The Role of HOX Transcription Factors in Cancer Predisposition and Progression


1
Shandong Provincial Key Laboratory of Animal Cell and Developmental Biology, School of Life Sciences, Shandong University, Qingdao 266237, China
2
Faculty of Biochemistry and Molecular Medicine, Biocenter Oulu, University of Oulu, 90220 Oulu, Finland
*
Authors to whom correspondence should be addressed.
Cancers 2019, 11(4), 528; https://doi.org/10.3390/cancers11040528
Submission received: 18 March 2019
/
Revised: 8 April 2019
/
Accepted: 10 April 2019
/
Published: 12 April 2019
(This article belongs to the Special Issue HOX Genes in Cancer)
Abstract
:Homeobox (HOX) transcription factors, encoded by a subset of homeodomain superfamily genes, play pivotal roles in many aspects of cellular physiology, embryonic development, and tissue homeostasis. Findings over the past decade have revealed that mutations in HOX genes can lead to increased cancer predisposition, and HOX genes might mediate the effect of many other cancer susceptibility factors by recognizing or executing altered genetic information. Remarkably, several lines of evidence highlight the interplays between HOX transcription factors and cancer risk loci discovered by genome-wide association studies, thereby gaining molecular and biological insight into cancer etiology. In addition, deregulated HOX gene expression impacts various aspects of cancer progression, including tumor angiogenesis, cell autophagy, proliferation, apoptosis, tumor cell migration, and metabolism. In this review, we will discuss the fundamental roles of HOX genes in cancer susceptibility and progression, highlighting multiple molecular mechanisms of HOX involved gene misregulation, as well as their potential implications in clinical practice.
1. Introduction
The homeobox genes encode a highly conserved family of transcription factors that play essential roles in embryonic development and tissue homeostasis. In humans, there are four HOX gene clusters, HOXA, HOXB, HOXC, and HOXD, located on different chromosomes, at 7p15, 17q21.2, 12q13, and 2q31 loci, respectively. Each cluster consists of 9 to 11 HOX genes arranged in order. A total of 39 transcription factors are encoded by HOX genes and regulate a series of downstream target genes in a precise manner. The spatial and temporal expression pattern of these HOX transcription factors and their controlled genes is the main mechanism defining the organogenesis of limbs and organs along the anterior-posterior (A-P) axis during embryonic development of flies and vertebrates [1,2]. Accumulating evidence shows that functional abnormalities of HOX transcription factors play critical roles in the development and progression of many types of cancers [1,2].
Both mutation and aberrant expression can alter the function of the HOX transcription factor by gene regulation, and subsequently affect downstream events of cancer development. Mutations in the HOX DNA binding domain and cofactor-interaction domain may alter the structure and function of protein, thereby leading to an aberrant capability of DNA binding [3] and protein–protein interaction [4], respectively. In cancer, the deregulated HOX gene expression has been widely recognized as a driving force in tumorigenesis [1]. Either up-regulation or down-regulation of HOX genes have both been reported to be associated with cancer under various conditions, where HOX genes act as tumor suppressors or proto-oncogenes depending on cancer type. For example, HOXA9 was found to be overexpressed in leukemia, but downregulated in breast cancer. As a proto-oncogene, high expression levels of HOXA9 are often associated with increased cancer risk [2]. Aberrant expression of HOXA9 was proven to play critical roles in the development of acute leukemia through reprogramming the epigenome or synergizing with other transcription factors and signaling pathways, thereby considered as one of the driving forces in leukemogenesis [5,6]. On the other hand, as a tumor suppressor, HOXA9 inhibits the tumor phenotype by regulating the expression of BRCA1 in breast cancer [7]. In clinical settings, lower expression of HOXA9 is greatly associated with elevated tumor invasion, metastasis, and patient mortality [7]. In addition to this, the deregulation of many HOX genes has been found in a variety of cancers, and often reported in association with an increased cancer risk and poor survival rate of cancer patients [1,2].
HOX genes also play increasingly important roles in genetic predisposition for many types of cancers. A previous large-scale twin study provided solid evidence that hereditary factors made a significant contribution to colorectal, breast, and prostate cancer. In addition, the research also revealed suggestive evidence of limited heritability with leukemia and cancers of the stomach, lung, pancreas, ovary, and bladder [8]. With a wide application of exome sequencing and genome-wide association study (GWAS), substantial independent susceptibility loci marked with single nucleotide polymorphism (SNP) have been discovered for nearly all types of cancers, such as colorectal [9], breast [10], prostate [11], lung [12], cervical cancer [13], acute lymphoblastic leukemia [14], chronic lymphocytic leukemia [15], and so forth. A combination of these risk mutations may define individuals with different inherited susceptibility to developing cancers [16]. These risk genetic alterations also lay the groundwork for personalized precision cancer medicine. Among the genes, somatic or germline mutations in HOX genes have proven to influence cancer susceptibility. Further, HOX transcription factors often recognize and execute genetic information buried in cancer risk loci. In this review, we will systematically introduce the progress in deciphering the roles of HOX genes in cancer susceptibility and progression as well as the underlying mechanisms. We also briefly discuss clinical implications of HOX proteins as cancer therapeutic targets.
2. HOX Transcription Factors in Cancer Predisposition
2.1. Coding Genetic Mutations in HOX Genes
Coding mutations in HOX genes have been widely observed in association with cancer predisposition. Current evidence indicates that this type of mutation mostly locate in the HOX transcription factors, HOXB13 and HOXD4, as well as a long non-coding RNAs (lncRNA), HOXA11-AS, from the antisense strand in the homeobox A cluster (Figure 1A).
2.1.1. HOXB13 Mutations
HOXB13 is a homeobox B transcription factor gene and is known to be important in prostate development and tumorigenesis. Multiple studies reported a significant association of HOXB13 with many types of cancers [17,18,19,20,21]. Remarkably, the inherited mutations in HOXB13 have been widely observed for a genetic contribution to prostate cancer risk. Specifically, several missense rare mutations in HOXB13, including G84E, Y88D, L144P, G216C, and R229G, have been identified in association with increased prostate cancer susceptibility through targeted germline DNA sequencing of the 17q21-22 region in a cohort of 94 prostate cancer patients [22]. In particular, the rare, but recurrent HOXB13 G84E mutation is strongly associated with an increased risk of familial prostate cancer in European descents and subsequently found to be highly associated with prostate cancer risk in additional populations [23,24,25,26,27,28,29,30]. Interestingly, the other HOXB13 mutation pattern differs among populations. Two variants, R229G and G216C, of HOXB13 were found in African descent [22]. The HOXB13 G135E mutation was discovered in association with increased prostate cancer risk of Chinese men [31]. Two additional HOXB13 F127C and G132E mutations were identified among Japanese men with prostate cancer [32]. The other two mutations, L144P and Y88D, were observed in the prostate cancer cell line, LNCaP and LAPC4, respectively [22].
Intriguingly, several studies report that the HOXB13 G84E mutation is not only associated with increased risk of prostate cancer, but also significantly associated with an elevated risk of leukemia, and cancers of the bladder, breast, and kidney [25,28,33,34,35], indicating that this prostate cancer susceptibility gene, HOXB13, may also play key roles in the predisposition to a variety of cancers.
Even though the underlying mechanisms of these mutations in promoting carcinogenesis are still unknown, their potential impact could be inferred based on the domain function in protein–DNA and protein–protein interactions. The HOXB13 gene has two highly conserved MEIS (myeloid ecotropic viral integration site) binding domains on exon 1, and one homeodomain located in exon 2 [36]. A functional HOXB13–MEIS1 interaction plays pivotal roles in modulating cellular proliferation and gene expression in prostate cancer [37]. Both G84E and Y88D mutations are located in the first MEIS interacting domain, while L144P and G135E mutations reside in the second MEIS interacting domain [22,31]. Thus, these mutations in MEIS interacting domains may affect the HOXB13 function through an alteration of its binding ability to MEIS cofactors or subsequent target DNA sequence recognition. Consequently, this will affect the expression of downstream target genes [31]. However, the most recent study shows that the G84E mutation does not influence the capability of the first MEIS interacting domain in HOXB13 to physically interact with MEIS1 in a pull-down assay [37], suggesting that G84E is not a loss-of-function mutation and might influence HOXB13 DNA binding specificity and cofactor interacting profiles in a subtle manner. The other mutations, R229G and G216C, are located in the N-terminal portion of the homeobox domain and affect highly conserved amino acid residues [22]. Two additional variants, F127C and G132E, are near the HOXB13 N-terminal domain [32]. Computational modeling analysis of the HOXB13 transcription factor indicates that these coding mutations might introduce structural changes in the protein. For example, the mutants, G84E and G135E, may lead to promoted protein stability and an increased half-life, thereby conferring increased cancer susceptibility [38]. Despite efforts devoted to elucidating the impact of HOXB13 mutations, investigation on clinical prostate tumor tissues shows that HOXB13 gene expression at both mRNA and protein levels does not differ between samples carrying the variant and wild-type allele [39].
Intriguingly, the cooperation between the rare HOXB13 mutation and other risk factors may play an important role in promoting cancer risk. Strong chromatin binding of HOXB13 at gene regulatory regions of CIP2A was observed, and the G84E mutation further promoted this chromatin binding in the immortalized benign prostate cell line, RWPE-1 [40]. RNA interference experiments further confirmed that HOXB13 functionally promotes CIP2A transcription. More importantly, the simultaneous presence of HOXB13 (G84E) and the common CIP2A (R229Q) variant confers higher prostate cancer risk and disease aggressiveness, as well as poor prognosis [40]. Nevertheless, detailed biological function and mechanism of these HOXB13 gene mutations still need to be explored.
2.1.2. HOXD4 Mutations
Germline missense mutation in another HOX transcription factor, HOXD4, was also detected to be associated with an increased risk of childhood acute lymphoblastic leukemia (ALL) [41]. The E81V mutation leads to a partial loss-of-function, defined by reduced transcriptional activity at the autoregulatory enhancer of the HOXD4 gene. This mechanism might be involved in the occurrence of childhood ALL [41].
2.1.3. HOX Locus lncRNAs
Except for the aforementioned protein-coding HOX genes, two highly conserved lncRNA, HOXA11-AS at the homeobox A region and HOTAIR at homeobox C region, have also been reported for cancer susceptibility. HOXA11-AS with a minor allele T of exonic variant, rs17427875, inhibits cell survival, proliferation, migration, and invasion to a greater extent than the common allele A does in epithelial ovarian cancer [42]. As revealed in a meta-analysis, three SNPs, rs4759314, rs902778, and rs1899663, in HOTAIR are also the genetic predisposition factors in breast cancer, cervical cancer, and ovarian cancer [43]. Two further SNPs in HOTAIR, rs12427129 and rs3816153, are associated with hepatocellular carcinoma susceptibility too [44]. It is worth mentioning that many lncRNAs have been frequently found to impact cancer susceptibility [45], raising the question of whether additional HOX locus lncRNAs are involved in cancer predisposition and progression.
Collectively, these findings highlight the difficulties in the functional investigation of cancer risk-associated HOX coding mutations, and raise a possibility to perturb these mutations using advanced genome-editing tools to limit predisposition to cancers in the clinical preventive settings.
2.2. Risk Loci Influencing HOX Gene Expression
GWASs have thus far identified hundreds of common variants associated with cancer predisposition. According to systems annotation of the GWAS catalog database, about 93% of the risk SNPs are located in the non-protein coding regions of the genome, including intronic and intergenic regions [46]. There is increasing evidence to show that these SNPs are significantly enriched in DNase I hypersensitive sites and cistromes of transcription factors, such as HOXB13 [47,48], and are likely to act as regulatory elements to alter the expression of target genes, such as the HOX family member, HOXA11 [48]. Thus, these SNPs function potentially as regulatory variants.
Until recently, several regulatory SNPs were reported to influence cancer risk through regulating the expression of HOX genes. Misregulated expression of HOX genes may lead to changes of the downstream gene expression and signaling pathways that play fundamental roles in cancers. This is in line with the observations that the expression levels of HOX genes are often found to be overexpressed or downregulated in many types of cancers due to various genetic and epigenetic mechanisms [1,49]. Here, we focus on several cancer susceptibility loci that may impact disease risk through misregulating the expression of HOX genes (Figure 1B).
2.2.1. 7p15.2 Locus
The 7p15.2 locus with three SNPs, rs10486567, rs67152137, and rs7808935, has been found in association with an increased susceptibility of prostate cancer [50,51,52]. Encouraged by our previous report [48], a recent functional study identified a long-range chromatin interaction between the risk region of the 7p15.2 locus and the HOXA13 gene, located ~873 kb away [53]. Deletion of the risk region harboring several prostate cancer risk-associated SNPs using CRISPR (clustered regularly interspaced palindromic repeats)-Cas (CRISPR-associated proteins)-mediated genome editing resulted in a loss of one anchor point of the repressive chromatin loop, which may subsequently alter the three-dimensional chromatin structure and cause upregulation of HOXA13 and HOTTIP in the HOXA locus, leading to genome-wide transcriptomic changes [53]. Together, this study demonstrated that HOXA13 is also a target gene transforming the roles of risk regulatory SNPs at the 7p15.2 locus that influences prostate cancer susceptibility.
2.2.2. 2q31.1 Locus
Another example comes from the multiple SNPs at the 2q31.1 locus that have been reported in GWAS analysis for an association with an increased risk of mucinous ovarian carcinoma (MOC) [54] and high-grade serous epithelial ovarian cancer (HGSOC) [55]. In the mechanistic studies, a chromatin loop spanned 31 to 55 kb of the genomic region with the HOXD9 promoter and SNPs at this locus identified using chromosome conformation capture analysis (3C) [54,55]. Subsequently, the risk SNP, rs711830, genotype was markedly associated with the expression of HOXD9 in an expression quantitative trait loci (eQTL) analysis. Ectopic expression of HOXD9 in MOC cells resulted in a significant increase in anchorage-independent growth [54]. In HGSOC, another SNP, rs2857532, located at this locus was defined as a leading causal variant, which may influence chromatin binding of the HOMEZ, BEN, and RelA-p65 transcription factors and subsequent alteration of HOXD9 expression [55]. Consistent with this, HOXD9 overexpression in the immortalized ovarian surface of epithelial cells significantly increases anchorage-independent growth, shortens population-doubling time, and reduces contact inhibition [55]. These studies together suggest that the HOXD9 gene mediates the function of risk SNPs at the 2q31.1 locus, conferring increased susceptibility and tumor cellular transformation of MOC and HGSOC.
2.2.3. 2q31 Allele rs2072590
The minor allele of rs2072590 at 2q31 was discovered in association with an increased risk of ovarian cancer (OC) [56]. This SNP lies in the non-coding DNA region downstream of HOXD3 and upstream of HOXD1 in the HOXD locus [56] and tags 19 genetic variants according to HaploReg analysis [57]. Both HOXD1 and HOXD3 genes have been reported for their involvement in cancer development [56]. Bioinformatics analysis plus functional annotation showed that rs2072590 together with tagged SNPs may lead to OC susceptibility through regulation of the expression of HOXD1 and HOXD3 [56,57]. Interestingly, the SNP, rs2072590, is also mapped within a lncRNA, namely HOXD-AS1, located between HOXD1 and HOXD3 in the HOXD cluster. Accumulating evidence reveals critical roles of HOXD-AS1 in cancer development and progression through different mechanisms [58]. Here, the 2q31 SNP, rs2072590, might play regulatory roles in fine-tuning the expression of HOXD-AS1, thereby contributing to OC predisposition and progression.
2.2.4. rs11614913 Locus
The SNP, rs11614913, in the miR-196a2 locus was reported to be associated with risk of childhood ALL [59] and glioma [60] in a Chinese population. HOXC8 is the potential target gene of hsa-miR-196a2 according to a comprehensive analysis using three bioinformatics methods [59]. Rs11614913 risk allele C increases the expression levels of mature mir-196a2 and may affect the binding of mature miR-196a2 to its target mRNA in childhood ALL [59]. However, in glioma, rs11614913 polymorphism does not significantly affect the expression of mature miR-196a2; rather, it takes effect through altering its target gene, HOXC8, expression [60]. This finding raises another layer of complexity for a risk SNP either directly or indirectly through a miRNA to influence the expression of a potential cancer susceptibility gene, HOXC8.
2.2.5. rs34631763 Locus
The last example is relevant to the SNP, rs34631763, within growth factor independence 1(Gfi1) that functions as a DNA binding transcriptional repressor. It is known that Gfi1 represses transcription by recruiting histone-modifying enzymes, such as lysine-specific histone demethylase 1A (LSD1), G9a (EHMT2, euchromatic histone lysine methyltransferase 2), and histone deacetylases (HDACs), to target gene promoters [61,62]. The SNP, rs34631763, in the Gfi1 gene exon was considered to be associated with acute myeloid leukemia (AML) risk in humans [63]. This missense variation introduces amino acid substitution from serine (GFI1-36S) to asparagine (GFI1-36N) at position 36 of protein Gfi1 [63]. In contrast to GFI1-36S, the GFI1-36N variant lacks the ability to bind its target gene that encodes the leukemia-associated transcription factor, HOXA9, and is unable to modify histone modifications that regulate HOXA9 expression [64,65]. Finally, the GFI1-36N variant depresses the HOXA9 expression by altering the epigenetic histone modification, which is consistent with the observation of frequently elevated HOXA9 expression levels in AML patients carrying the variant [64]. This study indicates a novel mechanism by which a cancer risk variant contributes to the HOXA9 overexpression, which may lead to epigenome reprogramming and protein–protein interaction to promote leukemogenesis [5,6].
2.2.6. rs920778 Locus
The SNP, rs920778, located within the intron 2 region of HOTAIR, was reported to have a significant association with an increased risk of esophageal squamous cell carcinoma (ESCC) in a Chinese population. It can act as an intronic enhancer element to regulate the expression of HOTAIR. Compared with normal allele C, the risk allele T of the SNP, rs920778, significantly increased ESCC risk by upregulating HOTAIR expression [66].
2.3. Risk SNPs Modulating Chromatin Binding of HOX Transcription Factors
Current evidence emerging from functional elucidation of regulatory risk SNPs show that transcription factors are usually involved in the recognition and execution of genetic information implicated in cancer risk-associated genetic variations, thereby leading to altered gene expression and increased cancer susceptibility [46,48]. Under most conditions, the risk allele influences the DNA-binding affinity of given transcription factors, resulting in altered enhancer or promoter activity and causing varied downstream gene expression, which may finally confer increased cancer susceptibility [47,48,67]. As described in the following sections, several transcription factors are altered in DNA binding by given causal risk variants conferring cancer susceptibility and progression [47,68] (Figure 1C).
2.3.1. 6q22 Allele rs339331
The SNP, rs339331, at the 6q22 locus has been reported to be associated with increased prostate cancer risk in multiple populations, including Japanese, African American, and European descent as well as Chinese men [47]. Functional studies demonstrated that this variant resides in a canonical HOXB13-binding site defined by bioinformatic and ChIP-seq (chromatin immunoprecipitation sequencing) analysis. The prostate cancer risk-associated T allele at rs339331 increases chromatin binding of HOXB13 to an active transcriptional enhancer, conferring allele-specific upregulation of the target gene, RFX6 [47,69] (Figure 1C). Epigenome and transcription activator-like effector nuclease (TALEN)-mediated genome editing assays further demonstrated the direct roles of rs339331 in regulating HOXB13 chromatin binding activity and the expression of RFX6 [69]. In the clinical setting, RFX6 upregulation in human prostate cancer correlates with tumor progression, metastasis, and risk of biochemical relapse [47]. Together, this study presented the first example of a regulatory risk SNP being responsible for prostate cancer pathogenesis through cooperation with the prostate-lineage-specific transcription factor, HOXB13, to regulate a novel oncogene, RFX6.
2.3.2. 19q13 Allele rs11672691
In contrast to indolent prostate cancer, the aggressive form of the disease usually indicates poor prognosis. The SNP, rs11672691, at the 19q13 locus was identified in association with aggressive prostate cancer risk in a European population [70] and prostate cancer specific mortality in a large US cohort [71]. Further genetic association analysis in a Finnish cohort of prostate cancer demonstrated that the allele G of rs11672691 is markedly associated with advanced tumor stage, prostate-specific antigen (PSA) progression, and the development of castration-resistant prostate cancer, the hallmark clinical features of aggressive prostate cancer susceptibility [68]. A follow-up functional study revealed that HOX transcription factor, HOXA2, plays essential roles in the causal actions and biological effects of this variation. The SNP, rs11672691, resides in an active enhancer element and the risk G allele increases the chromatin binding of HOXA2, which subsequently promotes the expression of PCAT19 and CEACAM21 (Figure 1C), which may contribute to the aggressive phenotype of prostate cancer [68]. Interestingly, an additional study discovered a rs11672691-mediated promoter-enhancer switching mechanism driving the expression of lncRNA PCAT19 and thus the initiation and progression of aggressive prostate cancer [72]. The transcription factors’, NKX3.1 and YY1, DNA binding are altered by the 19q13 alleles, including rs11672691 and rs887391. Thus, these results showed that HOXA2 and additional transcription factors mediate the regulatory effect of the risk SNP at the 19q13 locus on PCAT19 and CEACAM21 and eventually lead to aggressive prostate cancer susceptibility [73] and also raise new questions of how these transcription factors compete for the binding to the SNP region.
Together, these tumor-type-specific contributions of HOX transcription factors in cancer susceptibility may serve as potential targets for inventing new therapeutic interference in global cancer risk prevention. It is therefore evident that the deregulation of HOX genes across these cancer risk loci promotes cancer susceptibility, initiation, and progression to advanced stages.
3. HOX Genes Mediate Effects of Other Genetic and Epigenetic Variation
Beside the important roles in affecting cancer susceptibility caused by germline genetic variations, the HOX genes also mediate the effects of a wide variety of somatic variations at both genetic and epigenetic levels. These kinds of somatic variations mainly include an abnormal epigenetic status that alters HOX gene expression. Here, we will discuss how somatic gene mutations in DNMT3A, ASXL1, and NPMC1 regulate the expression of HOX genes through DNA methylation and histone modifications as well as gene fusions related to HOX transcription factors driving cancer progression (Figure 2).
3.1. Abnormal Epigenetic Alteration Affecting HOX Genes
DNA methylation is a pivotal epigenetic mechanism for defining cellular identity and regulating the activity of gene regulatory elements, including promoters and enhancers [74,75]. Aberrant DNA methylation patterns are critical in the development of all types of cancers [76]. In particular, changes in DNA methylation patterns influence HOX genes that commonly occur in many types of cancers [77,78,79,80,81]. Hypermethylation of the promoter-located CpG island usually leads to transcriptional inhibition and is involved in the inactivation of many tumor suppressor genes, such as HOXA4 in chronic lymphocytic leukemia (CLL) [81]. In addition, HOTAIR plays an important role in epigenetically remodeling chromatin states. HOTAIR can recruit polycomb repressive complex 2 (PRC2) to induce H3K27me3 modification in specific polycomb group target genes to decrease gene expression, such as HOXD gene clusters [82,83]. Hence, altered DNA methylation is likely to be a major potential mechanism in dysregulating HOX gene expression in cancer, thereby contributing to cancer development. For example, the tumor suppressor gene, HOXA4, was observed to be hypermethylated at promoter-associated CpG islands and correlated with low levels of HOXA4 expression in CLL [81]. In contrast, the most recent study of genome-wide DNA methylation profiles in 30 normal tissues and 35 solid tumors found that gene-body DNA hypermethylation is greatly associated with elevated expression of HOX oncogenes, thus providing additional epigenetic mechanism on the regulation of HOX genes in cancers [49]. Further, accumulating evidence suggests that methylation alteration in HOX genes could serve as a prognostic marker in cancer therapy [77,78,80]. For example, promoter methylation of HOXA9 has been associated with prognostication and can serve as potential predictive biomarker for cisplatin chemotherapy resistant bladder cancer [84].
3.2. Somatic Gene Mutations Deregulating HOX Transcription Factors
3.2.1. DNMT3A-R882H
Mutation R882H in DNA methyltransferase 3A (DNMT3A) was frequently found in hematological cancer, and up to 60% of patients with acute myeloid leukemia (AML) carry this mutation in heterozygous [85]. The DNMT3A-R882H variant acts in a dominant negative manner and results in a disrupted methylation function, which could subsequently upregulate both HOXA and HOXB cluster genes that are crucial for leukemogenesis [86]. Mechanically, DNMT3A-R882H directly binds to the HOXA gene cluster, therefore inducing DNA hypomethylation as well as H3K27 acetylation and promoting transcriptional activation of Meis1, Mn1, and HOXA that are required for DNMT3A-R882H-mediated AML progression [87]. In addition, DNMT3A-R882H significantly accelerated the progression of leukemia in the presence of other known mutations, such as NRAS-G12D, NPM1c, or IDH1R132H coexisting with the DNMT3A mutation in human AMLs, providing a susceptible genetic background in epigenetically misregulating the expression of HOX genes [87].
3.2.2. ASXL1 Mutation
Recurrent somatic mutations in the addition of sex combs-like 1 gene (ASXL1) was often found in patients of myeloproliferative neoplasms and AML [88,89]. ASXL1 physically interacts with EZH2, a core member of polycomb repressive complex 2 (PRC2), leading to genome-wide histone modification of H3K27me3, including the genomic region at the posterior HOXA cluster. Similar to the ASXL1 loss condition, these somatic mutations also result in the exclusion of H3K27me3 and EZH2 from the HOXA cluster [88]. Once the H3K27me3 epigenetic signature was lost at HOXA clusters, HOXA gene expression significantly increased, in particular for HOXA9 and HOXA10. Mechanistically, ASXL1 loss-of-function mutations upregulates the expression level of HOXA genes by altering their methylation profile, leading to the development of cancer [88].
3.2.3. NPM1 Mutation
Somatic mutations in the NPM1 gene that encodes nucleophosmin are commonly discovered in human AMLs, accounting for about 35% of AML patients [90]. These mutations named NPM1c destroy the N-terminal nucleolar localization signal of nucleophosmin and produce a novel nuclear export signal, resulting in an anomalous cytoplasmic localization of the mutant nucleophosmin. Expression of the most common form of NPM1c in a conditional knock-in mouse model causes overexpression of several HOXA genes [90]. Further evidences show that in AML cells, the anomalous cytoplasmic localization of NPM1c regulates HOXA gene expression though histone acetylation of H3K27 at HOX gene super-enhancers. Either nuclear relocalization or targeted degradation of NPM1c can result in immediate downregulation of HOX genes and promotes differentiation of AML cells [91], suggesting an alternative therapeutic way to HOX genes in human AMLs carrying NPM1 mutations.
3.3. Gene Fusions Cooperating with HOX Transcription Factors
3.3.1. TMPRSS2-ERG (T2E) Fusion
Structural rearrangements of TMPRSS2-ERG (T2E) are present in over 50% of human prostate cancer and lead to aberrant activation of the ERG transcription factor [92,93]. In addition, the occurrence of T2E fusion is significantly associated with aggressive prostate cancer [94]. In an elegant recent study, T2E was found to physically interact with HOXB13 and FOXA1, thereby inducing T2E-specific cis-regulatory landscape in T2E-positive prostate cancer compared with non-T2E cases. Furthermore, a T2E-specific epigenomic program leads to activation of NOTCH signaling, raising a possibility of targeting T2E-positive cancers through antagonization of the NOTCH pathway [93]. These results indicate that the HOX transcription factor, HOXB13, plays important roles in mediating the oncogenic effect of T2E initiating prostate cancer development.
3.3.2. NUP98 Gene Fusion
Another example of a HOX transcription factor cooperating gene fusion is observed in nucleoporin 98kDa (NUP98) gene fusions that result from chromosomal translocation associated with multiple hematoplastic malignancies [95,96,97]. NUP98 gene fusion usually encodes a fusion protein that retains the N-terminal of NUP98 with potential for transcriptional activation [96]. NUP98 could fuse to at least 28 different genes, including multiple HOX family members [96]. NUP98 fusion with HOXA9 was found to be co-localized with MLL1 on the chromatin of the HOX gene promoter region [97]. Furthermore, NUP98–HOXA9 (NHA9) could induce aberrant expression of dozens of genes playing roles in primary human CD34+ hematopoietic cell proliferation and differentiation [98]. Another fusion gene, NUP98–HOXD13 (NHD13) plays roles in inducing thymocyte self-renewal via Lmo2 and its critical cofactor, Lyl1 [99,100]. On the other hand, some NUP98 fusions with genes other than HOX influence the development of leukemia by affecting epigenetic landscapes across the HOX genes, and subsequently leads to aberrant HOX gene expression. For example, NUP98–PHF23 (NP23) fusion is associated with multiple hematological cancer [95]. Mechanically, NP23 binds to a specific subset of H3K4me3-enriched chromatin sites, including at HOXA, HOXB, and MEIS1, and drug-targeted inhibition of H3K4me3 downregulates the expression of these target genes, leading to rapid and selective cell death of NP23-expressing myeloblasts [95]. Another example of non-HOX relevant NUP98 fusion, NUP98–NSD1 (nuclear receptor-binding SET domain protein 1), can upregulate the expression of HOXA7, HOXA9, HOXA10, and MEIS1 as oncogenes in human AMLs [101]. Mechanically, NUP98–NSD1 binds directly to the regulatory genomic elements near HOXA7 and HOXA9, and subsequently maintains histone acetylation and methylation of H3K36, consequently preventing transcriptional suppression of the HOXA cluster mediated by EZH2 during differentiation [101]. In conclusion, HOX genes are frequently engaged in NUP98 fusion, thus mediating cancer progression, by acting as fusion partner genes of NUP98, such as HOXA9 and HOXD13, or by mediating the effect of other NUP98 fusions as their target genes in a wide range of hematologic malignancies.
3.3.3. MLL and Other Gene Fusions
The mixed-lineage leukemia (MLL) gene encodes a large histone methyltransferase possessing H3K4 methyltransferase activity, thereby actively regulating the expression of the genes, including HOX family members [102]. MLL translocation with its partner genes is highly involved in leukemogenesis through the regulation of HOX gene expression [102]. For example, in MLL-rearranged leukemia, the MLL oncogene promoted myeloid transformation genetically relies on HOXA7 and HOXA9 [103]. One of these fusions, MLL–ENL may cause leukemia by regulating the abnormal expression of HOXA4–A11 in the HOXA cluster [104]. Another two fusions, MLL–hDOT1L and MLL–AF10 induce H3K79 hypermethylation at the HOXA9 locus and subsequently upregulate HOXA9 expression [105]. Mechanically, the H3K79 hypermethylation and subsequent dysregulation of HOXA and MEIS1 expression caused by the MLL–AF10 fusion oncoprotein involves recruitment of DOT1L through direct physical interaction of DOT1L–AF10 to the HOXA gene cluster [106]. Similarly, CALM-AF10 fusion resulting from t (10; 11) (p12; q23) translocation causes H3K79 hypomethylation at the HOXA5 locus by recruiting DOT1L [107]. In addition to histone methylation at H3K79, forced dimerization of MLL also recruits accessory transcription factors to the assembly transcriptional activation complex for activation of HOX gene expression [108]. Overall, HOX gene deregulation plays an important role in MLL/AF10 fusion-induced leukemogenesis. Therefore, it can be appreciated that while these gene fusions relevant to HOX transcription factors are fundamental in key stages of cancer development, their mechanistic functions can be exploited to repress tumorigenesis.
3.3.4. EWS-FLI1 Fusion
EWS-FLI1 fusion is the hallmark of Ewing′s sarcoma and plays important oncogenic roles in malignant transformation. It was reported that EWS-FLI1 can reprogram the epigenome, in particular through recruitment of epigenetic regulators that facilitate chromatin opening and activate gene expression [109]. Interestingly, Ewing′s sarcoma indicates a unique HOX profile that includes aberrant upregulation of posterior HOXD genes. This aberrant elevation of HOXD gene expression is associated with loss of the H3K27me3 mark and gain of the H3K4me3 mark, which is mediated by EWS-FLI1 fusions. Thus, EWS-FLI1 can contribute to EWS-ETS-driven sarcoma genesis and maintenance by deregulating HOX gene expression in Ewing′s sarcoma, similar to MLL-fusion-driven leukemogenesis [110].
4. HOX Genes in Cancer Progression
As described above, the roles of HOX genes in cancer predisposition and development largely involve deregulation of the HOX gene as well as HOX transcription factor downstream target genes. The consequent effects of deregulated HOX genes in carcinogenesis can be explained as an expansion of their normal function. Based on numerous evidences about the HOX gene function in cancer progression, their roles can be classified into seven aspects, including angiogenesis, autophagy, differentiation, apoptosis, proliferation, invasion, and metastasis a well as metabolism that are briefly described in Table 1.
4.1. Angiogenesis
Angiogenesis plays key roles in the progression of solid tumors. Several HOX genes have been shown to function in promoting angiogenesis of solid tumors. HOXB7, HOXB9, and HOXA11 antisense RNA (HOXA11-AS) are involved in promoting angiogenesis by upregulating pro-angiogenic genes’ expression, including interleukin-8 and angiopoietin-2 [111,112,114,115,116]. HOXB7 overexpression is associated with enhanced expression of angiogenic genes in the breast cancer cell line, SKBR3, indicating that HOXB7 is a critical factor upstream of pro-angiogenic genes [111]. More evidences were observed in multiple myeloma expressing HOXB7 to regulate myeloma pro-angiogenic properties [112]. ChIP-seq assays have uncovered hundreds of HOXB7 chromatin binding sites in the breast cancer cell line, BT-474, with ectopic expression of HOXB7 [113], thus providing a new avenue to a deep understanding of the function of HOXB7 in driving breast cancer progression and maybe multiple myeloma. HOXB9 is another potent driver of angiogenesis, promoting angiogenic recruitment by tumor cells [115]. Suppression of EGR1 and HOXB9 could result in global downregulation of genes involved in angiogenesis pathways in multiple ovarian and renal tumor models [114]. Nanoliposome-mediated delivery of microRNA-192 is indicated as an effective therapeutic for suppressing tumor angiogenesis mechanistically through downregulation of EGR1 and HOXB9 expression in tumors [114]. Another example of HOX-involved angiogenic promotion is lncRNA HOXA11 antisense RNA, named as HOXA11-AS, which was significantly overexpressed in non-small cell lung cancer (NSCLC) [116]. Tumor formation experiments revealed that HOXA11-AS promotes angiogenesis in several lung cancer cell lines [116]. In contrast with these angiogenesis-promoting HOX genes, the other HOX family members, such as HOXA5, were considered as antiangiogenic genes [117]. The sustained expression of HOXA5 results in downregulation of many pro-angiogenic genes and upregulation of anti-angiogenic genes in stationary endothelial cells (ECs) [118]. Mechanistically, the presence of MicroRNA-130a could reduce the anti-angiogenic activity of ECs by directly targeting the 3′-UTR of HOXA5 [117]. Taking these observations together, in a clinical translational view, suppressing the expression of HOXB7, HOXB9, HOXA11-AS, and microRNA-130a, or maintaining the expression of HOXA5 and microRNA-192, provides a potential therapeutic strategy to restrain tumor-associated angiogenesis and thus inhibit the growth of tumors.
4.2. Autophagy
Autophagy is a survival-promoting biological process that recycles aged or malfunctioning intracellular proteins and organelles and provides substrates to sustain essential metabolism in starvation and stress. Autophagy also plays important roles in the development of tumors and has been shown to have two paradoxical functions in cancer [174]. Some cancers can be inhibited by autophagy, and some rely on autophagy for survival [175]. Multiple HOX genes are involved in the regulation of autophagy process in cancers. For example, in human glioblastoma cells, HOXC9 is an indicator of poor prognosis and inhibits transcription of the DAPK1 gene through direct binding to its promoter during autophagy process [119]. Silencing of HOXC9 could release the inhibitory effect on the DAPK1 gene and initiate autophagy by activating DAPK1-Beclin1 pathway [119]. MicroRNA-193a-3p was proven to suppress cancer development by silencing multiple genes, including HOXC9 [120]. Hence, promoting cell autophagy by directly silencing HOXC9 protein expression is a promising new cancer therapeutic strategy.
Another example of HOX genes in autophagy was observed in a direct inhibition of HOXC6 with miR-185, promoting apoptosis as well as autophagy through inhibition of the TGF-β1/mTOR pathway in nasopharyngeal carcinoma [121]. Besides HOX transcription factors, HOX transcript antisense RNA (HOTAIR) also plays a regulatory role in autophagy and is associated with the invasion and metastasis capacities of several types of cancers. Silencing HOTAIR inhibits cell autophagy, proliferation, and epithelial–mesenchymal transition (EMT) through suppression of the Wnt signaling pathway and an enhancement of the sensitivity to radiotherapy [122,123].
4.3. Differentiation
Various HOX genes play pivotal roles in cell differentiation and a less differentiated stage is strongly associated with more aggressive tumor behaviors [176]. All the HOXA genes except HOXA2 and HOXA5 induce delayed hematopoietic differentiation in primary hematopoietic cells [124]. For example, HOXA9 is extensively active in blocking differentiation of hematopoietic and lymphoid cancer, and participates in the characteristic myeloid differentiation block in MN1 (Meningioma 1) leukemia [125]. Moreover, NUP98–HOXA9 fusion can confer long-term proliferation and blockaded differentiation in human primary CD34+ hematopoietic cells [98]. In addition, HOXA9 can collaborate with MEIS1 to inhibit hematopoietic cell differentiation [126]. Similarly, cooperation of HOXA9 and FOXC1 can enhance the blockade of monocytic lineage and B-lineage differentiation [127]. Moreover, HOXA10 expression blocks tumor differentiation in prostate cancer while driving histotype differentiation and progression in ovarian endometrioid adenocarcinoma [128,129]. Another example comes from HOXB8 that incompletely blocks DMSO-induced granulocytic differentiation of HL-60 cells [130]. In comparison with these HOX genes, increased HOXC9 expression is associated with neuroblastoma differentiation and better prognosis in neuroblastoma patients. HOXC9 upregulation induced by retinoic acid (RA) can cause growth arrest and neuronal differentiation in neuroblastoma cells by upregulating neuronal differentiation genes and downregulating cell cycle promoting genes [131]. Intriguingly, another HOX transcription factor, HOXA5, can also be upregulated by RA and mediate retinoid differentiation therapy to block colon cancer progression. Elevated expression of HOXA5 strongly reduced tumor growth and prevents metastasis through forcing cancer stem cell differentiation [132,133]. It is worth mentioning that, in addition to HOX transcription factors, lncRNA HOTAIR overexpression may also affect differentiation and aggressiveness of urothelial carcinoma cells, but in a cell-type dependent manner [134].
4.4. Apoptosis
HOX genes can function both as an apoptosis-promoter and apoptosis-suppressor for cancer development. For example, accumulating evidence show that HOXA5 and HOXA10 are apoptosis-promoter genes. HOXA5 overexpression is associated with apoptosis in many cancers, including breast cancer [135,136], leukemia [137,138], osteosarcoma [139], lung [140], and cervical cancer [141]. In breast cancer cells, overexpression of HOXA5 promotes cell apoptosis by upregulating p53 expression [135] or activating caspase 2 and caspase 8 [136]. In addition, HOXA5 is also involved in RA-mediated apoptosis and cell growth inhibition [142]. Similarly, HOXA10 upregulation can also lead to increased p53 expression and induce apoptosis in ER positive breast cancer cell lines [143]. In contrast, HOXA9 and HOXC6 are considered to be apoptosis-suppressor genes. In T-cell acute lymphoblastic leukemia (T-ALL), HOXA9 acts as an oncogene by cooperating with JAK3/STAT5 at the transcriptional level through upregulating the expression of downstream genes, such as PIM1 and activator protein-1 (AP-1), thereby affecting cell survival and apoptosis [147]. Moreover, HOXA9 could also eliminate Meis1a-mediated apoptosis rather than Pbx1b-mediated apoptosis [126]. Notably, HOXC6 plays an important anti-apoptotic role by regulating BCL-2 expression [144,145] in human head and neck squamous cell carcinoma and cervical cancer, and suppressing NEP/MME and IGFBP-3 genes [146] in prostate cancer. Evidence that HOX genes could function differentially in cell apoptosis in certain cancers needs to be explored further in order to clearly demonstrate which cancer types will be suitable for HOX-targeted therapy in apoptotic signaling pathways.
4.5. Proliferation
Cancer cells are known as immortalized cells that could have unlimited proliferation and never die under appropriate conditions. Most of the HOX genes actively participate in cell proliferation in many cancers. In breast cancer, HOXA1 can stimulate cell proliferation and survival by activating the p44/42 MAPK (mitogen-activated protein kinase) signaling pathway [148] or the NF-κB pathway [149]. HOXA9 can directly drive the expression of IGF1 (insulin-like growth factor 1), which subsequently plays a key role in activating the insulin/IGF signaling pathway and other downstream signaling cascades. Functionally, proliferation and survival are preferentially affected in HOXA9-induced leukemia [150]. Besides, high expression of HOXB7 can promote cell proliferation and growth in colorectal [151] and hepatocellular carcinoma [152]. In colorectal cancer cells, HOXB7 is capable of inducing acceleration of G1-S transitions by activating PI3K/AKT and MAPK pathways, resulting in upregulation of p27Kip1 and cyclin D1 [151]. Moreover, upregulated HOXC6 expression enhances the proliferation of gastrointestinal carcinoids cells through activation of the oncogenic AP-1 signaling pathway [153]. In addition, overexpressed oncogene HOXB3 can lead to increased proliferation in NCI-H1437 and A549 cells [154,177]. HOXB3 could activate DNMT3B expression and subsequently lead to promoter methylation and repression of the gene, RASSFA1 [154], eventually eliminating the proliferation inhibition. Similarly, HOXD3 has also been shown to increase the proliferation and anti-apoptosis activity by activating genes associated with the MAPK/AKT cell signaling pathways in hepatocellular carcinoma [155]. HOXB9 expression could be upregulated by sustained ERK5 signal activity and actively participate in proliferation and anti-apoptosis in HL cell lines [156].
On the other hand, some HOX genes, including HOXC5, act as a proliferation inhibiting gene in cancer. In thymoma and testicular germ cell tumor (TGCT), expression of HOXC5 plays key roles in suppressing the activity of hTERT [157], a protein subunit of telomerase, which is often abnormally activated and involved in proliferation in cancer [158]. Thus, HOXC5 expression could prevent tumorigenesis by inhibiting the proliferation of adult somatic cells [157].
Intriguingly, the function of the HOXA10 gene is quite comprehensive, and acts as both a proliferation enhancer and proliferation inhibitor in cancers. HOXA10 overexpression could stimulate the proliferation of primitive myeloid progenitor cells, resulting in myeloid leukemia development [159], but also inhibit the proliferation during G2/M phases in testicular cancer cell models [160]. Collectively, these context-specific roles of HOXA10 in cancer cell proliferation serve as a cautionary reminder of therapeutic targeting of HOXA10. More evidence needs to be explored to determine which cancer types are suitable for therapeutic interference of cell proliferation with HOXA10 and other HOX proteins.
4.6. Invasion and Metastasis
Invasion and metastasis are the most common causes for mortality of cancer. HOX genes can function as invasion and metastasis-suppressor genes in cancer development. It has been revealed that multiple HOXA genes were involved in promoting invasion in breast cancer cells through the HMGA2/TET1/HOXA signaling pathway [161]. The expression of TET1 or HOXA9 significantly reduced the bone metastasis of breast cancer cells [161]. In addition, downregulation of HOXA10 expression in endometrial carcinoma cells is responsible for their invasive behavior, which might be due to the effect of HOXA10 on the inhibition of EMT by inducing the expression of epithelial cell adhesion molecule E-cadherin [162]. HOXB1 and HOXB3 expression downregulated by microRNA-10a could facilitate invasion and metastasis in pancreatic cancer cells [163]. While overexpression of microRNA-10b accounts for invasive and metastatic behavior in metastatic breast cancer by inhibiting synthesis of HOXD10 protein at the post-transcriptional level [156,164,165,166]. Downregulation of HOXD10 in cancer results in downregulation of microRNA-7 expression and upregulation of PAK1 expression, therefore promoting invasion and metastasis [167].
In contrast, other HOX genes, such as HOXB7, can function as invasion and metastasis-inducer genes in cancer development. HOXB7 overexpression contributes to tumorigenesis, tumor migration, and invasion through the induction of EMT in epithelial cells [168]. Also, HOXB7 overexpression induces invasive and metastatic breast cancer by activating the TGFβ signaling pathway [169]. In addition, increased metastases induced by mircoRNA-196b-5p in colorectal cancer is partially dependent on the regulation of HOXB7 and GALNT5 expression [170]. Besides the HOX transcription factors, lncRNA HOXA11-AS expression is positively correlated with the migration and invasion ability of gastric cancer cells [171]. Collectively, the mechanisms by which the HOX transcription factors described above regulate tumor invasion and metastasis are likely to be divergent, but the specific suppressive and inducing roles need to be further investigated as cancer-context-dependent therapeutic targets.
4.7. Metabolism
Metabolic pathways that support cell growth are altered and are not uniform in cancer cells [178]. Hypoxic tumor cells preferentially metabolize glucose to produce and release lactic acid through glycolysis, while other normal tumor cells use lactic acid as the substrate of mitochondrial oxidative phosphorylation (OXPHOS) [178]. It has been reported that HOXA9 [172] and HOXC8 [173] are involved in glycolysis and play important role in cancer metabolism. HOXA9 can function as a tumor suppressor gene though downregulation of the HIF-1α gene in cutaneous squamous cell carcinoma (cSCC) [172]. Given the essential role of HIF-1α in glucose metabolism [179], glycolysis is inhibited by the tumor suppressor, HOXA9 [172]. The expression of microRNA-365 can downregulate HOXA9 expression by directly binding to its 3′ UTR, thus raising a possibility to therapeutically target this microRNA in cSCC with low levels of HOXA9 [172]. Moreover, ectopic expression of HOXC8 can downregulate glycolysis-related genes, upregulate TCA cycle-related genes, and subsequently inhibit nasopharyngeal carcinoma progression [173]. Thus, sustained expression of HOXA9 and HOXC8 may provide a potential therapeutic strategy to inhibit tumor growth in glycolysis-exuberant cancer.
5. Conclusions
In this paper, the function and mechanism of HOX genes in cancer predisposition and progression were discussed. Briefly, several germline coding mutations in HOX genes, and common genetic variations in gene regulatory elements that regulate HOX expression or are recognized by given HOX transcription factors could lead to increased cancer susceptibility. By contrast, HOX genes also mediate the oncogenic effect of other genetic and epigenetic variations, including an abnormal epigenetic profile on HOX genes, somatic mutations in other genes, and gene fusions that can regulate the expression of HOX genes through the establishment of abnormal epigenetic modification. The deregulated HOX genes might subsequently cause cancer progression from seven tumor-relevant aspects, including angiogenesis, autophagy, differentiation, apoptosis, proliferation, invasion, and metastasis as well as metabolism as briefly summarized in Table 1. Notwithstanding the challenge of deciphering the cancer-context-specific roles of these HOX transcription factors and developing anti-HOX therapies in cancer settings, future cancer clinical treatment plans based on these findings can likely be identified. Thus, further studies are demanded to fully illustrate the function and mechanisms by which HOX genes contribute to cancer predisposition and progression before these efforts can be eventually translated into the development of new strategies for precision cancer medicine.
Author Contributions
Conceptualization, B.L, Q.H. and G.-H.W.; Writing—Original Draft Preparation, B.L, Q.H. and G.-H.W.; Writing—Review & Editing, Q.H. and G.-H.W.; Visualization, B.L, Q.H. and G.-H.W.; Funding Acquisition, Q.H. and G.-H.W.
Funding
This work was supported by the grants Shandong Provincial Natural Science Foundation, China, ZR2016CM50, the Academy of Finland, the Finnish Cancer Foundation, and the Jane and Aatos Erkkos Foundation.
Conflicts of Interest
The authors declare no conflict of interest. The funders played no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Shah, N.; Sukumar, S. The hox genes and their roles in oncogenesis. Nat. Rev. Cancer 2010, 10, 361–371. [Google Scholar] [CrossRef]
- Luo, Z.; Rhie, S.K.; Farnham, P.J. The enigmatic hox genes: Can we crack their code? Cancers 2019, 11, 323. [Google Scholar] [CrossRef]
- Berger, M.F.; Badis, G.; Gehrke, A.R.; Talukder, S.; Philippakis, A.A.; Pena-Castillo, L.; Alleyne, T.M.; Mnaimneh, S.; Botvinnik, O.B.; Chan, E.T.; et al. Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell 2008, 133, 1266–1276. [Google Scholar] [CrossRef]
- Mann, R.S.; Lelli, K.M.; Joshi, R. Hox specificity unique roles for cofactors and collaborators. Curr. Top. Dev. Biol. 2009, 88, 63–101. [Google Scholar] [PubMed]
- Sun, Y.; Zhou, B.; Mao, F.; Xu, J.; Miao, H.; Zou, Z.; Phuc Khoa, L.T.; Jang, Y.; Cai, S.; Witkin, M.; et al. Hoxa9 reprograms the enhancer landscape to promote leukemogenesis. Cancer Cell 2018, 34, 643–658. [Google Scholar] [CrossRef]
- Mohr, S.; Doebele, C.; Comoglio, F.; Berg, T.; Beck, J.; Bohnenberger, H.; Alexe, G.; Corso, J.; Strobel, P.; Wachter, A.; et al. Hoxa9 and meis1 cooperatively induce addiction to syk signaling by suppressing mir-146a in acute myeloid leukemia. Cancer Cell 2017, 31, 549–562. [Google Scholar] [CrossRef]
- Gilbert, P.M.; Mouw, J.K.; Unger, M.A.; Lakins, J.N.; Gbegnon, M.K.; Clemmer, V.B.; Benezra, M.; Licht, J.D.; Boudreau, N.J.; Tsai, K.K.; et al. Hoxa9 regulates brca1 expression to modulate human breast tumor phenotype. J. Clin. Investig. 2010, 120, 1535–1550. [Google Scholar] [CrossRef]
- Lichtenstein, P.; Holm, N.V.; Verkasalo, P.K.; Iliadou, A.; Kaprio, J.; Koskenvuo, M.; Pukkala, E.; Skytthe, A.; Hemminki, K. Environmental and heritable factors in the causation of cancer—Analyses of cohorts of twins from sweden, denmark, and finland. N. Engl. J. Med. 2000, 343, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Peters, U.; Jiao, S.; Schumacher, F.R.; Hutter, C.M.; Aragaki, A.K.; Baron, J.A.; Berndt, S.I.; Bezieau, S.; Brenner, H.; Butterbach, K.; et al. Identification of genetic susceptibility loci for colorectal tumors in a genome-wide meta-analysis. Gastroenterology 2013, 144, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Closas, M.; Chanock, S. Genetic susceptibility loci for breast cancer by estrogen receptor status. Clin. Cancer Res. 2008, 14, 8000–8009. [Google Scholar] [CrossRef] [PubMed]
- Eeles, R.A.; Olama, A.A.; Benlloch, S.; Saunders, E.J.; Leongamornlert, D.A.; Tymrakiewicz, M.; Ghoussaini, M.; Luccarini, C.; Dennis, J.; Jugurnauth-Little, S.; et al. Identification of 23 new prostate cancer susceptibility loci using the icogs custom genotyping array. Nat. Genet. 2013, 45, 385–391. [Google Scholar] [CrossRef]
- Brennan, P.; Hainaut, P.; Boffetta, P. Genetics of lung-cancer susceptibility. Lancet Oncol. 2011, 12, 399–408. [Google Scholar] [CrossRef]
- Chen, D.; Gyllensten, U. Lessons and implications from association studies and post-gwas analyses of cervical cancer. Trends Genet. 2015, 31, 41–54. [Google Scholar] [CrossRef]
- Sherborne, A.L.; Hemminki, K.; Kumar, R.; Bartram, C.R.; Stanulla, M.; Schrappe, M.; Petridou, E.; Semsei, A.F.; Szalai, C.; Sinnett, D.; et al. Rationale for an international consortium to study inherited genetic susceptibility to childhood acute lymphoblastic leukemia. Haematologica 2011, 96, 1049–1054. [Google Scholar] [CrossRef] [Green Version]
- Crowther-Swanepoel, D.; Houlston, R.S. Genetic variation and risk of chronic lymphocytic leukaemia. Semin. Cancer Biol. 2010, 20, 363–369. [Google Scholar] [CrossRef]
- Frank, S.A. Genetic predisposition to cancer—Insights from population genetics. Nat. Rev. Genet. 2004, 5, 764–772. [Google Scholar] [CrossRef]
- Edwards, S.; Campbell, C.; Flohr, P.; Shipley, J.; Giddings, I.; Te-Poele, R.; Dodson, A.; Foster, C.; Clark, J.; Jhavar, S.; et al. Expression analysis onto microarrays of randomly selected cdna clones highlights hoxb13 as a marker of human prostate cancer. Br. J. Cancer 2005, 92, 376–381. [Google Scholar] [CrossRef]
- Wang, Z.; Dahiya, S.; Provencher, H.; Muir, B.; Carney, E.; Coser, K.; Shioda, T.; Ma, X.J.; Sgroi, D.C. The prognostic biomarkers hoxb13, il17br, and chdh are regulated by estrogen in breast cancer. Clin. Cancer Res. 2007, 13, 6327–6334. [Google Scholar] [CrossRef]
- Miao, J.; Wang, Z.; Provencher, H.; Muir, B.; Dahiya, S.; Carney, E.; Leong, C.O.; Sgroi, D.C.; Orsulic, S. Hoxb13 promotes ovarian cancer progression. Proc. Natl. Acad. Sci. USA 2007, 104, 17093–17098. [Google Scholar] [CrossRef]
- Jung, C.; Kim, R.S.; Zhang, H.; Lee, S.J.; Sheng, H.; Loehrer, P.J.; Gardner, T.A.; Jeng, M.H.; Kao, C. Hoxb13 is downregulated in colorectal cancer to confer tcf4-mediated transactivation. Br. J. Cancer 2005, 92, 2233–2239. [Google Scholar] [CrossRef]
- Zhang, E.; Han, L.; Yin, D.; He, X.; Hong, L.; Si, X.; Qiu, M.; Xu, T.; De, W.; Xu, L.; et al. H3k27 acetylation activated-long non-coding rna ccat1 affects cell proliferation and migration by regulating spry4 and hoxb13 expression in esophageal squamous cell carcinoma. Nucleic Acids Res. 2017, 45, 3086–3101. [Google Scholar] [CrossRef]
- Ewing, C.M.; Ray, A.M.; Lange, E.M.; Zuhlke, K.A.; Robbins, C.M.; Tembe, W.D.; Wiley, K.E.; Isaacs, S.D.; Johng, D.; Wang, Y.; et al. Germline mutations in hoxb13 and prostate-cancer risk. N. Engl. J. Med. 2012, 366, 141–149. [Google Scholar] [CrossRef]
- Lynch, H.T.; Shaw, T.G. Familial prostate cancer and hoxb13 founder mutations: Geographic and racial/ethnic variations. Hum. Genet. 2013, 132, 1–4. [Google Scholar] [CrossRef]
- Karlsson, R.; Aly, M.; Clements, M.; Zheng, L.; Adolfsson, J.; Xu, J.; Gronberg, H.; Wiklund, F. A population-based assessment of germline hoxb13 g84e mutation and prostate cancer risk. Eur. Urol. 2014, 65, 169–176. [Google Scholar] [CrossRef]
- Hoffmann, T.J.; Sakoda, L.C.; Shen, L.; Jorgenson, E.; Habel, L.A.; Liu, J.; Kvale, M.N.; Asgari, M.M.; Banda, Y.; Corley, D.; et al. Imputation of the rare hoxb13 g84e mutation and cancer risk in a large population-based cohort. PLoS Genet. 2015, 11, e1004930. [Google Scholar] [CrossRef]
- Kote-Jarai, Z.; Mikropoulos, C.; Leongamornlert, D.A.; Dadaev, T.; Tymrakiewicz, M.; Saunders, E.J.; Jones, M.; Jugurnauth-Little, S.; Govindasami, K.; Guy, M.; et al. Prevalence of the hoxb13 g84e germline mutation in british men and correlation with prostate cancer risk, tumour characteristics and clinical outcomes. Ann. Oncol. 2015, 26, 756–761. [Google Scholar] [CrossRef]
- Storebjerg, T.M.; Hoyer, S.; Kirkegaard, P.; Bro, F.; LuCamp Study, G.; Orntoft, T.F.; Borre, M.; Sorensen, K.D. Prevalence of the hoxb13 g84e mutation in danish men undergoing radical prostatectomy and its correlations with prostate cancer risk and aggressiveness. BJU Int. 2016, 118, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Pilie, P.G.; Johnson, A.M.; Hanson, K.L.; Dayno, M.E.; Kapron, A.L.; Stoffel, E.M.; Cooney, K.A. Germline genetic variants in men with prostate cancer and one or more additional cancers. Cancer 2017, 123, 3925–3932. [Google Scholar] [CrossRef] [PubMed]
- Brechka, H.; Bhanvadia, R.R.; VanOpstall, C.; Vander Griend, D.J. Hoxb13 mutations and binding partners in prostate development and cancer: Function, clinical significance, and future directions. Genes Dis. 2017, 4, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ewing, C.M.; Zheng, S.; Grindedaal, E.M.; Cooney, K.A.; Wiley, K.; Djurovic, S.; Andreassen, O.A.; Axcrona, K.; Mills, I.G.; et al. Genetic factors influencing prostate cancer risk in norwegian men. Prostate 2018, 78, 186–192. [Google Scholar] [CrossRef]
- Lin, X.; Qu, L.; Chen, Z.; Xu, C.; Ye, D.; Shao, Q.; Wang, X.; Qi, J.; Chen, Z.; Zhou, F.; et al. A novel germline mutation in hoxb13 is associated with prostate cancer risk in chinese men. Prostate 2013, 73, 169–175. [Google Scholar] [CrossRef]
- Hayano, T.; Matsui, H.; Nakaoka, H.; Ohtake, N.; Hosomichi, K.; Suzuki, K.; Inoue, I. Germline variants of prostate cancer in japanese families. PLoS ONE 2016, 11, e0164233. [Google Scholar] [CrossRef]
- Beebe-Dimmer, J.L.; Hathcock, M.; Yee, C.; Okoth, L.A.; Ewing, C.M.; Isaacs, W.B.; Cooney, K.A.; Thibodeau, S.N. The hoxb13 g84e mutation is associated with an increased risk for prostate cancer and other malignancies. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1366–1372. [Google Scholar] [CrossRef]
- Alanee, S.; Couch, F.; Offit, K. Association of a hoxb13 variant with breast cancer. N. Engl. J. Med. 2012, 367, 480–481. [Google Scholar] [CrossRef]
- Akbari, M.R.; Anderson, L.N.; Buchanan, D.D.; Clendenning, M.; Jenkins, M.A.; Win, A.K.; Hopper, J.L.; Giles, G.G.; Nam, R.; Narod, S.; et al. Germline hoxb13 p.Gly84glu mutation and risk of colorectal cancer. Cancer Epidemiol. 2013, 37, 424–427. [Google Scholar] [CrossRef]
- Williams, T.M.; Williams, M.E.; Innis, J.W. Range of hox/tale superclass associations and protein domain requirements for hoxa13:Meis interaction. Dev. Biol. 2005, 277, 457–471. [Google Scholar] [CrossRef]
- Johng, D.; Torga, G.; Ewing, C.M.; Jin, K.; Norris, J.D.; McDonnell, D.P.; Isaacs, W.B. Hoxb13 interaction with meis1 modifies proliferation and gene expression in prostate cancer. Prostate 2019, 79, 414–424. [Google Scholar] [CrossRef]
- Chandrasekaran, G.; Hwang, E.C.; Kang, T.W.; Kwon, D.D.; Park, K.; Lee, J.J.; Lakshmanan, V.K. Computational modeling of complete hoxb13 protein for predicting the functional effect of snps and the associated role in hereditary prostate cancer. Sci. Rep. 2017, 7, 43830. [Google Scholar] [CrossRef]
- FitzGerald, L.M.; Raspin, K.; Marthick, J.R.; Field, M.A.; Malley, R.C.; Thomson, R.J.; Blackburn, N.B.; Banks, A.; Charlesworth, J.C.; Donovan, S.; et al. Impact of the G84E variant on HOXB13 gene and protein expression in formalin-fixed, paraffin-embedded prostate tumours. Sci. Rep. 2017, 7, 17778. [Google Scholar] [CrossRef] [Green Version]
- Sipeky, C.; Gao, P.; Zhang, Q.; Wang, L.; Ettala, O.; Talala, K.M.; Tammela, T.L.J.; Auvinen, A.; Wiklund, F.; Wei, G.H.; et al. Synergistic interaction of hoxb13 and cip2a predisposes to aggressive prostate cancer. Clin. Cancer Res. 2018, 24, 6265–6276. [Google Scholar] [CrossRef]
- van Scherpenzeel Thim, V.; Remacle, S.; Picard, J.; Cornu, G.; Gofflot, F.; Rezsohazy, R.; Verellen-Dumoulin, C. Mutation analysis of the hox paralogous 4-13 genes in children with acute lymphoid malignancies: Identification of a novel germline mutation of hoxd4 leading to a partial loss-of-function. Hum. Mutat. 2005, 25, 384–395. [Google Scholar] [CrossRef]
- Richards, E.J.; Permuth-Wey, J.; Li, Y.J.; Chen, Y.A.; Coppola, D.; Reid, B.M.; Lin, H.Y.; Teer, J.K.; Berchuck, A.; Birrer, M.J.; et al. A functional variant in hoxa11-as, a novel long non-coding rna, inhibits the oncogenic phenotype of epithelial ovarian cancer. Oncotarget 2015, 6, 34745–34757. [Google Scholar] [CrossRef]
- Li, J.; Liu, R.; Tang, S.; Feng, F.; Wang, X.; Qi, L.; Liu, C.; Yao, Y.; Sun, C. The effect of long noncoding rnas hox transcript antisense intergenic rna single-nucleotide polymorphisms on breast cancer, cervical cancer, and ovarian cancer susceptibility: A meta-analysis. J. Cell. Biochem. 2018. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, L.; Lin, Z.; Ji, X.; Pi, L.; Lin, X.; Tian, N.; Liu, G.; Liu, Q.; Lin, Z.; et al. Snp-snp and snp-environment interactions of potentially functional hotair snps modify the risk of hepatocellular carcinoma. Mol. Carcinog. 2018. [Google Scholar] [CrossRef]
- Gao, P.; Wei, G.H. Genomic insight into the role of lncrna in cancer susceptibility. Int. J. Mol. Sci. 2017, 18, 1239. [Google Scholar]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.Z.; Brody, J.; et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef]
- Huang, Q.; Whitington, T.; Gao, P.; Lindberg, J.F.; Yang, Y.; Sun, J.; Vaisanen, M.R.; Szulkin, R.; Annala, M.; Yan, J.; et al. A prostate cancer susceptibility allele at 6q22 increases rfx6 expression by modulating hoxb13 chromatin binding. Nat. Genet. 2014, 46, 126–135. [Google Scholar] [CrossRef]
- Whitington, T.; Gao, P.; Song, W.; Ross-Adams, H.; Lamb, A.D.; Yang, Y.H.; Svezia, I.; Klevebring, D.; Mills, I.G.; Karlsson, R.; et al. Gene regulatory mechanisms underpinning prostate cancer susceptibility. Nat. Genet. 2016, 48, 387–397. [Google Scholar] [CrossRef]
- Su, J.; Huang, Y.H.; Cui, X.; Wang, X.; Zhang, X.; Lei, Y.; Xu, J.; Lin, X.; Chen, K.; Lv, J.; et al. Homeobox oncogene activation by pan-cancer DNA hypermethylation. Genome Biol. 2018, 19, 108. [Google Scholar] [CrossRef]
- Prokunina-Olsson, L.; Fu, Y.P.; Tang, W.; Jacobs, K.B.; Hayes, R.B.; Kraft, P.; Berndt, S.I.; Wacholder, S.; Yu, K.; Hutchinson, A.; et al. Refining the prostate cancer genetic association within the jazf1 gene on chromosome 7p15.2. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1349–1355. [Google Scholar] [CrossRef]
- Chen, L.S.; Fann, J.C.Y.; Chiu, S.Y.H.; Yen, A.M.F.; Wahlfors, T.; Tammela, T.L.; Chen, H.H.; Auvinen, A.; Schleutker, J. Assessing interactions of two loci (rs4242382 and rs10486567) in familial prostate cancer: Statistical evaluation of epistasis. PLoS ONE 2014, 9, e89508. [Google Scholar] [CrossRef]
- Han, Y.; Hazelett, D.J.; Wiklund, F.; Schumacher, F.R.; Stram, D.O.; Berndt, S.I.; Wang, Z.; Rand, K.A.; Hoover, R.N.; Machiela, M.J.; et al. Integration of multiethnic fine-mapping and genomic annotation to prioritize candidate functional snps at prostate cancer susceptibility regions. Hum. Mol. Genet. 2015, 24, 5603–5618. [Google Scholar] [CrossRef]
- Luo, Z.; Rhie, S.K.; Lay, F.D.; Farnham, P.J. A prostate cancer risk element functions as a repressive loop that regulates hoxa13. Cell Rep. 2017, 21, 1411–1417. [Google Scholar] [CrossRef]
- Kelemen, L.E.; Lawrenson, K.; Tyrer, J.; Li, Q.; Lee, J.M.; Seo, J.H.; Phelan, C.M.; Beesley, J.; Chen, X.; Spindler, T.J.; et al. Genome-wide significant risk associations for mucinous ovarian carcinoma. Nat. Genet. 2015, 47, 888–897. [Google Scholar] [CrossRef] [Green Version]
- Lawrenson, K.; Li, Q.; Kar, S.; Seo, J.H.; Tyrer, J.; Spindler, T.J.; Lee, J.; Chen, Y.; Karst, A.; Drapkin, R.; et al. Cis-eqtl analysis and functional validation of candidate susceptibility genes for high-grade serous ovarian cancer. Nat. Commun. 2015, 6, 8234. [Google Scholar] [CrossRef]
- Goode, E.L.; Chenevix-Trench, G.; Song, H.; Ramus, S.J.; Notaridou, M.; Lawrenson, K.; Widschwendter, M.; Vierkant, R.A.; Larson, M.C.; Kjaer, S.K.; et al. A genome-wide association study identifies susceptibility loci for ovarian cancer at 2q31 and 8q24. Nat. Genet. 2010, 42, 874–879. [Google Scholar] [CrossRef]
- Guo, L.Y.; Peng, Y.; Sun, L.; Han, X.; Xu, J.; Mao, D.W. Ovarian cancer variant rs2072590 is associated with hoxd1 and hoxd3 gene expression. Oncotarget 2017, 8, 103410–103414. [Google Scholar] [CrossRef]
- Li, L.; Wang, Y.; Zhang, X.; Huang, Q.; Diao, Y.; Yin, H.; Liu, H. Long non-coding rna hoxd-as1 in cancer. Clin. Chim. Acta 2018, 487, 197–201. [Google Scholar] [CrossRef]
- Tong, N.; Xu, B.; Shi, D.; Du, M.; Li, X.; Sheng, X.; Wang, M.; Chu, H.; Fang, Y.; Li, J.; et al. Hsa-mir-196a2 polymorphism increases the risk of acute lymphoblastic leukemia in chinese children. Mutat. Res. 2014, 759, 16–21. [Google Scholar] [CrossRef]
- Sibin, M.K.; Harshitha, S.M.; Narasingarao, K.V.; Dhananjaya, I.B.; Dhaval, P.S.; Chetan, G.K. Effect of rs11614913 polymorphism on mature mir196a2 expression and its target gene hoxc8 expression in human glioma. J. Mol. Neurosci. 2017, 61, 144–151. [Google Scholar] [CrossRef]
- Duan, Z.; Zarebski, A.; Montoya-Durango, D.; Grimes, H.L.; Horwitz, M. Gfi1 coordinates epigenetic repression of p21cip/waf1 by recruitment of histone lysine methyltransferase g9a and histone deacetylase 1. Mol. Cell. Biol. 2005, 25, 10338–10351. [Google Scholar] [CrossRef]
- Saleque, S.; Kim, J.; Rooke, H.M.; Orkin, S.H. Epigenetic regulation of hematopoietic differentiation by gfi-1 and gfi-1b is mediated by the cofactors corest and lsd1. Mol. Cell 2007, 27, 562–572. [Google Scholar] [CrossRef]
- Khandanpour, C.; Thiede, C.; Valk, P.J.; Sharif-Askari, E.; Nuckel, H.; Lohmann, D.; Horsthemke, B.; Siffert, W.; Neubauer, A.; Grzeschik, K.H.; et al. A variant allele of growth factor independence 1 (gfi1) is associated with acute myeloid leukemia. Blood 2010, 115, 2462–2472. [Google Scholar] [CrossRef]
- Khandanpour, C.; Krongold, J.; Schutte, J.; Bouwman, F.; Vassen, L.; Gaudreau, M.C.; Chen, R.; Calero-Nieto, F.J.; Diamanti, E.; Hannah, R.; et al. The human gfi136n variant induces epigenetic changes at the hoxa9 locus and accelerates k-ras driven myeloproliferative disorder in mice. Blood 2012, 120, 4006–4017. [Google Scholar] [CrossRef]
- Botezatu, L.; Michel, L.C.; Helness, A.; Vadnais, C.; Makishima, H.; Hones, J.M.; Robert, F.; Vassen, L.; Thivakaran, A.; Al-Matary, Y.; et al. Epigenetic therapy as a novel approach for gfi136n-associated murine/human aml. Exp. Hematol. 2016, 44, 713–726. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, L.; Fu, G.; Sun, F.; Shi, J.; Wei, J.; Lu, C.; Zhou, C.; Yuan, Q.; Yang, M. The identification of an escc susceptibility snp rs920778 that regulates the expression of lncrna hotair via a novel intronic enhancer. Carcinogenesis 2014, 35, 2062–2067. [Google Scholar] [CrossRef]
- Deplancke, B.; Alpern, D.; Gardeux, V. The genetics of transcription factor DNA binding variation. Cell 2016, 166, 538–554. [Google Scholar] [CrossRef]
- Gao, P.; Xia, J.H.; Sipeky, C.; Dong, X.M.; Zhang, Q.; Yang, Y.; Zhang, P.; Cruz, S.P.; Zhang, K.; Zhu, J.; et al. Biology and clinical implications of the 19q13 aggressive prostate cancer susceptibility locus. Cell 2018, 174, 576–589. [Google Scholar] [CrossRef]
- Spisak, S.; Lawrenson, K.; Fu, Y.; Csabai, I.; Cottman, R.T.; Seo, J.H.; Haiman, C.; Han, Y.; Lenci, R.; Li, Q.; et al. Causel: An epigenome- and genome-editing pipeline for establishing function of noncoding gwas variants. Nat. Med. 2015, 21, 1357–1363. [Google Scholar] [CrossRef]
- Amin Al Olama, A.; Kote-Jarai, Z.; Schumacher, F.R.; Wiklund, F.; Berndt, S.I.; Benlloch, S.; Giles, G.G.; Severi, G.; Neal, D.E.; Hamdy, F.C.; et al. A meta-analysis of genome-wide association studies to identify prostate cancer susceptibility loci associated with aggressive and non-aggressive disease. Hum. Mol. Genet. 2013, 22, 408–415. [Google Scholar] [CrossRef]
- Shui, I.M.; Lindstrom, S.; Kibel, A.S.; Berndt, S.I.; Campa, D.; Gerke, T.; Penney, K.L.; Albanes, D.; Berg, C.; Bueno-de-Mesquita, H.B.; et al. Prostate cancer (pca) risk variants and risk of fatal pca in the national cancer institute breast and prostate cancer cohort consortium. Eur. Urol. 2014, 65, 1069–1075. [Google Scholar] [CrossRef]
- Hua, J.T.; Ahmed, M.; Guo, H.; Zhang, Y.; Chen, S.; Soares, F.; Lu, J.; Zhou, S.; Wang, M.; Li, H.; et al. Risk snp-mediated promoter-enhancer switching drives prostate cancer through lncrna pcat19. Cell 2018, 174, 564–575. [Google Scholar] [CrossRef]
- Xia, J.H.; Wei, G.H. Oncogenic regulatory circuits driven by 19q13 rs11672691 underlies prostate cancer aggressiveness. Mol. Cell. Oncol. 2018, 5, e1516451. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Schubeler, D. Function and information content of DNA methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef]
- Strathdee, G.; Brown, R. Aberrant DNA methylation in cancer: Potential clinical interventions. Expert Rev. Mol. Med. 2002, 4, 1–17. [Google Scholar] [CrossRef]
- Chen, L.N.; Rubin, R.S.; Othepa, E.; Cer, C.; Yun, E.; Agarwal, R.P.; Collins, B.T.; McGeagh, K.; Pahira, J.; Bandi, G.; et al. Correlation of hoxd3 promoter hypermethylation with clinical and pathologic features in screening prostate biopsies. Prostate 2014, 74, 714–721. [Google Scholar] [CrossRef]
- Pilato, B.; Pinto, R.; De Summa, S.; Lambo, R.; Paradiso, A.; Tommasi, S. Hox gene methylation status analysis in patients with hereditary breast cancer. J. Hum. Genet. 2013, 58, 51–53. [Google Scholar] [CrossRef]
- Pfeifer, G.P.; Rauch, T.A. DNA methylation patterns in lung carcinomas. Semin. Cancer Biol. 2009, 19, 181–187. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, A.; Jonsson, M.; Lauss, M.; Brunnstrom, H.; Jonsson, P.; Borg, A.; Jonsson, G.; Ringner, M.; Planck, M.; Staaf, J. Genome-wide DNA methylation analysis of lung carcinoma reveals one neuroendocrine and four adenocarcinoma epitypes associated with patient outcome. Clin. Cancer Res. 2014, 20, 6127–6140. [Google Scholar] [CrossRef]
- Strathdee, G.; Sim, A.; Parker, A.; Oscier, D.; Brown, R. Promoter hypermethylation silences expression of the hoxa4 gene and correlates with igvh mutational status in cll. Leukemia 2006, 20, 1326–1329. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Lee, S.H.; Jones, A.; Fiegl, H.; Kalwa, M.; Wagner, W.; Chindera, K.; Evans, I.; Dubeau, L.; Orjalo, A.; et al. Hotair and its surrogate DNA methylation signature indicate carboplatin resistance in ovarian cancer. Genome Med. 2015, 7, 108. [Google Scholar] [CrossRef]
- Liu, Y.W.; Sun, M.; Xia, R.; Zhang, E.B.; Liu, X.H.; Zhang, Z.H.; Xu, T.P.; De, W.; Liu, B.R.; Wang, Z.X. Linchotair epigenetically silences mir34a by binding to prc2 to promote the epithelial-to-mesenchymal transition in human gastric cancer. Cell Death Dis. 2015, 6, e1802. [Google Scholar] [CrossRef]
- Xylinas, E.; Hassler, M.R.; Zhuang, D.; Krzywinski, M.; Erdem, Z.; Robinson, B.D.; Elemento, O.; Clozel, T.; Shariat, S.F. An epigenomic approach to improving response to neoadjuvant cisplatin chemotherapy in bladder cancer. Biomolecules 2016, 6, 37. [Google Scholar] [CrossRef]
- Mayle, A.; Yang, L.; Rodriguez, B.; Zhou, T.; Chang, E.; Curry, C.V.; Challen, G.A.; Li, W.; Wheeler, D.; Rebel, V.I.; et al. Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood 2015, 125, 629–638. [Google Scholar] [CrossRef]
- Tan, Y.T.; Sun, Y.; Zhu, S.H.; Ye, L.; Zhao, C.J.; Zhao, W.L.; Chen, Z.; Chen, S.J.; Liu, H. Deregulation of hox genes by dnmt3a and mll mutations converges on bmi1. Leukemia 2016, 30, 1609–1612. [Google Scholar] [CrossRef]
- Lu, R.; Wang, P.; Parton, T.; Zhou, Y.; Chrysovergis, K.; Rockowitz, S.; Chen, W.Y.; Abdel-Wahab, O.; Wade, P.A.; Zheng, D.; et al. Epigenetic perturbations by arg882-mutated dnmt3a potentiate aberrant stem cell gene-expression program and acute leukemia development. Cancer Cell 2016, 30, 92–107. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Adli, M.; LaFave, L.M.; Gao, J.; Hricik, T.; Shih, A.H.; Pandey, S.; Patel, J.P.; Chung, Y.R.; Koche, R.; et al. Asxl1 mutations promote myeloid transformation through loss of prc2-mediated gene repression. Cancer Cell 2012, 22, 180–193. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef]
- Vassiliou, G.S.; Cooper, J.L.; Rad, R.; Li, J.; Rice, S.; Uren, A.; Rad, L.; Ellis, P.; Andrews, R.; Banerjee, R.; et al. Mutant nucleophosmin and cooperating pathways drive leukemia initiation and progression in mice. Nat. Genet. 2011, 43, 470–475. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, L.; Gundry, M.C.; Sorcini, D.; Guzman, A.G.; Huang, Y.H.; Ramabadran, R.; Gionfriddo, I.; Mezzasoma, F.; Milano, F.; Nabet, B.; et al. Mutant npm1 maintains the leukemic state through hox expression. Cancer Cell 2018, 34, 499–512. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of tmprss2 and ets transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef]
- Kron, K.J.; Murison, A.; Zhou, S.; Huang, V.; Yamaguchi, T.N.; Shiah, Y.J.; Fraser, M.; van der Kwast, T.; Boutros, P.C.; Bristow, R.G.; et al. Tmprss2-erg fusion co-opts master transcription factors and activates notch signaling in primary prostate cancer. Nat. Genet. 2017, 49, 1336–1345. [Google Scholar] [CrossRef]
- Wang, J.; Cai, Y.; Ren, C.; Ittmann, M. Expression of variant tmprss2/erg fusion messenger rnas is associated with aggressive prostate cancer. Cancer Res. 2006, 66, 8347–8351. [Google Scholar] [CrossRef]
- Gough, S.M.; Lee, F.; Yang, F.; Walker, R.L.; Zhu, Y.J.; Pineda, M.; Onozawa, M.; Chung, Y.J.; Bilke, S.; Wagner, E.K.; et al. Nup98-phf23 is a chromatin-modifying oncoprotein that causes a wide array of leukemias sensitive to inhibition of phd histone reader function. Cancer Discov. 2014, 4, 564–577. [Google Scholar] [CrossRef]
- Gough, S.M.; Slape, C.I.; Aplan, P.D. Nup98 gene fusions and hematopoietic malignancies: Common themes and new biologic insights. Blood 2011, 118, 6247–6257. [Google Scholar] [CrossRef]
- Xu, H.; Valerio, D.G.; Eisold, M.E.; Sinha, A.; Koche, R.P.; Hu, W.; Chen, C.W.; Chu, S.H.; Brien, G.L.; Park, C.Y.; et al. Nup98 fusion proteins interact with the nsl and mll1 complexes to drive leukemogenesis. Cancer Cell 2016, 30, 863–878. [Google Scholar] [CrossRef]
- Takeda, A.; Goolsby, C.; Yaseen, N.R. Nup98-hoxa9 induces long-term proliferation and blocks differentiation of primary human cd34+ hematopoietic cells. Cancer Res. 2006, 66, 6628–6637. [Google Scholar] [CrossRef]
- Shields, B.J.; Slape, C.I.; Vo, N.; Jackson, J.T.; Pliego-Zamora, A.; Ranasinghe, H.; Shi, W.; Curtis, D.J.; McCormack, M.P. The nup98-hoxd13 fusion oncogene induces thymocyte self-renewal via lmo2/lyl1. Leukemia 2019. [Google Scholar] [CrossRef]
- McCormack, M.P.; Shields, B.J.; Jackson, J.T.; Nasa, C.; Shi, W.; Slater, N.J.; Tremblay, C.S.; Rabbitts, T.H.; Curtis, D.J. Requirement for Lyl1 in a model of Lmo2-driven early T-cell precursor ALL. Blood 2013, 122, 2093–2103. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.G.; Cai, L.; Pasillas, M.P.; Kamps, M.P. Nup98-nsd1 links h3k36 methylation to hox-a gene activation and leukaemogenesis. Nat. Cell Biol. 2007, 9, 804–812. [Google Scholar] [CrossRef]
- Muntean, A.G.; Hess, J.L. The pathogenesis of mixed-lineage leukemia. Annu. Rev. Pathol. 2012, 7, 283–301. [Google Scholar] [CrossRef]
- Ayton, P.M.; Cleary, M.L. Transformation of myeloid progenitors by mll oncoproteins is dependent on hoxa7 and hoxa9. Genes Dev. 2003, 17, 2298–2307. [Google Scholar] [CrossRef]
- Horton, S.J.; Grier, D.G.; McGonigle, G.J.; Thompson, A.; Morrow, M.; De Silva, I.; Moulding, D.A.; Kioussis, D.; Lappin, T.R.; Brady, H.J.; et al. Continuous mll-enl expression is necessary to establish a “hox code” and maintain immortalization of hematopoietic progenitor cells. Cancer Res. 2005, 65, 9245–9252. [Google Scholar] [CrossRef]
- Okada, Y.; Feng, Q.; Lin, Y.; Jiang, Q.; Li, Y.; Coffield, V.M.; Su, L.; Xu, G.; Zhang, Y. Hdot1l links histone methylation to leukemogenesis. Cell 2005, 121, 167–178. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, B.; Qin, S.; Xu, J.; Harding, R.; Tempel, W.; Nayak, V.; Li, Y.; Loppnau, P.; Dou, Y.; et al. Structural and functional analysis of the dot1l-af10 complex reveals mechanistic insights into mll-af10-associated leukemogenesis. Genes Dev. 2018, 32, 341–346. [Google Scholar] [CrossRef]
- Lin, Y.H.; Kakadia, P.M.; Chen, Y.; Li, Y.Q.; Deshpande, A.J.; Buske, C.; Zhang, K.L.; Zhang, Y.; Xu, G.L.; Bohlander, S.K. Global reduction of the epigenetic h3k79 methylation mark and increased chromosomal instability in calm-af10-positive leukemias. Blood 2009, 114, 651–658. [Google Scholar] [CrossRef]
- Hsu, K.; Look, A.T. Turning on a dimer. Cancer Cell 2003, 4, 81–83. [Google Scholar] [CrossRef] [Green Version]
- Pfaltzgraff, E.R.; Apfelbaum, A.; Kassa, A.P.; Song, J.Y.; Jiang, W.; Suhan, T.K.; Wellik, D.M.; Lawlor, E.R. Anatomic origin of osteochondrogenic progenitors impacts sensitivity to ews-fli1-induced transformation. Cancers 2019, 11, 313. [Google Scholar] [CrossRef]
- Svoboda, L.K.; Harris, A.; Bailey, N.J.; Schwentner, R.; Tomazou, E.; von Levetzow, C.; Magnuson, B.; Ljungman, M.; Kovar, H.; Lawlor, E.R. Overexpression of hox genes is prevalent in ewing sarcoma and is associated with altered epigenetic regulation of developmental transcription programs. Epigenetics 2014, 9, 1613–1625. [Google Scholar] [CrossRef]
- Care, A.; Felicetti, F.; Meccia, E.; Bottero, L.; Parenza, M.; Stoppacciaro, A.; Peschle, C.; Colombo, M.P. Hoxb7: A key factor for tumor-associated angiogenic switch. Cancer Res. 2001, 61, 6532–6539. [Google Scholar]
- Storti, P.; Donofrio, G.; Colla, S.; Airoldi, I.; Bolzoni, M.; Agnelli, L.; Abeltino, M.; Todoerti, K.; Lazzaretti, M.; Mancini, C.; et al. Hoxb7 expression by myeloma cells regulates their pro-angiogenic properties in multiple myeloma patients. Leukemia 2011, 25, 527–537. [Google Scholar] [CrossRef]
- Heinonen, H.; Lepikhova, T.; Sahu, B.; Pehkonen, H.; Pihlajamaa, P.; Louhimo, R.; Gao, P.; Wei, G.H.; Hautaniemi, S.; Janne, O.A.; et al. Identification of several potential chromatin binding sites of hoxb7 and its downstream target genes in breast cancer. Int. J. Cancer 2015, 137, 2374–2383. [Google Scholar] [CrossRef]
- Wu, S.Y.; Rupaimoole, R.; Shen, F.; Pradeep, S.; Pecot, C.V.; Ivan, C.; Nagaraja, A.S.; Gharpure, K.M.; Pham, E.; Hatakeyama, H.; et al. A mir-192-egr1-hoxb9 regulatory network controls the angiogenic switch in cancer. Nat. Commun. 2016, 7, 11169. [Google Scholar] [CrossRef]
- Hayashida, T.; Takahashi, F.; Chiba, N.; Brachtel, E.; Takahashi, M.; Godin-Heymann, N.; Gross, K.W.; Vivanco, M.; Wijendran, V.; Shioda, T.; et al. Hoxb9, a gene overexpressed in breast cancer, promotes tumorigenicity and lung metastasis. Proc. Natl. Acad. Sci. USA 2010, 107, 1100–1105. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, W.J.; Gan, T.Q.; Zhang, X.L.; Xie, Z.C.; Ye, Z.H.; Deng, Y.; Wang, Z.F.; Cai, K.T.; Li, S.K.; et al. Clinical significance and effect of lncrna hoxa11-as in nsclc: A study based on bioinformatics, in vitro and in vivo verification. Sci. Rep. 2017, 7, 5567. [Google Scholar] [CrossRef]
- Chen, Y.; Gorski, D.H. Regulation of angiogenesis through a microrna (mir-130a) that down-regulates antiangiogenic homeobox genes gax and hoxa5. Blood 2007, 111, 1217–1226. [Google Scholar] [CrossRef]
- Rhoads, K.; Arderiu, G.; Charboneau, A.; Hansen, S.L.; Hoffman, W.; Boudreau, N. A role for hox a5 in regulating angiogenesis and vascular patterning. Lymphat. Res. Biol. 2005, 3, 240–252. [Google Scholar] [CrossRef]
- Xuan, F.; Huang, M.; Liu, W.; Ding, H.; Yang, L.; Cui, H. Homeobox c9 suppresses beclin1-mediated autophagy in glioblastoma by directly inhibiting the transcription of death-associated protein kinase 1. Neuro Oncol. 2016, 18, 819–829. [Google Scholar] [CrossRef]
- Tsai, K.W.; Leung, C.M.; Lo, Y.H.; Chen, T.W.; Chan, W.C.; Yu, S.Y.; Tu, Y.T.; Lam, H.C.; Li, S.C.; Ger, L.P.; et al. Arm selection preference of microrna-193a varies in breast cancer. Sci. Rep. 2016, 6, 28176. [Google Scholar] [CrossRef]
- Cheng, J.Z.; Chen, J.J.; Wang, Z.G.; Yu, D. Microrna-185 inhibits cell proliferation while promoting apoptosis and autophagy through negative regulation of tgf-beta 1/mtor axis and hoxc6 in nasopharyngeal carcinoma. Cancer Biomark. 2018, 23, 107–123. [Google Scholar] [CrossRef]
- Pawlowska, E.; Szczepanska, J.; Blasiak, J. The long noncoding rna hotair in breast cancer: Does autophagy play a role? Int. J. Mol. Sci. 2017, 18, 2317. [Google Scholar] [CrossRef]
- Guo, X.; Xiao, H.; Guo, S.; Li, J.; Wang, Y.; Chen, J.; Lou, G. Long noncoding rna hotair knockdown inhibits autophagy and epithelial-mesenchymal transition through the wnt signaling pathway in radioresistant human cervical cancer hela cells. J. Cell. Physiol. 2019, 234, 3478–3489. [Google Scholar] [CrossRef]
- Bach, C.; Buhl, S.; Mueller, D.; Garcia-Cuellar, M.P.; Maethner, E.; Slany, R.K. Leukemogenic transformation by hoxa cluster genes. Blood 2010, 115, 2910–2918. [Google Scholar] [CrossRef]
- Lai, C.K.; Norddahl, G.L.; Maetzig, T.; Rosten, P.; Lohr, T.; Sanchez Milde, L.; von Krosigk, N.; Docking, T.R.; Heuser, M.; Karsan, A.; et al. Meis2 as a critical player in mn1-induced leukemia. Blood Cancer J. 2017, 7, e613. [Google Scholar] [CrossRef]
- Wermuth, P.J.; Buchberg, A.M. Meis1-mediated apoptosis is caspase dependent and can be suppressed by coexpression of hoxa9 in murine and human cell lines. Blood 2005, 105, 1222–1230. [Google Scholar] [CrossRef]
- Somerville, T.D.; Wiseman, D.H.; Spencer, G.J.; Huang, X.; Lynch, J.T.; Leong, H.S.; Williams, E.L.; Cheesman, E.; Somervaille, T.C. Frequent derepression of the mesenchymal transcription factor gene foxc1 in acute myeloid leukemia. Cancer Cell 2015, 28, 329–342. [Google Scholar] [CrossRef]
- Hatanaka, Y.; de Velasco, M.A.; Oki, T.; Shimizu, N.; Nozawa, M.; Yoshimura, K.; Yoshikawa, K.; Nishio, K.; Uemura, H. Hoxa10 expression profiling in prostate cancer. Prostate 2019, 79, 554–563. [Google Scholar] [CrossRef]
- Tanwar, P.S.; Kaneko-Tarui, T.; Lee, H.J.; Zhang, L.H.; Teixeira, J.M. Pten loss and hoxa10 expression are associated with ovarian endometrioid adenocarcinoma differentiation and progression. Carcinogenesis 2013, 34, 893–901. [Google Scholar] [CrossRef]
- Krishnaraju, K.; Hoffman, B.; Liebermann, D.A. Lineage-specific regulation of hematopoiesis by hox-b8 (hox-2.4): Inhibition of granulocytic differentiation and potentiation of monocytic differentiation. Blood 1997, 90, 1840–1849. [Google Scholar]
- Mao, L.; Ding, J.; Zha, Y.; Yang, L.; McCarthy, B.A.; King, W.; Cui, H.; Ding, H.F. Hoxc9 links cell-cycle exit and neuronal differentiation and is a prognostic marker in neuroblastoma. Cancer Res. 2011, 71, 4314–4324. [Google Scholar] [CrossRef]
- Tan, S.H.; Barker, N. Stemming colorectal cancer growth and metastasis: Hoxa5 forces cancer stem cells to differentiate. Cancer Cell 2015, 28, 683–685. [Google Scholar] [CrossRef]
- Ordonez-Moran, P.; Dafflon, C.; Imajo, M.; Nishida, E.; Huelsken, J. Hoxa5 counteracts stem cell traits by inhibiting wnt signaling in colorectal cancer. Cancer Cell 2015, 28, 815–829. [Google Scholar] [CrossRef]
- Heubach, J.; Monsior, J.; Deenen, R.; Niegisch, G.; Szarvas, T.; Niedworok, C.; Schulz, W.A.; Hoffmann, M.J. The long noncoding rna hotair has tissue and cell type-dependent effects on hox gene expression and phenotype of urothelial cancer cells. Mol. Cancer 2015, 14, 108. [Google Scholar] [CrossRef]
- Raman, V.; Martensen, S.A.; Reisman, D.; Evron, E.; Odenwald, W.F.; Jaffee, E.; Marks, J.; Sukumar, S. Compromised hoxa5 function can limit p53 expression in human breast tumours. Nature 2000, 405, 974–978. [Google Scholar] [CrossRef]
- Chen, H.; Chung, S.; Sukumar, S. Hoxa5-induced apoptosis in breast cancer cells is mediated by caspases 2 and 8. Mol. Cell. Biol. 2003, 24, 924–935. [Google Scholar] [CrossRef]
- Liu, W.J.; Zhang, T.; Guo, Q.L.; Liu, C.Y.; Bai, Y.Q. Effect of atra on the expression of hoxa5 gene in k562 cells and its relationship with cell cycle and apoptosis. Mol. Med. Rep. 2016, 13, 4221–4228. [Google Scholar] [CrossRef]
- Huang, H.P.; Liu, W.J.; Guo, Q.L.; Bai, Y.Q. Effect of silencing hoxa5 gene expression using rna interference on cell cycle and apoptosis in jurkat cells. Int. J. Mol. Med. 2016, 37, 669–678. [Google Scholar] [CrossRef]
- Chen, Y.Q.; Yang, T.Q.; Zhou, B.; Yang, M.X.; Feng, H.J.; Wang, Y.L. Hoxa5 overexpression promotes osteosarcoma cell apoptosis through the p53 and p38alpha mapk pathway. Gene 2018, 689, 18–23. [Google Scholar] [CrossRef]
- Zhu, Q.; Lv, T.; Wu, Y.; Shi, X.; Liu, H.; Song, Y. Long non-coding rna 00312 regulated by hoxa5 inhibits tumour proliferation and promotes apoptosis in non-small cell lung cancer. J. Cell. Mol. Med. 2017, 21, 2184–2198. [Google Scholar] [CrossRef]
- Wang, Z.; Yu, C.; Wang, H. Hoxa5 inhibits the proliferation and induces the apoptosis of cervical cancer cells via regulation of protein kinase b and p27. Oncol Rep. 2019, 41, 1122–1130. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, H.; Lee, J.; Liang, X.; Wu, X.; Zhu, T.; Lo, P.K.; Zhang, X.; Sukumar, S. Hoxa5 acts directly downstream of retinoic acid receptor beta and contributes to retinoic acid-induced apoptosis and growth inhibition. Cancer Res. 2007, 67, 8007–8013. [Google Scholar] [CrossRef]
- Chu, M.C.; Selam, F.B.; Taylor, H.S. Hoxa10 regulates p53 expression and matrigel invasion in human breast cancer cells. Cancer Biol. Ther. 2014, 3, 568–572. [Google Scholar] [CrossRef]
- Moon, S.M.; Kim, S.A.; Yoon, J.H.; Ahn, S.G. Hoxc6 is deregulated in human head and neck squamous cell carcinoma and modulates bcl-2 expression. J. Biol. Chem. 2012, 287, 35678–35688. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, C.; Liu, N.; Hou, J.; Xiao, W.; Wang, H. Hoxc6 promotes cervical cancer progression via regulation of bcl-2. FASEB J. 2019, 33, 3901–3911. [Google Scholar] [CrossRef]
- Ramachandran, S.; Liu, P.; Young, A.N.; Yin-Goen, Q.; Lim, S.D.; Laycock, N.; Amin, M.B.; Carney, J.K.; Marshall, F.F.; Petros, J.A.; et al. Loss of hoxc6 expression induces apoptosis in prostate cancer cells. Oncogene 2005, 24, 188–198. [Google Scholar] [CrossRef]
- de Bock, C.E.; Demeyer, S.; Degryse, S.; Verbeke, D.; Sweron, B.; Gielen, O.; Vandepoel, R.; Vicente, C.; Vanden Bempt, M.; Dagklis, A.; et al. Hoxa9 cooperates with activated jak/stat signaling to drive leukemia development. Cancer Discov. 2018, 8, 616–631. [Google Scholar] [CrossRef]
- Brock, A.; Krause, S.; Li, H.; Kowalski, M.; Goldberg, M.S.; Collins, J.J.; Ingber, D.E. Silencing hoxa1 by intraductal injection of sirna lipidoid nanoparticles prevents mammary tumor progression in mice. Sci. Transl. Med. 2014, 6, 217ra2. [Google Scholar] [CrossRef]
- Taminiau, A.; Draime, A.; Tys, J.; Lambert, B.; Vandeputte, J.; Nguyen, N.; Renard, P.; Geerts, D.; Rezsohazy, R. Hoxa1 binds rbck1/hoil-1 and traf2 and modulates the tnf/nf-kappab pathway in a transcription-independent manner. Nucleic Acids Res. 2016, 44, 7331–7349. [Google Scholar]
- Steger, J.; Fuller, E.; Garcia-Cuellar, M.P.; Hetzner, K.; Slany, R.K. Insulin-like growth factor 1 is a direct hoxa9 target important for hematopoietic transformation. Leukemia 2015, 29, 901–908. [Google Scholar] [CrossRef]
- Liao, W.T.; Jiang, D.; Yuan, J.; Cui, Y.M.; Shi, X.W.; Chen, C.M.; Bian, X.W.; Deng, Y.J.; Ding, Y.Q. Hoxb7 as a prognostic factor and mediator of colorectal cancer progression. Clin. Cancer Res. 2011, 17, 3569–3578. [Google Scholar] [CrossRef]
- Huan, H.B.; Yang, D.P.; Wen, X.D.; Chen, X.J.; Zhang, L.; Wu, L.L.; Bie, P.; Xia, F. Hoxb7 accelerates the malignant progression of hepatocellular carcinoma by promoting stemness and epithelial-mesenchymal transition. J. Exp. Clin. Cancer Res. 2017, 36, 86. [Google Scholar] [CrossRef]
- Fujiki, K.; Duerr, E.M.; Kikuchi, H.; Ng, A.; Xavier, R.J.; Mizukami, Y.; Imamura, T.; Kulke, M.H.; Chung, D.C. Hoxc6 is overexpressed in gastrointestinal carcinoids and interacts with jund to regulate tumor growth. Gastroenterology 2008, 135, 907–916. [Google Scholar] [CrossRef]
- Palakurthy, R.K.; Wajapeyee, N.; Santra, M.K.; Gazin, C.; Lin, L.; Gobeil, S.; Green, M.R. Epigenetic silencing of the rassf1a tumor suppressor gene through hoxb3-mediated induction of dnmt3b expression. Mol. Cell 2009, 36, 219–230. [Google Scholar] [CrossRef]
- Wang, L.M.; Sun, H.F.; Wang, X.F.; Hou, N.; Zhao, L.Y.; Tong, D.D.; He, K.; Yang, Y.; Song, T.S.; Yang, J.; et al. Egr1 mediates mir-203a suppress the hepatocellular carcinoma cells progression by targeting hoxd3 through egfr signaling pathway. Oncotarget 2016, 7, 45302–45316. [Google Scholar] [CrossRef]
- Nagel, S.; Burek, C.; Venturini, L.; Scherr, M.; Quentmeier, H.; Meyer, C.; Rosenwald, A.; Drexler, H.G.; MacLeod, R.A. Comprehensive analysis of homeobox genes in hodgkin lymphoma cell lines identifies dysregulated expression of hoxb9 mediated via erk5 signaling and bmi1. Blood 2007, 109, 3015–3023. [Google Scholar] [CrossRef]
- Yan, T.; Ooi, W.F.; Qamra, A.; Cheung, A.; Ma, D.; Sundaram, G.M.; Xu, C.; Xing, M.; Poon, L.; Wang, J.; et al. Hoxc5 and mir-615-3p target newly evolved genomic regions to repress htert and inhibit tumorigenesis. Nat. Commun. 2018, 9, 100. [Google Scholar] [CrossRef]
- Hahn, W.C.; Stewart, S.A.; Brooks, M.W.; York, S.G.; Eaton, E.; Kurachi, A.; Beijersbergen, R.L.; Knoll, J.H.M.; Meyerson, M.; Weinberg, R.A. Inhibition of telomerase limits the growth of human cancer cells. Nat. Med. 1999, 5, 1164–1170. [Google Scholar] [CrossRef]
- Bjornsson, J.M.; Andersson, E.; Lundstrom, P.; Larsson, N.; Xu, X.F.; Repetowska, E.; Humphries, R.K.; Karlsson, S. Proliferation of primitive myeloid progenitors can be reversibly induced by hoxa10. Blood 2001, 98, 3301–3308. [Google Scholar] [CrossRef]
- Chen, R.; Li, H.; Li, Y.; Fazli, L.; Gleave, M.; Nappi, L.; Dong, X. Loss of nuclear functions of hoxa10 is associated with testicular cancer proliferation. Front. Oncol. 2018, 8, 594. [Google Scholar] [CrossRef]
- Sun, M.; Song, C.X.; Huang, H.; Frankenberger, C.A.; Sankarasharma, D.; Gomes, S.; Chen, P.; Chen, J.; Chada, K.K.; He, C.; et al. Hmga2/tet1/hoxa9 signaling pathway regulates breast cancer growth and metastasis. Proc. Natl. Acad. Sci. USA 2013, 110, 9920–9925. [Google Scholar] [CrossRef]
- Yoshida, H.; Broaddus, R.; Cheng, W.; Xie, S.; Naora, H. Deregulation of the hoxa10 homeobox gene in endometrial carcinoma: Role in epithelial-mesenchymal transition. Cancer Res. 2006, 66, 889–897. [Google Scholar] [CrossRef]
- Weiss, F.U.; Marques, I.J.; Woltering, J.M.; Vlecken, D.H.; Aghdassi, A.; Partecke, L.I.; Heidecke, C.D.; Lerch, M.M.; Bagowski, C.P. Retinoic acid receptor antagonists inhibit mir-10a expression and block metastatic behavior of pancreatic cancer. Gastroenterology 2009, 137, 2136–2145. [Google Scholar] [CrossRef]
- Ma, L.; Reinhardt, F.; Pan, E.; Soutschek, J.; Bhat, B.; Marcusson, E.G.; Teruya-Feldstein, J.; Bell, G.W.; Weinberg, R.A. Therapeutic silencing of mir-10b inhibits metastasis in a mouse mammary tumor model. Nat. Biotechnol. 2010, 28, 341–347. [Google Scholar] [CrossRef]
- Kim, J.; Siverly, A.N.; Chen, D.; Wang, M.; Yuan, Y.; Wang, Y.; Lee, H.; Zhang, J.; Muller, W.J.; Liang, H.; et al. Ablation of mir-10b suppresses oncogene-induced mammary tumorigenesis and metastasis and reactivates tumor-suppressive pathways. Cancer Res. 2016, 76, 6424–6435. [Google Scholar] [CrossRef] [PubMed]
- Gabriely, G.; Teplyuk, N.M.; Krichevsky, A.M. Context effect: Microrna-10b in cancer cell proliferation, spread and death. Autophagy 2011, 7, 1384–1386. [Google Scholar] [CrossRef]
- Reddy, S.D.; Ohshiro, K.; Rayala, S.K.; Kumar, R. Microrna-7, a homeobox d10 target, inhibits p21-activated kinase 1 and regulates its functions. Cancer Res. 2008, 68, 8195–8200. [Google Scholar] [CrossRef]
- Wu, X.; Chen, H.; Parker, B.; Rubin, E.; Zhu, T.; Lee, J.S.; Argani, P.; Sukumar, S. Hoxb7, a homeodomain protein, is overexpressed in breast cancer and confers epithelial-mesenchymal transition. Cancer Res. 2006, 66, 9527–9534. [Google Scholar] [CrossRef]
- Liu, S.; Jin, K.; Hui, Y.; Fu, J.; Jie, C.; Feng, S.; Reisman, D.; Wang, Q.; Fan, D.; Sukumar, S.; et al. Hoxb7 promotes malignant progression by activating the tgfbeta signaling pathway. Cancer Res. 2015, 75, 709–719. [Google Scholar] [CrossRef]
- Stiegelbauer, V.; Vychytilova-Faltejskova, P.; Karbiener, M.; Pehserl, A.M.; Reicher, A.; Resel, M.; Heitzer, E.; Ivan, C.; Bullock, M.; Ling, H.; et al. Mir-196b-5p regulates colorectal cancer cell migration and metastases through interaction with hoxb7 and galnt5. Clin. Cancer Res. 2017, 23, 5255–5266. [Google Scholar] [CrossRef]
- Sun, M.; Nie, F.; Wang, Y.; Zhang, Z.; Hou, J.; He, D.; Xie, M.; Xu, L.; De, W.; Wang, Z.; et al. Lncrna hoxa11-as promotes proliferation and invasion of gastric cancer by scaffolding the chromatin modification factors prc2, lsd1, and dnmt1. Cancer Res. 2016, 76, 6299–6310. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, Y.; Zhou, M.; Zhang, Y.; Wang, P.; Li, X.; Yang, J.; Wang, H.; Ding, Z. Hoxa9 inhibits hif-1alpha-mediated glycolysis through interacting with crip2 to repress cutaneous squamous cell carcinoma development. Nat. Commun. 2018, 9, 1480. [Google Scholar] [CrossRef]
- Jiang, Y.; Yan, B.; Lai, W.; Shi, Y.; Xiao, D.; Jia, J.; Liu, S.; Li, H.; Lu, J.; Li, Z.; et al. Repression of hox genes by lmp1 in nasopharyngeal carcinoma and modulation of glycolytic pathway genes by hoxc8. Oncogene 2015, 34, 6079–6091. [Google Scholar] [CrossRef] [PubMed]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, metabolism, and cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef] [PubMed]
- Jogi, A.; Vaapil, M.; Johansson, M.; Pahlman, S. Cancer cell differentiation heterogeneity and aggressive behavior in solid tumors. Ups. J. Med. Sci. 2012, 117, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Dammann, R.; Li, C.; Yoon, J.H.; Chin, P.L.; Bates, S.; Pfeifer, G.P. Epigenetic inactivation of a ras association domain family protein from the lung tumour suppressor locus 3p21.3. Nat. Genet. 2000, 25, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Peiris-Pages, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31. [Google Scholar] [CrossRef]
- Courtnay, R.; Ngo, D.C.; Malik, N.; Ververis, K.; Tortorella, S.M.; Karagiannis, T.C. Cancer metabolism and the warburg effect: The role of hif-1 and pi3k. Mol. Biol. Rep. 2015, 42, 841–851. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Involvement of HOX genes in cancer susceptibility. (A) Coding mutations in HOX transcription factors and HOX locus long non-coding RNAs (lncRNA) conferring increased cancer risk. (B) Risk loci conferring increased cancer predisposition through HOX deregulation. Several known cancer risk loci harboring transcriptional regulatory regions can subsequently regulate the expression of HOX genes. (C) HOX transcription factors deciphering altered regulatory genetic information of risk single nucleotide polymorphism (SNP). The HOX transcription factors, HOXB13 and HOXA2, bind the risk allele with higher affinity compared to the protective allele, thereby leading to the upregulated expression of RFX6 and PCAT19/CEACAM21, respectively, and conferring prostate cancer susceptibility.
Figure 1.
Involvement of HOX genes in cancer susceptibility. (A) Coding mutations in HOX transcription factors and HOX locus long non-coding RNAs (lncRNA) conferring increased cancer risk. (B) Risk loci conferring increased cancer predisposition through HOX deregulation. Several known cancer risk loci harboring transcriptional regulatory regions can subsequently regulate the expression of HOX genes. (C) HOX transcription factors deciphering altered regulatory genetic information of risk single nucleotide polymorphism (SNP). The HOX transcription factors, HOXB13 and HOXA2, bind the risk allele with higher affinity compared to the protective allele, thereby leading to the upregulated expression of RFX6 and PCAT19/CEACAM21, respectively, and conferring prostate cancer susceptibility.

Figure 2.
HOX gene-involved functional somatic variations in cancer development. Deregulation of HOX gene expression resulting from somatic variations plays an important role in cancer development. These somatic variations mainly include epigenetic alteration, gene mutations, and gene fusions induced by chromatin translocation.
Figure 2.
HOX gene-involved functional somatic variations in cancer development. Deregulation of HOX gene expression resulting from somatic variations plays an important role in cancer development. These somatic variations mainly include epigenetic alteration, gene mutations, and gene fusions induced by chromatin translocation.


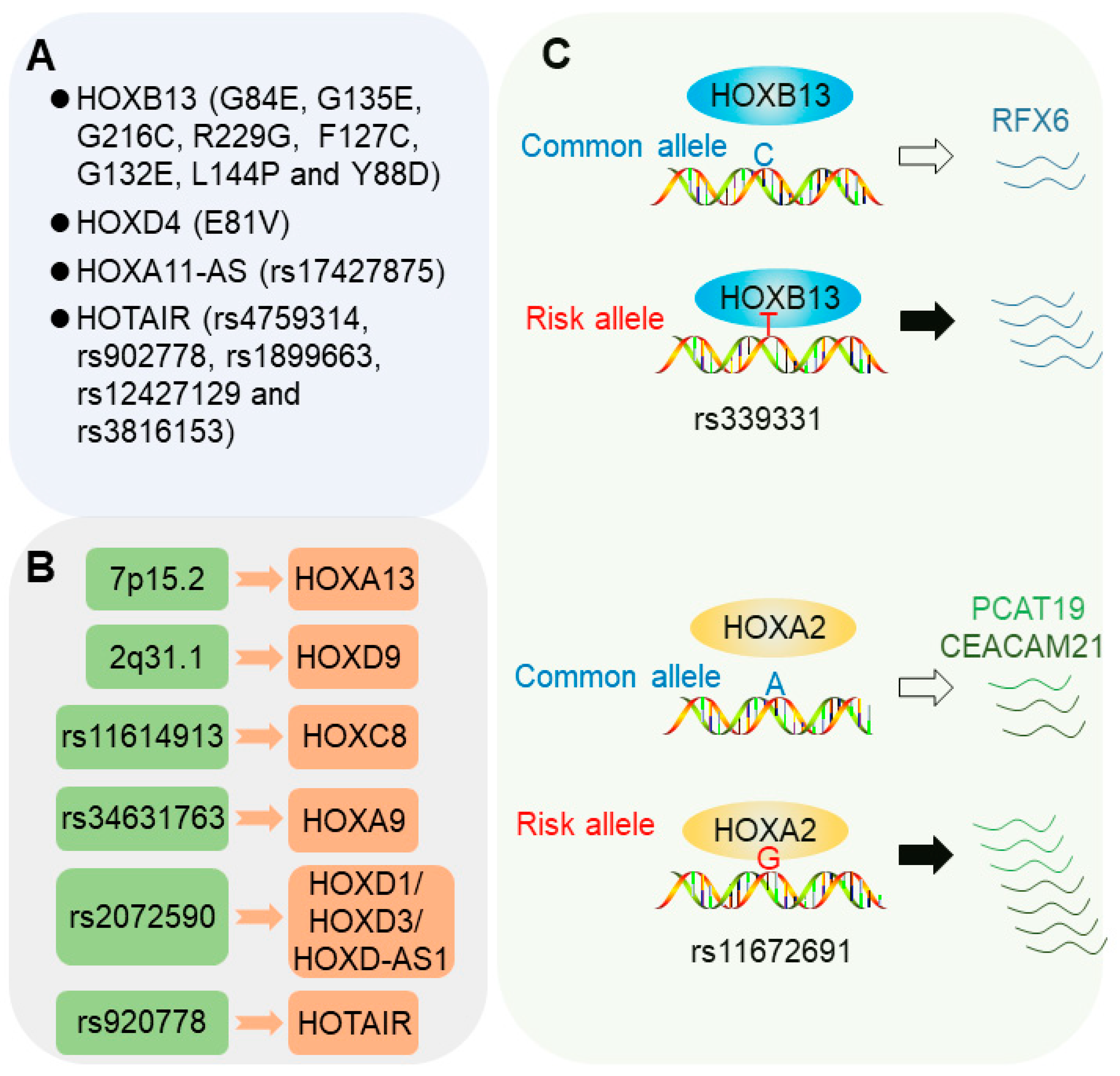{kind=link}
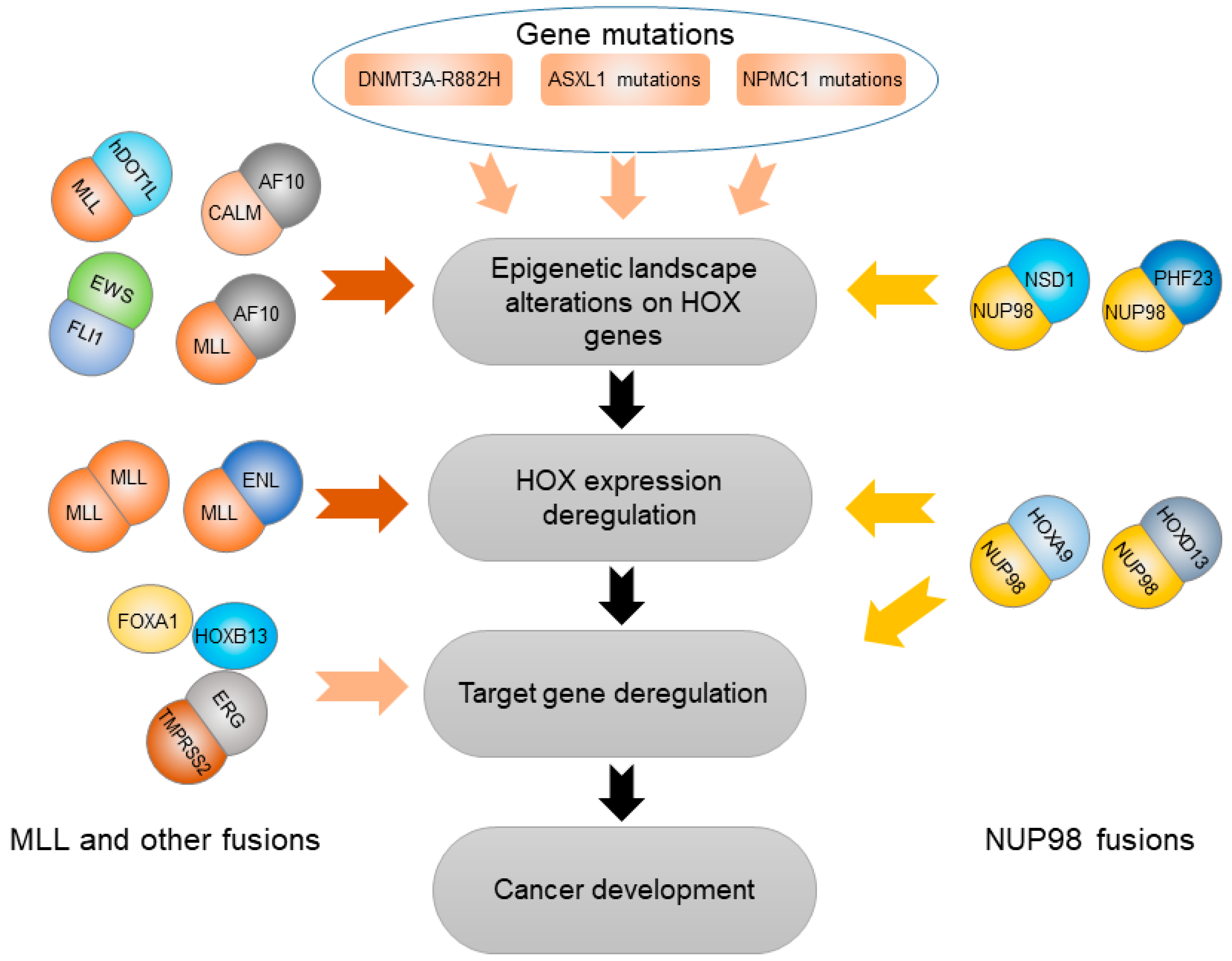{kind=link}
Table 1.
Overview of HOX genes that contribute to the seven aspects of cancer development and progression.
Table 1.
Overview of HOX genes that contribute to the seven aspects of cancer development and progression.
| Progression | HOX Gene | Tumor Cells Type | Function |
|---|---|---|---|
| Angiogenesis | HOXB7 [111,112,113] | Breast cancer Multiple myeloma | Upregulated HOXB7 drives angiogenic gene expression |
| HOXB9 [114,115] | Ovarian cancer Renal cancer Breast cancer | Downregulated HOXB9 attenuates angiogenic gene expression | |
| HOXA11-AS [116] | NSCLC | Upregulated HOXA11-AS promotes angiogenesis | |
| HOXA5 [117,118] | ECs | Sustained HOXA5 expression downregulates angiogenic genes and upregulates anti-angiogenic genes | |
| Autophagy | HOXC9 [119,120] | Glioblastoma | HOXC9 acts as a transcription inhibitor to directly binding to the promoter of DAPK1 |
| HOXC6 [121] | NPC | Downregulated HOXC6 promotes apoptosis and autophagy by inhibiting the TGF-β1/mTOR pathway | |
| HOTAIR [122,123] | Cervical cancer; Breast cancer; Chondrosarcoma | Downregulated HOTAIR inhibits autophagy | |
| Differentiation | HOXA clusters (except HOXA2 and HOXA5) [124] | Hematopoietic cells | HOXA genes except HOXA2 and HOXA5 induce delayed hematopoietic differentiation |
| HOXA9 [98,125,126,127] | Hematopoietic and lymphoid cancer. | HOXA9 involves in blocking differentiation NUP98–HOXA9 fusion, cooperation of HOXA9 with either Meis1 or FOXC1 inhibit differentiation | |
| HOXA10 [128,129] | Prostate cancer; OEA | HOXA10 blocks or promotes differentiation in a cancer-type-dependent manner | |
| HOXB8 [130] | HL-60 cells | HOXB8 blocks DMSO-induced granulocytic differentiation | |
| HOXC9 [131] | Neuroblastoma | HOXC9 promotes neuronal differentiation | |
| HOXA5 [132,133] | Colon cancer | Upregulated HOXA5 promotes differentiation of cancer stem cells | |
| HOTAIR [134] | Urothelial carcinoma | HOTAIR overexpression may affect differentiation state | |
| Apoptosis | HOXA5 [135,136,137,138,139,140,141,142] | Breast cancer; Leukemia; Osteosarcoma; Lung and cervical cancer | HOXA5 could activate apoptosis by upregulating p53 expression or activating caspase 2 and caspase 8; HOXA5 is involved in RA-mediated apoptosis |
| HOXA10 [143] | Breast cancer | HOXA10 could activate apoptosis by upregulating p53 expression | |
| HOXC6 [144,145,146] | HNSCC; Cervical cancer; Prostate cancer | HOXC6 plays an important anti-apoptotic role through regulating the expression of bcl-2 or suppressing NEP/MME and IGFBP-3 genes | |
| HOXA9 [126,147] | Leukemia | HOXA9 functions as an apoptosis suppressor by cooperating with JAK3/STAT5; HOXA9 could eliminate Meis1a-mediated apoptosis | |
| Proliferation | HOXA1 [148,149] | Breast cancer | HOXA1 promotes cell proliferation and survival by activating p44/42 MAPK signaling pathway or NF-κB pathway; |
| HOXA9 [150] | Leukemia | HOXA9 upregulates Igf1 to promote proliferation and survival | |
| HOXB7 [151,152] | Colorectal cancer Hepatocellular carcinoma | HOXB7 promotes cell proliferation and growth by accelerating G1-S transitions | |
| HOXC6 [153] | Gastrointestinal carcinoids cells | HOXC6 promotes cell proliferation by activating the oncogenic AP-1 signaling pathway | |
| HOXB3 [154] | NCI-H1437 cells A549 cells | HOXB3 promotes cell proliferation through silencing gene RASSFA1 | |
| HOXD3 [155] | Hepatocellular carcinoma | HOXD3 promotes proliferation and anti-apoptosis by activating MAPK/AKT cell signaling pathways | |
| HOXB9 [156] | HL cell lines | HOXB9 upregulated by ERK5 signal promotes proliferation and anti-apoptosis | |
| HOXC5 [157,158] | Thymoma; TGCT | HOXC5 inhibits proliferation by inhibiting hTERT expression | |
| HOXA10 [159,160] | Myeloid leukemia; Testicular cancer | Overexpressed HOXA10 stimulates the proliferation in myeloid leukemia; HOXA10 also inhibits cell proliferation during G2/M phases in testicular cancer cells | |
| Invasion and Metastasis | HOXA9 [161] | Breast cancer cell | HOXA9 expression could reduce bone metastasis |
| HOXA10 [162] | Endometrial carcinoma | HOXA10 suppresses invasion by inhibiting EMT | |
| HOXB1 and HOXB3 [163] | Pancreatic cancer | HOXB1 and HOXB3 downregulation facilitates invasion and metastasis | |
| HOXD10 [156,164,165,166,167] | Breast cancer | HOXD10 downregulation suppresses invasion | |
| HOXB7 [168,169,170] | Breast cancer | HOXB7 overexpression induces invasive and metastatic by activating the TGFβ signaling pathway | |
| HOXA11-AS [171] | Gastric cancer | HOXA11-AS expression promotes metastasis and invasion | |
| Metabolism | HOXA9 [172] | cSCC | HOXA9 inhibits glycolysis by negatively regulating HIF-1α |
| HOXC8 [173] | Nasopharyngeal carcinoma | HOXC8 downregulates glycolysis-related genes and upregulates TCA cycle-related genes |
Abbreviations: HOXA11-AS, HOXA11 antisense RNA; NSCLC, non-small cell lung cancer; ECs, endothelial cells; DAPK1,Death Associated Protein Kinase 1; NPC, nasopharyngeal carcinoma; TGF-β, transforming growth factor-β; mTOR, mammalian target of rapamycin; HOTAIR, HOX transcript antisense RNA; OEA, ovarian endometrioid adenocarcinoma; RA, retinoic acid; HNSCC, head and neck squamous cell carcinoma; MAPK, mitogen-activated protein kinase; Igf1, insulin-like growth factor 1; AP-1, activator protein-1; TGCT, testicular germ cell tumor; hTERT, telomerase reverse transcriptase; EMT, epithelial-mesenchymal transition; TGF-β, Transforming growth factor β; cSCC, cutaneous squamous cell carcinoma; HIF-1α, hypoxia inducible factor-1.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Li, B.; Huang, Q.; Wei, G.-H. The Role of HOX Transcription Factors in Cancer Predisposition and Progression. Cancers 2019, 11, 528. https://doi.org/10.3390/cancers11040528
AMA Style
Li B, Huang Q, Wei G-H. The Role of HOX Transcription Factors in Cancer Predisposition and Progression. Cancers. 2019; 11(4):528. https://doi.org/10.3390/cancers11040528
Chicago/Turabian StyleLi, Bo, Qilai Huang, and Gong-Hong Wei. 2019. "The Role of HOX Transcription Factors in Cancer Predisposition and Progression" Cancers 11, no. 4: 528. https://doi.org/10.3390/cancers11040528
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.