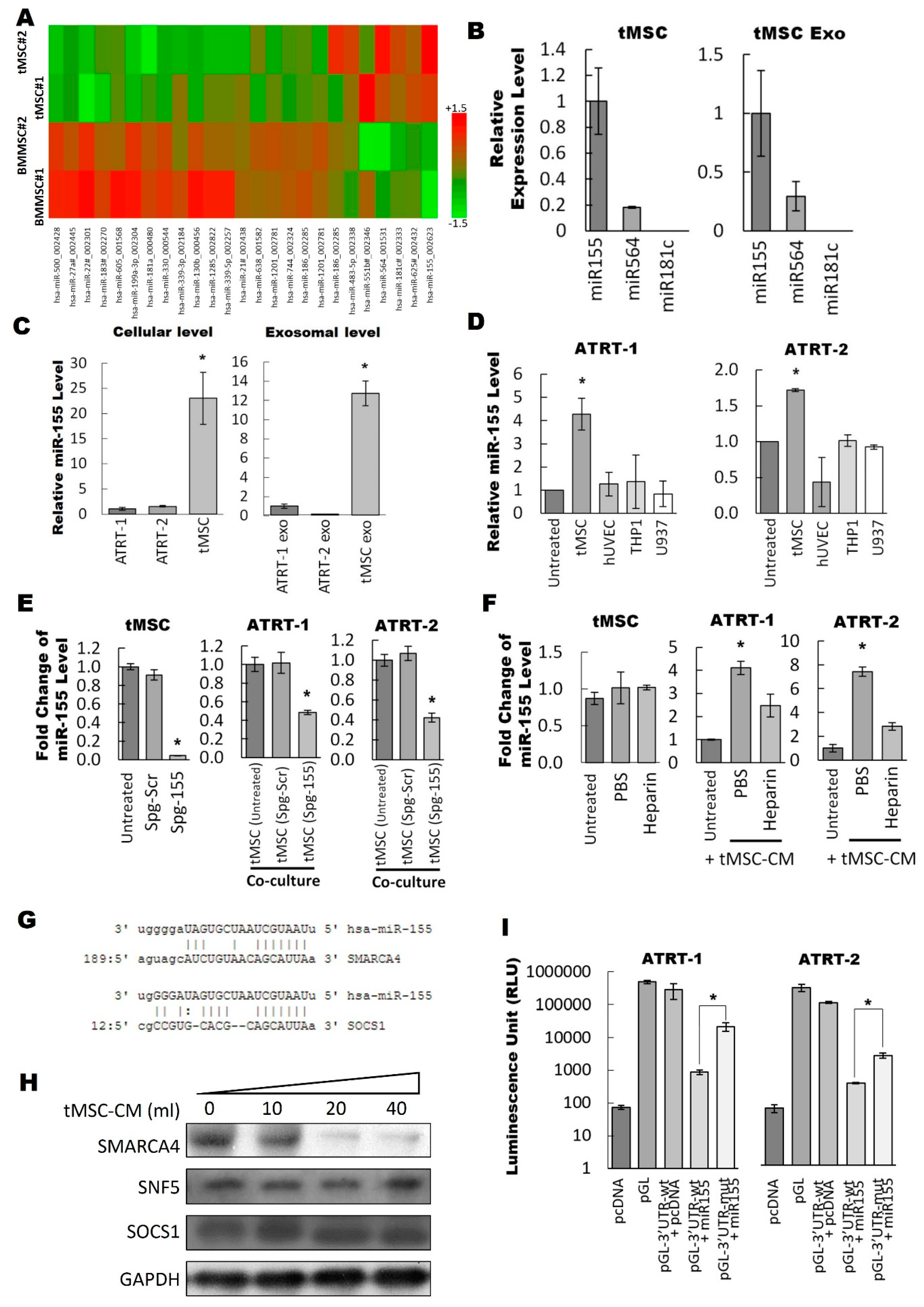
3.3. Exosomal miR-155 Directly Targets SMARCA4 in ATRT Cells
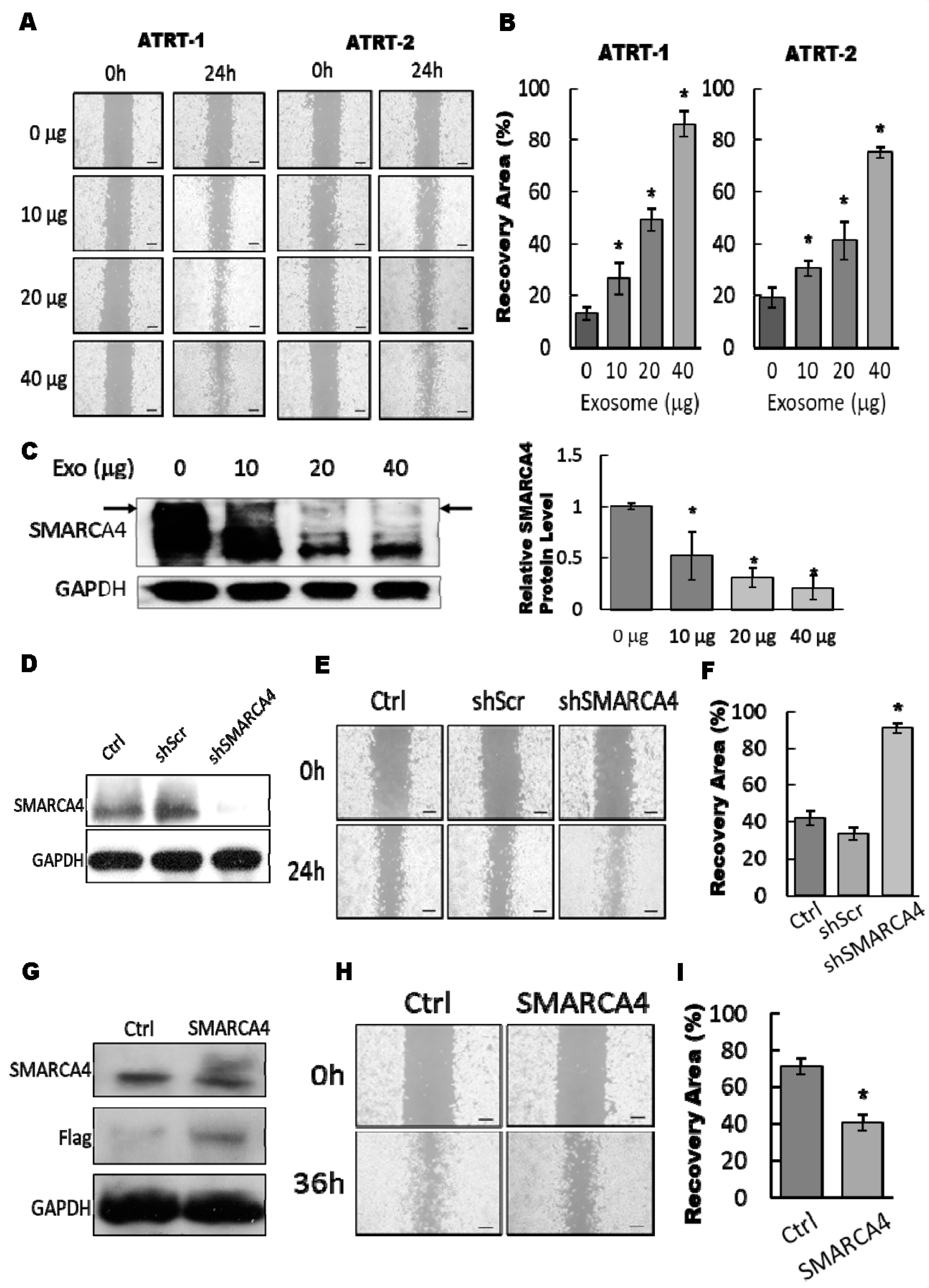
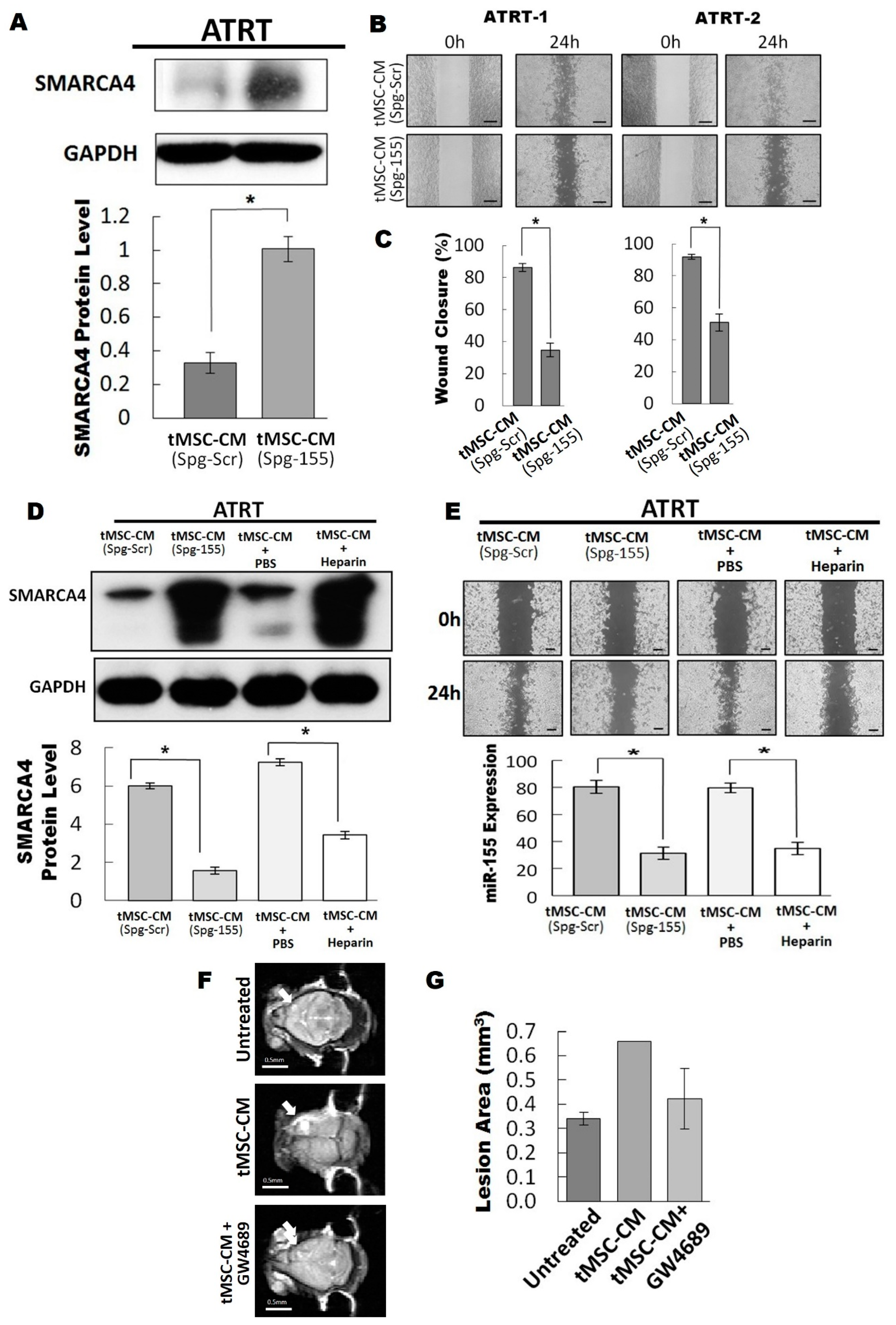
3.4. The Exosomal-miR155/SMARCA4 Pathway Regulates ATRT Migration Ability
3.5. Blocking of the Exosomal miR155/SMARCA4 Signaling Suppressed ATRT Migration and Tumor Growth
4. Discussion
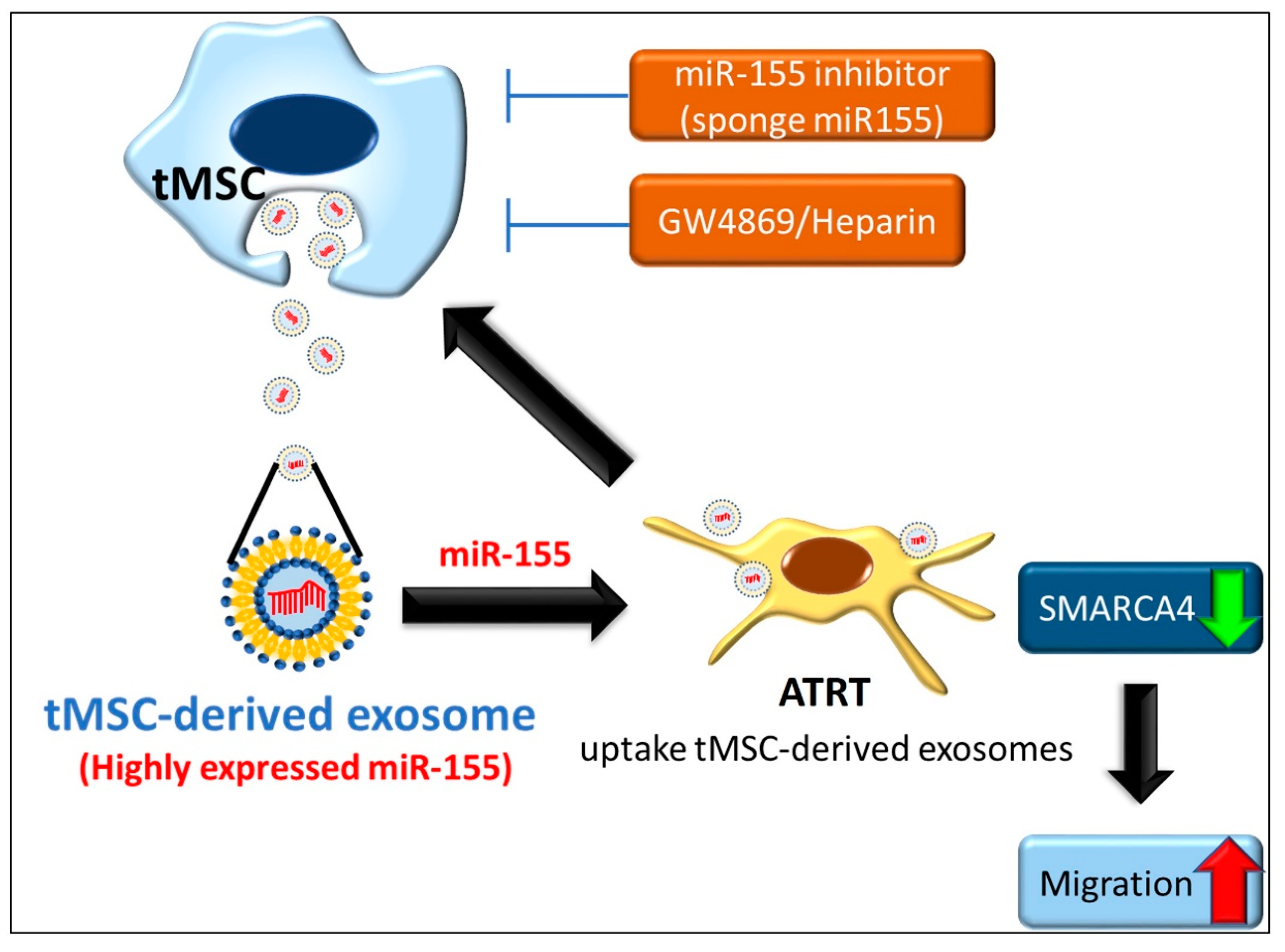
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kaderali, Z.; Lamberti-Pasculli, M.; Rutka, J.T. The changing epidemiology of paediatric brain tumours: A review from the hospital for sick children. Childs Nerv. Syst. 2009, 25, 787–793. [Google Scholar] [CrossRef]
- Chan, V.; Marro, A.; Findlay, J.M.; Schmitt, L.M.; Das, S. A systematic review of atypical teratoid rhabdoid tumor in adults. Front. Oncol. 2018, 8, 567. [Google Scholar] [CrossRef]
- Barresi, V.; Lionti, S.; Raso, A.; Esposito, F.; Cannavo, S.; Angileri, F.F. Pituitary atypical teratoid rhabdoid tumor in a patient with prolactinoma: A unique description. Neuropathology 2018, 38, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Bikowska, B.; Grajkowska, W.; Jozwiak, J. Atypical teratoid/rhabdoid tumor: Short clinical description and insight into possible mechanism of the disease. Eur. J. Neurol. 2011, 18, 813–818. [Google Scholar] [CrossRef]
- Athale, U.H.; Duckworth, J.; Odame, I.; Barr, R. Childhood atypical teratoid rhabdoid tumor of the central nervous system: A meta-analysis of observational studies. J. Pediatr. Hematol. Oncol. 2009, 31, 651–663. [Google Scholar] [CrossRef]
- Shetzer, Y.; Solomon, H.; Koifman, G.; Molchadsky, A.; Horesh, S.; Rotter, V. The paradigm of mutant p53-expressing cancer stem cells and drug resistance. Carcinogenesis 2014, 35, 1196–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Wu, D.; Wu, P.; Chen, Z.; Huang, J. The cancer stem cell niche: Cross talk between cancer stem cells and their microenvironment. Tumor Biol. 2014, 35, 3945–3951. [Google Scholar] [CrossRef]
- Albini, A.; Bruno, A.; Gallo, C.; Pajardi, G.; Noonan, D.M.; Dallaglio, K. Cancer stem cells and the tumor microenvironment: Interplay in tumor heterogeneity. Connect. Tissue Res. 2015, 56, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Papaccio, F.; Paino, F.; Regad, T.; Papaccio, G.; Desiderio, V.; Tirino, V. Concise review: Cancer cells, cancer stem cells, and mesenchymal stem cells: Influence in cancer development. Stem Cells Transl. Med. 2017, 6, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Melzer, C.; Yang, Y.; Hass, R. Interaction of msc with tumor cells. Cell Commun. Signal. 2016, 14, 20. [Google Scholar] [CrossRef]
- Mathivanan, S.; Fahner, C.J.; Reid, G.E.; Simpson, R.J. Exocarta 2012: Database of exosomal proteins, rna and lipids. Nucleic acids Res. 2012, 40, D1241–D1244. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, C.; Harikumar, K.B. The origin and functions of exosomes in cancer. Front. Oncol. 2018, 8, 66. [Google Scholar] [CrossRef]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor egfrviii by microvesicles derived from tumour cells. Nat. Cell Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef]
- Figueroa, J.; Phillips, L.M.; Shahar, T.; Hossain, A.; Gumin, J.; Kim, H.; Bean, A.J.; Calin, G.A.; Fueyo, J.; Walters, E.T.; et al. Exosomes from glioma-associated mesenchymal stem cells increase the tumorigenicity of glioma stem-like cells via transfer of mir-1587. Cancer Res. 2017, 77, 5808–5819. [Google Scholar] [CrossRef]
- Bartel, D.P. Micrornas: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Ferretti, E.; De Smaele, E.; Miele, E.; Laneve, P.; Po, A.; Pelloni, M.; Paganelli, A.; Di Marcotullio, L.; Caffarelli, E.; Screpanti, I.; et al. Concerted microrna control of hedgehog signalling in cerebellar neuronal progenitor and tumour cells. EMBO J. 2008, 27, 2616–2627. [Google Scholar] [CrossRef]
- Mattiske, S.; Suetani, R.J.; Neilsen, P.M.; Callen, D.F. The oncogenic role of mir-155 in breast cancer. Cancer Epidemiol. Prev. Biomark. 2012, 21, 1236–1243. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, J.; Wang, X.; Shi, S.; Wang, W.; Chen, Z. Appraising microrna-155 as a noninvasive diagnostic biomarker for cancer detection: A meta-analysis. Medicine 2016, 95, e2450. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.H.; Chen, M.T.; Wang, M.L.; Lee, Y.Y.; Chiou, G.Y.; Chien, C.S.; Huang, P.I.; Chen, Y.W.; Huang, M.C.; Chiou, S.H.; et al. Cisplatin-selected resistance is associated with increased motility and stem-like properties via activation of stat3/snail axis in atypical teratoid/rhabdoid tumor cells. Oncotarget 2015, 6, 1750–1768. [Google Scholar] [PubMed]
- Zhang, C.; Zhai, W.; Xie, Y.; Chen, Q.; Zhu, W.; Sun, X. Mesenchymal stem cells derived from breast cancer tissue promote the proliferation and migration of the mcf-7 cell line in vitro. Oncol. Lett. 2013, 6, 1577–1582. [Google Scholar] [CrossRef]
- Chiou, S.H.; Kao, C.L.; Chen, Y.W.; Chien, C.S.; Hung, S.C.; Lo, J.F.; Chen, Y.J.; Ku, H.H.; Hsu, M.T.; Wong, T.T. Identification of cd133-positive radioresistant cells in atypical teratoid/rhabdoid tumor. PLoS ONE 2008, 3, e2090. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.L.; Chiou, S.H.; Chen, Y.J.; Singh, S.; Lin, H.T.; Liu, R.S.; Lo, C.W.; Yang, C.C.; Chi, C.W.; Lee, C.H.; et al. Increased expression of osteopontin gene in atypical teratoid/rhabdoid tumor of the central nervous system. Mod. Pathol. 2005, 18, 769–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.Y.; Yang, Y.P.; Huang, M.C.; Wang, M.L.; Yen, S.H.; Huang, P.I.; Chen, Y.W.; Chiou, S.H.; Lan, Y.T.; Ma, H.I.; et al. Microrna142-3p promotes tumor-initiating and radioresistant properties in malignant pediatric brain tumors. Cell Transpl. 2014, 23, 669–690. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.L.; Chiou, S.H.; Ho, D.M.; Chen, Y.J.; Liu, R.S.; Lo, C.W.; Tsai, F.T.; Lin, C.H.; Ku, H.H.; Yu, S.M.; et al. Elevation of plasma and cerebrospinal fluid osteopontin levels in patients with atypical teratoid/rhabdoid tumor. Am. J. Clin. Pathol. 2005, 123, 297–304. [Google Scholar] [CrossRef]
- Torchia, J.; Golbourn, B.; Feng, S.; Ho, K.C.; Sin-Chan, P.; Vasiljevic, A.; Norman, J.D.; Guilhamon, P.; Garzia, L.; Agamez, N.R.; et al. Integrated (epi)-genomic analyses identify subgroup-specific therapeutic targets in cns rhabdoid tumors. Cancer cell 2016, 30, 891–908. [Google Scholar] [CrossRef] [PubMed]
- Chiou, G.Y.; Cherng, J.Y.; Hsu, H.S.; Wang, M.L.; Tsai, C.M.; Lu, K.H.; Chien, Y.; Hung, S.C.; Chen, Y.W.; Wong, C.I.; et al. Cationic polyurethanes-short branch pei-mediated delivery of mir145 inhibited epithelial-mesenchymal transdifferentiation and cancer stem-like properties and in lung adenocarcinoma. J. Control. Release 2012, 159, 240–250. [Google Scholar] [CrossRef]
- Ebert, M.S.; Neilson, J.R.; Sharp, P.A. Microrna sponges: Competitive inhibitors of small rnas in mammalian cells. Nat. Methods 2007, 4, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Xu, H.; Han, H.; Song, S.; Zhang, X.; Ouyang, L.; Qian, C.; Hong, Y.; Qiu, Y.; Zhou, W.; et al. Exosome-mediated transfer of lncrunx2-as1 from multiple myeloma cells to mscs contributes to osteogenesis. Oncogene 2018, 37, 5508–5519. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, W.Q.; Zhu, Y.; Han, X.Q.; Liu, N. Exosomes derived from mesenchymal stromal cells pretreated with advanced glycation end product-bovine serum albumin inhibit calcification of vascular smooth muscle cells. Front. Endocrinol. 2018, 9, 524. [Google Scholar] [CrossRef]
- Xiao, C.; Wang, K.; Xu, Y.; Hu, H.; Zhang, N.; Wang, Y.; Zhong, Z.; Zhao, J.; Li, Q.; Zhu, D.; et al. Transplanted mesenchymal stem cells reduce autophagic flux in infarcted hearts via the exosomal transfer of mir-125b. Circ. Res. 2018, 123, 564–578. [Google Scholar] [CrossRef]
- Christianson, H.C.; Svensson, K.J.; van Kuppevelt, T.H.; Li, J.P.; Belting, M. Cancer cell exosomes depend on cell-surface heparan sulfate proteoglycans for their internalization and functional activity. Proc. Natl. Acad. Sci. USA 2013, 110, 17380–17385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Ridzon, D.A.; Broomer, A.J.; Zhou, Z.; Lee, D.H.; Nguyen, J.T.; Barbisin, M.; Xu, N.L.; Mahuvakar, V.R.; Andersen, M.R.; et al. Real-time quantification of micrornas by stem-loop rt-pcr. Nucleic Acids Res. 2005, 33, e179. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.L.; Huang, P.I.; Tsai, P.H.; Tsai, M.L.; Lo, J.F.; Lee, Y.Y.; Chen, Y.J.; Chen, Y.W.; Chiou, S.H. Resveratrol-induced apoptosis and increased radiosensitivity in cd133-positive cells derived from atypical teratoid/rhabdoid tumor. Int. J. Radiat. Oncol. Biol. Phys. 2009, 74, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Bussard, K.M.; Mutkus, L.; Stumpf, K.; Gomez-Manzano, C.; Marini, F.C. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016, 18, 84. [Google Scholar] [CrossRef]
- Bhome, R.; Del Vecchio, F.; Lee, G.H.; Bullock, M.D.; Primrose, J.N.; Sayan, A.E.; Mirnezami, A.H. Exosomal micrornas (exomirs): Small molecules with a big role in cancer. Cancer Lett. 2018, 420, 228–235. [Google Scholar] [CrossRef]
- Guerrero-Martinez, J.A.; Reyes, J.C. High expression of smarca4 or smarca2 is frequently associated with an opposite prognosis in cancer. Sci. Rep. 2018, 8, 2043. [Google Scholar] [CrossRef]
- Ramos, P.; Karnezis, A.N.; Craig, D.W.; Sekulic, A.; Russell, M.L.; Hendricks, W.P.; Corneveaux, J.J.; Barrett, M.T.; Shumansky, K.; Yang, Y.; et al. Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in smarca4. Nat. Genet. 2014, 46, 427–429. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Su, Y.; Fa, X. Restoration of brg1 inhibits proliferation and metastasis of lung cancer by regulating tumor suppressor mir-148b. Onco Targets Ther. 2015, 8, 3603–3612. [Google Scholar]
- Jubierre, L.; Soriano, A.; Planells-Ferrer, L.; Paris-Coderch, L.; Tenbaum, S.P.; Romero, O.A.; Moubarak, R.S.; Almazan-Moga, A.; Molist, C.; Roma, J.; et al. Brg1/smarca4 is essential for neuroblastoma cell viability through modulation of cell death and survival pathways. Oncogene 2016, 35, 5179–5190. [Google Scholar] [CrossRef]
- Beaurivage, C.; Champagne, A.; Tobelaim, W.S.; Pomerleau, V.; Menendez, A.; Saucier, C. Socs1 in cancer: An oncogene and a tumor suppressor. Cytokine 2016, 82, 87–94. [Google Scholar] [CrossRef]
- Lazennec, G.; Lam, P.Y. Recent discoveries concerning the tumor—mesenchymal stem cell interactions. Biochim. Biophys. Acta 2016, 1866, 290–299. [Google Scholar] [CrossRef]
- Hwang, W.L.; Yang, M.H.; Tsai, M.L.; Lan, H.Y.; Su, S.H.; Chang, S.C.; Teng, H.W.; Yang, S.H.; Lan, Y.T.; Chiou, S.H.; et al. Snail regulates interleukin-8 expression, stem cell-like activity, and tumorigenicity of human colorectal carcinoma cells. Gastroenterology 2011, 141, 279–291. [Google Scholar] [CrossRef]
- Tsai, K.S.; Yang, S.H.; Lei, Y.P.; Tsai, C.C.; Chen, H.W.; Hsu, C.Y.; Chen, L.L.; Wang, H.W.; Miller, S.A.; Chiou, S.H.; et al. Mesenchymal stem cells promote formation of colorectal tumors in mice. Gastroenterology 2011, 141, 1046–1056. [Google Scholar] [CrossRef]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mrnas and micrornas is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef]
- Zhou, W.; Fong, M.Y.; Min, Y.; Somlo, G.; Liu, L.; Palomares, M.R.; Yu, Y.; Chow, A.; O’Connor, S.T.; Chin, A.R.; et al. Cancer-secreted mir-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 2014, 25, 501–515. [Google Scholar] [CrossRef]
- Aleckovic, M.; Kang, Y. Welcoming treat: Astrocyte-derived exosomes induce pten suppression to foster brain metastasis. Cancer Cell 2015, 28, 554–556. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hingorani, S.R. Intercepting cancer communiques: Exosomes as heralds of malignancy. Cancer Cell 2015, 28, 151–153. [Google Scholar] [CrossRef]
- Zhang, J.; Li, S.; Li, L.; Li, M.; Guo, C.; Yao, J.; Mi, S. Exosome and exosomal microrna: Trafficking, sorting, and function. Genom. Proteom. Bioinform. 2015, 13, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, S.; Yao, J.; Lowery, F.J.; Zhang, Q.; Huang, W.C.; Li, P.; Li, M.; Wang, X.; Zhang, C.; et al. Microenvironment-induced pten loss by exosomal microrna primes brain metastasis outgrowth. Nature 2015, 527, 100–104. [Google Scholar] [CrossRef]
- Chiu, C.F.; Chang, Y.W.; Kuo, K.T.; Shen, Y.S.; Liu, C.Y.; Yu, Y.H.; Cheng, C.C.; Lee, K.Y.; Chen, F.C.; Hsu, M.K.; et al. Nf-kappab-driven suppression of foxo3a contributes to egfr mutation-independent gefitinib resistance. Proc. Natl. Acad. Sci. USA 2016, 113, E2526–E2535. [Google Scholar] [CrossRef]
- Santos, J.C.; Lima, N.D.S.; Sarian, L.O.; Matheu, A.; Ribeiro, M.L. Exosome-mediated breast cancer chemoresistance via mir-155 transfer. Sci. Rep. 2018, 8, 829. [Google Scholar] [CrossRef]
- Liu, F.; Kong, X.; Lv, L.; Gao, J. Tgf-beta1 acts through mir-155 to down-regulate tp53inp1 in promoting epithelial-mesenchymal transition and cancer stem cell phenotypes. Cancer Lett. 2015, 359, 288–298. [Google Scholar] [CrossRef]
- Gao, S.; Wang, Y.; Wang, M.; Li, Z.; Zhao, Z.; Wang, R.X.; Wu, R.; Yuan, Z.; Cui, R.; Jiao, K.; et al. Microrna-155, induced by foxp3 through transcriptional repression of brca1, is associated with tumor initiation in human breast cancer. Oncotarget 2017, 8, 41451–41464. [Google Scholar] [PubMed]
- Mikamori, M.; Yamada, D.; Eguchi, H.; Hasegawa, S.; Kishimoto, T.; Tomimaru, Y.; Asaoka, T.; Noda, T.; Wada, H.; Kawamoto, K.; et al. Microrna-155 controls exosome synthesis and promotes gemcitabine resistance in pancreatic ductal adenocarcinoma. Sci. Rep. 2017, 7, 42339. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.-P.; Nguyen, P.N.N.; Ma, H.-I.; Ho, W.-J.; Chen, Y.-W.; Chien, Y.; Yarmishyn, A.A.; Huang, P.-I.; Lo, W.-L.; Wang, C.-Y.; et al. Tumor Mesenchymal Stromal Cells Regulate Cell Migration of Atypical Teratoid Rhabdoid Tumor through Exosome-Mediated miR155/SMARCA4 Pathway. Cancers 2019, 11, 720. https://doi.org/10.3390/cancers11050720
Yang Y-P, Nguyen PNN, Ma H-I, Ho W-J, Chen Y-W, Chien Y, Yarmishyn AA, Huang P-I, Lo W-L, Wang C-Y, et al. Tumor Mesenchymal Stromal Cells Regulate Cell Migration of Atypical Teratoid Rhabdoid Tumor through Exosome-Mediated miR155/SMARCA4 Pathway. Cancers. 2019; 11(5):720. https://doi.org/10.3390/cancers11050720
Chicago/Turabian StyleYang, Yi-Ping, Phan Nguyen Nhi Nguyen, Hsin-I Ma, Wen-Jin Ho, Yi-Wei Chen, Yueh Chien, Aliaksandr A. Yarmishyn, Pin-I Huang, Wen-Liang Lo, Chien-Ying Wang, and et al. 2019. "Tumor Mesenchymal Stromal Cells Regulate Cell Migration of Atypical Teratoid Rhabdoid Tumor through Exosome-Mediated miR155/SMARCA4 Pathway" Cancers 11, no. 5: 720. https://doi.org/10.3390/cancers11050720